

Abstract
It is thought that cancer cells engage in Warburg metabolism to meet intrinsic biosynthetic requirements of cell growth and proliferatsion. Papers by Chang et al. and Ho et al. show that Warburg metabolism enables tumor cells to restrict glucose availability to T cells, suppressing anti-tumor immunity.
In the presence of oxygen, most differentiated cells utilize mitochondrial oxidative phosphorylation to generate energy in the form of adenosine triphosphate (ATP) that can be used to sustain cellular processes. In the absence of oxygen, such cells revert to much less efficient glycolysis as a means of ATP production. Cancer cells often utilize glycolysis despite the presence of oxygen (aerobic glycolysis or the “Warburg effect”) (Warburg, 1956). While less efficient at producing energy, it is thought that this form of metabolism supports the macromolecular requirements of cell growth and proliferation. Thus, the field has primarily focused on Warburg metabolism as an adaptation that confers intrinsic growth advantages to tumor cells themselves. However, cancer cells may consume nutrients, particularly glucose, in excess of their requirement to sustain proliferation and cell growth (Vander Heiden et al., 2009). This raises the possibility that nutrient consumption serves additional roles to meeting the intrinsic bioenergetic and biosynthetic requirements of cancer cells. In this issue of Cell, Ho et al. (2015) and Chang et al. (2015) show that Warburg metabolism provides tumor cells with a cell-extrinsic advantage, promoting depletion of extracellular glucose which renders tumor-infiltrating T cells dysfunctional.
In both studies, glycolysis within tumor cells is shown to cause depletion of extracellular glucose which restricts glucose availability to T cells. Decreased glucose availability causes suppression of glycolytic metabolism within T cells, and this is associated with decreased effector function (Figure 1, left). Ho et al. identify a mechanism by which glucose metabolism directly controls effector function. The authors find that T cell receptor (TCR)-induced Ca2+ flux is markedly dependent upon extracellular glucose and glucose metabolism by T cells. Sarco/endoplasmic reticulum (ER) Ca2+-ATPase (SERCA) is an ATP-dependent Ca2+ channel that pumps Ca2+ from the cytoplasm into the ER. Extracellular glucose is shown to promote accumulation of the glycolytic metabolite, phosphoenolpyruvate (PEP), which inhibits SERCA-dependent evacuation of Ca2+ from the cytosol into the ER, thereby increasing TCR-induced Ca2+ flux and effector function (Figure 1, right). This observation adds to a growing list of examples whereby metabolic processes directly control the outcome of T cell activation (Chang et al., 2013; Maclver et al., 2013).
Figure 1. Nutrient Competition between Tumor Cells and T Cells Controls Immune Function within Tumors.
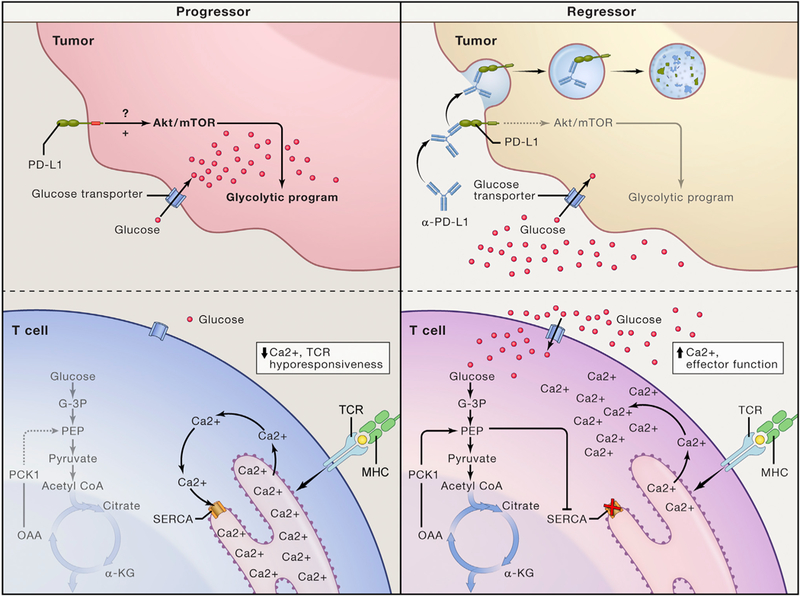
Schematic depicting glucose metabolism and cellular signaling In highly glycolytic progressor tumors and repressor tumors undergoing therapy. In the progressor tumor (left), constitutive activation of the Akt/mTOR pathway by PD-L1 expressed on tumor cells causes high levels of tumor cell glycolysis and absorption of extracellular glucose. Decreased extracellular glucose levels causes impaired glycolysis in T cells, wherein depletion of the glycolytic metabolite PEP causes unrestrained SERCA activity, sequestration of cytoplasmic Ca2+ into the ER and impairment of TCR-induced Ca2+ flux and effector function. In the regressor tumor (right), therapeutic anti-PD-L1 antibodies bind to PD-L1 causing its endocytosis and inactivation. Loss of constitutive PD-L1 signaling leads to decreased activation of the Akt-mTOR pathway decreased tumor cell glycolysis and increased extracellular glucose concentrations. Increased extracellular glucose drives T cell glycolysis, replenishing PEP levels, inhibiting SERCA-dependent sequestration of cytoplasmic Ca2+ and promoting TCR-induced Ca2+ flux and anti-tumor effector functions. Alternatively, constitutive overexpression of PCK1 in adoptively transferred T cells increases availability of PEP leading to inhibition of SERCA, increased anti-tumor effector function and tumor regression.
That tumor cell glycolysis directly suppresses T cells raises the possibility that tumor metabolism can be therapeutically manipulated to improve immune function within tumors. Checkpoint blockade immunotherapy with anti-PD-L1 antibodies is thought to work by limiting inhibitory PD-1 signaling received by tumor-specific T cells (Keir et al., 2008). Chang et al. made the surprising observation that PD-1 ligand (PD-L1) expressed by tumor cells provides a constitutive “reverse signal” that promotes tumor cell glycolysis through activation of the AKT/mTOR pathway (Figure 1, left). Treatment of tumor cells with therapeutic anti-PD-L1 antibodies attenuates glycolysis by triggering PD-L1 endocytosis (Figure 1, right). Remarkably, two other checkpoint-blockade antibodies, anti-PD-1 and anti-CTLA-4, are also shown to cause changes in extracellular glucose concentrations within tumors, though mechanisms for these observations are unclear. That PD-L1 expression causes constitutive activation of the Akt/mTOR pathway has important implications for understanding tumor cell biology and tumor-host interactions, and it will be important to characterize precise molecular mechanisms by which PD-L1 constitutively activates the Akt/mTOR pathway. Given that immune checkpoint blockade elicits durable clinical responses and improves survival in patients with certain metastatic cancers (Larkin et al., 2015; Topalian et al., 2012), it is relevant to measure the effect of checkpoint blockade antibodies on intratumoral nutrient availability and T cell metabolism in patients and correlate this with clinical outcomes. Further, it will be important to dissect the effects of checkpoint blockade on inhibitory T cell signaling versus tumor cell metabolism.
Instead of manipulating tumor cell metabolism, Ho et.al. suggest an alternate approach to improve T cell function by mimicking nutrient availability within transferred T cells during adoptive cell therapy (ACT). Phosphoenolpyruvate Carboxykinase (PCK1) converts oxaloacetate into PEP. By overexpressing Pck1 in transferred T cells, Ho et al. are able to artificially increase PEP levels, restoring TCR-induced Ca2+ flux and anti-tumor T cell function despite the presence of low environmental glucose levels within tumors. Intriguingly, blocking glucose metabolism during expansion of T cells for adoptive immunotherapy withholds effector differentiation and promotes differentiation of memory cells which mediate superior tumor clearance (Sukumar et al., 2013). These findings provide striking examples of how modulating T cell metabolism can improve the outcome of adoptive cell therapy for cancer.
Taken together, the two new studies provide compelling evidence that cancer cells subvert the metabolic characteristics of the tumor microenvironment to shape immune responses within tumors. The results also provide an explanation of how nutrient consumption in excess of the bioenergetic and biosynthetic requirements may benefit cancer cells. As Warburg’s original observation is revisited in ever new reincarnations, it remains to be seen whether insights from the field of immunometabolism will change the game at this new front in our war against cancer.
REFERENCES
- Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MW, and van der Windt GJW (2015). Cell 162, this issue, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho P-C, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui Y-C, Cui G, Micevic G, Perales JC, et al. (2015). Cell 162, this issue, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, and Sharpe AH (2008). Annu. Rev. Immunol. 26, 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. (2015). N. Engl. J. Med. 373, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Michalek RD, and Rathmell JC (2013). Annu. Rev. Immunol. 31, 259–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. (2013). J. Clin. Invest. 123, 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). N. Engl. J. Med. 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1956). Science 123, 309–314. [DOI] [PubMed] [Google Scholar]