

Abstract
The major histocompatibility complex (MHC) is a highly polymorphic and polygenic genomic region that plays a crucial role in immune-related diseases. Given the need for comparative studies on the variability of immunologically important genes among wild populations and species, we investigated the allelic variation of MHC class II DRB among three congeneric true lemur species: the red-fronted lemur (Eulemur rufifrons), red-bellied lemur (Eulemur rubriventer), and black lemur (Eulemur macaco). We noninvasively collected hair and faecal samples from these species across different regions in Madagascar. We assessed DRB exon 2 polymorphism with a newly developed primer set, amplifying nearly all non-synonymous codons of the antigen-binding sites. We defined 26 DRB alleles from 45 individuals (17 alleles from E. rufifrons (N = 18); 5 from E. rubriventer (N = 7); and 4 from E. macaco (N = 20). All detected alleles are novel and show high levels of nucleotide (26.8%) and non-synonymous codon polymorphism (39.4%). In these lemur species, we found neither evidence of a duplication of DRB genes nor a sharing of alleles among sympatric groups or allopatric populations of the same species. The non-sharing of alleles may be the result of a geographical separation over a long time span and/or different pathogen selection pressures. We found dN/dS rates > 1 in the functionally important antigen recognition sites, providing evidence for balancing selection. Especially for small and isolated populations, quantifying and monitoring DRB variation are recommended to establish successful conservation plans that mitigate the possible loss of immunogenetic diversity in lemurs.
Electronic supplementary material
The online version of this article (10.1007/s00251-018-1085-z) contains supplementary material, which is available to authorized users.
Keywords: Major histocompatibility complex, Eulemur, Genetic diversity, Balancing selection, Polymorphism
Introduction
The major histocompatibility complex (MHC) is a highly polymorphic and polygenic genomic region and diversity at this complex is considered an important measure of immunocompetence (Klein 1986; Kelley et al. 2005; Piertney and Oliver 2006). The MHC class I and II genes play a crucial role in innate and adaptive immunity, having a major impact on disease resistance (Oliver and Piertney 2006; Schwensow et al. 2007; Rioux et al. 2009; Savage and Zamudio 2011). These genes encode cell surface receptors that present antigens derived from intra- and extracellular parasites and pathogens to T lymphocytes that may consequently initiate an immune response (Germain 1994; Rammensee et al. 1995). Therefore, the MHC genotype determines the diversity of parasites and pathogens that can be recognised, and correlations between particular MHC alleles, high allelic diversity, and number of MHC genes on the one hand and disease resistance on the other have been demonstrated across vertebrate taxa (Briles et al. 1983; Langefors et al. 2001; Schad et al. 2005). For this reason, genetic variation in functionally important MHC gene families plays a central role in vertebrate immunity and in the viability and long-term survival of wildlife populations (Piertney and Oliver 2006; Siddle et al. 2007; Radwan et al. 2010).
The MHC class II region varies between species in the number and presence of genes (Kelley et al. 2005). Some MHC gene families are highly variable, not only in number of alleles but also in the extent of sequence variation between alleles. Variation in the MHC is generated at multiple levels. Animals interact with their immediate environment and are exposed continuously to parasites and pathogens. Selection processes increase resistance to such pressures by generating allelic variation through mutation and recombination, which is well reflected in the diversity patterns of MHC genes (Penn et al. 2002; Kurtz et al. 2006). MHC polymorphism can be similar among species due to their co-ancestry (Klein 1987; Figueroa et al. 1988; McConnell et al. 1988).
The high allelic variation is maintained by balancing selection (Sommer 2005; Piertney and Oliver 2006; Spurgin and Richardson 2010; Grogan et al. 2016), with an increased ratio of non-synonymous over synonymous substitutions at the functionally important antigen-binding sites (ABS) (Garrigan and Hedrick 2003; Fijarczyk and Babik 2015). Balancing selection can be attributed to two processes. First, it can occur when heterozygous individuals are favoured, likely as a result of their ability to respond to a broader array of pathogens than homozygotes (Doherty and Zinkernagel 1975). Second, frequency-dependent selection assumes a co-evolutionary arms race between hosts and pathogens (Takahata and Nei 1990). Polymorphism is maintained when rare alleles are more resistant to pathogens, and are consequently favoured and spread through the population. As soon as parasites have developed antigenicity for these antigens, new, rare alleles will have selective advantage and lead to genetic diversity in populations (Takahata and Nei 1990; Borghans et al. 2004).
Gene duplications and deletions can occur in MHC regions in many primate species (Klein et al. 1993; Nei et al. 1997; Kulski et al. 1999), which result in copy number variation (CNV) among individuals (Slierendregt et al. 1994; Kulski et al. 1999; Doxiadis et al. 2010) and between species (Adams and Parham 2001; Kelley et al. 2005). However, selection against deleterious gene duplications in the MHC operates as well (Shiina et al. 2006), and CNV can also be lost due to genetic drift (Schrider and Hahn 2010; Eimes et al. 2011).
For decades, much has been known about MHC variation, structure, and evolution in humans, captive non-human primates, and other model organisms (Klein 1986; Root-Bernstein 2005). Most of these previous studies focused on the second exon of the MHC II DRB gene(s), because this exon encodes functionally important peptides of the antigen-binding site (ABS) (Harf and Sommer 2005). Since exon 2 has been described to be the most polymorphic part in many class II genes, it is therefore assumed to be involved in the susceptibility to specific pathogens (Brown et al. 1993). The class II genes are physically linked, and alleles on these genes are in strong linkage disequilibrium (Marsh et al. 1999). Therefore, DRB gene diversity patterns can be a good indicator for the genetic variation in other class II genes, and even for other less closely linked MHC genes (Kelley et al. 2005). In addition, the MHC system is one of the few genetic systems where balancing selection has been revealed in humans and rodents under laboratory conditions and where studies on various captive or semi-captive breeding primate populations exist (Lafont et al. 2007; Schwensow et al. 2007), although comparatively little research has been done on wild populations of mammals, and on primates in particular (e.g., Tung et al. 2015). Captive populations usually guarantee an easy access to blood or other tissue types, which results in the high quality and quantity of DNA extracts. In contrast, studies on the variation in the MHC system of free-ranging wild animal populations, including lemurs, are still rare compared to those on captive animals (Bernatchez and Landry 2003; Kaesler et al. 2017). For animal welfare or technical reasons, such studies often rely on noninvasive sampling for genetic and molecular ecology research. This involves challenges to error-free genotyping from a low quantity of low-quality materials.
Over the past century, lemurs have experienced major declines in range size, and nearly half of all lemur species in Madagascar are threatened with extinction as a result of anthropogenic habitat disturbance and unsustainable hunting (Mittermeier et al. 2010). This study focuses on three species of true lemurs, genus Eulemur, family Lemuridae: the red-fronted lemur (Eulemur rufifrons), the red-bellied lemur (Eulemur rubriventer), and the black lemur (Eulemur macaco), each of which diverged about 4.5 million years ago (mya) (Yoder 2007; Markolf et al. 2013). Two of these species (E. rufifrons and E. rubriventer) live sympatrically but do not hybridise, and all other Eulemur populations are geographically isolated (Markolf et al. 2013). Owing to their highly ecological flexibility, different species of the genus Eulemur occupy most biogeographic regions of Madagascar (Johnson 2006), including some of the smaller peripheral islands (Colquhoun 1993). At the same time, they show many similarities in morphology and physiology, as they are genetically closely related (Markolf et al. 2013). Therefore, this genus provides an opportunity to assess the importance of environmental differences in MHC variation as well as the role of balancing selection, which needs to be clarified in Eulemurs.
The specific objective of this study was to investigate the allelic variation of the DRB gene of the three species of wild true lemurs in different congeneric species across the island Madagascar. In this study, we used a new set of primers that amplify elongated fragments including amino acids 9–13 of exon 2, which represent one of the most important antigen-binding motifs of the beta chain of DRB. As a result, this study provides a baseline from which to expand further exploration of lemur MHC in conjunction with wildlife diseases, demographic processes, and other selective forces.
Materials and methods
Study species
True lemurs (genus Eulemur, family Lemuridae) are morphologically much alike and are medium-sized (body and tail length 30–50 cm, 2–4 kg) arboreal primates that occasionally move quadrupedally on the ground. Their diet consists primarily of fruits, flowers, and leaves (Markolf 2013), although they are all capable of adding alternative food sources such as invertebrates to their diet. This study focuses on three Eulemur species: the red-fronted lemur (Eulemur rufifrons), the red-bellied lemur (E. rubriventer), and the black lemur (E. macaco). The main difference between these Eulemur species is their social organisation, including group size: Eulemur rufifrons and E. macaco live in multi-male, multi-female groups of 4 to 18 individuals (Overdorff 1996; Erhart and Overdorff 2008), whereas E. rubriventer lives in small monogamous groups from two to five individuals (Overdorff 1996). All three species are listed in the IUCN Red List of Threatened Species: Eulemur rufifrons as ‘near threatened’, and E. rubriventer and E. macaco as ‘vulnerable’ (Andriaholinirina et al. 2014).
Study site
We collected biological materials from three different lemur species in four different field sites across Madagascar. We collected samples from Eulemur rufifrons and E. rubriventer in Ranomafana National Park (NP) (N, − 21.32; E, 47.40), samples from E. rufifrons in Isalo NP (N, − 22.47; E, 45.26) and Kirindy Forest (N, − 20.07; E, 44.66), and samples from E. macaco on Nosy Komba (N, − 13.46; E, 48.35) (Fig. 1). Kirindy Forest and Isalo are located on the western side of Madagascar and consist of dry deciduous forest with pronounced seasonality. These western regions have a higher annual mean temperature than the eastern rainforests and receive less rainfall (Goodman et al. 2005). Ranomafana NP is a humid rainforest located on the eastern side of Madagascar. The island Nosy Komba is located in the north-west of Madagascar and is covered with tropical vegetation.
Fig. 1.
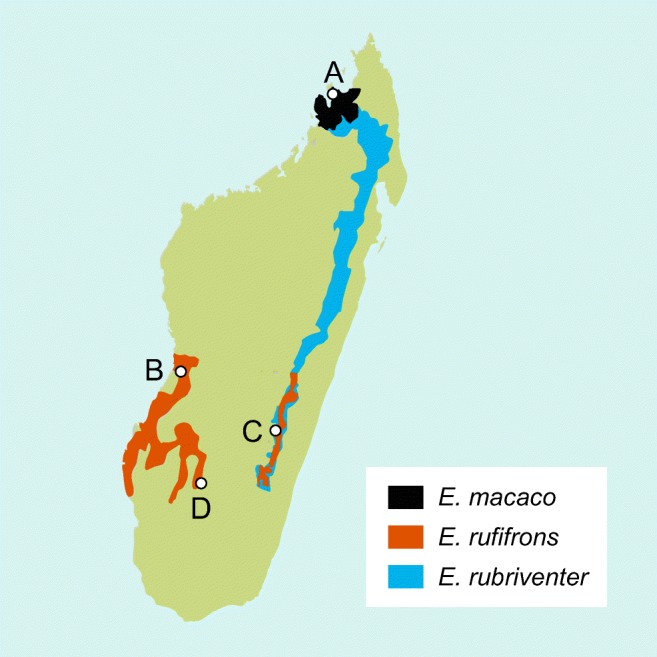
Study sites and geographic ranges of the study species. Map of Madagascar with the geographic ranges of the three study species, Eulemur macaco, E. rufifrons, and E. rubriventer, and the corresponding sites where samples were collected, (A) Nosy Komba, (B) Kirindy Forest, (C) Ranomafana NP, and (D) Isalo NP
Sample collection
In a noninvasive manner, we collected samples (N = 45 individuals; N = 51 faecal; and N = 20 hair samples) from individuals between October 2013 and May 2014. Immediately after animals had defecated, fresh faecal samples (3–4 g) of adult lemurs were collected with a pincer that we cleaned with ethanol to avoid contamination. We aimed to sample all adult individuals within a social group and prevented duplication by identifying each individual on the basis of its morphology. From some individuals, both hair and faecal samples were collected. Within 12 h after collection, we stored the collected samples in 15 mL tubes containing 15 g of silica beads to desiccate the faeces (Wasser et al. 1997). We stored these samples in the shade at ambient temperatures until further analyses by colleagues in the Department of Comparative Genetics & Refinement, Biomedical Primate Research Centre (BPRC) in the Netherlands. In addition to faecal samples, hair was collected opportunistically when possible and stored in small ziplock bags. When the lemurs approached closely enough, a tuft of hair was removed from the hip region. Sample collection and export were approved by the trilateral commission (CAFF/CORE) in Madagascar (permits 297/13 and 143/14/MEF/SG/DGF/DCB.SAP/SCBSE).
DNA extraction
Total DNA was extracted from faeces by using the QIAamp® DNA Stool kit (Qiagen) according to the manufacturer’s guidelines, with a few modifications (Nsubuga et al. 2004), which include the following. At the start of the extraction, (1) we covered the silica beads in 15-ml tubes containing the dried faecal samples with 1.5 to 2 mL of ASL buffer; (2) the tubes were shaken for 12 to 16 h at 25 °C; (3) the supernatant was fully removed from the preservation tubes to extract the DNA; (4) at the end of the extraction, the recommended step of 1-min centrifugation at full speed in a new collection tube was applied; (5) 50 μL of AE buffer was used for elution; and (6) incubation at room temperature for 20 min was followed by centrifugation at full speed for 2 min. DNA from hair was extracted by using the Gentra Puregene tissue kit (Qiagen) according to the manufacturer’s guidelines. DNA quality and quantity were estimated by absorbance at 260/280 nm on the ND-1000 NanoDrop®.
PCR reaction for DRB exon 2
A 213-bp fragment of DRB exon 2 was amplified by PCR using a generic 5′DRB-exon 2 primer CGT GTC CCC ACA GCA CGT TTC (Doxiadis et al. 2006) together with the 3′JS2 primer GAT CCC GTA GTT GTG TCT GCA (Schad et al. 2004). The PCR reactions were performed in a 50 μL volume containing 5 units of Platinum Taq polymerase (Invitrogen, Paisley, Scotland) with 0.2 μM of each primer, 5 mM MgCl2, 0.2 mM of each dNTP, 1X PCR buffer (Invitrogen, Paisley, Scotland), and 50–200 ng DNA. The cycling parameters were a 2-min initial denaturation step at 94 °C, followed by 3 cycles of 90 s at 94 °C, 90 s at 60 °C, and 90 s at 74 °C. This programme was followed by 32 cycles of 30 s at 94 °C, 30 s at 60 °C, and 30 s at 74 °C. A final extension step was performed at 72 °C for 7 min.
Cloning and sequencing
PCR products were purified using a GeneJet Gel Extraction Kit (Thermo Scientific™), and the purified amplicons were cloned into the pJET vector using the CloneJET PCR cloning kit, both according to the manufacturer’s guidelines (Thermo Scientific™). Next, the cloned amplicons were transformed in Escherichia coli XL1-blue cells by using the TransformAid Bacterial Transformation Kit (Thermo Scientific™). Per animal, 24 to 48 bacterial clones were picked, and plasmid DNA was isolated using a standard mini-preparation procedure. The purified plasmid DNA was sequenced on the ABI 3500 genetic analyser (Applied Biosystems, Foster City, USA). The sequencing reaction was performed by using 2 μM pJET primer, 1 μL BigDye terminator, and 2 μL of 5 × sequencing buffer in a total volume of 10 μL (Thermo Scientific™). The resulting sequences were analysed using the Sequence Navigator programme (Applied Biosystems, Foster City, USA). MHC sequences were revised manually by applying the Lasergene 12 SeqMan Pro Sequence Alignment Editor.
Allele discovery and nomenclature
All sequences were compared in BLAST at GenBank (National Centre for Biotechnology Information, NCBI) and turned out to be novel and related to Eulemur DRB exon 2. Only sequences with an identity higher than 95% to already-published lemur DRB alleles were considered to be of lemur origin and were selected for further analysis. Furthermore, only sequences that were detected at least two times, either in two different PCRs of the same sample or in two different animals, were accepted as being true and new alleles. The alleles were named numerically based on general principles used for the IPD-MHC 2.0 database (Maccari et al. 2017). We deposited all alleles in GenBank, and they were given the accession numbers MF682987-MF683012.
Phylogenetic analysis
We constructed a neighbour-joining phylogenetic tree to show phylogenetic relationships among DRB alleles of E. rufifrons, E. rubriventer, and E. macaco, with evolutionary distances computed according to the Kimura-2-parameter method (Saitou and Nei 1987). We used a bootstrap consensus tree inferred from 2000 replicates and included both transitions and transversions, assuming rates among sites to have a gamma distribution, with a gamma parameter set to 1. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates were collapsed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test is shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The analysis involved 26 nucleotide sequences. All positions containing gaps and missing data were eliminated. There was a total of 213 positions in the final dataset. Evolutionary analyses were conducted in MEGA V.5 (Tamura et al. 2011).
dN/dS calculations
We calculated the relative rate of non-synonymous (dN) and synonymous (dS) substitutions (Nei and Gojobori 1986) with the Jukes-Cantor correction (Jukes et al. 1969) for multiple hits in MEGA. Substitution rates were calculated for the overall sequences and then separately for ABS and non-ABS. Concordance with ABS in human MHC molecules was assumed, with the following beta chain residues: 9, 11, 13, 28, 30, 32, 37, 38, 47, 56, 60, 61, 65, 68, 70, 71, 74, and 78. Therefore, six other residues (i.e., 81, 82, 85, 86, 88, 89) that were identified as ABS (Brown et al. 1993) are not part of the amplicons derived in this study. Statistical differences in dN/dS rates were tested with a Z-test. For all calculations, the alpha level was set at 0.05.
Results
DRB allele definition and variation
A total of 26 DRB alleles could be identified among 45 individuals of E. rufifrons (N = 18), E. rubriventer (N = 7), and E. macaco (N = 20, Tables 1, 2). Eulemur rufifrons showed the highest allelic variation, with 17 different DRB alleles (Eufr-DRB*01–17) defined in 18 animals. Most individuals of this species lived in Kirindy Forest (Table 2, Fig. 1), and, accordingly, most alleles are defined in these animals (Table 1). In three animals from Isalo NP, three other DRB alleles were determined along with another two alleles in the two individuals from Ranomafana NP. Eulemur rubriventer showed the second highest allelic variation, with five alleles (Euru-DRB*01–05) among eight animals, whereas E. macaco was least polymorphic for its DRB gene, with only four alleles (Euma-DRB*01–17) defined in the 20 individuals genotyped (Tables 1, 2).
Table 1.
All detected alleles (N = 26) and the specific individual samples
| A. Eulemur rufifrons, Kirindy Forest | ||
| # | Allele | Animal ID |
| 1 | Eufr-DRB*01 | K1RF, K5RF |
| 2 | Eufr-DRB*02 | K7RF, K2RF |
| 3 | Eufr-DRB*03 | K10RF |
| 4 | Eufr-DRB*04 | K1RF, K25RF, K4RF, K9RF |
| 5 | Eufr-DRB*05 | K25RF |
| 6 | Eufr-DRB*06 | K6RF |
| 7 | Eufr-DRB*07 | K27RF, K8RF, K5RF |
| 8 | Eufr-DRB*08 | K10RF |
| 9 | Eufr-DRB*09 | K7RF, K2RF |
| 10 | Eufr-DRB*10 | K11RF, K4RF, K3RF |
| 11 | Eufr-DRB*11 | K8RF |
| 12 | Eufr-DRB*12 | K3RF, K11RF |
| B. Eulemur rufifrons, Isalo NP | ||
| 13 | Eufr-DRB*13 | I7RF, I4RF |
| 14 | Eufr-DRB*14 | I2RF |
| 15 | Eufr-DRB*15 | I4RF, I7RF, I2RF |
| C. Eulemur rufifrons, Ranomafana NP | ||
| 16 | Eufr-DRB*16 | R90RF |
| 17 | Eufr-DRB*17 | R105RF, R90RF |
| D. Eulemur rubriventer, Ranomafana NP | ||
| # | Allele | Animal ID |
| 1 | Euru-DRB*01 | R43RB, R29RB, R27RB |
| 2 | Euru-DRB*02 | R30RB, R27RB |
| 3 | Euru-DRB*03 | R33RB, R29RB, R35RB |
| 4 | Euru-DRB*04 | R30RB |
| 5 | Euru-DRB*05 | R33RB, R34RB, R55RB, R35RB |
| E. Eulemur macaco, Nosy Komba | ||
| # | Allele | Animal ID |
| 1 | Euma-DRB*01 | NK7, NK13, NK19, NK9, NK14, NK2 |
| 2 | Euma-DRB*02 | NK21, NK16, NK7, NK17, NK9 NK3, NK1EMA, NK4, NK12, NK1 |
| 3 | Euma-DRB*03 | NB1EMA, NK3, NK4, NK16, NK22, NK2, NK12 |
| 4 | Euma-DRB*04 | NK19 |
Table 2.
Individual MHC class II DRB exon 2 genotypes for 45 different lemurs
| A. Eulemur rufifrons, samples from Kirindy Forest, Isalo NP, and Ranomafana NP | |||
| Kirindy Forest | Allele 1 | Allele 2 | |
| 1 | K1RF | Eufr-DRB 01 | Eufr-DRB 04 |
| 2 | K2RF | Eufr-DRB 02 | Eufr-DRB 09 |
| 3 | K3RF | Eufr-DRB 10 | Eufr-DRB 12 |
| 4 | K4RF | Eufr-DRB 04 | Eufr-DRB 10 |
| 5 | K5RF | Eufr-DRB 01 | Eufr-DRB 07 |
| 6 | K6RF | Eufr-DRB 06 | |
| 7 | K7RF | Eufr-DRB 02 | Eufr-DRB 09 |
| 8 | K8RF | Eufr-DRB 07 | Eufr-DRB 11 |
| 9 | K9RF | Eufr-DRB 04 | |
| 10 | K10RF | Eufr-DRB 03 | Eufr-DRB 08 |
| 11 | K11RF | Eufr-DRB 10 | Eufr-DRB 12 |
| 12 | K25RF | Eufr-DRB 04 | Eufr-DRB 05 |
| 13 | K27RF | Eufr-DRB 07 | |
| Isalo NP | Allele 1 | Allele 2 | |
| 14 | I2RF | Eufr-DRB 14 | Eufr-DRB 15 |
| 15 | I4RF | Eufr-DRB 13 | Eufr-DRB 15 |
| 16 | I7RF | Eufr-DRB 13 | Eufr-DRB 15 |
| Ranomafana NP | Allele 1 | Allele 2 | |
| 17 | R105RF | Eufr-DRB 17 | |
| 18 | R90RF | Eufr-DRB 16 | Eufr-DRB 17 |
| B. Eulemur rubriventer, samples from Ranomafana NP | |||
| Ranomafana NP | Allele 1 | Allele 2 | |
| 1 | R43RB | Euru-DRB 01 | |
| 2 | R33RB | Euru-DRB 03 | Euru-DRB 05 |
| 3 | R34RB | Euru-DRB 05 | |
| 4 | R55RB | Euru-DRB 05 | |
| 5 | R29RB | Euru-DRB 01 | Euru-DRB 03 |
| 6 | R30RB | Euru-DRB 02 | Euru-DRB 04 |
| 7 | R27RB | Euru-DRB 01 | Euru-DRB 02 |
| C. Eulemur macaco, samples from Nosy Komba | |||
| Nosy Komba | Allele 1 | Allele 2 | |
| 1 | NK21 | Euma-DRB 02 | |
| 2 | NK16 | Euma-DRB 02 | Euma-DRB 03 |
| 3 | NK7 | Euma-DRB 01 | Euma-DRB 02 |
| 4 | NK17 | Euma-DRB 02 | |
| 5 | NK3 | Euma-DRB 02 | Euma-DRB 03 |
| 6 | NK1EMA | Euma-DRB 02 | |
| 7 | NB1EMA | Euma-DRB 03 | |
| 8 | NK22 | Euma-DRB 03 | |
| 9 | NK13 | Euma-DRB 01 | |
| 10 | NK4 | Euma-DRB 02 | Euma-DRB 03 |
| 11 | NK12 | Euma-DRB 02 | Euma-DRB 03 |
| 12 | NK1 | Euma-DRB 02 | |
| 13 | NK2 | Euma-DRB 01 | Euma-DRB 03 |
| 14 | NK14 | Euma-DRB 01 | |
| 15 | NK9 | Euma-DRB 01 | Euma-DRB 02 |
| 16 | NK10 | Euma-DRB 02 | Euma-DRB 03 |
| 17 | NK19 | Euma-DRB 01 | Euma-DRB 04 |
| 18 | NK23 | Euma-DRB 02 | Euma-DRB 03 |
| 19 | NK5 | Euma-DRB 02 | Euma-DRB 03 |
| 20 | NK15 | Euma-DRB 02 | |
Eufr-DRB was characterised by high polymorphism, with most alleles being observed in just one or two animals, whereas Euru-DRB but especially Euma-DRB alleles were detected in far more animals. Five to ten E. macaco individuals shared the same allele, with the exception of Euma-DRB*04, which was observed in one animal only (Table 1). Most individuals were heterozygous (Table 2; observed heterozygosity: Eulemur rufifrons, 0.78; E. rubriventer, 0.57; E. macaco, 0.55). None of the animals showed more than two DRB alleles, indicating that we did not detect a sign of DRB duplication in these species.
To visualise the phylogenetic affinities among species, we built a neighbour-joining tree, including the 26 different DRB alleles (Fig. 2). The branches in the resulting phylogenetic tree may indicate different DRB lineages (Fig. 2A–G). Each of the three species possesses one allele that clusters separately from the others, indicating evolutionarily long divergence times. The allele Euru-DRB04-RNP, found in E. rufifrons in Ranomafana NP, forms a single branch (Fig. 2E), whereas Eufr-DRB01-KIR and Euma-DRB02-NK, found in E. rufifrons in Kirindy Forest and E. macaco on Nosy Komba, respectively, form separate branches within one cluster (Fig. 2G). The other clusters were present in all three lemur species. Only a few DRB alleles within a species appear to be closely related. For example, the alleles Eufr-DRB02 and Eufr-DRB05 showed only two nucleotide differences and were isolated from animals from the same location, Kirindy Forest (Fig. 2B). In contrast, we identified closely related alleles with just two nucleotide differences from two different populations of the species Eulemur rufifrons: Eufr-DRB12-KIR from Kirindy Forest and Eufr-DRB15-IS from Isalo NP (Fig. 2C). The four alleles of E. macaco are located very distantly from each other in three different branches of the phylogenetic tree.
Fig. 2.
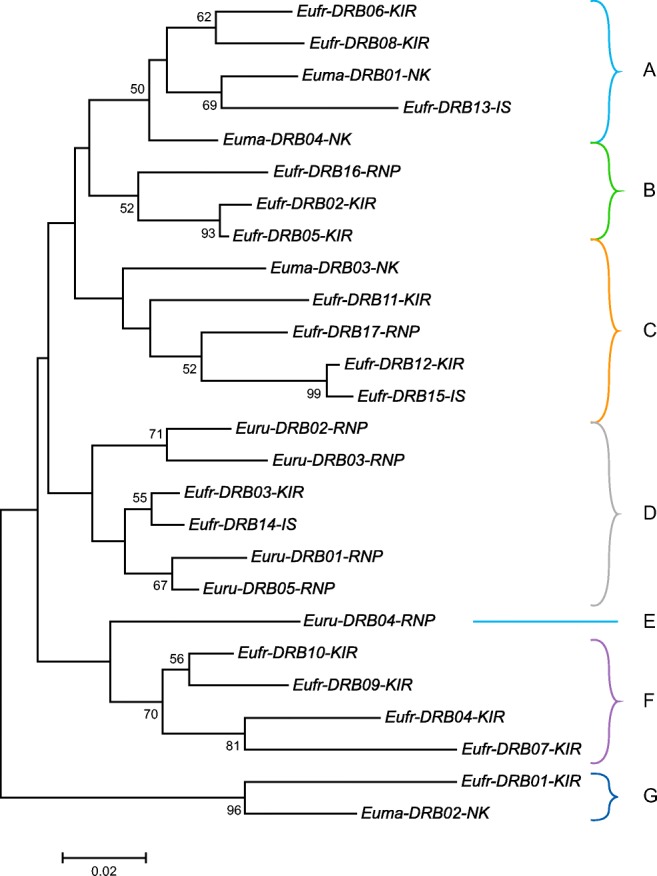
Neighbour-joining tree of exon 2 DRB sequences of Eulemur macaco, E. rufifrons, and E. rubriventer. Neighbour-joining tree constructed from 26 MHC II DRB exon 2 alleles in Eulemur macaco, E. rufifrons, and E. rubriventer. The tree was constructed in accordance with the Kimura-2-parameter model (Kimura 1980). The percentages of replicate trees in which the associated taxa cluster together in the bootstrap test are depicted in front of a node. Cluster designation is shown next to the branches (letters A–G). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used. An abbreviation of the location where the DRB allele has been detected is given in the allele name: Nosy Komba = NK; Kirindy Forest = KIR; Ranomafana NP = RNP; and Isalo NP = IS
Amino acid variation of the DR beta chain
All amplicons represent different DRB alleles, which appear to encode for unique peptides. They are composed of 71 amino acids, indicating a functional DRB molecule, with 28 variable amino acids (39.4%). Of the 18 ABS (Fig. 3, indicated by an asterisk), 15 sites (83.3%) are variable, while of the 53 non-ABS, only 13 (24.5%) are variable. The ratio of non-synonymous and synonymous substitution rates (dN/dS) at the complete fragments was elevated and was indicative of positive selection (Z = 3.99, P < 0.001). This can be attributed to the ABS (Z = 4.36, P < 0.001), as non-ABS did not show a significant positive selection (Z = 1.47, P = 0.07).
Fig. 3.

Deduced amino acid alignment of the DRB exon 2 of Eulemur macaco, E. rufifrons, and E. rubriventer. The sequence alignment starts at amino acid position 8 of exon 2. Dashes indicate identity with the first sequence. Asterisks indicate amino acids that are involved in peptide binding. Allele names are given as described for Fig. 2
Discussion
In this study, we obtained elongated DRB exon 2 fragments, which allowed us to define the nucleotides encoding nearly all non-synonymous codons of the antigen-binding sites. Especially amino acids 9 to 13 are of importance in encoding a peptide motif, which defines the relatedness of DRB alleles in humans and non-human primates (e.g., macaques), and are therefore used for lineage definition (Sommer 2005). These sequences were missing in the DRB amplicons of lemur DRB sequences published earlier in a mouse lemur species (Microcebus murinus) (Schad et al. 2004). Indeed, in the three lemur species analysed in this study, these amino acids were highly polymorphic, and they encode many different motifs that may be useful for phylogenetic purposes when more individuals will have been analysed (Fig. 3). Although the species in our study were all congeneric and some species lived in sympatry, alleles were neither shared between species nor between allopatric populations of a species (i.e., between the three geographically separated E. rufifrons populations). DRB allele sharing of evolutionarily related species with a divergence time of less than 1.5 mya has been observed: for instance, between rhesus macaques (Macaca mulatta) and cynomolgus macaques (Macaca fascicularis) (Doxiadis et al. 2006). The absence of allele sharing among the E. rufifrons populations may indicate that these populations have been separated for more than 1.5 million years, and different parasite loads may have led to a different DRB repertoire. Allele sharing of the three Eulemur species with a far higher divergence time of ~ 4.5 mya was therefore not expected. However, as the three E. rufifrons populations are assumed to belong to the same species, it is remarkable that they do not share identical DRB alleles. Owing to the low sample size, however, the most plausible explanation would be that not all DRB alleles have yet been defined, and therefore, low-frequency shared alleles may have been missed. Furthermore, a relatively high number of DRB homozygous animals may indicate that due to primer inconsistencies, not all alleles have been defined. More animals need to be sampled and analysed, and calculations of observed versus expected heterozygosity are needed to evaluate whether all DRB alleles have been detected.
In a comparison of the different lemur species and populations, DRB polymorphism was largest in E. rufifrons sampled in a dry deciduous forest (Kirindy Forest). In the lemurs in this forest, the prevalence, species richness, and infection intensities of gastrointestinal parasites are high when compared to other lemur populations in Madagascar (Clough 2010). Higher and more diverse parasite loads can lead to increased individual MHC diversity (Summers et al. 2003; Harf and Sommer 2005; Clough 2010; Eizaguirre et al. 2011). Furthermore, the number of unique alleles present in the population of E. macaco on the island Nosy Komba was more than four times lower than in the E. rufifrons population in Kirindy Forest. This might be the effect of a low pathogen pressure on this island, as was also observed on the nearby island Nosy Be (Junge and Louis 2007). In addition, the low allele diversity within this isolated island population may also be the result of a small founder population, the impact of inbreeding due to the small population size, and the lack of any influx of new individuals (Frankham 2015). In contrast to various other non-human primate species, we found no evidence of duplication of the DRB gene in true lemurs. We cannot exclude, however, that future studies with more sensitive techniques, such as next generation sequencing, may reveal duplication(s) of the DRB gene. DRB duplication and copy number variation (CNV) of MHC genes are a common phenomenon in vertebrates and have been reported in many primate species, including chimpanzees (Pan troglodytes), orangutans (Pongo pygmaeus), macaques (Doxiadis et al. 2006), dusky titis (Callicebus moloch) (Trtková et al. 1993), green monkeys (Chlorocebus sabaeus) (Rosal-Sánchez et al. 1998), and the Senegal bushbaby (Galago senegalensis) (Figueroa et al. 1994). Additionally, other Strepsirrhini primates, including the northern greater galago (Otolemur garnettii) and the Senegal bushbaby (Galago senegalensis) (Figueroa et al. 1994), have a duplicated DRB gene. One lemur species, the grey mouse lemur (Microcebus marinus), also shows DRB duplication (Go et al. 2002; Huchard et al. 2012). However, gene duplication is rare in lemurs (Go et al. 2002; Averdam et al. 2011), and therefore, our results confirm those of previous studies, suggesting that DRB duplication is relatively rare in this primate group.
With the exception of two clusters, phylogenetic analysis of the DRB alleles of the three Eulemur species genotyped in this study does not show clustering of the alleles per species (Fig. 2 B, F). This may be an indication that most DRB alleles are older than the species’ divergence time. Instead, we saw an intermingling of alleles from different species. The long branch lengths, at least for some clusters (e.g., Fig. 2G), also indicates long evolutionary distances. As a consequence, this finding suggests that these branches represent DRB lineages that are shared between the three Eulemur species and are older than the divergence of these species. The only four alleles of E. macaco are located at a considerable distance from each other in three branches, and therefore appear to belong to different lineages. Two animals of E. rufifrons and all E. rubriventer individuals tested in this study were from Ranomafana NP, which represents a special location. It is situated on the eastern side of Madagascar and has been isolated from the rest of the island by a major mountainous geographic barrier that runs in a north-south direction over the island. The geographic separation of the populations may explain why DRB alleles from animals in Ranomafana NP seem less closely related to other Eulemur DRB alleles. The potential different external infection pressures for the separated populations may have played a role in this observation as well. However, to substantiate this suggestion, far more samples have to be analysed in the future. Additionally, when more samples become available, intron sequences should be analysed that are under less selection pressure than coding sequences in order to gain a better insight into the phylogeny of DRB in lemurs (Doxiadis et al. 2012).
The DRB genes of all three true lemur species in our study express higher rates of non-synonymous substitutions than expected under the neutrality, similar to other findings on lemurs and other primate species (reviewed in Go et al. 2002). We demonstrate a higher dN/dS ratio in the ABS compared to the dN/dS ratio of the non-ABS in the respective domains, leading to different amino acid sequences. As expected and confirmed in many other studies (Schad et al. 2004, 2005; Harf and Sommer 2005), the rate of non-synonymous substitutions did not exceed the rate of synonymous substitutions in the non-ABS. These results indicate balancing selection, leading to high levels of DRB diversity and polymorphism. Some studies suggest that this selection pattern is driven by parasites, for example, in grey mouse lemurs (Schad et al. 2004, 2005) and mandrills (Mandrillus sphinx) (Abbott et al. 2006). As the diversity in MHC II may be linked to the diversity of parasites and pathogens that can be recognised by the host (Briles et al. 1983; Langefors et al. 2001; Schad et al. 2005), the role of parasites in driving DRB variation needs to be investigated further.
High polymorphism levels of MHC II genes are considered critical to the long-term survival of animal populations (Edwards and Potts 1996; Grogan et al. 2017), although species with low diversity could also be viable (Sommer et al. 2002). Like all lemurs, true lemurs face significant anthropogenic threats, including disease pressures, changing climatic conditions, and habitat loss and fragmentation (Schwitzer et al. 2013; Reuter et al. 2017). Many populations have become isolated (Irwin et al. 2010), and we indicate that an isolated population in our study shows a loss of genetic diversity. Studies quantifying DRB alleles can assess a species’ ability to respond to the many anthropogenic threats they are facing. Especially when comparing different populations and populations with rare or unevenly distributed alleles, a greater sampling effort is needed to detect most of the DRB diversity. Sampling within different areas that experience anthropogenic pressures would be very interesting from a conservational perspective. We recommend conservation management to include the analysis of DRB polymorphism as a key to the long-term survival of endangered species, such as lemurs in Madagascar. We also recommend investigating the association between DRB variation and disease resistance as well as other fitness parameters in threatened populations.
Electronic supplementary material
(DOCX 18 kb)
Acknowledgements
For their advice in the field, the authors want to thank Dr. P. Wright, Dr. E. Larney, and John E. Cadle, and the Centre Valbio staff (CVB), the Institute for the Conservation of Tropical Environments (ICTE), and the Madagascar Institute pour la Conservation des Ecosystèmes Tropicaux (MICET) for their logistical support, especially B. Andriamihaja, who facilitated arrangements regarding necessary permits. Thanks go to the Malagasy government and Madagascar National Parks (MNP) as well as Centre de Formation Professionelle Forestière (CFPF) for their formal permission. For their fieldwork assistance and indispensable knowledge, we want to thank all MSc students, field technicians, and guides that helped in collecting data at the various national parks. This work is part of the PhD thesis of IdW and was supported by an NWO/ALW and PE&RC grant (Grant No. 1208; Project No. 5120873001). We are grateful to other institutions that enabled the fieldwork needed for this study: the KNAW Academy Ecology Fund, the Dr. J. L. Dobberke Foundation, the Treub Foundation, the Dutch Fund for Research on Nature Conservation, and the Dutch Royal Zoological Society. Additionally, the authors wish to thank D. Devine for editing the manuscript and F. van Hassel for preparing the figures.
References
- Abbott KM, Wickings EJ, Knapp LA. High levels of diversity characterize mandrill (Mandrillus sphinx) Mhc-DRB sequences. Immunogenetics. 2006;58:628–640. doi: 10.1007/s00251-006-0132-3. [DOI] [PubMed] [Google Scholar]
- Adams EJ, Parham P. Species-specific evolution of MHC class I genes in the higher primates. Immunol Rev. 2001;183:41–64. doi: 10.1034/j.1600-065x.2001.1830104.x. [DOI] [PubMed] [Google Scholar]
- Andriaholinirina N, Baden A, Blanco M et al (2014) Eulemurs. IUCN Red List Threat Species e.T8207A16117505
- Averdam A, Kuschal C, Otto N, Westphal N, Roos C, Reinhardt R, Walter L. Sequence analysis of the grey mouse lemur (Microcebus murinus) MHC class II DQ and DR region. Immunogenetics. 2011;63:85–93. doi: 10.1007/s00251-010-0487-3. [DOI] [PubMed] [Google Scholar]
- Bernatchez L, Landry C. MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol. 2003;16:363–377. doi: 10.1046/j.1420-9101.2003.00531.x. [DOI] [PubMed] [Google Scholar]
- Borghans JA, Beltman JB, De Boer RJ. MHC polymorphism under host-pathogen coevolution. Immunogenetics. 2004;55:732–739. doi: 10.1007/s00251-003-0630-5. [DOI] [PubMed] [Google Scholar]
- Briles WE, Briles RW, Taffs Rolf E, Stone HA. Resistance to a malignant lymphoma in chickens is mapped to subregion of major histocompatibility (B) complex. Science. 1983;219:977–979. doi: 10.1126/science.6823560. [DOI] [PubMed] [Google Scholar]
- Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature. 1993;364:33–39. doi: 10.1038/364033a0. [DOI] [PubMed] [Google Scholar]
- Clough D. Gastro-intestinal parasites of red-fronted lemurs in Kirindy Forest, western Madagascar. J Parasitol. 2010;96:245–251. doi: 10.1645/GE-2258.1. [DOI] [PubMed] [Google Scholar]
- Colquhoun IC (1993) The socioecology of Eulemur macaco: a preliminary report. In: Lemur social systems and their ecological basis. Springer, Boston 11–24
- Doherty PC, Zinkernagel RM. Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature. 1975;256:50–52. doi: 10.1038/256050a0. [DOI] [PubMed] [Google Scholar]
- Doxiadis GG, Rouweler AJ, de Groot NG, et al. Extensive sharing of MHC class II alleles between rhesus and cynomolgus macaques. Immunogenetics. 2006;58:259–268. doi: 10.1007/s00251-006-0083-8. [DOI] [PubMed] [Google Scholar]
- Doxiadis GGM, de Groot N, de Groot NG, et al. Extensive DRB region diversity in cynomolgus macaques: recombination as a driving force. Immunogenetics. 2010;62:137–147. doi: 10.1007/s00251-010-0422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxiadis GG, Hoof I, de Groot N, Bontrop RE. Evolution of HLA-DRB genes. Mol Biol Evol. 2012;29:3843–3853. doi: 10.1093/molbev/mss186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SV, Potts WK (1996) Polymorphism of genes in the major histocompatibility complex (MHC): implications. Molecular Genetic Approaches in Conservation 214
- Eimes JA, Bollmer JL, Whittingham LA, et al. Rapid loss of MHC class II variation in a bottlenecked population is explained by drift and loss of copy number variation. J Evol Biol. 2011;24:1847–1856. doi: 10.1111/j.1420-9101.2011.02311.x. [DOI] [PubMed] [Google Scholar]
- Eizaguirre C, Lenz TL, Sommerfeld RD, Harrod C, Kalbe M, Milinski M. Parasite diversity, patterns of MHC II variation and olfactory based mate choice in diverging three-spined stickleback ecotypes. Evol Ecol. 2011;25:605–622. doi: 10.1007/s10682-010-9424-z. [DOI] [Google Scholar]
- Erhart EM, Overdorff DJ. Population demography and social structure changes in Eulemur fulvus rufus from 1988 to 2003. Am J Phys Anthropol. 2008;136:183–193. doi: 10.1002/ajpa.20793. [DOI] [PubMed] [Google Scholar]
- Figueroa F, Gúnther E, Klein J. MHC polymorphism pre-dating speciation. Nature. 1988;335:265–267. doi: 10.1038/335265a0. [DOI] [PubMed] [Google Scholar]
- Figueroa F, O’hUigin C, Tichy H, Klein J. The origin of the primate Mhc-DRB genes and allelic lineages as deduced from the study of prosimians. J Immunol. 1994;152:4455–4465. [PubMed] [Google Scholar]
- Fijarczyk A, Babik W. Detecting balancing selection in genomes: limits and prospects. Mol Ecol. 2015;24:3529–3545. doi: 10.1111/mec.13226. [DOI] [PubMed] [Google Scholar]
- Frankham R. Genetic rescue of small inbred populations: meta-analysis reveals large and consistent benefits of gene flow. Mol Ecol. 2015;24:2610–2618. doi: 10.1111/mec.13139. [DOI] [PubMed] [Google Scholar]
- Garrigan D, Hedrick PW. Perspective: detecting adaptive molecular polymorphism: lessons from the MHC. Evolution. 2003;57:1707–1722. doi: 10.1111/j.0014-3820.2003.tb00580.x. [DOI] [PubMed] [Google Scholar]
- Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- Go Y, Satta Y, Kawamoto Y, Rakotoarisoa G, Randrianjafy A, Koyama N, Hirai H. Mhc-DRB genes evolution in lemurs. Immunogenetics. 2002;54:403–417. doi: 10.1007/s00251-002-0480-6. [DOI] [PubMed] [Google Scholar]
- Goodman SM, Andriafidison D, Andrianaivoarivelo R, Cardiff SG, Ifticene E, Jenkins RKB, Kofoky A, Mbohoahy T, Rakotondravony D, Ranivo J, Ratrimomanarivo F, Razafimanahaka J, Racey PA. The distribution and conservation of bats in the dry regions of Madagascar. Anim Conserv. 2005;8:153–165. doi: 10.1017/S136794300500199X. [DOI] [Google Scholar]
- Grogan KE, McGinnis GJ, Sauther ML, et al. Next-generation genotyping of hypervariable loci in many individuals of a non-model species: technical and theoretical implications. BMC Genomics. 2016;17:1–16. doi: 10.1186/s12864-016-2503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grogan KE, Sauther ML, Cuozzo FP, Drea CM. Genetic wealth, population health: major histocompatibility complex variation in captive and wild ring-tailed lemurs (Lemur catta) Ecol Evol. 2017;7:7638–7649. doi: 10.1002/ece3.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harf R, Sommer S. Association between major histocompatibility complex class II DRB alleles and parasite load in the hairy-footed gerbil, Gerbillurus paeba, in the southern Kalahari. Mol Ecol. 2005;14:85–91. doi: 10.1111/j.1365-294X.2004.02402.x. [DOI] [PubMed] [Google Scholar]
- Huchard E, Albrecht C, Schliehe-Diecks S, Baniel A, Roos C, Peter PMK, Brameier M. Large-scale MHC class II genotyping of a wild lemur population by next generation sequencing. Immunogenetics. 2012;64:895–913. doi: 10.1007/s00251-012-0649-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin MT, Junge RE, Raharison JL, Samonds KE. Variation in physiological health of diademed sifakas across intact and fragmented forest at Tsinjoarivo, eastern Madagascar. Am J Primatol. 2010;72:1013–1025. doi: 10.1002/ajp.20847. [DOI] [PubMed] [Google Scholar]
- Johnson SE (2006) Evolutionary divergence in the brown lemur species complex. In: Lemurs. Springer US, pp 187–210
- Jukes TH, Cantor CR, Munro HN. Evolution of protein molecules. Mamm Protein Metab. 1969;3:132. [Google Scholar]
- Junge RE, Louis EE. Biomedical evaluation of black lemurs (Eulemur macaco macaco) in Lokobe Reserve, Madagascar. J Zoo Wildl Med. 2007;38:67–76. doi: 10.1638/06-006.1. [DOI] [PubMed] [Google Scholar]
- Kaesler E, Kappeler PM, Brameier M, Demeler J, Kraus C, Rakotoniaina JH, Hämäläinen AM, Huchard E. Shared evolutionary origin of major histocompatibility complex polymorphism in sympatric lemurs. Mol Ecol. 2017;26:5629–5645. doi: 10.1111/mec.14336. [DOI] [PubMed] [Google Scholar]
- Kelley J, Walter L, Trowsdale J. Comparative genomics of major histocompatibility complexes. Immunogenetics. 2005;56:683–695. doi: 10.1007/s00251-004-0717-7. [DOI] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Klein J. Natural history of the major histocompatibility complex. New York: Wiley; 1986. [Google Scholar]
- Klein J. Origin of major histocompatibility complex polymorphism: the trans-species hypothesis. Hum Immunol. 1987;19:155–162. doi: 10.1016/0198-8859(87)90066-8. [DOI] [PubMed] [Google Scholar]
- Klein J, O’huigin C, Figueroa F, et al. Different modes of Mhc evolution in primates. Mol Biol Evol. 1993;10:48–59. doi: 10.1093/oxfordjournals.molbev.a039999. [DOI] [PubMed] [Google Scholar]
- Kulski JK, Gaudieri S, Martin A, Dawkins RL. Coevolution of PERB11 (MIC) and HLA class I genes with HERV-16 and retroelements by extended genomic duplication. J Mol Evol. 1999;49:84–97. doi: 10.1007/PL00006537. [DOI] [PubMed] [Google Scholar]
- Kurtz J, Wegner KM, Kalbe M, Reusch TBH, Schaschl H, Hasselquist D, Milinski M. MHC genes and oxidative stress in sticklebacks: an immuno-ecological approach. Proc Biol Sci. 2006;273:1407–1414. doi: 10.1098/rspb.2005.3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafont BA, McGraw CM, Stukes SA, et al. The locus encoding an oligomorphic family of MHC-A alleles (Mane-A*06/Mamu-A*05) is present at high frequency in several macaque species. Immunogenetics. 2007;59:211–223. doi: 10.1007/s00251-007-0190-1. [DOI] [PubMed] [Google Scholar]
- Langefors A, Lohm J, Grahn M, Andersen O, Schantz T. Association between major histocompatibility complex class IIB alleles and resistance to Aeromonas salmonicida in Atlantic salmon. Proc R Soc B Biol Sci. 2001;268:479–485. doi: 10.1098/rspb.2000.1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccari G, Robinson J, Ballingall K, Guethlein LA, Grimholt U, Kaufman J, Ho CS, de Groot NG, Flicek P, Bontrop RE, Hammond JA, Marsh SGE. IPD-MHC 2.0: an improved inter-species database for the study of the major histocompatibility complex. Nucleic Acids Res. 2017;45:D860–D864. doi: 10.1093/nar/gkw1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markolf M (2013) Biodiversity of true lemurs (Eulemur spp.):-species delimitation and phylogeography in the brown lemur complex
- Markolf M, Rakotonirina H, Fichtel C, von Grumbkow P, Brameier M, Kappeler PM. True lemurs…true species - species delimitation using multiple data sources in the brown lemur complex. BMC Evol Biol. 2013;13:233. doi: 10.1186/1471-2148-13-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh SGE, Parham P, Barber LD. The HLA factsbook. San Diego: Academic Press; 1999. [Google Scholar]
- McConnell TJ, Talbot WS, McIndoe RA, Wakeland EK. The origin of MHC class II gene polymorphism within the genus Mus. Nature. 1988;332:651–654. doi: 10.1038/332651a0. [DOI] [PubMed] [Google Scholar]
- Mittermeier RA, Louis Jr EE, Richardson M et al (2010) Lemurs of Madagascar, 3rd edn, tropical field guide series. Conserv Int Arlington, VA
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nei M, Gu X, Sitnikova T. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc Natl Acad Sci. 1997;94:7799–7806. doi: 10.1073/pnas.94.15.7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nsubuga AM, Robbins MM, Roeder AD, Morin PA, Boesch C, Vigilant L. Factors affecting the amount of genomic DNA extracted from ape faeces and the identification of an improved sample storage method. Mol Ecol. 2004;13:2089–2094. doi: 10.1111/j.1365-294X.2004.02207.x. [DOI] [PubMed] [Google Scholar]
- Oliver MK, Piertney SB. Isolation and characterization of a MHC class II DRB locus in the European water vole (Arvicola terrestris) Immunogenetics. 2006;58:390–395. doi: 10.1007/s00251-006-0121-6. [DOI] [PubMed] [Google Scholar]
- Overdorff DJ. Ecological correlates to social structure in two lemur species in Madagascar. Am J Phys Anthr. 1996;40:327–342. doi: 10.1002/(SICI)1096-8644(199608)100:4<487::AID-AJPA4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Penn DJ, Damjanovich K, Potts WK. MHC heterozygosity confers a selective advantage against multiple-strain infections. Proc Natl Acad Sci. 2002;99:11260–11264. doi: 10.1073/pnas.162006499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piertney SB, Oliver MK. The evolutionary ecology of the major histocompatibility complex. Heredity. 2006;96:7–21. doi: 10.1038/sj.hdy.6800724. [DOI] [PubMed] [Google Scholar]
- Radwan J, Demiaszkiewicz AW, Kowalczyk R, Lachowicz J, Kawałko A, Wójcik JM, Pyziel AM, Babik W. An evaluation of two potential risk factors, MHC diversity and host density, for infection by an invasive nematode Ashworthius sidemi in endangered European bison (Bison bonasus) Biol Conserv. 2010;143:2049–2053. doi: 10.1016/j.biocon.2010.05.012. [DOI] [Google Scholar]
- Rammensee H-G, Friede T, Stevanović S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- Reuter KE, LaFleur M, Clarke TA. Endangered species: illegal lemur trade grows in Madagascar. Nature. 2017;541:157. doi: 10.1038/541157d. [DOI] [PubMed] [Google Scholar]
- Rioux JD, Goyette P, Vyse TJ, et al. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc Natl Acad Sci. 2009;106:18680–18685. doi: 10.1073/pnas.0909307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root-Bernstein R. Antigenic complementarity between HIV and other AIDS-associated infections results in idiotype–antiidiotype antibody complexes that cross react with lymphocyte proteins. Vaccine. 2005;23:2160–2163. doi: 10.1016/j.vaccine.2005.01.049. [DOI] [PubMed] [Google Scholar]
- Rosal-Sánchez M, Paz-Artal E, Moreno-Pelayo MA, et al. Polymorphism of Mhc-DRB alleles in Cercopithecus aethiops (green monkey): generation and functionality. HLA. 1998;51:541–548. doi: 10.1111/j.1399-0039.1998.tb02989.x. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Savage AE, Zamudio KR. MHC genotypes associate with resistance to a frog-killing fungus. Proc Natl Acad Sci. 2011;108:16705–16710. doi: 10.1073/pnas.1106893108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schad J, Sommer S, Ganzhorn JU. MHC variability of a small lemur in the littoral forest fragments of southeastern Madagascar. Conserv Genet. 2004;5:299–309. doi: 10.1023/B:COGE.0000031137.50239.d3. [DOI] [Google Scholar]
- Schad J, Ganzhorn JU, Sommer S. Parasite burden and constitution of major histocompatibility complex in the Malagasy mouse lemur, Microcebus smurinus. Evolution. 2005;59:439–450. doi: 10.1111/j.0014-3820.2005.tb01002.x. [DOI] [PubMed] [Google Scholar]
- Schrider DR, Hahn MW. Gene copy-number polymorphism in nature. Proc Biol Sci. 2010;277:3213–3221. doi: 10.1098/rspb.2010.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwensow N, Fietz J, Dausmann KH, Sommer S. Neutral versus adaptive genetic variation in parasite resistance: importance of major histocompatibility complex supertypes in a free-ranging primate. Heredity. 2007;99:265–277. doi: 10.1038/sj.hdy.6800993. [DOI] [PubMed] [Google Scholar]
- Schwitzer C, Mittermeier RA, Davies N et al (2013) Lemurs of Madagascar: a strategy for their conservation 2013–2016. Bristol, UK IUCN SSC Primate Specialist Group, Bristol Conservation and Science Foundation, and Conservation International 185
- Shiina T, Ota M, Shimizu S, et al. Rapid evolution of major histocompatibility complex class I genes in primates generates new disease alleles in humans via hitchhiking diversity. Genetics. 2006;173:1555–1570. doi: 10.1534/genetics.106.057034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddle HV, Kreiss A, Eldridge MDB, Noonan E, Clarke CJ, Pyecroft S, Woods GM, Belov K. Transmission of a fatal clonal tumor by biting occurs due to depleted MHC diversity in a threatened carnivorous marsupial. Proc Natl Acad Sci. 2007;104:16221–16226. doi: 10.1073/pnas.0704580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slierendregt BL, Otting N, van Besouw N, et al. Expansion and contraction of rhesus macaque DRB regions by duplication and deletion. J Immunol. 1994;152:2298–2307. [PubMed] [Google Scholar]
- Sommer S. The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool. 2005;2:16. doi: 10.1186/1742-9994-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer S, Schwab D, Ganzhorn JU. MHC diversity of endemic Malagasy rodents in relation to geographic range and social system. Behav Ecol Sociobiol. 2002;51:214–221. doi: 10.1007/s00265-001-0432-4. [DOI] [Google Scholar]
- Spurgin LG, Richardson DS (2010) How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proceeding of the Royal Society of London B Biological Sciences: 2009-2084 [DOI] [PMC free article] [PubMed]
- Summers K, McKeon SEA, Sellars JON, et al. Parasitic exploitation as an engine of diversity. Biol Rev. 2003;78:639–675. doi: 10.1017/S146479310300616X. [DOI] [PubMed] [Google Scholar]
- Takahata N, Nei M. Allelic genealogy under overdominant and frequency-dependent selection and polymorphism of major histocompatibility complex loci. Genetics. 1990;124:967–978. doi: 10.1093/genetics/124.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trtková K, Kupfermann H, Grahovac B, et al. Mhc-DRB genes of platyrrhine primates. Immunogenetics. 1993;38:210–222. doi: 10.1007/BF00211521. [DOI] [PubMed] [Google Scholar]
- Tung J, Barreiro LB, Burns MB, Grenier JC, Lynch J, Grieneisen LE, Altmann J, Alberts SC, Blekhman R, Archie EA. Social networks predict gut microbiome composition in wild baboons. Elife. 2015;2015:1–18. doi: 10.7554/eLife.05224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasser S, Houston C, Koehler G, et al. Techniques for application of faecal DNA methods to field studies of Ursids. Mol Ecol. 1997;6:1091–1097. doi: 10.1046/j.1365-294X.1997.00281.x. [DOI] [PubMed] [Google Scholar]
- Yoder AD. Lemurs. Curr Biol. 2007;17:R866–R868. doi: 10.1016/j.cub.2007.07.050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 18 kb)