

Abstract
Upon stimulation, small numbers of naive CD8+ T cells proliferate and differentiate into a variety of memory and effector cell types. CD8+ T cells can persist for years and kill tumour cells and virally infected cells. The functional and phenotypic changes that occur during CD8+ T cell differentiation are well characterized, but the epigenetic states that underlie these changes are incompletely understood. Here, we review the epigenetic processes that direct CD8+ T cell differentiation and function. We focus on epigenetic modification of DNA and associated histones at genes and their regulatory elements. We also describe structural changes in chromatin organization that affect gene expression. Finally, we examine the translational potential of epigenetic interventions to improve CD8+ T cell function in individuals with chronic infections and cancer.
The adaptive immune system exhibits remarkable phenotypic and functional plasticity during immune responses. Activation of naive CD8+ T cells triggers widespread alterations in cell cycle, metabolism and protein expression, resulting in the generation of cells with distinct cellular phenotypes. While this cellular plasticity is encoded in our DNA, cells themselves are genotypically identical. The ability of cells to use identical underlying genomes to generate diverse phenotypes is, in part, accounted for by epigenetics. It has become clear that epigenetic mechanisms, acting in conjunction with transcription factors, play a critical role in orchestrating the transcriptional changes associated with CD8+ T cell differentiation. Specifically, they allow signal transduction cascades acting through common transcription factors to drive cell type-specific transcriptional responses, and they provide a mechanism for the heritable maintenance of cell type-specific gene expression after inciting signals have dissipated. Understanding the epigenetic mechanisms regulating CD8+ T cell differentiation will have implications for both basic T cell biology and translational immunotherapy. In this Review, we summarize our current understanding of the epigenetics of CD8+ T cell differentiation, specifically exploring the influence of progressive changes in DNA methylation, histone modification and chromatin architecture on gene expression and lineage specification. We highlight technical advances that have facilitated this new understanding and examine the translational potential of therapies aimed at manipulating T cell epigenetic programmes.
CD8+ T cell differentiation states
A number of CD8+ T cell lineage relationship models have been proposed to account for the predominance of effector T cells during the acute phase of immune responses and memory T cells at later stages after an antigenic challenge. According to the On–Off–On, or circular, differentiation model1, naive T cells differentiate into effector T cells upon antigen encounter. Upon pathogen clearance, effector T cells either undergo apoptosis or differentiate into memory T cells2. Thus, according to this model, a proportion of T cells differentiates from naive cells to effector cells and finally to memory cells, where they await secondary antigen encounter before beginning the cycle again. The circular nature of this model would result in an on–off–on or off–on–off pattern of transcriptional and epigenetic changes over time1 and would require cycles of dedifferentiation and redifferentiation3,4 (FIG. 1a), a process not known to occur in adult somatic tissues5. Conversely, according to the developmental, or linear, differentiation model6 (FIG. 1b), the strength and duration of antigenic and inflammatory signals are key determinants of T cell differentiation, with strong or repetitive signals progressively driving the acquisition of effector characteristics and terminal effector differentiation7,8. By contrast, weak signals fail to drive full effector differentiation and, instead, result in the differentiation of memory cells6,8–10. Thus, although there is a predominance of effector cells during early stages of immune responses, these cells represent the final stage of T cell differentiation and die upon antigen withdrawal. Left behind is the comparatively smaller population of memory T cells that failed to fully differentiate into effector T cells but that persist to establish long-lived immunological memory. The linear model, therefore, places memory T cells as an intermediate step within CD8+ T cell differentiation. This reflects the transcriptional profiles of CD8+ T cell subsets, as memory T cells harbour transcriptional, phenotypic and epigenetic similarities with both effector and naive T cells10–15. Consequently, the linear model would result in gene expression and epigenetic patterns that change in a less cyclical manner (for example, on–off or off–on), instead resulting in gradual alterations to the epigenetic landscape as cells progress towards a terminally differentiated state, as seen in other developmental systems6.
Figure 1 |. Different CD8 + T cell differentiation models result in unique transcriptional and epigenetic patterns over time.

a | In the On–Off–On, or circular, model of CD8+ T cell differentiation, effector T (TEFF) cells represent biological intermediaries that either undergo apoptosis or differentiate into memory T cell subsets following antigen withdrawal. This sets up a recurring cycle of T cell differentiation (Naive→TEFF→TSCM→TCM→TEM→TEFF) that would result in an oscillating — on–off–on or off–on–off — pattern of transcriptional and epigenetic changes over time. b | In the developmental, or linear, differentiation model, the progressive acquisition of effector function during CD8+ T cell differentiation (Naive→TEFF→TSCM→TCM→TEM→TEFF) depends on the strength and duration of antigenic signalling and results in the gradual loss of memory-associated gene expression and gain of effector-associated gene expression. These transcriptional changes are accompanied by similar changes in the epigenetic landscape, which are illustrated by the gradual, or progressive, gain or loss of activating and repressive histone modifications. TCM, central memory T; TEM, effector memory T; TSCM, stem cell memory T.
In addition to terminal differentiation, strong and/or chronic antigen stimulation can also drive T cell exhaustion16,17, as can self-antigens in the absence of co-stimulatory signals18. However, in contrast to terminally differentiated effector cells, exhausted T cells have a hypofunctional phenotype associated with decreased antigen-driven induction of effector cytokines18,19 and elevated expression of inhibitory cell surface receptors, including programmed cell death protein 1 (PD1)20, which is also a marker of T cell activation21. Although the exhausted state may have evolved to protect against detrimental immunogenicity and autoimmunity, cancer cells exploit this adaptive trait to promote immunosuppression22–24. T cell exhaustion, therefore, represents a major roadblock to endogenous immune-mediated clearance of cancer cells and the burgeoning field of immunotherapy. Clarifications of where exhausted T cells reside within the canonical CD8+ T cell differentiation programme, as well as the epigenetic changes underlying their establishment and maintenance, will be crucial for future therapeutic endeavours.
Epigenetic changes during differentiation
In both the circular and linear differentiation models, antigenic signalling plays an important role in driving the transcriptional changes that underlie acquisition of effector cell characteristics10,12. As we discuss, epigenetic changes occurring during this time provide a means for T cells to both initiate the signal-driven transcriptional changes that drive differentiation and maintain these expression patterns following signal withdrawal. Our understanding of epigenetic changes, as described in this Review, is mostly derived from investigations of pooled cell populations. Therefore, although the presence of an epigenetic modification is a binary phenomenon at the level of an individual allele, signals obtained from these studies reflect changes in the frequency of a particular epigenetic modification across a pool of alleles for a given locus within investigated cell populations.
Epigenetic modifications at genic loci regulate current and future transcription.
DNA methylation (FIG. 2a) and histone post-translational modifications (FIG. 2b) represent two extensively studied epigenetic mechanisms. Most analyses of DNA methylation focus predominantly on CG dinucleotide (CpG)-dense regions, termed CpG islands (CGIs), which are located primarily at transcriptional start sites (TSSs). Generally, DNA methylation is associated with transcriptional repression, although it is associated with transcriptional activation when found within gene bodies25,26. These trends hold true during CD8+ T cell differentiation, where genome-wide DNA methylation profiles indicate that methylation marks are lost at the promoters of genes whose expression increases during differentiation and are gained at the promoters of genes whose expression decreases27–30. Importantly, although studies of DNA methylation are often focused on promoters, methylation occurs throughout the genome to affect gene expression, including at neighbouring CGI shores and shelves, intergenic non-coding regions and gene bodies26. Therefore, when investi gating DNA methy lation patterns in a cellular system, it is important to expand beyond traditional promoter and/or TSS-biased analyses. Indeed, DNA methylation patterns at distal enhancer elements and CGI shores exhibit dynamic lineage-specific changes during cellular differentiation that negatively correlate with both gene expression and activating histone modifications31–34, and the presence of the intermediary base 5-hydroxy methylcytosine (5hmC) is found at both genic and intergenic regions of the genome, where it correlates with transcriptional activation35.
Figure 2 |. Features of DNA methylation and histone modifications.

a | The DNA methylation cycle of cytosine nucleotide bases is depicted. DNA (cyto-sine-5)-methyltransferase 3A (DNMT3A) and DNMT3B add methylation (m) modifications to unmodified cytosines in a de novo manner, whereas DNMT1 acts to maintain established patterns during DNA replication. Passive demethylation results from a lack of maintenance methylation and is replication dependent, whereas active demethylation is directed by Ten-eleven translocation (TET) proteins and can be either replication dependent or independent. TET proteins mediate the serial oxidation of methylated cytosines (red bases), resulting in multiple intermediary bases, including 5-hydroxymethylcytosine (green bases). Eventually, modified bases are returned to an unmodified state via the base excision repair (BER) pathway. b | Nucleosomes consist of two copies each of the histone proteins H2A, H2B, H3 and H4 encircled by DNA. The amino-terminal tails of histone proteins protrude from the nucleosome structure and can be post-translationally modified. The amino acid sequence of the histone H3 N-terminal tail is shown160 along with the position of select lysines (K) that are subject to methylation (me) and/or acetylation (ac); however, this is not an exhaustive list of all H3 post-translational modifications. c | Chromatin can be broadly categorized as either euchromatin or heterochromatin on the basis of its accessibility level. Euchromatin is characterized by an open chromatin conformation that is less compact and more accessible by regulatory proteins (yellow oval). Heterochromatin is categorized as either facultative or constitutive. Both are more compact and inaccessible relative to euchromatin; however, constitutive heterochromatin is generally more compacted and is associated with gene-poor repetitive regions that largely remain in a closed state. Facultative heterochromatin, by contrast, is associated with regions that often transition to an open conformation as transcriptional requirements change. Each chromatin state has specific histone post-translational modifications that are associated with it, as depicted.
Histone modification profiles in CD8+ T cell subsets, obtained using chromatin immunoprecipitation followed by sequencing (ChIP–seq), indicate that, as with DNA methylation, promoters and gene bodies undergo progressive changes in the distribution and accumulation of histone modifications during differentiation that correlate with gene expression patterns15,28,36–44. Within the nucleus, DNA is organized into structural units termed nucleosomes that consist of eight histone subunits (two copies each of H2A, H2B, H3 and H4) that are subject to covalent post-translational modification of the amino acid residues of their amino-terminal tails (FIG. 2b). Histone modifications are found at intergenic and genic regions of the genome and have various effects on transcription (TABLE 1). Generally speaking, histone acetylation has an activating effect on transcription, whereas histone methylation can have either activating or repressive effects45,46. Multiple studies in mice and humans investigating epigenetic patterns during the differentiation of naive CD8+ T cells into memory and effector T cells found that activation-associated modifications, such as H3K4me3 and H3K9ac, are lost and that repressive DNA methylation and H3K27me3 modifications are gained at gene loci whose expression is reduced in effector cells. This includes memory-cell-associated transcription factors such as FOXO1, KLF2, LEF1 and TCF7, as well as genes that are highly expressed in memory cell subsets, including IL2RA, CD27, TNF, CCR7 and SELL. Alternatively, effector-cell-associated transcription factors (EOMES, TBX21 and PRDM1) and functional effector genes (GZMA, GZMB, PRF1, IFNG and KLRG1) demonstrate decreased repressive and increased activating epigenetic modifications at these loci in effector cells15,27–30,36–44. The initiation of these epigenetic changes in CD8+ T cells following antigenic recognition is illustrative of one of the functions of epigenetics, which is to facilitate transcriptional changes in response to external stimuli. Additionally, epigenetic marks serve to maintain these transcriptional profiles following stimulus withdrawal. This is illustrated in work from Abdelsamed and colleagues, who observed the previously mentioned progressive changes in DNA methylation as cells differentiate from naive cells into memory subsets: stem cell memory T (TSCM) cells, central memory T (TCM) cells and effector memory T (TEM) cells29 EM. Importantly, when sorted memory cell subsets are grown ex vivo in an antigen-free environ ment, subset-specific methylation patterns are stable at effector-associated loci (such as IFNG and PRF1), which remain poised for expression following secondary antigen encounter. However, selected memory-associated loci (such as CCR7 and SELL) appear to gain methy lation during short-term ex vivo culture, which is likely to be indicative of homeostatic memory cell differentiation observed in this study29 and others47. In this way, epigenetic regulation is flexible, allowing for memory cell differentiation driven by homeostatic cytokines while maintaining a poised state at genes required for rapid initiation of effector function.
Table 1 |.
Histone post-translational modifications and associated regulatory proteins
| Modification | Primary location | Writers | Erasers | Readers |
|---|---|---|---|---|
| Activating histone modifications | ||||
| H3K4me1 | Enhancers and gene body | SETD1A, SETD1B, KMT2A, KMT2B, KMT2CandKMT2D161 | KDM1A and KDMlB162 | KAT5 (REF. 163) |
| H3K4me3 | Promoter and TSS | SETD1A, SETD1B, KMT2A, KMT2B, KMT2C and KMT2D161 | KDM2B, KDM5A, KDM5B, KDM5C and KDM5D162 | NURF (BPTF)164 |
| H3K36me3 | Gene body | SETD2 and NSD1 (REF. 162) | KDM4A, KDM4B and KDM4C162 | PSIP1 (REF. 165) and MORF4L1 (REF 64) |
| H3K9ac | Promoter and TSS | KAT2A and KAT2B166 | HDACs167, SIRT1 and SIRT6 (REF 168) | BET family (BRD4)169 |
| H3K27ac | Enhancers | p300 and CREBBP166 | HDACs167 | BET family (BRD4)169 |
| Repressive histone modifications | ||||
| H3K9me2 | Constitutive and facultative heterochromatin | EHMT1 and EHMT2170 | KDM1A, KDM1B, KDM3A, KDM3B, JMJD1C, KDM4A, KDM4B, KDM4C, KDM4D, KDM7A,JHDM1E and JHDMlF162 | CBX5 (REF. 171) |
| H3K9me3 | Constitutive heterochromatin | SUV39H1, SUV39H2 (REFS 172,173) and SETDB1 (REF. 58) | KDM4A, KDM4B, KDM4C and KDM4D162 | CBX5 (REF. 171) |
| H3K27me3 | Facultative heterochromatin | PRC2 (EZH2)63 | KDM6B and KDM6A162 | PRC1 (CBX proteins, BMI1) and PRC2 (EED)63 |
BET, bromodomain and extra-terminal domain; BMI1, Polycomb complex protein BMI1; BPTF, nucleosome-remodelling factor subunit BPTF; BRD4, bromodomain-containing protein 4; CBX, chromobox protein homologue; CREBBP, CREB-binding protein; EED, embryonic ectoderm development protein; EHMT1, histone-lysine N-methyltransferase EHMT1; EZH2, histone-lysine N-methyltransferase EZH2; HDACs, histone deacetylases; JHDM1, Jumonji domain-containing histone demethylation protein 1; JMJD1C, Jumonji domain-containing protein 1C; KAT, histone acetyltransferase; KDM, lysine-specific demethylase; KMT2A, histone-lysine N-methyltransferase 2A; MORF4L1, mortality factor 4-like protein 1; NURF, nucleosome-remodelling factor; p300, histone acetyltransferase p300; PRC, Polycomb repressive complex; PSIP1, PC4 and SF2-interacting protein 1; SETD1A, histone-lysine N-methyltransferase SETD1A; SETDB1, histone-lysine N-methyltransferase SETDB1; SIRT, sirtuin; SUV39H, histone-lysine N-methyltransferase SUV39H; TSS, transcriptional start site.
A transcriptionally poised state can also be seen in the bivalent chromatin signature observed in T cells. Although a single histone modification is associated with a particular transcriptional state, these modifications also act in combination to control gene expression. For instance, H3K27ac and H3K4me1 are often found together at active enhancer regions, whereas the presence of only H3K4me1 is often found at enhancers associated with transcriptionally poised genes46. In addition, the presence of activating H3K4me3 and repressive H3K27me3 marks at gene promoters is referred to as a bivalent chromatin signature. Bivalency is believed to mark genes as transcriptionally poised, and resolution to a monovalent state is often observed during development48–50. Early in T cell development, the CD8A locus is bivalently marked in CD4–CD8α– (double-negative) thymocytes, in which it is transcriptionally silent. In CD4+CD8α+ (double-positive) cells, the locus resolves to a monovalent H3K4me3 state and cells express CD8α. Upon further differentiation into CD8α single-positive cells, H3K4me3 is maintained, whereas H3K27me3 reappears at the CD8A locus in CD4 single-positive cells, in which transcription is repressed51. Naive T cells, TCM cells and TEM cells also exhibit bivalent signatures specifically at genes whose expression is induced following T cell activation, including BMI1, TBX21, EOMES and IRF4. In these studies, most bivalent genes resolve to a monovalent H3K4me3 state and increase transcriptional activity37,38. This suggests that bivalency in mature T cells is used in multipotent cells predominantly to epi-genetically mark transcriptionally silenced genes that are destined for future activation following an encounter with the appropriate initiating signals.
Epigenetic modifications at enhancers enable cell type-specific transcription.
In contrast to the progressive changes in epigenetic modifications observed over time at promoters and gene bodies in naive, memory and effector CD8+ T cells, histone marks delineating the enhancer repertoires of these subsets are strikingly distinct. Enhancers function together with promoters to regulate gene expression and, importantly, although a single gene isoform is regulated by one promoter, it can be regulated by multiple enhancers. Genome-wide ana lyses in multiple cell types indicate that lineage-specific enhancer activity corresponds to lineage-specific gene expression15,52–57. Therefore, the establishment of distinct sets of enhancers in different cell types enables cell type-specific transcriptional responses to be initiated by similar signal transduction and transcription factor pathways. Specific histone modifications and histone modifying proteins are associated with active enhancers, including H3K4me1, H3K27ac and the histone acetyltransferase p300, and genome-wide profiles of these markers enable annotation of putative enhancers and super enhancers54,57. This approach has been used to map putative regulatory elements across CD8+ T cell subsets, identifying over 24,000 enhancers and 1,200 super enhancers15. In contrast to progressive changes in the distribution and accumulation of epigenetic modifications observed at genic regions, striking specificity of enhancer repertoires in naive, memory and effector CD8+ T cells is observed, with 77% of enhancers and 62% of super enhancers corresponding to a single subset. Motif analysis of subset-specific enhancers identifies an enrichment of subset-specific transcription factor binding sites, including T-bet (also known as TBX21) and eomeso dermin homologue (EOMES) enrichment at effector-specific enhancers, forkhead box protein O1 (FOXO1) and transcription factor 1 (TCF1; also known as HNF1A) enrichment at memory-specific enhancers and TCF1 enrichment at naive-specific enhancers. Active enhancers are also positively associated with the expression of bioinformatically predicted target genes15. These data support the involvement of lineage-specific enhancer repertoires in directing lineage-specific transcriptional responses during CD8+ T cell differentiation.
Functional consequences of epigenetic modifications.
Epigenetic modifications influence transcriptional activity via direct and indirect mechanisms. DNA methy lation, for instance, can influence gene expression directly by affecting transcription factor binding affinity26 and indirectly by engaging methyl-CpG-binding proteins, which can recruit repressive histone modifying enzymes58–60. Among CD8+ T cells, demethylated regions identified in effector cells are enriched for binding sites of effector-associated transcription factor families27, which is suggestive of direct regulatory effects of DNA methylation. In addition, knockout of the gene encoding methyl-CpG-binding domain protein 2 (MBD2) results in differentiation defects in CD8+ T cells61, suggesting the existence of indirect gene regulation by DNA methylation mediated via MBD2. However, further characterization of the proteins mediating the transcriptional effects of DNA methylation in CD8+ T cells is needed.
Histone modifications can also affect gene transcription via direct or indirect methods, often by initiating changes to chromatin compaction. Histone acetylation reduces DNA–nucleosome binding affinity, as well as internucleosome interactions, thereby directly contributing to chromatin decompaction. Alternatively, histone methylation and acetylation modifications are recognized by specific epigenetic modifying proteins termed ‘readers’. Reader proteins can influence gene expression in a variety of ways, including by interacting with the transcriptional machinery, facilitating mRNA processing and recruiting chromatin-remodelling proteins; recruitment affects chromatin compaction62–64. Chromatin decompaction results in an open chromatin state, also referred to as euchromatin (FIG. 2c), where the DNA of regulatory regions and genes is more accessible for binding by regulatory factors and the transcriptional machinery, facilitating gene expression. Conversely, compacted and closed chromatin, also called hetero chromatin, is less accessible and is associated with regions of the genome that are transcriptionally silenced46,62 (FIG. 2c). Techniques such as the assay for transposase-accessible chromatin-sequencing65 (ATAC–seq) (BOX 1) have enabled genome-wide measurement of open, accessible chromatin in low numbers of cells, enabling the identification of potential enhancers and regions of active transcription. Global changes in chromatin accessibility during CD8+ T cell differentiation, as measured by ATAC–seq, identify numerous differentially accessible regions between naive, memory and effector T cells in mice. Most of these regions are observed at intergenic loci and positively correlate with the expression of neighbouring genes, suggesting that they contain enhancer elements and further highlighting the importance of subset-specific epigenetic patterns14,66,67. ATAC–seq profiles from human CD8+ naive T cells, TCM cells and TEM cells similarly indicate differentially accessible regions at putative enhancers corresponding to differential expression of subset-specific genes. Motif analysis at differentially accessible regions in TEM cells identifies enrichment of transcription factors that are known to be involved in effector cell differentiation, including basic leucine zipper transcriptional factor ATF-like (BATF), EOMES, activator protein 1 (AP-1) and T-bet68. Altogether, both progressive and subset-specific epigenetic patterns at gene loci and active enhancers, respectively, are responsible for regulating the transcriptional changes observed during CD8+ T cell differentiation.
Box 1 |. Tools for unravelling the epigenome.
DNA methylation and histone modifications
Bisulfite conversion of genomic DNA can be used in conjunction with whole-genome sequencing or increasingly comprehensive arrays to identify DNA methylation patterns. Importantly, conventional bisulfite techniques do not distinguish between 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC), although additional modifications can be used to profile 5hmC patterns151. Chromatin immunoprecipitation followed by sequencing (ChIP–seq) is commonly used to profile histone modifications, and ChIP–seq-based techniques can also probe for 5mC and 5hmC151.
Chromatin accessibility
DNase–seq152 and formaldehyde-assisted isolation of regulatory elements (FAIRE)–seq153 enable the identification of open chromatin regions, although these techniques require large starting cell populations (≥10 million cells). Alternatively, assay for transposase-accessible chromatin-sequencing (ATAC–seq)65 enables accessibility profiling with ≤50,000 cells, allowing for the investigation of rare or precious samples. While supplying a wealth of information on cellular chromatin state, accessibility assays fail to definitively ascribe functionality and must be combined with other assays to functionally annotate open regions. Often, proximity-based analyses are used to assign target genes to active regulatory regions; however, this may misassign enhancers that act over long distances and ignore complex interactions.
3D chromatin interactions
Hi-C, a chromosome conformation capture (3C)-based technique, is capable of identifying physical interactions between loci in a genome-wide manner at very high resolution93,97. This type of investigation allows an unbiased look at chromatin interactions across the genome. However, alterations in the technique, including promoter capture Hi-C154 and the ChIP-based chromatin interaction analysis with paired-end tag (ChIA-PET) sequencing155, enable targeted genome-wide investigations.
Altogether, multiple genome-wide tools exist for comprehensive profiling of the epigenome in T cells. Systematic profiling of the epigenome in CD8+ T cell differentiation states, when combined with transcriptome data, would provide a powerful resource for understanding T cell differentiation. Furthermore, ongoing technical advances, particularly in single-cell profiling, will no doubt facilitate exciting progress in the field.
Epigenetic regulators of differentiation
The placement, turnover and activity of DNA and histone modifications are dependent on epigenetic modifying proteins (TABLE 1). Specifically, ‘writers’ and ‘erasers’ are responsible for adding and removing epigenetic modifications, respectively, whereas readers contain protein domains that specifically recognize certain epigenetic modifications45,46,62. For instance, readers containing bromodomains, such as bromodomain-containing protein 4 (BRD4), typically recognize acetylated lysines, whereas chromodomain-containing proteins, including chromobox protein homologue 5 (CBX5), recognize trimethylated lysines62. As we discuss, the directional activities and asymmetric expression of epigenetic modifying proteins throughout CD8+ T cell differentiation regulate subset-specific cellular functions and may even contribute to fate decisions at the earliest stages of naive T cell activation (BOX 2). The development of pharmacological and genetic approaches to target the activity of epigenetic modifying enzymes, therefore, provides the potential for manipulation of immune cell function and differentiation.
Box 2 |. Asymmetric differentiation and the role of epigenetics.
Single-cell RNA sequencing analysis in mice has identified heterogeneity in Ezh2 (which encodes histone-lysine N-methyltransferase EZH2) expression arising as early as the first cell division following stimulation of naive T cells, in which it appears to silence memory-associated gene expression in daughter cells hypothesized to develop into effector T cells77. This raises the compelling possibility that epigenetic modifying proteins act in an asymmetric manner following naive CD8+ T cell activation to mark cells destined for specific cell fates. Ezh2 expression is low in naive T cells and increases in only a subset of daughter cells following the first division; therefore, another factor is probably responsible for Ezh2 induction in these cells. This could be achieved either by asymmetric cell division (see the figure, left panel) or population asymmetry (see the figure, right panel)156. The former involves the asymmetric apportionment of a regulatory factor (or factors) during cytokinesis, in this instance, resulting in the activation of Ezh2 transcription and EZH2-dependent silencing of memory-associated genes in one daughter cell, predisposing it to an effector cell fate. The other daughter cell would not receive these regulatory signals and would become predisposed to a memory cell fate. Instances of asymmetric apportionment of cellular factors have been observed during CD8+ T cell division157–159. Alternatively, with population asymmetry, differences in Ezh2 transcription may be initiated by stochastic extrinsic and/or intrinsic factors (for example, weak versus strong antigenic signalling) encountered before cell division that lead to uniform fates for both daughter cells dependent on the extrinsic and/or intrinsic factors encountered by the parental cell. In this way, divergent cell fates are achieved at the population level. Importantly, population-based analyses do not discern between these two models, confounding the true nature of asymmetric mechanisms contributing to CD8+ T cell differentiation. Lineage-tracking studies performed at the single-cell level will be needed to dissect these related mechanisms, as well as to investigate the involvement of epigenetic regulation.
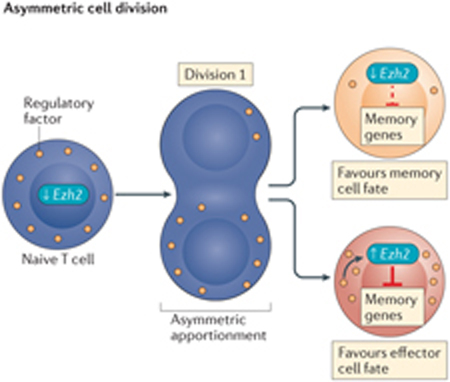
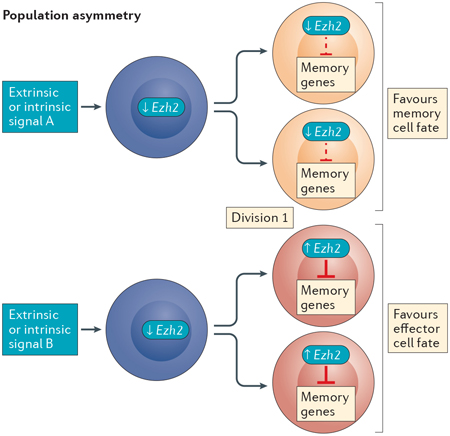
Epigenetic modifying proteins play unique roles at multiple stages of differentiation.
The effects on cellular activity of histone-modifying enzymes are highly dependent on the cellular environment. As a result, modifying enzymes can have differential activity depending on the differentiation state of the cell. This is especially apparent in the activity of Polycomb repressive complex 1 (PRC1) and PRC2. The expression of Polycomb complex protein BMI1, a component of the H3K27me3 reader complex PRC1, is induced by T cell receptor (TCR) stimulation in both naive CD8+ T cells and antigen-experienced memory precursor T cells (killer cell lectin-like subfamily G member 1 (KLRG1)–CD44hi). However, this induction of BMI1 expression is lost in terminally differentiated effector T cells (KLRG1+CD44hi)63,69, a subset associated with replicative senescence and terminal differentiation. In mouse embryonic fibroblasts, BMI1 represses expression of the Cdkn2a locus, the gene products (p16INK4A and p14ARF) of which promote cellular senescence and apoptosis70,71. Consistent with this, BMI1 expression in KLRG1–CD8+ T cells is associated with reduced p16INK4A and p14ARF expression compared with KLRG1+ cells, and reduced BMI1 expression results in defects in T cell population expansion69. These data support subset-specific activities of BMI1, wherein memory-like KLRG1− T cell subsets utilize BMI1-mediated repression to maintain proliferative capacity whereas, as cells become terminally differentiated, this activity is lost (FIG. 3a).
Figure 3 |. Mechanisms of epigenetic-mediated control of CD8 + T cell differentiation.

a | Repressive histone-modifying enzymes Polycomb complex protein BMI1 and histone-lysine N-methyltransferase EZH2, as part of Polycomb repressive complex 1 (PRC1) and PRC2, respectively, contribute to the functional phenotypes of memory-like killer cell lectin-like subfamily G member 1 (KLRG1)–CD8+ T cells and terminally differentiated KLRG1+CD8+ T cells via subset-specific activity. Differential targeting of Notch repressors Numb and Fbxw7 and cell cycle repressors, including Cdkn2a, results in activation or repression of Notch and retinoblastoma-associated protein 1 (RB1)–p53 pathways, ultimately affecting polyfunctionality and apoptosis. Similarly, differential repression of memory-associated loci affects the memory-associated transcriptional programme. In memory-like subsets, active transcription facilitated by forkhead box protein O1 (FOXO1) binding may inhibit EZH2-mediated repression. b | The left panel illustrates a bromodomain-containing protein 4 (BRD4)-dependent regulatory cascade that is critical for normal effector cell differentiation. The recognition of lysine acetylation marks (yellow pentagon) by the histone reader protein BRD4 increases expression of the transcription factor Batf (which encodes basic leucine zipper transcriptional factor ATF-like (BATF)). BATF, together with activator protein 1 (AP-1), transcriptionally represses the histone deacetylase gene Sirt1 (which encodes protein deacetylase sirtuin 1 (SIRT1)). Decreased activity of SIRT1 increases acetylation at the Tbx21 (which encodes T-bet) locus, resulting in increased expression of Tbx21 and T-bet target genes. The addition of the bromodomain inhibitor JQ1 upends this pathway (right panel), with the lack of Sirt1 repression contributing to decreased histone acetylation and the reduced expression of Tbx21 and T-bet targets. Ultimately, JQ1 inhibits effector differentiation, which results in increased stem cell memory T (TSCM) and central memory T (TCM) cells. Ac, acetylation; TF, transcription factor.
Similar subset-specific activities are observed for histone-lysine N-methyltransferase EZH2, which is part of the H3K27me3 writer complex PRC2 (FIG. 3a). Phenotypic characterization of EZH2+CD8+ T cells in peripheral blood by surface marker expression identifies them as predominantly effector memory cells. These cells do not express KLRG1 or other markers of senescence and have increased polyfunctionality and resistance to spontaneous and induced apoptosis72. Similar to BMI1, EZH2 expression is induced upon TCR stimulation, and genetic or pharmacological inhibition of its activity inhibits cytokine production, increases apoptosis and reduces TCR-dependent T cell population expansion72,73. These phenotypes are attributed to EZH2-dependent repression of the Notch signalling repressors NUMB and FBXW7, whose loci display reduced H3K27me3 following EZH2 knockdown. Notch inhibition phenocopies the polyfunctional and apoptotic defects seen with EZH2 inhibition, supporting EZH2-directed regulation of Notch signalling as an important regulatory pathway in TEM cells72.
In terminally differentiated effector T cells (KLRG1hiinterleukin-7 receptor (IL-7R)low), memory-associated loci have increased H3K27me3 marks concomitant with transcriptional silencing compared with TEM cells (KLRG1lowIL-7Rhi)74. Interestingly, effector-associated loci do not display increased H3K27me3 marks in TEM cells despite low gene expression, suggesting that H3K27me3-dependent repression is specific to effector differentiation. Indeed, conditional knockout of Ezh2 in effector cells reduces clonal expansion and generates T cells with memory-like characteristics, including increased expression of TCF1, CD27, L-selectin and FOXO1, as well as decreased KLRG1 expression. The development of memory T cells is unaffected; however, recall responses to a secondary infection are impaired, reinforcing the importance of EZH2 activity during effector differentiation74. These studies demonstrate similar subset-dependent activity of BMI1 and EZH2 during the differentiation of CD8+ effector T cells. In effector memory and memory precursor cells that lack expression of KLRG1, BMI1 and EZH2 act to maintain proliferative capacity and protect against apoptosis in response to TCR stimulation69,72,73. In KLRG1+ terminally differentiated effector cells, BMI1 induction is reduced, and EZH2 silences memory-associated loci, enabling effector differentiation (FIG. 3a). Deciphering the specific mechanisms regulating such switches in function remains an active area of research. Memory-associated genes subject to H3K27me3 modification in effector cells show increased levels of FOXO1 binding in KLRG1− memory cells74. This suggests that FOXO1 protects against EZH2-dependent repression and that this protection is absent in effector cells that express less FOXO1 (FIG. 3a). Additionally, EZH2 is regulated post-transcriptionally by microRNAs72 and post-translationally by AKT-mediated phosphorylation75, representing multiple mechanisms by which T cell subsets can regulate EZH2 activity.
Indirect and direct control of epigenetic information by transcription factors.
Complex interactions exist between epigenetic modifying proteins and lineage-specific transcription factors that establish and maintain the lineage-specific histone modifications and DNA methylation profiles that regulate T cell differentiation and function. Genetic disruption of the effector-associated transcription factor Prdm1 (which encodes PR domain zinc finger protein 1 (PRDM1)) in CD8+ T cells results in increased numbers of memory cells, as well as reduced apoptosis and increased proliferative responses to cytokines, including IL-2 (REF. 43). PRDM1 recruits repressive histone modifiers histone-lysine N-methyltransferase EHMT2 and histone deacetylase 2 (HDAC2) to the Il2ra and Cd27 loci, the expression of which is associated with memory T cells43. Prdm1 deletion decreases repressive histone marks and increases activating histone marks at these loci, resulting in increased expression following viral infection43. These data are in keeping with a model in which PRDM1 promotes effector cell differentiation by facilitating epi-genetic repression of memory-specific genes in a manner similar to EZH2 and other repressive epigenetic modifiers5,76,77,78. The link between transcription factors and epigenetics was extended by a study showing that TCF1 and lymphoid enhancer-binding factor 1 (LEF1) have intrinsic histone deacetylase activity important for lineage specification during thymocyte development79. Further work is needed to determine what role, if any, this activity may have during mature CD8+ T cell activation, particularly during memory cell differentiation, when TCF1 and LEF1 expression are highest.
Pharmacological disruption of epigenetic modifiers to affect differentiation.
The ability to manipulate T cell function through exogenous interventions has important therapeutic implications. Targeting epigenetic modifying enzymes represents an intriguing possibility, as their regulatory actions, although substantial, are inherently reversible. Effective manipulation of T cell regulatory mechanisms will necessitate a thorough understanding of the interactions between epi-genetic modifiers and regulatory transcription factors. Canonical effector cell differentiation depends on the activity of multiple effector-associated transcription factors, including BATF and T-bet42 (FIG. 3b), whereas BACH2 restrains AP-1-driven effector cell programmes to promote memory cell differentiation80. Also involved in regulating effector cell differentiation are the epi-genetic modifying proteins BRD4 (REF. 81), a reader of acetylated lysines, and the histone deacetylase sirtuin 1 (SIRT1)42. These proteins represent attractive, druggable epigenetic targets for manipulating effector cell differentiation. JQ1 is a pharmacological inhibitor of the BET family of bromodomain-containing proteins, including BRD4. Mechanistically, treatment of mouse T cells with JQ1 reduces BATF expression, which increases the transcriptionally repressive activity of SIRT1 at the Tbx21 locus42,81 (FIG. 3b). Phenotypically, JQ1 treatment of naive CD8+ T cells upon stimulation in vitro results in increased TSCM and TCM cell populations and increased polyfunctionality and in vivo, JQ1-treated cells demonstrate greater cell persistence, proliferation and cytokine production81. These data demonstrate the power of pharmacological targeting of epigenetic modifiers to impact CD8+ T cell differentiation and function.
Targeting DNA methylation impacts T cell function.
As previously discussed, DNA methylation is correlated with gene expression changes during CD8+ T cell differentiation. The addition, maintenance and removal of methylation marks is achieved by replication-dependent and replication-independent processes, which are facilitated by multiple DNA (cytosine-5)-methyltransferases (DNMTs) and hydroxylases (FIG. 2a). DNMT3A and DNMT3B establish de novo methylation patterns, whereas DNMT1 is responsible for the maintenance of methylation during DNA replication. Lack of DNMT1 activity results in the loss of methylation marks via replication-dependent passive demethylation26. There is convincing evidence supporting lineage-specific roles for DNA methyltransferases in the development and function of T cells. DNMT1-deficient thymocytes exhibit reduced viability82, and disruption of Dnmt3a in mature CD8+ T cells results in increased memory cell differentiation and reduced terminal effector differentiation76. Differentially hypomethylated regions in Dnmt3a-knockout T cells are observed at genes that are generally downregulated during effector cell differentiation, including the memory cell-specific transcription factor Tcf7 (REF. 76). Thus, a role of DNMT3A during T cell differentiation appears to be the establishment of inhibitory methylation patterns at memory-specific loci.
Active demethylation provides a targeted way for cells to control DNA demethylation and allows for additional layers of regulation via the intermediary bases produced in the process. Active DNA demethylation is achieved through enzymatic oxidation of methylated cytosines to intermediary bases, including 5hmC, by the Ten-eleven translocation proteins (methylcytosine dioxygenases TET1, TET2 and TET3). This can result in either replication-dependent demethylation, owing to reduced DNMT1 activity towards hemi-5hmC, or replication-independent demethylation, where intermediary bases are further modified by TET proteins and eventually removed by the base excision repair pathway35,83 (FIG. 2a). Mice lacking TET proteins display differentiation and functional defects in various developmental systems35, demonstrating the importance of the active demethylation pathway in development. Evidence suggests that active demethylation plays a role in T cell differentiation, as 5hmC is enriched at active enhancers and genes during thymocyte differentiation84 and conditional deletion of Tet2 in T cells impacts CD8+ T cell differentiation following viral infection85. This suggests that targeting the DNA methylation cycle may be an attractive avenue for manipulating T cell function; however, current DNMT inhibitors have substantial toxicity and nonspecific inhibitory activity86, which diminishes their therapeutic effectiveness. A greater understanding of the activity of DNMTs and TET proteins in T cells, together with improved pharmacological options, may improve the therapeutic viability of targeting the DNA methylation cycle.
Higher-order chromatin organization
DNA packaging within the nucleus represents a monumental level of compaction and organization. At the most basic level, chromatin can be organized into open or closed regions on the basis of chromatin accessibility. ATAC–seq studies have identified unique chromatin accessibility patterns within CD8+ T cell subsets14,68. Pairwise analysis of differentially accessible regions between distinct subsets in mice indicates that naive and effector T cells have the most dissimilar accessibility profiles14, consistent with similar differences in global gene expression10. ATAC–seq profiles from human CD8+ T cell subsets similarly show that naive and TEM cells have the most dissimilar profiles and that TCM and TEM cell populations are most similar68. These data suggest that chromatin organization patterns during CD8+ T cell differentiation are comparable to those observed for DNA methylation and histone modifications (FIG. 1).
3D chromatin architecture.
Increasingly, it is apparent that 3D chromatin architecture contributes substantially to gene regulation. Genome-wide chromatin interaction maps in other tissues reveal a hierarchy of 3D chromatin organization within the nucleus, which begins with the appreciation that chromosomes are organized into discrete topologically associating domains (TADs)87,88. These megabase-sized regions are highly self-interacting, and they restrict the spread of heterochromatic H3K9me3 marks, which end at TAD boundaries. The boundaries themselves are enriched for the transcriptional repressor CTCF. CTCF is a chromatin-remodelling protein and insulating factor, and although TADs are generally stable throughout development, intra-TAD interactions are dynamic87–90. Within TADs, cell type-specific chromatin loops between distant loci bring distal enhancer elements into contact with the genes they regulate, and lineage-specific chromatin interactions correlating with lineage-specific gene expression in a variety of cell types and during haematopoietic, neuronal and epidermal cell differentiation87–95. These interactions can be detected using chromosome conformation capture96 (3C) or derivative techniques97. In T cells, 3C has identified developmentally relevant enhancer–promoter loops that regulate Vα–Jα recombination in thymocytes, cytokine expression in CD4+ T cells and CD8A and CD8B expression in CD8+ T cells98–101.
Technical advances and reduced sequencing costs support genome-wide investigations of chromatin interactions with increasing resolution using Hi-C and other high-throughput methods (BOX 1). A recent study used a variation of Hi-C, known as promoter capture Hi-C, to specifically enrich for such enhancer–promoter interactions in 17 primary human haematopoietic cell types, including sorted naive CD8+ T cells and a heterogeneous population of total CD8+ T cells94. Although not able to distinguish between the various T cell differentiation subsets, this work identifies numerous cell type-specific promoter-interacting regions that are enriched for enhancer-associated marks, including H3K27ac and H3K4me1 and an open chromatin conformation. Cell type-specific interactions are associated with target gene expression and, for genes targeted by multiple putative enhancers, there is an additive effect on expression. Importantly, researchers observe a median distance of 331 kilobases between putative enhancer regions and their target genes. These types of long-distance acting enhancers would be difficult to assign functionality to without the aid of chromatin interaction profiles. Genome-wide characterization of chromatin interactions in specific CD8+ T cell subsets, therefore, will be invaluable for the identification and interpretation of enhancer activity.
Chromatin-remodelling proteins.
Cell-type-specific chromatin interactions are mediated by a combination of chromatin-remodelling factors, including CTCF and mediator and cohesin complexes88,92,95. Loss of these factors disrupts chromatin architecture, particularly functional enhancer–promoter loops, and leads to altered differentiation patterns92,102,103. This is observed in thymo cytes following ablation of CTCF and cohesin, which impairs T cell differentiation, disrupts looping interactions and results in aberrant gene expression52,98,104,105. Chromatin remodellers also interact with epigenetic readers, writers and erasers to facilitate gene expression106,107. TCR signalling in mouse CD8+ T cells activates expression of the chromatin-organizer SATB1, which recruits a repressive histone deacetylase complex to a Pdcd1 (which encodes PD1) enhancer element, resulting in gene repression108. Detailing the changes in chromatin organization that occur throughout CD8+ T cell differentiation will contribute substantially to our understanding of regulatory changes during differentiation. Altogether, chromatin organization plays an essential role in the epigenetic regulatory network, particularly in regulating the establishment and maintenance of enhancer activity.
Using epigenetics to improve immunotherapy
Cancer immunotherapies, including checkpoint inhibitor therapy and adoptive cell therapy (ACT), can induce striking clinical responses in patients with metastatic cancer, although responses are limited to a subset of individuals within specific cancer subtypes109,110. Although multiple factors are responsible for incomplete effectiveness, two major roadblocks include the development of T cell exhaustion and the functional impairment of cells used for ACT. As highlighted in this Review, epigenetic processes direct a substantial portion of the T cell differentiation programme together with lineage-specific transcription factors. We propose that alterations to gene expression affecting T cell differentiation and function can be achieved by targeting epigenetic enzymes or the functionally relevant genomic loci that they regulate. This targeted approach would eliminate the negative side effects of more indiscriminate genetic or pharmacological interventions and has the potential to substantially improve current immunotherapies by promoting memory cell differentiation or by preventing or reversing T cell exhaustion.
Arrested effector model of exhaustion.
Although exhausted T cells are generally believed to develop from effector cells, experimental evidence supports a more nuanced version of this long-held model. KLRG1low memory precursor cells transferred to a mouse with chronic lymphocytic choriomeningitis virus infection can persist in vivo and subsequently develop characteristics of exhaustion, in contrast to KLRG1hi terminally differentiated effector cells111. Additionally, in a tumour model of exhaustion, the transfer of tumour-specific memory cells to tumour-bearing mice results in the acquisition of tumour-infiltrating lymphocytes (TILs) displaying an exhausted phenotype112. Phenotypic studies have identified extensive heterogeneity within the exhausted population on the basis of the expression of multiple cellular markers112–117. These distinct cellular subsets display divergent functional capabilities, particularly regarding their proliferative potential. Phenotypically, exhausted cells with greater proliferative potential often display characteristics associated with memory cells, including decreased PD1 expression and increased TCF1 and CXC-chemokine receptor 5 (CXCR5) expression in addition to the capacity for self-renewal and differentiation112–117. Importantly, these cells are observed early during the establishment of exhausted responses112,118. Therefore, we propose an arrested effector model of T cell exhaustion whereby exhausted T cells arise from memory or effector T cells that have diverged from the canonical differentiation path at a point before the terminal effector state (FIG. 4). The functional heterogeneity of exhausted T cells may, therefore, reflect the different stages at which cells initiate the exhaustion programme, with cells arrested at early stages of differentiation (such as memory precursors) displaying memory-like characteristics and cells arrested at later stages exhibiting a terminally differentiated exhausted state (FIG. 4).
Figure 4 |. The arrested model of CD8 + T cell exhaustion.

The arrested model of CD8+ T cell exhaustion represents a branchpoint of the linear differentiation model, at which strong and/or repetitive antigenic stimulation, often accompanied by a lack of co-stimulatory signals, arrests canonical differentiation. Increasingly, it has been shown that the exhausted state is heterogeneous, with a subset of exhausted T (TEX) cells exhibiting memory-like phenotypes and characterized by specific cell surface markers (left side, light orange box). Conversely, more differentiated TEX cells exhibit their own unique cell surface marker expression (right side, dark orange box). The stage along canonical T cell differentiation at which cells become arrested may determine their TEX cell phenotype (light blue arrows); however, continued differentiation within the exhausted state may also occur. Memory-like TEX cells appear to be more responsive to checkpoint inhibitor therapy, which we hypothesize releases arrested cells, returning them to the canonical differentiation path. Importantly, in the linear model, this would ultimately result in terminal differentiation and apoptosis of TEX cells that had reversed the arrested state. CXCR5, CXC-chemokine receptor 5; EOMES, eomesodermin homologue; PD1, programmed cell death protein 1; TCF1, transcription factor 1; TIM3, T cell immunoglobulin mucin receptor 3; TM, memory T; TEFF, effector T.
Memory-like exhausted T cell subsets respond best to checkpoint inhibition.
Checkpoint inhibitor therapy targeting PD1 or PD1 ligand 1 (PDL1) results in rapid reversal of CD8+ T cell hypofunctionality among exhausted viral antigen-specific cells119 and can provoke antitumour responses from functionally exhausted TILs in mice and humans22–24,67,119,120. The exhausted cells that are most amenable to checkpoint inhibitor blockade appear to be the less-differentiated memory-like subsets. These cells exhibit greater restoration of effector function following anti-PD1 or homeostatic cytokine treatment112,113,116,117; however, the removal of less-differentiated exhausted subsets is associated with defects in maintaining the exhaustion programme. Genetic deletion of Pdcd1 decreases levels of the proliferation-competent T-bethi exhausted T cell subset and increases levels of the terminally differentiated EOMEShi subset, resulting in long-term defects in proliferation and failed persistence of antigen-specific cells121. Similarly, genetic deletion of Tcf7, which is itself associated with increased exhausted T cell persistence115, abrogates the development of cells positive for CXCR5 and negative for T cell immunoglobulin mucin receptor 3 (TIM3; also known as HAVCR2) in favour of CXCR5–TIM3+ cells, which have decreased long-term proliferative capacity and persistence following chronic infection116. Therefore, proliferation-competent, less-differentiated exhausted cells are more responsive to checkpoint inhibition, yet their removal is detrimental to the persistence of antigen-specific T cells. This observation is consistent with the linear differentiation model, in which exhausted T cells responding to checkpoint inhibitor therapy would resume canonical differentiation, becoming functional effector T cells followed by terminal differentiation and apoptosis (FIGS 1b, 4). By contrast, the circular differentiation model predicts that rejuvenation of effector function would result in the formation of memory cells (FIG. 1a); however, memory formation is not observed following successful restoration of effector function via PDL1 blockade67. Ultimately, these data suggest that an unexpected consequence of checkpoint blockade is the promotion of terminal differentiation and removal of functional antigen-specific T cells from the circulation, which leaves behind a more differentiated population of exhausted cells that are resistant to treatment.
Epigenetic modulation of exhausted T cell differentiation subsets.
Epigenetic interventions may help to circumvent proposed negative side effects of checkpoint inhibition. Transcriptome analysis of early-emerging exhausted cells indicates comparatively increased expression of multiple repressive DNA and histone modifying enzymes, including Dnmt1, Dnmt3b and Ezh2 (REF. 118), suggesting epigenetic regulation of exhaustion. Indeed, work from Ghoneim and colleagues122 demonstrates a role for DNMT3A in establishing an exhaustion-specific DNA methylation programme. Although conditional loss of Dnmt3a in CD8+ effector T cells did not abrogate the development of exhaustion, it did alter the phenotypic composition of exhausted cells. Specifically, an increase in the frequency of less-differentiated exhausted T cell subsets was observed in conditional knockout (cKO) mice as characterized by high levels of T-bet and TCF1 expression and decreased TIM3 and EOMES expression. This shift in differentiation state of the exhausted population might partially explain the increased persistence and cytokine production of viral-specific CD8+ T cells observed in cKO mice treated with anti-PDL1 antibodies compared with wild-type treated mice122. Thus, modulation of exhausted cell subsets through Dnmt3a loss acts synergistically with checkpoint inhibition, presenting an exciting opportunity for combination immunotherapy regimens.
Targeted manipulation of gene expression.
Investigations of gene expression, DNA methylation and chromatin accessibility in exhausted T cells have established unique exhaustion-specific transcriptional and epigenetic profiles that are distinct from those observed for memory and effector populations14,66,67,122–124. These subset-specific differences can be exploited to differentially affect gene expression, particularly for genes expressed in multiple subsets or genes whose expression has divergent subset-specific functional effects. This would be particularly advantageous for T cell exhaustion, where uncoupling cellular dysfunction from T cell activation would have substantial therapeutic benefits. Single-cell RNA sequencing analyses suggest that metallothionein 1 (MT1) and GATA3 function as specific contributors to T cell dysfunction125, and ATAC–seq analysis identifies multiple exhaustion-specific chromatin accessibility loci126. These loci represent putative exhaustion-specific enhancers that act independently of activation-specific enhancers to regulate genes associated with exhaustion. This includes Pdcd1 and Lag3, which display both exhaustion-specific and activation-specific enhancers. Motif analysis at putative exhaustion-specific enhancers identifies an enrichment of nuclear receptor subfamily 4 group A (NR4A) and nuclear factor of activated T cells (NFAT) binding sites, suggesting that these transcription factors specifically regulate the exhausted state14,112,126. Indeed, in a tumour-driven model of exhaustion, drug-mediated reduction of NFAT activity results in decreased expression of PD1 and lymphocyte activation gene 3 protein (LAG3) and increased expression of TCF1 in adoptively transferred cells, in addition to increasing polyfunctionality after ex vivo culture with IL-15 (REF. 112). Together, these studies suggest that T cell dysfunction is affected independent of activation by targeting exhaustion-specific transcription factors.
Genetic manipulation of exhaustion-specific non-coding regulatory regions, such as enhancers, provides an alternative approach to achieve subset-specific functional alterations in T cells. T cell subsets each contain unique, therapeutically desirable functional attributes, and transferring such characteristics into a singular functional cell via genetic manipulation has traditionally been achieved by gene knockout or over-expression. These techniques, however, permanently alter the coding genome sequence, resulting in sustained transcriptional changes over the life of the cell, a process at odds with the coordinated changes in gene expression observed in normal T cell development. Targeting non-coding regulatory regions can circumvent such broad transcriptional changes. It has been experimentally demonstrated in vitro that removal of an exhaustion-specific Pdcd1 enhancer decreases Pdcd1 expression66. In vivo examples of the impact of non-coding, targeted genetic manipulation are limited. Importantly, CRISPR-mediated systemic deletion of a non-coding Il2ra enhancer element in vivo reduced IL-2Rα expression specifically in CD4+ effector T cells but not in regulatory T cells127. These studies demonstrate the feasibility of using subset-specific regulatory elements to manipulate gene expression in a targeted manner. Such an approach has the potential to create discrete changes in gene expression and, therefore, cell function within specific T cell subsets. This may include increasing the reprogrammability of exhausted subsets, uncoupling exhaustion from activation or preventing the loss of effector pools following treatment with checkpoint inhibitors. Thus, the genetic manipulation of subset-specific non-coding regulatory elements might enable the conditional expression and repression of therapeutically desirable and undesirable genes, increasing the effectiveness of immunotherapy.
T cell expansion without differentiation.
Tumour-reactive T cells used for ACT can be obtained from patient-derived TILs or peripheral blood mono-nuclear cells (PBMCs) genetically engineered with tumour-targeting TCRs or chimeric antigen receptors (CARs). Studies in mice and humans demonstrate the benefits of using less-differentiated CD8+ T cell subsets for ACT128–131. However, TILs and PBMCs harvested for clinical use often exhibit terminally differentiated and exhausted phenotypes22–24,132, and the ACT protocol requires substantial ex vivo expansion, which results in T cell differentiation and loss of proliferative potential133 (FIG. 5). Manipulation of specific signalling pathways (such as WNT, the serine/threo-nine protein kinase AKT and CD95) during cellular stimulation can enable uncoupling of expansion and differentiation132,134–136. Given the fundamental role of epigenetics in regulating CD8+ T cell differentiation, it is likely that manipulating epigenetic patterns during T cell expansion may also prove to be therapeutically viable. Indeed, ex vivo culture with the bromodomain inhibitor JQ1 or the metabolic by-product S-2-hydroxyglutarate (S-2HG) results in increased memory formation and greater T cell persistence and antitumour activity upon adoptive transfer81,137. Functionally, both treatments appear to inhibit effector differentiation, in part, by targeting epigenetic modifying proteins. JQ1 directly inhibits the histone acetylation reader BRD4 and indirectly inhibits the histone deacetylase SIRT1 (REF. 81) (FIG. 3b), and S-2HG competitively inhibits α-ketoglutarate-dependent proteins, including the Jumonji family of histone demethylases and the TET family of DNA hydroxylases138. These data demonstrate that pharmacological manipulation of epigenetic mechanisms can alter T cell differentiation in a clinically relevant manner.
Figure 5 |. Interventions for improving clinical outcomes of adoptive cell therapy.

Currently, T cells used for adoptive cell therapy (ACT) are either obtained directly from tumours in the form of tumour-infiltrating lymphocytes (TILs) or isolated from patient peripheral blood mononuclear cells (PBMCs). These cells are tested for tumour reactivity or transduced with a tumour-reactive chimeric antigen receptor (CAR) or T cell receptor (TCR) followed by an extensive ex vivo expansion step before reinfusion into the patient. Although clinically effective in some cases, there are major challenges associated with current protocols. In both the starting T cell population and the population obtained following expansion, cells exhibit a more differentiated and/or exhausted phenotype, which may hinder in vivo effectiveness. Additionally, following transfer, cells will encounter an immunosuppressive tumour microenvironment that may further promote exhaustion. There are many potential fixes for the challenges currently affecting ACT. Cellular reprogramming of PBMCs or TILs would obtain a younger, less differentiated starting population, and pharmacological interventions to target relevant signalling pathways and/or epigenetic modifying proteins could allow for T cell expansion without differentiation. Additionally, T cells could be genetically manipulated to remain impervious to the exhaustion-inducing effects of the tumour microenvironment. Targeting non-coding regulatory regions is of therapeutic interest for its ability to alter gene expression in a subset-specific manner. These potential solutions will require a thorough understanding of the proteins and epigenetic landscapes regulating the differentiation process. iPSCs, induced pluripotent stem cells; TEFF, effector T; TEX, exhausted T; TM, memory T.
Differentiation state impacts effectiveness of pharmacological interventions.
Although, as described above, pharmacological interventions can impact T cell differentiation and function, their effect can depend on the differentiation state of the target cell. Increasingly, the observation that CD8+ T cell subsets exhibit distinct metabolic profiles139 has made metabolic pathways an attractive druggable target owing to their influence on the epigenetic landscape140. Indeed, short-term treatment of naive cells with S-2HG or a glycolytic inhibitor during cellular expansion increases memory differentiation and improves ACT in mouse models137,141. However, limited glycolytic metabolism or oxygen availability, which increases intra cellular S-2HG137, is immunosuppressive in the tumour micro environment, as effector function is impaired. Furthermore, S-2HG-driven expression of memory-associated genes is observed only when administered to naive cells together with TCR stimulation and is not seen when administered 7 days after TCR stimulation, when cells are in a more differentiated state137. This suggests that the epigenetic programme of differentiated cells has already been established and that S-2HG alone is not capable of epigenetically rewriting cells to a memory state. Thus, not only is the effect of pharmacological interventions dependent on T cell differentiation state but effects that are desirable in one subset may be prohibitive in another. Given the heterogeneous and often differentiated nature of cells used for ACT, it is unclear how effective broad, extrinsic interventions would be for current clinical approaches.
Epigenetic reprogramming of T cells.
To overcome the terminally differentiated nature of cells used for ACT, cellular reprogramming of T cell populations to a naive state has emerged as an attractive therapeutic avenue (FIG. 5). T cell reprogramming strategies include either pluripotent reprogramming of cells into induced pluri-potent stem cells (iPSCs) followed by differentiation into naive T cells or direct reprogramming of differentiated subsets into less-differentiated ones142. While T cell reprogramming into iPSCs is possible via transient expression of the transcription factors octamer-binding protein 4 (OCT4; also known as POU5F1), SOX2, Krüppel-like factor 4 (KLF4) and MYC (collectively referred to as OSKM)143, their differentiation into fully functional naive CD8+ T cells remains elusive144. To achieve pluripotent reprogramming, OSKM act as pioneer factors, initiating extensive epi-genetic changes by interacting with stage-specific transcription factors, histone-modifying proteins and chromatin-remodelling enzymes145–150. Distal enhancers, especially, are subject to extensive chromatin reorganization causing widespread changes to enhancer repertoires during the reprogramming process145,148. Therefore, direct reprogramming of T cells will require similar epigenetic changes, in particular, reversing the silenced state of stemness and memory genes that is associated with T cell differentiation5. These changes could potentially be initiated by T cell-specific pioneer factors. Analysing the changing enhancer repertoires in CD8+ T cell subsets may aid in uncovering such factors, enabling direct reprogramming of T cells.
Concluding remarks
DNA methylation, histone modification and chromatin architecture collectively form the epigenetic landscape that contributes to the transcriptional regulation of CD8+ T cell differentiation and function. These epigenetic mechanisms enable cellular responses to initiating signals that are heritable and reversible and allow for common signalling pathways to drive cell-type-specific transcriptional responses. According to the linear differentiation model, a progressive pattern of epigenetic changes is observed at genic loci over time, whereas regulatory regions exhibit distinct lineage specificity. Although there has been substantial progress in characterizing the epigenetic patterns associated with T cell subsets, additional functional analyses are needed to expand on these mechanisms and further elucidate the role of epigenetic modifying proteins and transcription factors. Increasing our understanding of CD8+ T cell epigenetics will enable a greater understanding of T cell biology, will assist with targeted pharmacological or genetic interventions to impact subset differentiation and function, and has enormous therapeutic potential for improving cancer immunotherapy.
Chromatin architecture.
The 3D organization of chromatin within the nucleus, which contributes to DNA packaging and protection and is also instrumental for gene regulation via the formation of discrete chromatin interactions.
Terminal effector differentiation.
The final stage of CD8+ T cell differentiation, which follows the acquisition of effector function, precedes apoptosis and is characterized by cells that have lost stem-like characteristics, including pluripotency, self-renewal and persistence.
CpG islands.
(CGIs). DNA regions that are commonly found at gene promoters and consist of a higher than average density of CG dinucleotide bases. Hypermethylation of these regions is associated with transcriptional repression.
Bivalent chromatin.
Chromatin containing both activating H3K4me3 and repressive H3K27me3 modifications; often found at genes that are thought to be poised for future transcriptional activation or repression.
Super enhancers.
Large regulatory loci with numerous clustered enhancer elements and multiple transcription factor binding sites. Super enhancers have been associated with cell identity and disease-associated genes.
Checkpoint inhibitor therapy.
Therapy targeting either inhibitory cell surface receptors on T cells or their ligands expressed on cancer cells to circumvent tumour immunosuppression and boost antitumour immunity.
Adoptive cell therapy.
(ACT). The administration of naturally occurring or genetically engineered tumour-reactive T cells to patients for cancer therapy.
Arrested effector model.
An addendum to the developmental, or linear, differentiation model hypothesizing that CD8+ T cell exhaustion arises from T cells that become arrested before terminal effector differentiation. The stage at which cells arrest within canonical differentiation impacts their functionality as exhausted cells.
Cellular reprogramming.
The manipulation of one cell type into another by altering the transcriptional, epigenetic and functional characteristics of the cell in a way that does not occur physiologically.
Pluripotent reprogramming.
A type of cellular reprogramming that involves the conversion of a mature somatic cell into a less-differentiated, pluripotent cell type, referred to as an induced pluripotent stem cell.
Direct reprogramming.
A type of cellular reprogramming that involves the conversion of a mature, differentiated somatic cell type into another mature cell type without passing through an intermediate induced pluripotent stem cell state.
Pioneer factors.
Transcription factors that have the capacity to bind both open and closed chromatin. These proteins contribute to gene regulation by recruiting additional transcription factors and epigenetic modifying proteins and are critically important during cellular reprogramming.
Stemness.
Having characteristics associated with stem cells, specifically, the ability to self-renew and give rise to more differentiated progeny.
Acknowledgements
A.N.H. and N.P.R. are supported by the Cancer Moonshot program for the Center for Cell-based Therapy at the Center for Cancer Research, NCI/NIH (ZIA BC010763). This work was also supported by the Milstein Family Foundation. R.R is sup-ported by the Wellcome Trust and Royal Society (grant 105663/Z/14/Z), the Lister Institute, the UK Biotechnology and Biological Sciences Research Council (grant BB/N007794/1) and Cancer Research UK (grant C52623/A22597). The authors thank L. Gattinoni, D. Palmer, M. Sukumar, D. Gurusamy, C. Klebanoff, D. Clever, R. Eil, F. Grant, R. Nasrallah, D. Gyori, C. Imianowski, F. Sadiyah, K. Okkenhaug, M. Turner, W. Reik, R. Vizcardo, G. Butcher and S. Rosenberg for ideas and discussion.
Footnotes
Competing interests statement
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Youngblood B, Hale JS & Ahmed R T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology 139, 277–284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Opferman JT, Ober BT & Ashton-Rickardt PG Linear differentiation of cytotoxic effectors into memory T lymphocytes. Science 283, 1745–1748 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Akondy RS et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552, 362–367 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Youngblood B et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 552, 404–409 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henning AN, Klebanoff CA & Restifo NP Silencing stemness in T cell differentiation. Science 359, 163–164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Restifo NP & Gattinoni L Lineage relationship of effector and memory T cells. Curr. Opin. Immunol 25, 556–563 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teixeiro E et al. Different T cell receptor signals determine CD8+ memory versus effector development. Science 323, 502–505 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Wirth TC et al. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8+ T cell differentiation. Immunity 33, 128–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaech SM & Cui W Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol 12, 749–761 (2012).This is a review of the transcriptional pathways involved in CD8+ T cell differentiation and function during an acute immune response.
- 10.Roychoudhuri R et al. Transcriptional profiles reveal a stepwise developmental program of memory CD8+ T cell differentiation. Vaccine 33, 914–923 (2015).This study analyses the transcriptional profiles of CD8+ T cell subsets during a vaccine-induced immune response, identifying progressive changes consistent with the linear model.
- 11.Gattinoni L et al. A human memory T cell subset with stem cell-like properties. Nat. Med 17, 1290–1297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willinger T, Freeman T, Hasegawa H, McMichael AJ & Callan MF Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J. Immunol 175, 5895–5903 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Sarkar S et al. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J. Exp. Med 205, 625–640 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott-Browne JP et al. Dynamic changes in chromatin accessibility occur in CD8+ T cells responding to viral infection. Immunity 45, 1327–1340 (2016).This study identifies unique chromatin accessibility patterns in CD8+ T cell subsets during acute and chronic viral infections.
- 15.He B et al. CD8+ T cells utilize highly dynamic enhancer repertoires and regulatory circuitry in response to infections. Immunity 45, 1341–1354 (2016).In this study, the authors perform comprehensive mapping of enhancers and super enhancers in CD8+ T cell subsets, uncovering highly specific enhancer repertoires.
- 16.Gallimore A et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J. Exp. Med 187, 1383–1393 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zajac AJ et al. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med 188, 2205–2213 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schietinger A & Greenberg PD Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 35, 51–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pauken KE & Wherry EJ Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 36, 265–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeman GJ et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med 192, 1027–1034 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hokey DA et al. Activation drives PD-1 expression during vaccine-specific proliferation and following lentiviral infection in macaques. Eur. J. Immunol 38, 1435–1445 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmadzadeh M et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114, 1537–1544 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fourcade J et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med 207, 2175–2186 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuzaki J et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl Acad. Sci. USA 107, 7875–7880 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ball MP et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol 27, 361–368 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones PA Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet 13, 484–492 (2012).This is a review of the mechanisms and functions of DNA methylation in mammals.
- 27.Scharer CD, Barwick BG, Youngblood BA, Ahmed R & Boss JM Global DNA methylation remodeling accompanies CD8 T cell effector function. J. Immunol 191, 3419–3429 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez RM et al. Epigenetic networks regulate the transcriptional program in memory and terminally differentiated CD8+ T cells. J. Immunol 198, 937–949 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Abdelsamed HA et al. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J. Exp. Med 214, 1593–1606 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin MS et al. DNA methylation regulates the differential expression of CX3CR1 on human IL-7Rαlow and IL-7Rαhigh effector memory CD8+ T cells with distinct migratory capacities to the fractalkine. J. Immunol 195, 2861–2869 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stadler MB et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Meissner A et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Irizarry RA et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet 41, 178–186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji H et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 467, 338–342 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Branco MR, Ficz G & Reik W Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet 13, 7–13 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Araki Y, Fann M, Wersto R & Weng NP Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B). J. Immunol 180, 8102–8108 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Araki Y et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity 30, 912–925 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russ BE et al. Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8+ T cell differentiation. Immunity 41, 853–865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crompton JG et al. Lineage relationship of CD8+ T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell. Mol. Immunol 13, 502–513 (2016).This study identifies progressive, genome-wide changes in histone modifications in CD8+ T cell subsets, consistent with the linear model.
- 40.Juelich T et al. Interplay between chromatin remodeling and epigenetic changes during lineage-specific commitment to granzyme B expression. J. Immunol 183, 7063–7072 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Denton AE, Russ BE, Doherty PC, Rao S & Turner SJ Differentiation-dependent functional and epigenetic landscapes for cytokine genes in virus-specific CD8+ T cells. Proc. Natl Acad. Sci. USA 108, 15306–15311 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuroda S et al. Basic leucine zipper transcription factor, ATF-like (BATF) regulates epigenetically and energetically effector CD8 T-cell differentiation via Sirt1 expression. Proc. Natl Acad. Sci. USA 108, 14885–14889 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shin HM et al. Epigenetic modifications induced by Blimp-1 regulate CD8+ T cell memory progression during acute virus infection. Immunity 39, 661–675 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen ML et al. Dynamic regulation of permissive histone modifications and GATA3 binding underpin acquisition of granzyme A expression by virus-specific CD8+ T cells. Eur. J. Immunol 46, 307–318 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Strahl BD & Allis CD The language of covalent histone modifications. Nature 403, 41–45 (2000).This is a review on histone modifications and their general function.
- 46.Kimura H Histone modifications for human epigenome analysis. J. Hum. Genet 58, 439–445 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Geginat J, Lanzavecchia A & Sallusto F Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood 101, 4260–4266 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Bernstein BE et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Pan G et al. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1, 299–312 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Zhao XD et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 1, 286–298 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Harker N et al. Pre-TCR signaling and CD8 gene bivalent chromatin resolution during thymocyte development. J. Immunol 186, 6368–6377 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Ing-Simmons E et al. Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res. 25, 504–513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vahedi G et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature 520, 558–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whyte WA et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hnisz D et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kieffer-Kwon KR et al. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell 155, 1507–1520 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heinz S, Romanoski CE, Benner C & Glass CK The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell. Biol 16, 144–154 (2015).This is a review on the characteristics and function of enhancers and their role in regulating signal-driven transcriptional programmes.
- 58.Sarraf SA & Stancheva I Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell 15, 595–605 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Agarwal N et al. MeCP2 interacts with HP1 and modulates its heterochromatin association during myogenic differentiation. Nucleic Acids Res. 35, 5402–5408 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fujita N et al. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J. Biol. Chem 278, 24132–24138 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Kersh EN Impaired memory CD8 T cell development in the absence of methyl-CpG-binding domain protein 2. J. Immunol 177, 3821–3826 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Musselman CA, Lalonde ME, Cote J & Kutateladze TG Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol 19, 1218–1227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Di Croce L & Helin K Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol 20, 1147–1155 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Luco RF et al. Regulation of alternative splicing by histone modifications. Science 327, 996–1000 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sen DR et al. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pauken KE et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moskowitz DM et al. Epigenomics of human CD8 T cell differentiation and aging. Sci. Immunol 2, eaag0192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heffner M & Fearon DT Loss of T cell receptor-induced Bmi-1 in the KLRG1+ senescent CD8+ T lymphocyte. Proc. Natl Acad. Sci. USA 104, 13414–13419 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jacobs JJ, Kieboom K, Marino S, DePinho RA & van Lohuizen M The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397, 164–168 (1999). [DOI] [PubMed] [Google Scholar]
- 71.Sherr CJ Principles of tumor suppression. Cell 116, 235–246 (2004). [DOI] [PubMed] [Google Scholar]
- 72.Zhao E et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat. Immunol 17, 95–103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kato K et al. Identification of stem cell transcriptional programs normally expressed in embryonic and neural stem cells in alloreactive CD8+ T cells mediating graft-versus-host disease. Biol. Blood Marrow Transplant. 16, 751–771 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gray SM, Amezquita RA, Guan T, Kleinstein SH & Kaech SM Polycomb repressive complex 2-mediated chromatin repression guides effector CD8+ T cell terminal differentiation and loss of multipotency. Immunity 46, 596–608 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.He S et al. Ezh2 phosphorylation state determines its capacity to maintain CD8+ T memory precursors for antitumour immunity. Nat. Commun 8, 2125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ladle BH et al. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T-cell fate decisions following activation. Proc. Natl Acad. Sci. USA 113, 10631–10636 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kakaradov B et al. Early transcriptional and epigenetic regulation of CD8+ T cell differentiation revealed by single-cell RNA sequencing. Nat. Immunol 18, 422–432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pace L et al. The epigenetic control of stemness in CD8+ T cell fate commitment. Science 359, 177–186 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Xing S et al. Tcf1 and Lef1 transcription factors establish CD8+ T cell identity through intrinsic HDAC activity. Nat. Immunol 17, 695–703 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roychoudhuri R et al. BACH2 regulates CD8+ T cell differentiation by controlling access of AP-1 factors to enhancers. Nat. Immunol 17, 851–860 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kagoya Y et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Invest 126, 3479–3494 (2016).This is a functional study examining the effect of the bromodomain inhibitor JQ1 on T cell differentiation and antitumour activity and its underlying mechanism.
- 82.Lee PP et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774 (2001). [DOI] [PubMed] [Google Scholar]
- 83.Pastor WA, Aravind L & Rao A TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell. Biol 14, 341–356 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsagaratou A et al. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc. Natl Acad. Sci. USA 111, E3306–3315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carty SA et al. The loss of TET2 promotes CD8+ T cell memory differentiation. J. Immunol 200, 82–91 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lyko F & Brown R DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J. Natl Cancer Inst. 97, 1498–1506 (2005). [DOI] [PubMed] [Google Scholar]
- 87.Nora EP et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fraser J et al. Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol. Syst. Biol 11, 852 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schmitt AD et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 17, 2042–2059 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li G et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148, 84–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Phillips-Cremins JE et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rao SS et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).This study describes the application of Hi-C to multiple human and mouse cell lines, acquiring high-resolution data that enable the characterization of chromatin looping patterns in exquisite detail.
- 94.Javierre BM et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 167, 1369–1384.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lin YC et al. Global changes in the nuclear positioning of genes and intra- and interdomain genomic interactions that orchestrate B cell fate. Nat. Immunol 13, 1196–1204 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dekker J, Rippe K, Dekker M & Kleckner N Capturing chromosome conformation. Science 295, 1306–1311 (2002). [DOI] [PubMed] [Google Scholar]
- 97.Dekker J, Marti-Renom MA & Mirny LA Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet 14, 390–403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shih HY et al. Tcra gene recombination is supported by a Tcra enhancer- and CTCF-dependent chromatin hub. Proc. Natl Acad. Sci. USA 109, E3493–E3502 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tsytsykova AV et al. Activation-dependent intrachromosomal interactions formed by the TNF gene promoter and two distal enhancers. Proc. Natl Acad. Sci. USA 104, 16850–16855 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Spilianakis CG & Flavell RA Long-range intrachromosomal interactions in the T helper type 2 cytokine locus. Nat. Immunol 5, 1017–1027 (2004). [DOI] [PubMed] [Google Scholar]
- 101.Ktistaki E et al. CD8 locus nuclear dynamics during thymocyte development. J. Immunol 184, 5686–5695 (2010). [DOI] [PubMed] [Google Scholar]
- 102.Muto A et al. Nipbl and mediator cooperatively regulate gene expression to control limb development. PLOS Genet. 10, e1004671 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Watson LA et al. Dual effect of CTCF loss on neuroprogenitor differentiation and survival. J. Neurosci 34, 2860–2870 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Heath H et al. CTCF regulates cell cycle progression of alphabeta T cells in the thymus. EMBO J. 27, 2839–2850 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Seitan VC et al. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 23, 2066–2077 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ding N et al. Mediator links epigenetic silencing of neuronal gene expression with x-linked.mental retardation. Mol. Cell 31, 347–359 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tomaz RA et al. Jmjd2c facilitates the assembly of essential enhancer-protein complexes at the onset of embryonic stem cell differentiation. Development 144, 567–579 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stephen TL et al. SATB1 expression governs epigenetic repression of PD-1 in tumor-reactive T cells. Immunity 46, 51–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rosenberg SA & Restifo NP Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015).This is a review of current ACT practices for cancer treatment and the challenges and future directions of the field.
- 110.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Angelosanto JM, Blackburn SD, Crawford A & Wherry EJ Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol 86, 8161–8170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Philip M et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017).This comprehensive analysis of chromatin accessibility during the early and later stages of T cell exhaustion demonstrates the therapeutic relevance of this type of analysis via pharmacological inhibition of a putative. exhaustion-associated transcription factor.
- 113.Blackburn SD, Shin H, Freeman GJ & Wherry EJ Selective expansion of a subset of exhausted CD8 T cells by αPD-L1 blockade. Proc. Natl Acad. Sci. USA 105, 15016–15021 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Paley MA et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu T et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immuno 1, eaai8593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Im SJ et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.He R et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 537, 412–428 (2016). [DOI] [PubMed] [Google Scholar]
- 118.Schietinger A et al. Tumor-specific T cell dysfunctionis a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401 (2016).This is an exhaustive study on the transcriptome of early-stage and late-stage exhausted T cells in a tumour-driven model demonstrating unique profiles at these stages and including comparisons with transcriptomes of viral-driven and self-tolerant models of T cell dysfunction.
- 119.Barber DL et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006). [DOI] [PubMed] [Google Scholar]
- 120.Duraiswamy J, Freeman GJ & Coukos G Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 73, 6900–6912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Odorizzi PM, Pauken KE, Paley MA, Sharpe A & Wherry EJ Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med 212, 1125–1137 (2015).This is a functional study showing that PD1 is not required for virally initiated T cell exhaustion but is needed to maintain the exhausted cell pool and prevent terminal differentiation.
- 122.Ghoneim HE et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 170, 142–157.e19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wherry EJ et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]
- 124.Doering TA et al. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 37, 1130–1144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Singer M et al. A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell 166, 1500–1511.e9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mognol GP et al. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc. Natl Acad. Sci. USA 114, E2776–E2785 (2017).This study demonstrates the innovative use of ATAC–seq to identify exhaustion-specific and activation-specific regulatory regions in a tumour-driven model of T cell exhaustion.
- 127.Simeonov DR et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gattinoni L et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Invest 115, 1616–1626 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rosenberg SA et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 17, 4550–4557 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rufer N, Dragowska W, Thornbury G, Roosnek E & Lansdorp PM Telomere length dynamics in human lymphocyte subpopulations measured by flow cytometry. Nat. Biotechnol 16, 743–747 (1998). [DOI] [PubMed] [Google Scholar]
- 131.Sommermeyer D et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30, 492–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Klebanoff CA et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J. Clin. Invest 126, 318–334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Powell DJ Jr., Dudley ME, Robbins PF & Rosenberg SA Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood 105, 241–250 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gattinoni L et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med 15, 808–813 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hinrichs CS et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 111, 5326–5333 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Crompton JG et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 75, 296–305 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tyrakis PA et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 540, 236–241 (2016).This is a functional study demonstrating the impact that the metabolic intermediary S-2HG can have on CD8+ T cell differentiation via epigenetic modulations.
- 138.Losman JA & Kaelin WG Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 27, 836–852 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.O’Neill LA, Kishton RJ & Rathmell J A guide to immunometabolism for immunologists. Nat. Rev. Immunol 16, 553–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Etchegaray JP & Mostoslavsky R Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol. Cell 62, 695–711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sukumar M et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest 123, 4479–4488 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Crompton JG, Clever D, Vizcardo R, Rao M & Restifo NP Reprogramming antitumor immunity. Trends Immunol. 35, 178–185 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Takahashi K & Yamanaka S Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006). [DOI] [PubMed] [Google Scholar]
- 144.Themeli M, Riviere I & Sadelain M New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell 16, 357–366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Soufi A, Donahue G & Zaret KS Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang H et al. Intrachromosomal looping is required for activation of endogenous pluripotency genes during reprogramming. Cell Stem Cell 13, 30–35 (2013). [DOI] [PubMed] [Google Scholar]
- 147.Ang YS et al. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell 145, 183–197 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chronis C et al. Cooperative binding of transcription factors orchestrates reprogramming. Cell 168, 442–459.e20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mansour AA et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature 488, 409–413 (2012). [DOI] [PubMed] [Google Scholar]
- 150.Wei Z et al. Klf4 organizes long-range chromosomal interactions with the Oct4 locus in reprogramming and pluripotency. Cell Stem Cell 13, 36–47 (2013). [DOI] [PubMed] [Google Scholar]
- 151.Yong WS, Hsu FM & Chen PY Profiling genome-wide DNA methylation. Epigenetics Chromatin 9, 26 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Crawford GE et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 16, 123–131 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Giresi PG, Kim J, McDaniell RM, Iyer VR & Lieb JD FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17, 877–885 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schoenfelder S et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 25, 582–597 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li G et al. Chromatin Interaction Analysis with Paired-End Tag (ChIA-PET) sequencing technology and application. BMC Genomics 15 (Suppl. 12), S11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Simons BD & Clevers H Strategies for homeostatic stem cell self-renewal in adult tissues. Cell 145, 851–862 (2011). [DOI] [PubMed] [Google Scholar]
- 157.Lin WH et al. Asymmetric PI3K signaling driving developmental and regenerative cell fate bifurcation. Cell Rep. 13, 2203–2218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pollizzi KN et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8+ T cell differentiation. Nat. Immunol 17, 704–711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Verbist KC et al. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature 532, 389–393 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nicholson JM et al. Histone structures: targets for modifications by molecular assemblies. Ann. NY Acad. Sci 1030, 644–655 (2004). [DOI] [PubMed] [Google Scholar]
- 161.Del Rizzo PA & Trievel RC Substrate and product specificities of SET domain methyltransferases. Epigenetics 6, 1059–1067 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fueyo R, Garcia MA & Martinez-Balbas MA Jumonji family histone demethylases in neural development. Cell Tissue Res. 359, 87–98 (2015). [DOI] [PubMed] [Google Scholar]
- 163.Jeong KW et al. Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat. Struct. Mol. Biol 18, 1358–1365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li H et al. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442, 91–95 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pradeepa MM, Sutherland HG, Ule J, Grimes GR & Bickmore WA Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLOS Genet. 8, e1002717 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jin Q et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sengupta N & Seto E Regulation of histone deacetylase activities. J. Cell. Biochem 93, 57–67 (2004). [DOI] [PubMed] [Google Scholar]
- 168.Santos L, Escande C & Denicola A Potential modulation of sirtuins by oxidative stress. Oxid Med. Cell Longev 2016, 9831825 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Taniguchi Y The Bromodomain and Extra-Terminal Domain (BET) family: functional anatomy of BET paralogous proteins. Int. J. Mol. Sci 17, E1849 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Benevento M, van de Molengraft M, van Westen R, van Bokhoven H & Kasri NN The role of chromatin repressive marks in cognition and disease: a focus on the repressive complex GLP/G9a. Neurobiol. Learn. Mem 124, 88–96 (2015). [DOI] [PubMed] [Google Scholar]
- 171.Azzaz AM et al. Human heterochromatin protein 1α promotes nucleosome associations that drive chromatin condensation. J. Biol. Chem 289, 6850–6861 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rea S et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599 (2000). [DOI] [PubMed] [Google Scholar]
- 173.Rice JC et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell 12, 1591–1598. [DOI] [PubMed] [Google Scholar]