

Abstract
The heart manifests hypertrophic growth in response to elevation of afterload pressure. Cardiac myocyte growth involves new protein synthesis and membrane expansion, of which a number of cellular quality control machineries are stimulated to maintain function and homeostasis. The unfolded protein response (UPR) is potently induced during cardiac hypertrophy to enhance protein-folding capacity and eliminate terminally misfolded proteins. However, whether the UPR directly regulates cardiac myocyte growth remains to be fully determined. Here, we show that GRP78 (glucose-regulated protein of 78 kDa), an endoplasmic reticulum-resident chaperone and a critical UPR regulator, is induced by cardiac hypertrophy. Importantly, overexpression of GRP78 in cardiomyocytes is sufficient to potentiate hypertrophic stimulus-triggered growth. At the in vivo level, transgenic hearts overexpressing GRP78 mount elevated hypertrophic growth in response to pressure overload. We went further to show that GRP78 increases GATA4 (GATA binding protein 4) level, which may stimulate Anf (atrial natriuretic factor) expression and promote cardiac hypertrophic growth. Silencing of GATA4 in cultured neonatal rat ventricular myocytes significantly diminishes GRP78-mediated growth response. Our results therefore reveal that protein-folding chaperone GRP78 may directly enhance cardiomyocyte growth by stimulating cardiac-specific transcriptional factor GATA4.
Keywords: GRP78, cardiac hypertrophy, GATA4, pressure overload, hypertension
Summary
Cardiac GRP78 enhances hypertrophic growth in response to pressure overload in the heart, which may represent a novel role of GRP78 in regulating cardiac pathophysiology in addition to the unfolded protein response.
INTRODUCTION
Approximately one-third of adults 18 years or older are affected by high blood pressure.1 This number is even higher among elderly. Numerous epidemiological studies have shown that hypertension causes severe complications and represents a major risk factor of heart disease, stroke and death.2 In response to elevated blood pressure, the heart manifests hypertrophic growth to ameliorate ventricular wall stress and maintains cardiac performance to ensure homeostatic perfusion of the body.3 Due to limited proliferating capacity in adult stage, the heart does so by enlarging individual cardiomyocytes. This concentric growth leads to increases in cardiac mass and enhancement of pumping function.4 Hypertrophic growth however is above and beyond simple addition of sarcomeres. A myriad of signaling molecules need to coordinate and orchestrate the growth response for normal cardiac performance.4 Over the years, a complex network of pathways have been identified in this process, including calcium handling,5 metabolism,6 excitation-contraction coupling,7 growth signaling,8 etc.
Increases in protein biosynthesis during cardiac hypertrophic growth lead to activation of the unfolded protein response (UPR), a cellular adaptive process to accommodate the augmented demands in protein folding.9 Under resting conditions, minimal protein-folding requirement is maintained in the endoplasmic reticulum (ER) by a host of chaperones, foldases, oxidoreductases, isomerases, etc.10 Glucose-regulated protein of 78 kDa (GRP78), the most abundant ER-resident chaperone, interacts with luminal domains of the three signaling transducers, protein kinase R-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcriptional factor 6 (ATF6). These interactions sequester the transducers from activation and downstream actions. Upon accumulation of misfolded proteins in the ER, GRP78 preferentially binds the exposed hydrophobic domains in client proteins and therefore liberates the three signaling branches. As a result, the UPR transiently reduces protein translation to decrease ER load, upregulates protein chaperones, and stimulates ER-associated protein degradation to eliminate terminally misfolded proteins. In so doing, the balance of protein folding capacity and demands is regained, thereby maintaining ER homeostasis.
The UPR plays critical roles in the initiation and development of cardiovascular disease, including myocardial infarction, ischemia/reperfusion (I/R), diabetic cardiomyopathy, etc.11–13 In hypertensive heart disease, most prior studies focus on the protein quality control action of the UPR. However, emerging evidence points to additional layers of regulation above and beyond protein folding. Early work by Okada et al shows that excessive activation of CHOP (C/EBP homologous protein), a downstream effector of the UPR, by pressure overload leads to apoptotic cardiac cell death and heart failure.14 Indeed, ablation of CHOP ameliorates pathological cardiac remodeling and regresses disease progression.15 In contrast, PERK deficiency in the heart exacerbates cardiac dysfunction by pressure overload.16 On the other hand, Glembotski and colleagues found that ATF6 may counteract cardiac hypertrophic growth by directly stimulating Rcan1 (regulator of calcineurin 1) in cultured cardiac myocytes.17 Considering the versatile role of the UPR in metabolism, cell growth and apoptosis in addition to protein quality control, it is possible that the UPR directly regulate cardiac hypertrophic growth in response to hemodynamic stress.
GRP78 is an essential, ER-resident protein chaperone.18 Accumulating studies have shown that GRP78 plays critical roles in the heart under both physiological and pathological conditions.19 Using genetically engineered animal models, we found that GRP78 deficiency in cardiomyocytes leads to cardiac dilation, heart failure, and early mortality.20 GRP78 expression is augmented by myocardial infarction21 and cardiac I/R.22 We recently showed that overexpression of GRP78 in cardiomyocytes protects the heart from reperfusion injury via activation of the pro-surviving AKT signal.23 Despite these findings, our understanding of GRP78 in hypertensive heart disease remains vacant. Here, we sought to elucidate the role of GRP78 in the heart in response to pressure overload using both animal models and primary cardiac myocyte culture.
MATERIALS AND METHODS
The data, methods, and materials that support the findings of this study are available from the corresponding author on reasonable request.
Animals
All mice were on C57BL/6 background. Animals were maintained on a 12 hours dark/light cycle (6 AM to 6 PM) and housed in a barrier facility with unlimited access to chow food (2916, Teklad) and water. The Institutional Animal Care and the Use Committee of University of Texas Southwestern Medical Center has approved all animal experiments. Genotyping primers are provided in Table S1.
Statistics
Statistical analysis was performed using GraphPad Prism 7. Data are expressed as mean ± SEM The Student’s t test (2-tailed) was performed to compare difference between two groups. Additionally, one-way or two-way ANOVA was used for multiple groups, followed by Tukey’s test. P < 0.05 was considered statistically significant.
RESULTS
Induction of GRP78 by pressure overload in the heart.
GRP78 is an essential protein chaperone localized in the ER and the master regulator of the UPR, which plays critical roles in physiology and pathophysiology.18 In the heart, GRP78 expression is upregulated during cardiogenesis and elimination of GRP78 in cardiomyocytes is not compatible with life.24, 25 Here, we set out to investigate the role of GRP78 during cardiac hypertrophic growth under pressure overload.
We subjected wild type (WT) adult mice to thoracic aortic constriction (TAC), which creates aortic stenosis and mimics clinical situations of high blood pressure. The pressure gradient across aortic constriction site was determined 24 hours post surgery (Figure S1A). We have shown previously that TAC leads to significant cardiac hypertrophic growth in a time-dependent manner.26 The early phase of adaptive growth may progress into decompensation and heart failure. We harvested the hearts 7 days post TAC and subjected to immunoblotting. Rcan1.4 (regulator of calcineurin 1.4) is a transcriptional target of calcineurin signaling and commonly used as a molecular marker of hypertrophic growth.27 As positive controls, we found significant increases in the expression of βMHC, Anf and Rcan1.4 (Figure 1A and 1B), indicating robust and prominent hypertrophic growth.26 Importantly, GRP78 expression was strongly induced. We next turned to cultured cardiac myocytes to validate GRP78 induction by hypertrophic growth. Neonatal rat ventricular myocytes (NRVMs) were isolated from day 1–2 newly born rats. We treated the cells with phenylephrine (PE, 50 μM) to stimulate cellular hypertrophy for 24 hours. We found that GRP78 was significantly elevated (Figure 1C and 1D). Taken together, these results suggest that GRP78 as an UPR marker is upregulated in response to cardiac cell hypertrophic growth.
Figure 1. Induction of GRP78 by pressure overload in the heart.
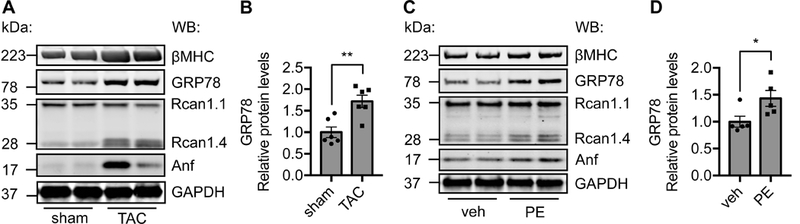
A. GRP78 protein expression was increased in the heart in response to pressure overload. Thoracic aortic constriction (TAC) was performed to induce cardiac hypertrophy. Note that multiple protein makers of cardiac hypertrophy show strong upregulation. GAPDH was used as a loading control. B. Quantification of A supported significant elevation of GRP78 by pressure overload. N = 6. C. GRP78 was upregulated in cardiac myocytes by hypertrophy stimulation. Cultured neonatal rat ventricular myocytes (NRVMs) were treated with phenylephrine (50 μM) for 24 hours. GRP78 was detected by western blotting. PE treatment stimulated cellular hypertrophic growth as indicated by increases in βMHC, Rcan1.4, and Anf. D. Quantification of C showed significant upregulation of GRP78. N = 5. *, p < 0.05; **, p < 0.01.
Overexpression of GRP78 promotes hypertrophic growth in the heart.
We next took an in vivo approach to investigate the role of GRP78 in cardiac hypertrophy. We generated a transgenic mouse model with GRP78 under control of the universal CAG promoter.23 A loxP site-flanked transcriptional/translational stop cassette was inserted between the promoter and GRP78. We crossed this transgenic mouse model with the cardiomyocyte-restricted αMHC-Cre transgenic animal. Cre-mediated cleavage of the stop cassette led to expression of GRP78 only in cardiac myocytes (Figure S1B).
We subjected double transgenic (TG) mice, along with littermate single transgenic controls, to TAC surgery for a week. Pressure gradient was measured that showed no detectable difference between control and TG mice (Figure S1C), indicating the surgery was similarly performed. We found that transgenic animals manifested significantly more severe cardiac hypertrophy and elevated fibrosis (Figure 2A and S1D). Individual cardiac myocytes were strongly enlarged (Figure 2B and 2C). Indeed, the ratio of heart weight to body weight was higher in transgenic TAC mice compared to controls (Figure 2D and 2E). Collectively, these data suggest that overexpression of GRP78 in cardiomyocytes leads to more profound cardiac hypertrophic response to pressure overload in vivo.
Figure 2. Overexpression of GRP78 promotes hypertrophic growth in the heart.
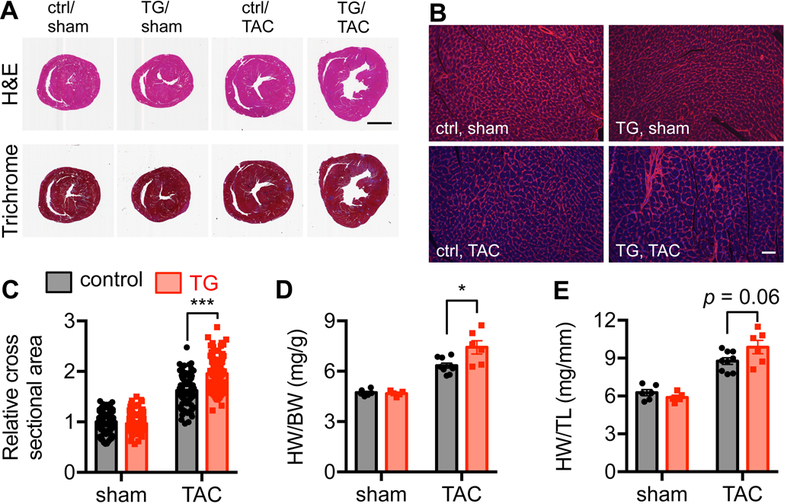
A. Control and transgenic (TG) mice were subjected to sham or TAC surgery. Hematoxylin and Eosin (H&E) staining showed more enlargement of the transgenic heart compared to control. Masson’s Trichrome staining suggested an increase in fibrosis of the TG heart. Scale bar, 2 mm. B. Cardiac sections were used for wheat germ agglutinin (WGA) staining to visualize individual cells in the heart. Scale bar, 50 μm. C. GRP78 overexpression led to significant increases in cardiac cell size after TAC. N = 120–133. D. Overexpression of GRP78 in cardiomyocytes caused elevation in the ratio of heart weight to body weight, indicating more profound cardiac hypertrophic growth. N = 5–9. E. Quantification of heart weight/tibia length showed a trend of increase in the transgenic hearts compared to controls. N = 5–9. *, p < 0.05; ***, p < 0.001.
Overexpression of GRP78 exacerbates cardiac response to pressure overload.
We next set up to examine cardiac function using echocardiography with gently constrained, conscious mice. Transgenic animals showed increased cardiac chamber size (Figure 3A). Cardiac function was impaired as indicated by depressed fractional shortening and ejection fraction (Figure 3B and S2A), while heart rate was maintained. The αMHC-Cre transgenics alone did not affect cardiac function (Figure S2B). Further analysis showed increases in LVID (left ventricular internal diameter) at both systole and diastole (Figure 3B), indicating a decline for systolic and diastolic performance. Consistently, diastolic and systolic IVS (interventricular septum) and LVPW (left ventricular posterior wall) were reduced. Moreover, cardiac function showed further deterioration 3 weeks post TAC (Figure S2C). In aggregate, these results indicate that GRP78 overexpression causes an exacerbated response to pressure overload.
Figure 3. Overexpression of GRP78 exacerbates cardiac response to pressure overload.
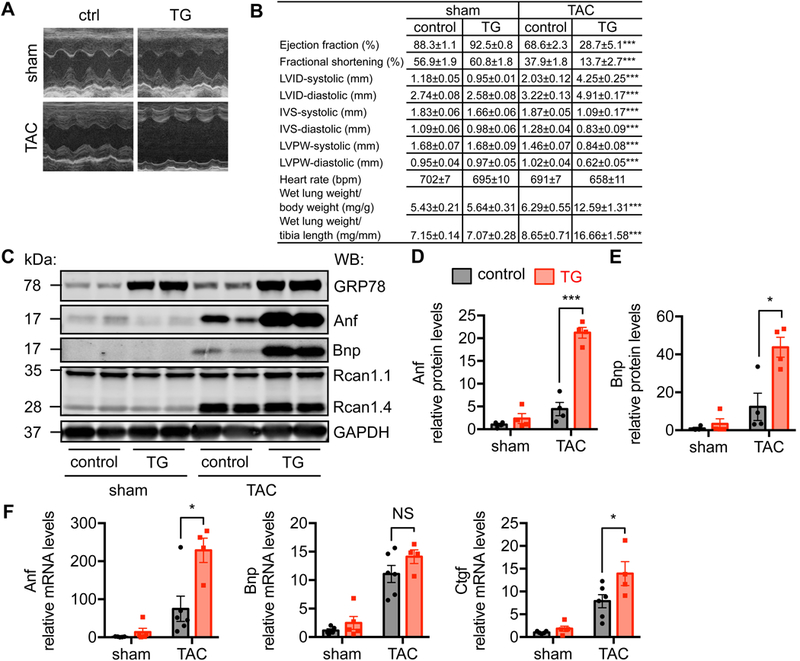
A. Representative M-mode images of echocardiography from conscious animals. B. GRP78 overexpression in the heart led to cardiac dysfunction and heart failure as shown by echocardiographic parameters and relative lung weight. N = 5–17. Comparison was done between control and TG groups after TAC. C. Transgenic mice showed elevation of Anf and Bnp, molecular markers of heart failure. D. Quantification of C indicated significant augmentation of Anf protein level in the transgenic hearts. N = 4. E. Bnp protein level was significantly upregulated in the transgenic mice compared to controls. N = 4. F. Anf and Ctgf mRNA levels were strongly increased in the transgenic hearts in response to TAC, while Bnp mRNA showed a trend of upregulation. N = 4–6. *, p < 0.05; ***, p < 0.001; NS, not significant.
Heart failure is presented clinically by the inability of the heart to pump blood and provide sufficient perfusion to the body. Here, we found that overexpression of GRP78 in cardiomyocytes caused heart failure after pressure overload. As a signature, ratio of wet lung weigh to either body weight or tibia length (Figure 3B) was significantly elevated in GRP78 transgenics. At the cellular level, molecular markers of heart failure were strongly induced, including Anf and Bnp (Figure 3C-3E). Quantitative PCR analysis confirmed the upregulation of these genes at the mRNA level (Figure 3F). Consistent with an aggravated response to pressure overload, Ctgf (connective tissue growth factor) as a marker of fibrosis was significantly increased (Figure 3F). In contrast, inflammatory genes were not significantly elevated in the TG hearts after TAC compared with controls (Figure S2D). No detectable increase of apoptosis was discovered (Figure S2E). Under overexpression of GRP78 at this level in the heart, the ER stress response was not altered (Figure S3A-S3C). Interestingly, we did not find significant difference in mTOR, AKT, or ERK between control and transgenic mice after TAC (Figure S3B and S3C), suggesting these signaling may not contribute to the enhanced hypertrophic growth in GRP78 transgenic mice. On the other hand, phosphorylation of NFATc3 and p38 only showed a trend of decrease in the transgenic heart after TAC (Figure S3B and S3C), which may not play a major role in the pro-hypertrophic response in GRP78 transgenic mice. Our results collectively show that GRP78 overexpression in cardiomyocytes in vivo leads to exacerbation in cardiac response to pressure overload and heart failure.
Overexpression of GRP78 increases cardiomyocyte hypertrophic growth in vitro.
We next turned to cell culture to address whether the pro-hypertrophic action of GRP78 in cardiomyocytes was a cell-autonomous phenomenon. We overexpressed GRP78 by adenovirus infection in NRVMs. We then treated the cells with PE for 24 hours. We found that overexpression GRP78 per se in NRVMs did not affected cardiac cell size, which however potentiated the response to PE (Figure 4A and 4B). To further corroborate this finding, we performed a leucine incorporation assay to assess protein synthesis. Consistently, overexpression of GRP78 in NRVMs led to a higher increase in protein synthesis after PE treatment (Figure 4C). At the cellular level, we found that Anf expression was further increased at both protein (Figure 4D and 4E) and mRNA (Fig. 4F) levels. Consistent with the in vivo data, no differences were found in mTOR, AKT, ERK, p38 or NFAT (Figure S4A-S4B). Furthermore, we tested another hypertrophic stimulus, angiotensin II. Likewise, overexpression of GRP78 led to more profound increases in cell size after angiotensin II treatment (Figure S5A-S5B), which was not accompanied by elevation of p38 phosphorylation (Figure S5C-S5D). Taken together, these results suggest that overexpression of GRP78 enhances growth response to hypertrophic stimuli at cellular level.
Figure 4. Overexpression of GRP78 increases cardiomyocyte hypertrophic growth in vitro.
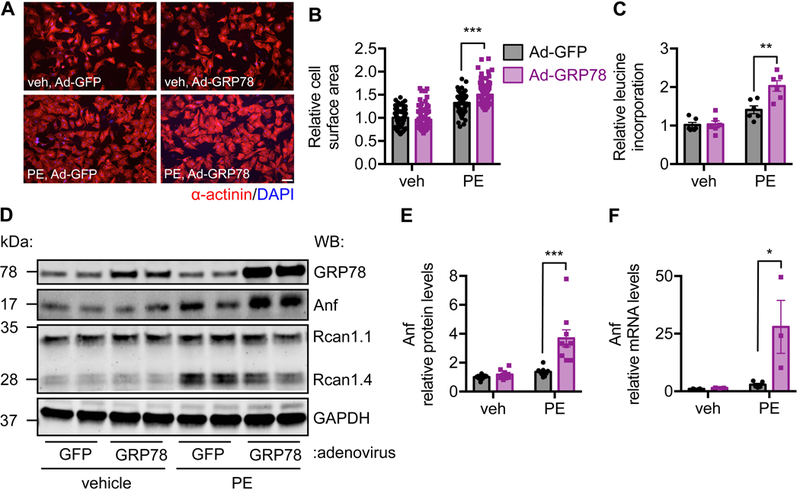
A. GRP78 was overexpressed by adenovirus infection in NRVMs. After PE treatment for 24 hours, cells were harvested for immunofluorescent staining of α-done between control and TG groups after.B. GRP78 overexpression in NRVMs led to more profound hypertrophic growth as shown by increase in cardiac cell size. N = 91–93. C. Overexpression of GRP78 led to increase in protein synthesis, assessed by radioactive leucine incorporation assay. N = 6. D. Anf protein level was augmented by GRP78 overexpression in NRVMs after PE treatment. E. Quantification of D showed significant upregulation of Anf protein. N = 9. F. Anf mRNA level was significantly increased by GRP78 overexpression in NRVMs. N = 3–5. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Knockdown of GRP78 inhibits cardiomyocyte hypertrophic response in vitro.
We next asked the question about requirement of GRP78 in the hypertrophic response. We silenced GRP78 by siRNA transfection. After PE treatment, cells were harvested for cell size determination. Knockdown of GRP78 significantly diminished hypertrophic response to PE (Figure 5A and 5B), which was consistent with decreased protein synthesis rate (Figure 5C). At the molecular level, we found that induction of Anf by PE was strongly inhibited at both protein (Figure 5D and 5E) and mRNA (Figure 5F) levels. Collectively, these data lend further support that GRP78 is critical to promote cardiac cell growth in response to hypertrophy stimulation.
Figure 5. Knockdown of GRP78 inhibits cardiomyocyte hypertrophic growth in vitro.
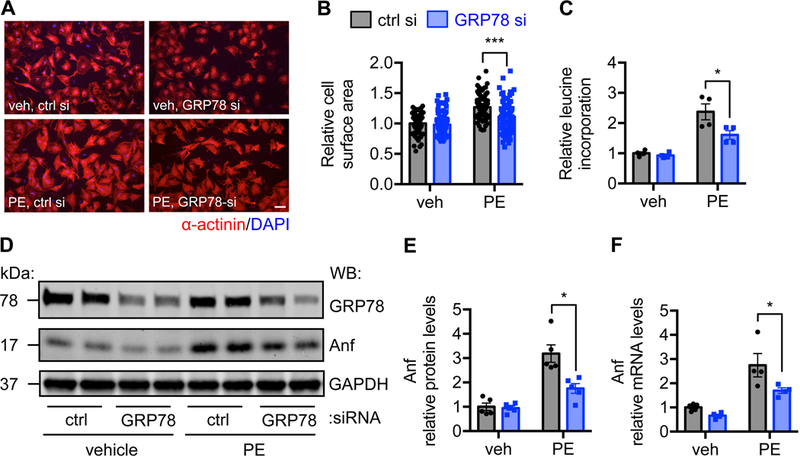
A. GRP78 expression was reduced by siRNA-mediated knockdown. PE treatment was conducted for 24 hours. Immunofluorescent staining for α-actinin was performed. B. Quantification of A showed significant decrease in cell size after GRP78 knockdown. N = 80–106. C. Leucine incorporation assay showed decrease in protein synthesis after GRP78 silencing. N = 4. D. GRP78 knockdown caused reduction in Anf protein expression. E. Quantification of D showed a significant decrease in Anf protein level after GRP78 knockdown. N = 5. F. GRP78 silencing reduced Anf mRNA expression as assessed by qPCR. N = 4–5. *, p < 0.05; ***, p < 0.001.
GATA4 is required for GRP78-mediated promotion of hypertrophic growth.
GATA4 belongs to a family of transcriptional factors binding to “GATA” DNA motif. GATA4 plays an essential role in cardiogenesis during embryonic development.28 Deficiency of GATA4 leads to severe congenital heart disease and heart failure. Furthermore, GATA4 is a key regulator of hypertrophic growth in response to pressure overload.18, 29 We therefore asked the question whether GATA4 was involved in GRP78-mediated cardiac hypertrophic growth.
We found that GATA4 protein level was significantly elevated in the GRP78 transgenic mice after TAC (Figure 6A). In NRVMs, GATA4 was increased by GRP78 overexpression after PE treatment (Figure 6B), indicating a cell-autonomous phenomenon. Importantly, silencing of GRP78 decreased the protein level of GATA4 after PE administration (Figure 6C). These results indicate GATA4 protein level is correlated with GRP78 expression in cardiac myocytes in response to hypertrophy stimulation.
Figure 6. GATA4 is required for GRP78-mediated promotion of hypertrophic growth.
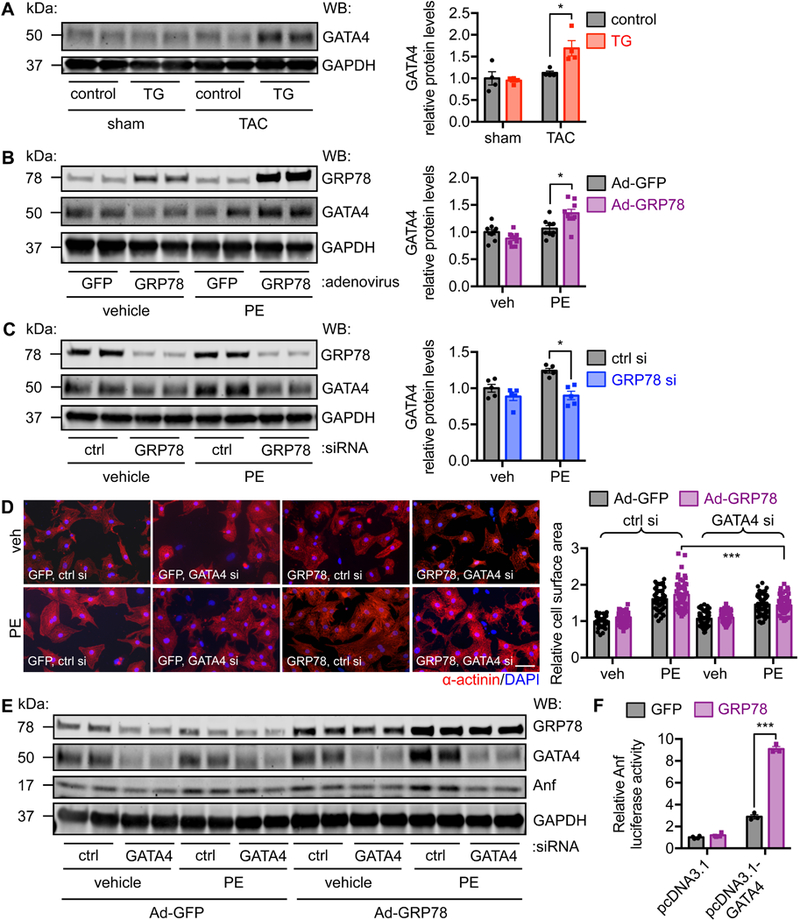
A. GATA4 protein expression was increased in the GRP78 transgenic hearts in response to pressure overload. Control and TG mice of 8–10 weeks old were subjected to TAC and western blotting was performed a week later. N = 4. B. GATA4 protein level was upregulated by GRP78 overexpression in NRVMs. GRP78 was overexpressed by adenovirus-mediated infection. The cells were then treated by PE for 24 hours. N = 9. C. GRP78 knockdown led to decrease in GATA4 protein expression. N = 5. D. Knockdown of GATA4 in NRVMs diminished GRP78-mediated hypertrophic response. GRP78 was overexpressed by adenoviral infection and GATA4 was silenced by siRNA transfection. PE treatment was conducted for 24 hours. Immunostaining was done to detect α-actinin. N = 50–80. E. Knockdown of GATA4 reduced GRP78-induced Anf upregulation. F. GRP78 expression enhanced GATA4-dependent Anf promoter activity. An Anf promoter-driven luciferase plasmid was transfected to HEK-293T cells, along with pcDNA3.1 expressing GATA4. Co-transfection of GRP78 led to more profound elevation of luciferase activity, indicating GRP78 potentiates GATA4-dependent Anf induction. N = 3–4. *, p < 0.05; ***, p < 0.001.
To further define the relationship between GRP78 and GATA4, we overexpressed GRP78 in NRVMs by adenovirus infection. We then silenced GATA4 by siRNA transfection. After PE treatment, we found that GRP78 overexpression led to more profound hypertrophic growth, which was significantly suppressed by GATA4 knockdown (Figure 6D). Consistent with these findings, Anf expression was diminished by GATA4 silencing (Figure 6E). We next set out to examine whether GRP78 could enhance the transcriptional activity of GATA4 on Anf. We performed a luciferase assay in HEK-293T cells. We transfected the Anf-luciferase plasmid along with either control pcDNA3.1 or pcDNA3.1-GATA4 construct. Expression of GATA4 stimulated Anf-luciferase activity (Figure 6F). Importantly, overexpression of GRP78 under this condition led to further augmentation of the Anf promoter activity. Importantly, MEF2a-mediated upregulation of Anf-luciferase activity was not potentiated by GRP78 overexpression (Figure S6A), indicating specific relationship between GRP78 and GATA4. Further, we set up to examine whether GRP78 overexpression affected GATA4 protein stability. NRVMs were infected by adenovirus expressing either GFP or GRP78. Cycloheximide was used to suppress new protein translation. The cells were then harvested at different time to determine GATA4 protein levels. We found that GATA4 protein stability was significantly increased by GRP78 overexpression (Figure S6B-S6C). Taken together, these data support a model that GRP78 induction during hypertrophic growth increases GATA4 protein and enhances its transcriptional activity in cardiac myocytes, which may be one of the underlying mechanisms of GRP78-mediated hypertrophic growth in response to pressure overload.
DISCUSSION
GRP78, one of the most abundant protein-folding chaperones,30, 31 was also discovered as a glucose-regulated protein.32 At the resting condition, GRP78 binds ER luminal domains of the three signaling transducers of the UPR and governs the downstream signaling events.9 Upon accumulation of misfolded protein clients, GRP78 exerts its protein-folding action and liberates the sequestration of UPR effectors. As a result, the ER stress response ensues to regain homeostasis. GRP78 is therefore not only a master chaperone for protein folding, but also a key regulator of the UPR.
Hypertension is one of the most important risk factors of heart failure. In response to high blood pressure, the heart shows extraordinary plasticity and manifests hypertrophic growth to ameliorate ventricular wall stress.33 It is still a matter of debate whether hypertrophic growth is adaptive or detrimental. Early studies show that suppression of this pressure overload-induced hypertrophic growth by Cyclosporine A does not affect cardiac performance.34 Consistently, treatment with rapamycin, an inhibitor of mTOR signaling, leads to diminishment of cardiac hypertrophic growth and suppression of heart failure development.35 Even treatment after the establishment of cardiac hypertrophy, rapamycin can rescue cardiomyopathy from pressure overload.36 These studies together support that cardiac hypertrophic growth is not required to maintain homeostatic function in response to pressure overload. Hypertrophy may therefore be a therapeutic target in treatment of hypertensive heart disease.
Accumulating evidence however suggests the existence of adaptive hypertrophic growth. Cardiac-specific deletion of mTOR, therefore eliminating growth in response to pressure overload, is detrimental.37 The knockout heart progresses into cardiac dilation and failure after TAC. Similarly, cardiomyocyte-restricted knockout of RheB or Raptor leads to impaired adaptive growth and cardiomyopathy.38, 39 The underlying reasons for this discrepancy are not entirely known. However, pharmaceutical inhibition of growth response, by targeting either NFAT or mTOR, may only partially suppress hypertrophy. In contrast, genetic deletion of mTOR signaling components leads to full elimination of growth response. The different degree of growth inhibition may play a role in these opposite responses.8 Future work is warranted to dissect the underlying mechanisms and provide novel targets for therapeutic gain.
To maintain homeostatic performance, hypertrophic heart needs to orchestrate various signaling, more than mere increase in size. Over the past years, numerous pathways and molecules have been identified, including calcium signaling, metabolism, protein synthesis, inflammation, etc.4 Most of these events are potent inducers of the UPR.40 Indeed, we show that GRP78, as a master regulator and marker of the UPR, is strongly upregulated. Upstream signal to stimulate the UPR may involve accumulation of misfolded proteins in the ER. On one hand, cardiac cell growth poses a higher demand of protein synthesis/folding, which may trigger the UPR. On the other hand, pathological processes during hypertrophic growth, such as hypoxia and calcium abnormalities, may exacerbate protein misfolding, which adds another layer of burden on ER homeostasis. The UPR and GRP78 upregulation may therefore be elicited during cardiac hypertrophic growth.
At both in vivo and in vitro levels, forced overexpression of GRP78 leads to more profound hypertrophic growth of cardiomyocytes. On the other hand, knockdown of GRP78 decreases the growth response in cultured cardiomyocytes. Our further data suggest this response may not involve mTOR and several other established pathways of hypertrophic growth, such as NFAT, ERK, p38 and MEF2. Further, in contrast to the action of GRP78 in cardiac I/R,23 GRP78 overexpression does not activate AKT signaling. Interestingly, we show that GATA4 is strongly correlated with GRP78 manipulations and GRP78 overexpression significantly potentiates GATA4-dependent Anf promoter activity. These findings support our hypothesis that GRP78 may stimulate GATA4 signaling to promoter hypertrophic growth. Previous studies have shown a direct interaction between MEF2 and GATA4 to stimulate Anf expression.41 Since we did not detect appreciable changes in MEF2, it likely plays a minor role. Questions however remain about the molecular mechanisms between GRP78 and GATA4 interaction. GRP78 is mainly localized in the ER, which likely precludes its interaction with transcriptional factor GATA4. However, GRP78 has been discovered in other compartments in the cells, such as plasma membrane and mitochondria.42 Although nuclear localization has not been validated, it may not be entirely impossible considering the versatile roles of GRP78 recently discovered.
PERSPECTIVES
Under hypertensive condition, the heart manifests hypertrophic growth to accommodate ventricular wall stress. We found GRP78, a molecular maker of the UPR, is potently induced by hypertrophic stimuli in cardiomyocytes. Overexpression of GRP78 leads to a higher degree of growth at both in vivo and in vitro levels. Mechanistically, GRP78 may corroborate with GATA4 and promote more profound hypertrophic growth in response to pressure overload.
Supplementary Material
NOVELTY AND SIGNIFICANCE
What Is New?
We show that GRP78 (glucose-regulated protein of 78 kDa) potentiates hypertrophic response in the heart by activating GATA4 (GATA binding protein 4).
GRP78 stimulates transcriptional activity of GATA4.
What Is Relevant?
GRP78 is potently upregulated in the heart during hypertrophic growth.
GRP78 is a master regulator of the unfolded protein response and an endoplasmic reticulum resident chaperone.
Acknowledgments
SOURCES OF FUNDING
We thank the UTSW Molecular Pathology Core (John Shelton) for help with histology. We thank the Animal Resource Center of UTSW for mouse generation, breeding, and maintenance. This work was supported by grants from AHA (14SDG18440002 to Z.V.W.), NIH (R01-HL137723 to Z.V.W.), and Natural Science Foundation of Zhejiang Province (Q19H020043 to X.B.). This study was also supported, in part, by research grants to R.C.A. from the Heart and Stroke Foundation of Canada (G-15–0009389), the Canadian Institutes of Health Research (MOP-286787), and a Heart and Stroke Foundation of Ontario Program Grant (PRG6502). Financial support from St. Joseph’s Healthcare Hamilton is acknowledged. R.C.A. is a Career Investigator of the Heart and Stroke Foundation of Ontario and holds the Amgen Canada Research Chair in the Division of Nephrology at St. Joseph’s Healthcare and McMaster University.
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Nwankwo T, Yoon SS, Burt V and Gu Q. Hypertension among adults in the United States: National Health and Nutrition Examination Survey, 2011–2012. NCHS data brief. 2013:1–8. [PubMed] [Google Scholar]
- 2.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS and Muntner P. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 3.Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327–34. [DOI] [PubMed] [Google Scholar]
- 4.Heineke J and Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. [DOI] [PubMed] [Google Scholar]
- 5.Balke CW and Shorofsky SR. Alterations in calcium handling in cardiac hypertrophy and heart failure. Cardiovasc Res. 1998;37:290–9. [DOI] [PubMed] [Google Scholar]
- 6.Ritterhoff J and Tian R. Metabolism in cardiomyopathy: every substrate matters. Cardiovasc Res. 2017;113:411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen-Izu Y, Chen L, Banyasz T, McCulle SL, Norton B, Scharf SM, Agarwal A, Patwardhan A, Izu LT and Balke CW. Hypertension-induced remodeling of cardiac excitation-contraction coupling in ventricular myocytes occurs prior to hypertrophy development. Am J Physiol Heart Circ Physiol. 2007;293:H3301–10. [DOI] [PubMed] [Google Scholar]
- 8.Sciarretta S, Volpe M and Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114:549–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walter P and Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. [DOI] [PubMed] [Google Scholar]
- 10.Austin RC. The unfolded protein response in health and disease. Antioxid Redox Signal. 2009;11:2279–87. [DOI] [PubMed] [Google Scholar]
- 11.Minamino T, Komuro I and Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107:1071–82. [DOI] [PubMed] [Google Scholar]
- 12.Groenendyk J, Sreenivasaiah PK, Kim do H, Agellon LB and Michalak M. Biology of endoplasmic reticulum stress in the heart. Circ Res. 2010;107:1185–97. [DOI] [PubMed] [Google Scholar]
- 13.Glembotski CC. The role of the unfolded protein response in the heart. J Mol Cell Cardiol. 2008;44:453–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M and Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–12. [DOI] [PubMed] [Google Scholar]
- 15.Fu HY, Okada K, Liao Y, Tsukamoto O, Isomura T, Asai M, Sawada T, Okuda K, Asano Y, Sanada S, Asanuma H, Asakura M, Takashima S, Komuro I, Kitakaze M and Minamino T. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010;122:361–9. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Kwak D, Lu Z, Xu X, Fassett J, Wang H, Wei Y, Cavener DR, Hu X, Hall J, Bache RJ and Chen Y. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension. 2014;64:738–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belmont PJ, Tadimalla A, Chen WJ, Martindale JJ, Thuerauf DJ, Marcinko M, Gude N, Sussman MA and Glembotski CC. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J Biol Chem. 2008;283:14012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Xu L, Gillette TG, Jiang X and Wang ZV. The unfolded protein response in ischemic heart disease. J Mol Cell Cardiol. 2018;117:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Bi X, Zhang G, Deng Y, Luo X, Xu L, Scherer PE, Ferdous A, Fu G, Gillette TG, Lee AS, Jiang X and Wang ZV. Glucose-regulated protein 78 is essential for cardiac myocyte survival. Cell Death Differ. 2018. doi: 10.1038/s41418-018-0109-4. Epub 2018 Apr 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA and Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–82. [DOI] [PubMed] [Google Scholar]
- 22.Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, Criollo A, Luo X, Tan W, Jiang N, Lehrman MA, Rothermel BA, Lee AH, Lavandero S, Mammen PP, Ferdous A, Gillette TG, Scherer PE and Hill JA. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell. 2014;156:1179–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bi X, Zhang G, Wang X, Nguyen C, May HI, Li X, Al-Hashimi AA, Austin RC, Gillette TG, Fu G, Wang ZV and Hill JA. Endoplasmic Reticulum Chaperone GRP78 Protects Heart From Ischemia/Reperfusion Injury Through Akt Activation. Circ Res. 2018;122:1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo S, Mao C, Lee B and Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mao C, Tai WC, Bai Y, Poizat C and Lee AS. In vivo regulation of Grp78/BiP transcription in the embryonic heart: role of the endoplasmic reticulum stress response element and GATA-4. J Biol Chem. 2006;281:8877–87. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Zhang Y, Ding G, May HI, Xu J, Gillette TG, Wang H and Wang ZV. Temporal dynamics of cardiac hypertrophic growth in response to pressure overload. Am J Physiol Heart Circ Physiol. 2017;313:H1119–h1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vega RB, Rothermel BA, Weinheimer CJ, Kovacs A, Naseem RH, Bassel-Duby R, Williams RS and Olson EN. Dual roles of modulatory calcineurin-interacting protein 1 in cardiac hypertrophy. Proc Natl Acad Sci U S A. 2003;100:669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molkentin JD, Lin Q, Duncan SA and Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–72. [DOI] [PubMed] [Google Scholar]
- 29.van Berlo JH, Elrod JW, Aronow BJ, Pu WT and Molkentin JD. Serine 105 phosphorylation of transcription factor GATA4 is necessary for stress-induced cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A. 2011;108:12331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26:504–10. [DOI] [PubMed] [Google Scholar]
- 31.Little E, Ramakrishnan M, Roy B, Gazit G and Lee AS. The glucose-regulated proteins (GRP78 and GRP94): functions, gene regulation, and applications. Crit Rev Eukaryot Gene Expr. 1994;4:1–18. [DOI] [PubMed] [Google Scholar]
- 32.Kozutsumi Y, Segal M, Normington K, Gething MJ and Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–4. [DOI] [PubMed] [Google Scholar]
- 33.Hill JA and Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–80. [DOI] [PubMed] [Google Scholar]
- 34.Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE and Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–9. [DOI] [PubMed] [Google Scholar]
- 35.Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ and Izumo S. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107:1664–70. [DOI] [PubMed] [Google Scholar]
- 36.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T and Izumo S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–5. [DOI] [PubMed] [Google Scholar]
- 37.Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, Dalton ND, Peterson KL, Guan KL, Brown JH, Chen J, Sonenberg N and Condorelli G. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120:2805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, Krishnan J, Lerch R, Hall MN, Ruegg MA, Pedrazzini T and Brink M. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123:1073–82. [DOI] [PubMed] [Google Scholar]
- 39.Tamai T, Yamaguchi O, Hikoso S, Takeda T, Taneike M, Oka T, Oyabu J, Murakawa T, Nakayama H, Uno Y, Horie K, Nishida K, Sonenberg N, Shah AM, Takeda J, Komuro I and Otsu K. Rheb (Ras homologue enriched in brain)-dependent mammalian target of rapamycin complex 1 (mTORC1) activation becomes indispensable for cardiac hypertrophic growth after early postnatal period. J Biol Chem. 2013;288:10176–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arrieta A, Blackwood EA and Glembotski CC. ER Protein Quality Control and the Unfolded Protein Response in the Heart. Curr Top Microbiol Immunol. 2018;414:193–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morin S, Charron F, Robitaille L and Nemer M. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 2000;19:2046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ni M, Zhang Y and Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434:181–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.