

Abstract
Endoplasmic reticulum (ER)-associated degradation (ERAD) and the unfolded protein response (UPR) are two key quality-control machineries in the cell. ERAD is responsible for the clearance of misfolded proteins in the ER for cytosolic proteasomal degradation, while UPR is activated in response to the accumulation of misfolded proteins. It has long been thought that ERAD is an integral part of UPR because expression of many ERAD genes is controlled by UPR; however, recent studies have suggested that ERAD has a direct role in controlling the protein turnover and abundance of IRE1α, the most conserved UPR sensor. Here, we review recent advances in our understanding of IRE1α activation and propose that UPR and ERAD engage in an intimate crosstalk to define folding capacity and maintain homeostasis in the ER.
Introduction
The ER is a major protein folding compartment that actively monitors the biosynthesis, assembly, and trafficking of most secretory and membrane proteins [1]. Thousands of distinct gene products flux through the ER, many of which control normal physiology, and are linked to human health and disease [2]. To maintain protein homeostasis, cells must ensure the fidelity of protein folding and maturation. However, certain physiological and pathological conditions can lead to an imbalance between protein-folding demand and the folding capacity, resulting in an accumulation of misfolded proteins in the ER, with resultant ER stress [2,3]. To maintain ER homeostasis, cells have evolved protein quality-control systems through the action of different pathways, including the UPR, ERAD, and autophagy.
ER-resident proteins are mainly cleared by ERAD for proteasomal degradation in the ER, while protein aggregates in the ER may be cleared by autophagy [4–6]. The SEL1L-HRD1 protein complex represents the most conserved ERAD machinery in mammals with SEL1L being the cofactor for the E3 ligase HRD1 (reviewed in [4,5,7]). Ubiquitinated ERAD substrates by the action of HRD1 and SEL1L are extracted from the ER membrane to the cytosol, a process referred to as ‘retrotranslocation’ or ‘dislocation’. This process is coupled to the energy derived from ATP hydrolysis by the AAA ATPase VCP/p97 (Cdc48 in yeast) and its cofactors [8–10]. The accumulation of misfolded proteins in the ER triggers UPR by activating three main UPR sensors, IRE1α, PERK and ATF6, on the ER membrane (reviewed in [3,11]). They subsequently initiate signaling pathways to enhance protein folding and degradation, and attenuate protein translation. While the functions of UPR and autophagy have been extensively characterized both in vitro and in vivo, we are only just beginning to appreciate the physiological importance of ERAD in vivo [7].
While the ER quality-control systems are integrated to assist and monitor protein folding and/or degradation, thereby maintaining ER homeostasis under different physiological cues, the crosstalk among these quality control systems has not yet been fully investigated. Here, we first review the history and major advances in the ERAD and UPR fields, and then highlight recent advances in our understanding of the functions of IRE1α and SEL1L-HRD1, with a focus on their crosstalk.
Historic Perspectives
During the early 1990s, Peter Walter at University of California, San Francisco and Kazutoshi Mori at Kyoto University independently identified an ER-to-nucleus signaling pathway known as the UPR, mediated by an ER membrane protein called ERN1 or inositol requiring enzyme 1 (IRE1) in budding yeast [12–14]. Soon after, they reported identification of a gene encoding the IRE1-specific substrate HAC1, a transcription factor [15–17]. Later, the Kaufman group at the University of Michigan and the Ron group at New York University cloned the mammalian homolog IRE1α [18,19]. In addition to IRE1α, two other major sensors of ER stress, the pancreatic eIF2α kinase (PEK) or protein kinase R (PKR)-like ER kinase (PERK), and the activating transcription factor 6 (ATF6), were later identified in worms and mammals by the Shi/Wek and Mori groups, respectively [20,21]. Among the three UPR signaling branches, IRE1α is the most conserved ER stress sensor, from budding yeast to humans.
Around the same time when yeast IRE1 was identified, several laboratories were in a race to identify the mysterious proteolytic system(s) for ER proteins. In 1988–1991 Jennifer Lippincott-Schwartz and Richard Klausner at the National Institutes of Health showed that the degradation of unassembled subunits of T cell receptor (TCR) occurs at a site closely associated with the ER, through a lysosome-independent mechanism [22,23]. Shortly after, work by the Dieter Wolf group helped to define a set of misfolded proteins, including a mutated version of a soluble protein carboxypeptidase Y (CPY*), as the first ERAD substrate in yeast [24]. These findings later contributed to the establishment of a concept of protein quality control in the ER. During the period 1992–1997, several laboratories showed that the degradation of ER proteins, including TCR subunits [25], cystic fibrosis transmembrane conductance regulator [26,27], mutant α1-antitrypsin [28], yeast carboxypeptidase [29], prepro-alpha factor [30], and MHC class I heavy chains [31,32], all required the ubiquitin-proteasome pathway in the cytoplasm. In 1996, Ardythe McCracken and Jeffrey Brodsky at University of Pittsburgh termed this specialized destruction process ‘ERAD’ [33]. Around the same time, Randy Hampton at University of California, San Diego performed a genetic screen in budding yeast of the turnover of HMG-CoA reductase (HMGCR), a rate-limiting enzyme involved in cholesterol biosynthesis, which led to the identification a group of proteins termed ‘Hrd’ (HMG-CoA reductase degradation) [34]. This family includes the E3 ligase Hrd1p and its cofactor Hrd3p [35]. Dieter Wolf at Universität Stuttgart independently performed a similar screen for mutants defective in ERAD using CPY* in budding yeast, which led to discovery of the genes in the Hrd1 pathway, such as Der1 and Der3/Hrd1 [36,37]. These two nonbiased studies indicated that HRD1 has a role in the degradation of both membrane and ER luminal proteins.
Notably, before the identification of yeast Hrd1p/Hrd3p, the Greenwald group at Columbia University identified Caenorhabditis elegans SEL-11 (Hrd1p homolog) and SEL-1 (Hrd3p homolog) as negative regulators of LIN12/NOTCH-like protein, which were later proposed to regulate LIN12 turnover [38–40]. In 1997, the mammalian homolog of SEL-1 was cloned and named ‘SEL1L’ by Ida Biunno [41]; in 2002, the human HRD1 gene was first cloned from HEK293 cDNA as a homolog to yeast Hrd1p by the Nomura group at Hokkaido University [42]. Concurrently, HRD1 was also termed as Synoviolin by the Nakajima group in 2003 because it was cloned from rheumatoid synovial cells [43]. Its upregulation in synovial tissues from patients with rheumatoid arthritis has implicated HRD1 in arthropathy [43]. However, neither HRD1 nor SEL1L were not recognized as ERAD components until 1–2 years later, when the Wiertz and Ploegh groups at Leiden University Medical Center and Harvard Medical School demonstrated the requirement of human HRD1 and SEL1L, respectively, in ERAD complex formation and model substrate degradation [44,45].
Key breakthroughs and major events related to IRE1α UPR and SEL1L-HRD1 ERAD are shown in Figure 1. Here, we highlight recent advances in our understanding of the function of IRE1α and the SEL1L-HRD1 protein complex, focusing on their crosstalk. Readers are referred to several recent excellent reviews on the topics related to ERAD [5,7,46,47] and UPR [3,11] in general.
Figure 1. Timeline for Key Discoveries in the SEL1L-HRD1 Endoplasmic Reticulum (ER)-Associated Degradation (ERAD) and Inositol-Requiring Enzyme 1 (IRE1)-α Unfolded Protein Response (UPR) Fields.

A chronology of notable events throughout the history of IRE1α UPR (A) and HRD1-SEL1L ERAD (B) over the past three decades [8,12–22,24,26–35,37,42,44,45,50–52,55,56,58,67,80,96,101,111,112,120,131–139].
IRE1α of UPR
IRE1α is a type I transmembrane protein with molecular weight of 110 KDa, containing an N-terminal luminal sensor domain as well as cytosolic kinase and RNase domains. Under basal conditions, IRE1α is present largely in an inactive state, and is found associated with the ER chaperone (see Glossary) BiP/Grp78 (reviewed in [11]). Upon activation, IRE1α self-associates into dimers orhigher-orders oligomers,[48,49], leadingto trans-autophosphorylationof the IRE1α kinase domain and, subsequently, the activation of the RNase domain. Activated IRE1α initiates the unconventional mRNA splicing of X-box binding protein (XBP1), generating a frameshift that encodes a potent transcription factor, XBP1s. XBP1s activates the transcription of a subset of UPR-related genes linked to protein folding, secretion, ERAD, and lipid synthesis [50–54]. In addition, IRE1α may be involved in a promiscuous RNA degradation process known as regulated IRE1-dependent decay (RIDD), for ER-localized mRNAs, ribosomal RNA, and miRNAs [55].
Below, we discuss recent findings on the newly identified IRE1α binding proteins HSP47 and ERdj4 (Figure 2). Other IRE1α interactors or regulators, such as BAX, BAK, BI-1, AIP1, NMIIB, RACK1, PKA, PP2A, PARP16, and others, have been reviewed extensively elsewhere and will not be discussed here [11].
Figure 2. Regulators of Inositol-Requiring Enzyme 1 (IRE1)-α Activation.
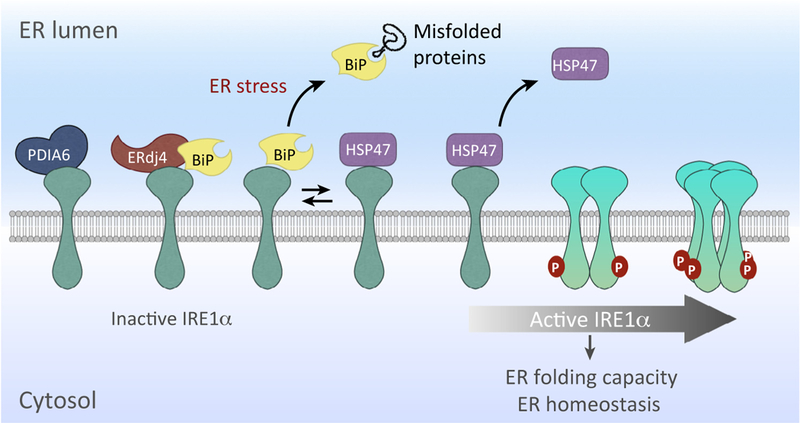
The activity of IRE1α is regulated through the binding of several cofactors that modulate its inhibition [BiP, ERdj4, and protein disulfide isomerase (PDI)-A6] and activation (HSP47). IRE1α is maintained as a monomer in a repressed state under basal conditions through an association with BiP. The endoplasmic reticulum (ER) chaperone HSP47 binds to IRE1, enhancing its activation by displacing BiP, which facilitates IRE1α dimerization/oligomerization for activation. Following activation, IRE1α oligomers are converted to monomers by association with PDIA6. ERdj4 represses IRE1α by recruiting BiP.
HSP47
In resting cells, inactive IRE1α is found associated with BiP, one of several abundant ER chaperones in the ER [56]. During ER stress, BiP is recruited to misfolded proteins, accompanied by IRE1α activation (Figure 2). While BiP was the first protein found to interact with the luminal domain of inactive IRE1α, the detailed events regulating IRE1α activation in the ER lumen have remained unclear.
A recent study by Sepulveda et al. [57] uncovered ER chaperone HSP47 as a novel IRE1α regulator. The authors identified HSP47 as a binding partner of IRE1α in a mass spectrometry-based immunoprecipitation proteomics screen of epitope-tagged IRE1α in mouse embryonic fibroblasts (MEFs). Overexpression of HSP47 in MEFs not only enhanceed XBP1 mRNA splicing and transcription of several XBP1s-dependent UPR target genes, but also increased the RIDD activity of IRE1α during ER stress. Indeed, further analyses demonstrated that HSP47 positively regulates IRE1α activity (as shown by its oligomerization and phosphorylation), while not altering the total IRE1α protein levels. Conversely, downregulation of HSP47 delays IRE1α activation and attenuates XBP1 mRNA splicing.
Mechanistically, the authors showed that HSP47 directly competes with BiP for binding to the luminal domain of IRE1α. Under ER stress, BiP dissociates from IRE1α with a concurrent increase in IRE1α-HSP47 association (Figure 2). Once disengaged from IRE1α to interact with misfolded proteins, BiP may have only limited ability to return due to IRE1α occupancy by HSP47. Similarly, HSP47 binding may accelerate and stabilize IRE1α oligomerization, leading to rapid IRE1α activation. Analysis of the binding kinetics of HSP47 and BiP with the dimerization-defective IRE1α-D123P mutant (aspartic acid at position 123 mutated to proline [58]) revealed that the IRE1α-D123P mutant constitutively interacts with BiP but not HSP47 and, hence, is largely unresponsive to ER stress. Furthermore, in vivo analysis of Drosophila melanogaster with HSP47 knockdown challenged by pharmacological ER stress demonstrated the importance of HSP47 in IRE1α activation and signaling. Future studies are needed to further dissect the competitive relationship between BiP and HSP47 in IRE1α activation and to delineate the physiological significance of HSP47 in the activation of the other UPR sensors, PERK and ATF6.
ERdj4
The Ron laboratory reported the identification of the ER co-chaperone ERdj4 as a cofactor for BiP to cooperatively regulate IRE1 activation [59]. When ATP is bound, BiP exhibits low affinity for IRE1α [59]. ATP hydrolysis then switches BiP into a closed conformation to stabilize the binding. Similar to all HSP70s, the intrinsic ATPase activity of BiP is weak, but J-protein co-chaperones, such as ERdj4, accelerate ATP hydrolysis to augment the efficiency of substrate recognition and BiP binding [60,61]. Biochemical studies using purified recombinant proteins showed that ERdj4 recruits BiP and stimulates the ATPase activity of BiP while directly binding to IRE1α. Moreover, the initial weak binding of BiP to IRE1α luminal domain (IRE1LD) became stronger and more stable following ERdj4 J-domain-stimulated ATP hydrolysis, leading to the disruption of IRE1LD dimer to its monomeric form. Given the report that ERdj4 is a downstream target of XBP1s [62], the ‘ERdj4-IRE1α’ axis may represent a negative feedback to fine-tune IRE1α activation and signaling. In line with this notion, ERdj4 deficiency has been shown to cause constitutive ER stress with increased XBP1 mRNA splicing in mice [63], mimicking XBP1 overexpression [64].
Previously, it has been reported that protein disulfide isomerase family 6 (PDIA6) limits IRE1α activity by converting oligomeric IRE1α back to the monomeric form through the cycle of formation and breakage of disulfide bonds between IRE1α and PDIA6 [65]. Thus, we speculate that ERdj4 attenuates IRE1α activation by facilitating BiP binding, following IRE1α deoligomerization by PDIA6 (Figure 2).
Outstanding Questions
All three IRE1 regulators discussed in this section, HSP47, ERdj4, and PDIA6, are IRE1α-XBP1 targets, which raises interesting questions of how these factors work together (cooperatively or competitively) or independently, to regulate the activation state of IRE1α during basal and stress conditions. Given that the IRE1α-BiP complex is still detected in ERdj4-depleted cells, other ERdj proteins may function similarly to help repress IRE1α activation [59]. Indeed, similar to ERdj4 deficiency, deletion of ERdj2 in Chinese hamster ovary (CHO) cells results in an induction of IRE1α RNase activity as measured by a flow cytometry-based reporter assay [59]. Moreover, deletion of ERdj2 also strongly activates the CHOP reporter (a downstream effector of PERK pathway), suggesting that ERdj proteins also regulate PERK activation. It remains to be tested whether the actions of ERdj proteins are conserved for all or only some branches of the UPR.
SEL1L-HRD1 ERAD
ERAD has a critical role in the maintenance of the integrity of the ER proteome by constantly monitoring the folding of polypeptides under normal and stress conditions [4]. Proteins that fail to achieve, or lose, proper folding status are directed for clearance (‘quality’ control), and this mechanism has also been deployed to eliminate excessive copies of certain proteins (‘quantity’ control) [66]. ERAD occurs in a multistep process comprising recognition, extraction, and ubiquitination of ER proteins for cytosolic proteasomal degradation. To accomplish such coordinated processes, several ERAD components are organized into distinct functional networks [67,68]. In a simplified model of ERAD, chaperones and lectins (e.g., BiP, EDEM, ERdj, OS9, XTP3B, etc.) within the ER lumen recognize unfolded nascent polypeptide sub-strates [69–74] and hand them over to membrane-embedded ERAD adaptors (e.g., SEL1L, Erlins, Insigs, etc.) [45,75–79], when then undergo retrotranslocation through a putative channel (candidates includes HRD1, DER1, and DFM1) followed by polyubiquitination [80–82]. Once exposed in the cytosol, VCP/p97 and its helper proteins provide the energy source for substrate extraction from the ER membrane [8,83–86].
A dozen of ERAD E3 ligases that have been identified to date, including HRD1, GP78, TRC8, TEB4/MARCH6, RNF5/Rma1, RNF170, RNF185, and TMEM129. HRD1 (also known as SYVN1) is the best characterized [44,87–95]. SEL1L is an indispensable adaptor of HRD1 because it has an essential role in HRD1 stability and likely also substrate recruitment [74– 76,96–98]. In yeast, Hrd3p is also indispensable for Hrd1p function by stabilizing Hrd1p, but Hrd1p overexpression can bypass Hrd3 requirement [84,99]. In mammals, the relationship between SEL1L and HRD1 is likely to be more complicated depending on cell types and substrates [67], and requires further investigation.
In recent years, the generation and characterization of cell-type specific SEL1L-or HRD1-deficient mouse and cellular models have provided unprecedented insights into the physiological significance of ERAD, and have led to the identification of several endogenous substrates, including IRE1α, B cell development-specific pre-B cell receptor (pre-BCR) and Fas, peroxi-some proliferator activator receptor γb coactivator β (PGC1β), B lymphocyte-induced maturation protein 1 (BLIMP1), p53, NF-E2-related factor 2 (NRF2), and, recently, the prohormones, pro-arginine-vasopressin (proAVP) and pro-opiomelanocortin (POMC) in neuroendocrine cells [96,100–110].
Germline deletion of SEL1L or HRD1 in mice leads to embryonic lethality around embryonic day 11–14 [111,112], while acute deletion of either gene in adult mice leads to premature death within ~3 weeks [96,105]. Subsequent characterization of cell type-specific SEL1L [96,100–102,106,107] and HRD1 [103–105,110,113] deletion in adipocytes, immune cells, enterocytes, and AVP and POMC neurons has revealed the significance of SEL1L-HRD1 ERAD in a cell type-and substrate-specific manner in vivo [7]. For example, mice with SEL1L or HRD1 deficiency in adipocytes exhibit lipodystrophy and postprandial hyperlipidemia [100,105]. Mice with SEL1L ablation in AVP neurons progressively develop polyuria and polydipsia, both characteristic of diabetes insipidus [107]. Mice with SEL1L ablation in POMC neurons develop hyperphagia and obesity even when fed a low-fat chow diet [108]. These studies underscore the pathophysiological importance of SEL1L-HRD1 ERAD in health and disease. For a detailed discussion of this topic, readers are referred to a recent review [7].
Similarly, germline deletion of some ERAD components, such as Derlin-1 [114], Derlin-2 [115], or Ube2j1 [116], but not others (Derlin-3 and HERP [114]), leads to embryonic or perinatal lethality. These findings demonstrate the importance of ERAD in development and normal physiology.
IRE1α Degradation by SEL1L-HRD1 ERAD
In a proteomic analysis of isolated microsomes, IRE1α was found to accumulate in SEL1L-deficient MEFs without any change in gene transcription [101]. Further studies revealed that IREα protein stability is controlled by ERAD both in vivo and in vitro under basal conditions, which occurs in an HRD1-, SEL1L-and OS9-dependent manner (Figure 3, Key Figure). Interestingly, BiP is also required for IRE1α ERAD because depletion of BiP causes dissociation of IRE1α from ERAD components, stabilizing IRE1α. Under ER stress, both BiP and SEL1L-HRD1 are released from IRE1α, attenuating IRE1α degradation and promoting IRE1α stabilization and/or activation. It remains unclear whether dissociation of BiP and ERAD components from IRE1α under stress conditions is sequentially linked or coordinated. Whether ERAD-dependent proteasomal degradation of IRE1α requires cytosolic ERAD apparatus, such as p97, remains to be experimentally tested. However, given the well-established role of p97 in substrate retrotranslocation and extraction from the ER membrane [5], SEL1L-HRD1-mediated IRE1α degradation is likely p97 dependent.
Figure 3.
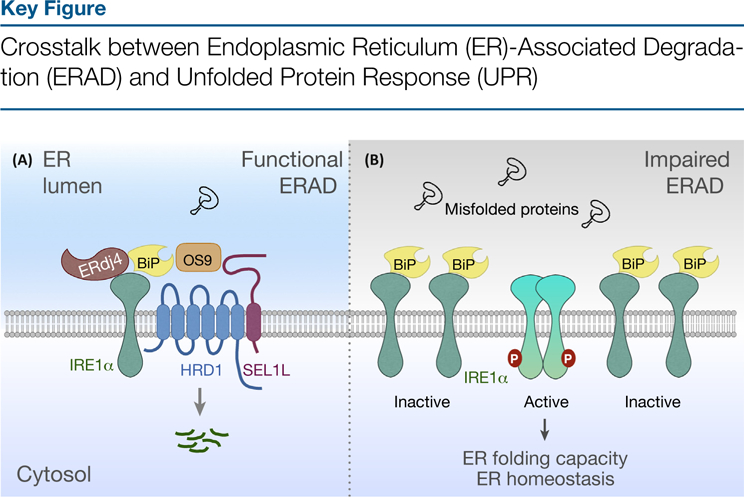
Inositol-requiring enzyme 1 (IRE1)-a is degraded via SEL1L-HRD1 ERAD, a process that depends on HRD1, SEL1L, OS9, and BiP (A). ERAD deficiency results in IRE1α accumulation, leading to its activation under basal conditions (B).
Deletion of either SEL1L or HRD1 in various tissues and cell types, including enterocytes, pancreas, adipose tissue, and MEFs, results in a profound 10–100-fold increase in IRE1α protein, while its mRNA levels are largely unchanged [101]. Moreover, the level of phosphory-lated IRE1α as well as its RNase enzymatic activity as measured by Xbp1 mRNA splicing are moderately elevated in ERAD-deficient cells under basal conditions, pointing to a key role of ERAD in the restraint of IRE1α hyperactivation. Although protein levels are highly elevated in the absence of SEL1L-HRD1 ERAD, IRE1α protein does not form detergent-insoluble aggregates as has been seen with some other ERAD substrates, and the IRE1α branch of the UPR remains responsive to ER stress [101]. This finding suggests that, in ERAD-deficient cells, a majority of accumulated IRE1α remains in a BiP-bound form. Whether accumulated IRE1α in ERAD-deficient models triggers XBP1s-independent effect(s) remains to be determined.
It is not clear whether IRE1α is subjected to ERAD-mediated quality control (due to an intrinsic propensity to misfold) or quantity control (when its protein level becomes disproportionately high). These two events are not necessarily mutually exclusive because SEL1L-HRD1 ERAD is unlikely to sense the abundance of ER proteins, but rather the folding or maturation state of its substrates. Together with the observation that IRE1α interacts extensively with ER chaperones, as discussed above, we propose that newly synthesized IRE1α protein may be misfolding prone and may be recognized and targeted for proteasomal degradation by SEL1L-HRD1 ERAD machinery (Figure 3). Under stress conditions, dimerization or oligomerization of IRE1α may result in the stabilization of IRE1α, which is accompanied by the dissociation of BiP. Alternatively, the presence of an excessive amount of misfolded proteins during stress out-competes IRE1α for ERAD, leading to its stabilization. The regulated degradation of IRE1α by ERAD adds another example of proteins subjected to a regulated quality and/or quantity control, in addition to the best-studied precedent of HMG-CoA reductase (HMGCR), the turnover of which is controlled in response to cellular sterol levels [34,79].
It remains mysterious how endogenous ERAD substrates are recognized and selected. Molecular chaperones and ER lectins are essential components of the process because of their ability to directly bind folding intermediates. Analysis of chaperone-peptide interactions using a peptide library in vivo has revealed that ERdj4 (and ERdj5) specifically binds to aggregation-prone sequences exhibiting substrate-binding patterns distinct from that of BiP [117]. Speculatively, the IRE1α luminal domain may expose a putative aggregation-prone region (probably the same or adjacent region that engages IRE1α dimerization and/or oligomerization) to serve as a recognition element for ERdj4. Given that previous studies have implicated ERdj4 in substrate recruitment for ERAD (e.g., epithelial sodium channel and surfactant protein C [118,119]), we speculate that ERdj4, together with BiP and other chaperones, may be involved in IRE1α ERAD. Hence, ERdj4 may develop two strategies to restrain IRE1α signaling, one through ERAD and the other through BiP-mediated inactivation. Proteo-mics-based identification and validation of additional IRE1α adaptors and modulators in the context of IRE1α ERAD will further advance our understanding of this fundamental degradative event. It is unclear whether ERAD controls the IRE1 protein stability in yeast. If the pathway is conserved, yeast genetic screens will be useful to identify genes involved in IRE1 ERAD.
Feedback Loop between UPR and ERAD in Health and Disease
SEL1L and HRD1 are transcriptionally regulated by the IRE1α-XBP1 signaling pathway [120]. An increase in ERAD activity can alleviate ER stress and restore ER homeostasis by promoting the clearance of unfolded and/or unwanted proteins. Thus, the discovery of regulated IRE1α ERAD represents a unique feedback mechanism. Limiting the amplitude and duration of IRE1α activity at basal levels is critical to maintain cellular homeostasis because aberrant activation of IRE1α can have deleterious outcomes, such as cell death [121]. Impaired ERAD function may alter ER homeostasis not only by affecting the clearance of misfolded proteins in general, but also by upregulating IRE1α activation. Abnormal accumulation and signaling of IRE1α due to impaired ERAD function may be causally linked to the disease pathogenesis.
IRE1α heterozygosity in the intestinal epithelium partially rescued the colitis caused by treatment with the dextran sodium sulfate in enterocyte-specific SEL1L-deficient mice, suggesting that ERAD deficiency in the pathogenesis of experimental colitis is due, at least in part, to a failure to limit excessive IRE1α (level and activity) [101]. However, defects associated with SEL1L-deficient Paneth cells, a secretary cell type in the small intestine, were not rescued by the loss of IRE1α [102]. In addition, a mutation S658P (serine at position 658 to proline) in the SEL1L gene (a mutation located in one of the Sel1-like-repeats in the ER lumen close to the transmembrane domain) has been identified in canines with early-onset cerebellar ataxia [122]. Although the molecular mechanism underlying the SEL1L-S658P mutation remains unclear, the increased expression of several UPR genes, including XBP1, ERdjs, ATF6, CHOP, and ERAD components, suggests that UPR is be elevated in response to a SEL1L defect. It will be of interest to determine the extent to which the SEL1L-S658P mutation leads to loss of function of SEL1L-HRD1 ERAD and, consequently, elevated IRE1α protein levels and signaling.
A loss-of-function mutation within Fam8A1, a component of the SEL1L-HRD1 protein complex [67,123], was identified during the analysis of whole-exome sequencing in 238 families (928 individuals) with autism spectrum disorder (ASD) [124]. It would be interesting to determine whether the Fam8A1 mutation associated with ASD is linked to SEL1L-HRD1 ERAD and, thus, possibly leads to dysregulated IRE1α signaling. Since dysfunction of the UPR and ERAD pathways independently has been reported in association with many human diseases, including neurodegenerative diseases, cancer, diabetes mellitus, ischemia, and infectious diseases, it will be important to determine whether the two pathways contribute cooperatively or synergistically contribute to disease initiation and progression [6]. Even more interesting would be to determine whether IRE1α inhibitors, such as 8866 and MKC-3946, could have beneficial effects in ERAD-associated animal models and human diseases [125,126].
By contrast, loss of function of IRE1α may also contribute to disease pathogenesis. The human somatic cancer-associated IRE1α-P830L mutant (proline at position 830 to leucine at the kinase/RNase junction) [127] has a shorter biological half-life than that of wild-type IRE1α because the mutant protein is structurally unstable [128]. The mutant is unable to form oligomers and autophosphorylates upon ER stress, and, thus, is defective in UPR activation. Although the biochemical mechanism underlying the action of IRE1α-P830L in cancer has not been systematically examined, no-to-low activation of IRE1α protein can cause disease, as shown in the case of diffuse large B cell lymphoma, in which impaired IRE1α gene expression is linked to cancer development [129]. Further investigation of the interplay between IRE1α-P830L, ERAD, and the role of BiP in this process will provide new insights into the importance of crosstalk between ERAD and UPR in disease pathogenesis.
Concluding Remarks and Future Perspectives
Recent studies have revealed that IRE1α and SEL1L-HRD1 ERAD, the two principal ER quality-control machineries, function in an independent and yet cooperative manner in maintaining a healthy ER proteome and cellular function. The notion that IRE1α is subject to negative feedback regulation by SEL1L-HRD1 ERAD in vivo provides an enlightening perspective into the maintenance of ER homeostasis by ER quality-control machineries in health and disease. Maintaining an optimal physiological concentration of IRE1α by ERAD is an important regulatory checkpoint that fine-tunes ERfolding capacity and ER homeostasis. The effect of ERAD on UPRmaynotbe limited to IRE1α, because an in vitro study has shown that SEL1L-HRD1 ERAD is also responsible for the turnover of ATF6 [130]; however, the extent to which this process occurs in vivo and its physiological significance remain unknown (see Outstanding Questions). Future studies will help gain a better understanding of the crosstalk between ERAD and UPR in different physiological and pathological settings, such as aging, cancer, metabolic syndrome, and neurodegeneration.
Outstanding Questions.
What are pathological conditions causally linked to aberrant UPR and/ or compromised ERAD function?
What are the physiological functions and significance of the crosstalk between SEL1L-HRD1 ERAD and IRE1α UPR in a tissue-and cell type-specific manner?
How does the crosstalk between ERAD and UPR affect ER homeostasis in health and disease? Does an imbalance in the crosstalk contribute to disease initiation and progression?
Does the effect of ERAD on UPR go beyond of IRE1α?
Highlights.
The UPR sensor IRE1α and SEL1L-HRD1 ERAD are the two most conserved branches of ER quality-control mechanisms.
IRE1α activation is controlled by different modulators, such as newly identified ERdj4, HSP47, and SEL1L-HRD1 ERAD.
SEL1L-HRD1 ERAD targets IRE1α for proteasomal degradation to restrain IRE1α signaling under basal condition.
Crosstalk between UPR and ERAD is critical for the maintenance of ER homeostasis under physiological and pathological conditions.
Acknowledgments
We apologize to colleagues whose work were not cited due to the space limitations. We thank Peter Arvan, Randy Hampton, and three anonymous reviewers for insightful comments on this manuscript, and members of the Qi laboratory for comments and technical assistance. This work is supported by NIH R01 GM113188, R01 DK105393, R01 DK111174, University of Michigan Protein Folding Diseases Initiative, Juvenile Diabetes Research Foundation (JDRF)2-SRA-2018–539-A-B, and American Diabetes Association (ADA) 1–12-CD-04. L.Q. is the recipient of the Junior Faculty and Career Development Awards from ADA.
Glossary
- BiP/GRP78:
an ER-resident HSP70 chaperone that regulates both protein folding and quality control of unfolded proteins.
- Chaperones:
a functionally related group of proteins assisting protein folding in the cell under physiological and stress conditions. They recognize and bind non-native proteins, thus preventing aggregation, and couple ATP binding/hydrolysis to the folding process. Repeated cycles of client binding and release ensure proper client folding. Typical chaperone members include HSP70 and HSP90.
- Co-chaperones:
assist chaperones in protein folding and other functions. Co-chaperones catalyze the hydrolysis of ATP to ADP on their respective chaperones, which then undergo a large conformational change to either bind to their substrates with higher affinity or aid in the release of the substrate following protein folding.
- HSP47:
an ER luminal chaperone involved in collagen maturation and trafficking as well as the activation of the UPR sensor IRE1α.
- J-protein (also called DnaJ):
defined by the presence of a J domain that can regulate the activity of the HSP70 family, including BiP; is known to be involved in the ERAD. ERdj4 may also be involved in the regulation of IRE1α activity.
- Protein disulfide isomerase (PDI):
a member of the thioredoxin superfamily of redox proteins with three catalytic activities: thiol-disulfide oxidoreductase, disulfide isomerase, and a redox-dependent chaperone. The disulfide isomerase function of PDIA6 has been reported to be important for limiting UPR signaling.
References
- 1.Braakman I and Hebert DN (2013) Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol 5, a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerriero CJ and Brodsky JL (2012) The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev 92, 537–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol 13, 89–102 [DOI] [PubMed] [Google Scholar]
- 4.Olzmann JA et al. (2013) The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol 5, a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christianson JC and Ye Y (2014) Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol 21, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Senft D and Ronai ZA (2015) UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci 40, 141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qi L et al. (2017) New insights into the physiological role of endoplasmic reticulum-associated degradation. Trends Cell. Biol 27, 430–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye Y et al. (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656 [DOI] [PubMed] [Google Scholar]
- 9.Ye Y et al. (2005) Inaugural Article: Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. U. S. A 102, 14132–14138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jarosch E et al. (2002) Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol 4, 134–139 [DOI] [PubMed] [Google Scholar]
- 11.Hetz C and Papa FR (2018) The unfolded protein response and cell fate control. Mol. Cell 69, 169–181 [DOI] [PubMed] [Google Scholar]
- 12.Mori K et al. (1992) A 22 bp cis-acting element is necessary and sufficient for the induction of the yeast KAR2 (BiP) gene by unfolded proteins. EMBO J 11, 2583–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mori K et al. (1993) A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74, 743–756 [DOI] [PubMed] [Google Scholar]
- 14.Cox JS et al. (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206 [DOI] [PubMed] [Google Scholar]
- 15.Cox JS and Walter P (1996) A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87, 391–404 [DOI] [PubMed] [Google Scholar]
- 16.Mori K et al. (1996) Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells 1, 803–817 [DOI] [PubMed] [Google Scholar]
- 17.Sidrauski C and Walter P (1997) The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039 [DOI] [PubMed] [Google Scholar]
- 18.Wang XZ et al. (1998) Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J 17, 5708–5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tirasophon W et al. (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev 12, 1812–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haze K et al. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Y et al. (1998) Identification and characterization of pan-creatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol 18, 7499–7509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lippincott-Schwartz J et al. (1988) Degradation from the endo-plasmic reticulum: disposing of newly synthesized proteins. Cell 54, 209–220 [DOI] [PubMed] [Google Scholar]
- 23.Bonifacino JS and Lippincott-Schwartz J (1991) Degradation of proteins within the endoplasmic reticulum. Curr. Opin. Cell Biol 3, 592–600 [DOI] [PubMed] [Google Scholar]
- 24.Finger A et al. (1993) Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur. J. Biochem 218, 565–574 [DOI] [PubMed] [Google Scholar]
- 25.Yu H et al. (1997) Cytosolic degradation of T-cell receptor alpha chains by the proteasome. J. Biol. Chem 272, 20800–20804 [DOI] [PubMed] [Google Scholar]
- 26.Ward CL et al. (1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 [DOI] [PubMed] [Google Scholar]
- 27.Jensen TJ et al. (1995) Multipleproteolyticsystems, including the proteasome, contribute to CFTR processing. Cell 83, 129–135 [DOI] [PubMed] [Google Scholar]
- 28.Qu D et al. (1996) Degradation of a mutant secretory protein, alpha1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J. Biol. Chem 271, 22791–22795 [DOI] [PubMed] [Google Scholar]
- 29.Hiller MM et al. (1996) ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science 273, 1725–1728 [DOI] [PubMed] [Google Scholar]
- 30.Werner ED et al. (1996) Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc. Natl. Acad. Sci. U. S. A 93, 13797–13801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wiertz EJ et al. (1996) The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 84, 769–779 [DOI] [PubMed] [Google Scholar]
- 32.Wiertz EJ et al. (1996) Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 384, 432–438 [DOI] [PubMed] [Google Scholar]
- 33.McCracken AA and Brodsky JL (1996) Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol 132, 291–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hampton RY et al. (1996) Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell 7, 2029–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bays NW et al. (2001) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol 3, 24–29 [DOI] [PubMed] [Google Scholar]
- 36.Bordallo J et al. (1998) Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell 9, 209–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knop M et al. (1996) Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J 15, 753–763 [PMC free article] [PubMed] [Google Scholar]
- 38.Sundaram M and Greenwald I (1993) Suppressors of a lin-12 hypomorph define genes that interact with both lin-12 and glp-1 in Caenorhabditis elegans. Genetics 135, 765–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grant B and Greenwald I (1997) Structure, function, and expression of SEL-1, a negative regulator of LIN-12 and GLP-1 in C. elegans. Development 124, 637–644 [DOI] [PubMed] [Google Scholar]
- 40.Choi MS et al. (2010) sel-11 and cdc-42, two negative modulators of LIN-12/Notch activity in C. elegans. PLoS One 5, e11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biunno I et al. (1997) Isolation of a pancreas-specific gene located on human chromosome 14q31: expression analysis in human pancreatic ductal carcinomas. Genomics 46, 284–286 [DOI] [PubMed] [Google Scholar]
- 42.Kaneko M et al. (2002) Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation. FEBS Lett 532, 147–152 [DOI] [PubMed] [Google Scholar]
- 43.Amano T et al. (2003) Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel pathogenic factor for arthropathy. Genes Dev 17, 2436–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kikkert M et al. (2004) Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem 279, 3525–3534 [DOI] [PubMed] [Google Scholar]
- 45.Lilley BN and Ploegh HL (2005) Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. U. S. A 102, 14296–14301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruggiano A et al. (2014) Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol 204, 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevenson J et al. (2016) Endoplasmic reticulum-associated degradation and lipid homeostasis. Annu. Rev. Nutr 36, 511–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ron D and Hubbard SR (2008) How IRE1 reacts to ER stress. Cell 132, 24–26 [DOI] [PubMed] [Google Scholar]
- 49.Halbleib K et al. (2017) Activation of the unfolded protein response by lipid bilayer stress. Mol. Cell 67, 673–684 e8 [DOI] [PubMed] [Google Scholar]
- 50.Calfon M et al. (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 [DOI] [PubMed] [Google Scholar]
- 51.Shen X et al. (2001) Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107, 893–903 [DOI] [PubMed] [Google Scholar]
- 52.Yoshida H et al. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- 53.Acosta-Alvear D et al. (2007) XBP1 controls diverse cell type-and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66 [DOI] [PubMed] [Google Scholar]
- 54.Lee AH et al. (2011) Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. U. S. A 108, 8885–8890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hollien J and Weissman JS (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107 [DOI] [PubMed] [Google Scholar]
- 56.Korennykh A et al. (2009) The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sepulveda D et al. (2018) Interactome screening identifies the ER luminal chaperone Hsp47 as a regulator of the unfolded protein response transducer IRE1alpha. Mol. Cell 69, 238–252 e7 [DOI] [PubMed] [Google Scholar]
- 58.Zhou J et al. (2006) The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. U. S. A 103, 14343–14348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amin-Wetzel N et al. (2017) A J-protein co-chaperone recruits BiP to monomerize IRE1 and repress the unfolded protein response. Cell 171, 1625–1637 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mayer MP (2013) Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci 38, 507–514 [DOI] [PubMed] [Google Scholar]
- 61.Awad W et al. (2008) BiP mutants that are unable to interact with endoplasmic reticulum DnaJ proteins provide insights into interdomain interactions in BiP. Proc. Natl. Acad. Sci. U. S. A 105, 1164–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee AH et al. (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol 23, 7448–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fritz JM et al. (2014) Deficiency of the BiP cochaperone ERdj4 causes constitutive endoplasmic reticulum stress and metabolic defects. Mol. Biol. Cell 25, 431–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deng Y et al. (2013) The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. J. Clin. Invest 123, 455–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eletto D et al. (2014) Protein disulfide isomerase A6 controls the decay of IRE1alpha signaling via disulfide-dependent association. Mol. Cell 53, 562–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hegde RS and Ploegh HL (2010) Quality and quantity control at the endoplasmic reticulum. Curr. Opin. Cell Biol 22, 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Christianson JC et al. (2012) Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol 14, 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hwang J et al. (2017) Characterization of protein complexes of the endoplasmic reticulum-associated degradation E3 ubiquitin ligase Hrd1. J. Biol. Chem 292, 9104–9116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bhamidipati A et al. (2005) Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol. Cell 19, 741–751 [DOI] [PubMed] [Google Scholar]
- 70.Kim W et al. (2005) Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol. Cell 19, 753–764 [DOI] [PubMed] [Google Scholar]
- 71.Molinari M et al. (2003) Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299, 1397–1400 [DOI] [PubMed] [Google Scholar]
- 72.Oda Y et al. (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299, 1394–1397 [DOI] [PubMed] [Google Scholar]
- 73.Cormier JH et al. (2009) EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol. Cell 34, 627–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Christianson JC et al. (2008) OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol 10, 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mueller B et al. (2006) SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol 175, 261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mueller B et al. (2008) SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. U. S. A 105, 12325–12330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pearce MM et al. (2007) SPFH2 mediates the endoplasmic reticulum-associated degradation of inositol 1,4,5-trisphos-phate receptors and other substrates in mammalian cells. J. Biol. Chem 282, 20104–20115 [DOI] [PubMed] [Google Scholar]
- 78.Theesfeld CL and Hampton RY (2013) Insulin-induced gene protein (INSIG)-dependent sterol regulation of Hmg2 endoplasmic reticulum-associated degradation (ERAD) in yeast. J. Biol. Chem 288, 8519–8530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Song BL et al. (2005) Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell 19, 829–840 [DOI] [PubMed] [Google Scholar]
- 80.Schoebel S et al. (2017) Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature 548, 352–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Neal S et al. (2018) The Dfm1 derlin is required for ERAD retrotranslocation of integral membrane proteins. Mol. Cell 69, 915. [DOI] [PubMed] [Google Scholar]
- 82.Mehnert M et al. (2014) Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol 16, 77–86 [DOI] [PubMed] [Google Scholar]
- 83.Baldridge RD and Rapoport TA (2016) Autoubiquitination of the Hrd1 ligase triggers protein retrotranslocation in ERAD. Cell 166, 394–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.arvalho P et al. (2010) Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 143, 579–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stein A et al. (2014) Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 158, 1375–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stolz A et al. (2011) Cdc48: a power machine in protein degradation. Trends Biochem. Sci 36, 515–523 [DOI] [PubMed] [Google Scholar]
- 87.Fang S et al. (2001) The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. U. S. A 98, 14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hassink G et al. (2005) TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J 388, 647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Younger JM et al. (2006) Sequential quality-control check-points triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 126, 571–582 [DOI] [PubMed] [Google Scholar]
- 90.Morito D et al. (2008) Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol. Biol. Cell 19, 1328–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stagg HR et al. (2009) The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J. Cell Biol 186, 685–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu JP et al. (2011) RNF170 protein, an endoplasmic reticulum membrane ubiquitin ligase, mediates inositol 1,4,5-trisphosphate receptor ubiquitination and degradation. J. Biol. Chem 286, 24426–24433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van de Weijer ML et al. (2014) A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nat. Commun 5, 3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van den Boomen DJ et al. (2014) TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proc. Natl. Acad. Sci. U. S. A 111, 11425–11430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.El Khouri E et al. (2013) RNF185 is a novel E3 ligase of endoplasmic reticulum-associated degradation (ERAD) that targets cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem 288, 31177–31191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sun S et al. (2014) Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. U. S. A 111, E582–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hosokawa N et al. (2008) Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem 283, 20914–20924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Iida Y et al. (2011) SEL1L protein critically determines the stability of the HRD1-SEL1L endoplasmic reticulum-associated degradation (ERAD) complex to optimize the degradation kinetics of ERAD substrates. J. Biol. Chem 286, 16929–16939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gardner RG et al. (2000) Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J. Cell Biol 151, 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sha H et al. (2014) The ER-associated degradation adaptor protein Sel1L regulates LPL secretion and lipid metabolism. Cell Metab 20, 458–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sun S et al. (2015) IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol 17, 1546–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sun S et al. (2016) Epithelial Sel1L is required for the maintenance of intestinal homeostasis. Mol. Biol. Cell 27, 483–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wu T et al. (2014) Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev 28, 708–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang H et al. (2014) Hrd1-mediated BLIMP-1 ubiquitination promotes dendritic cell MHCII expression for CD4 T cell priming during inflammation. J. Exp. Med 211, 2467–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fujita H et al. (2015) The E3 ligase synoviolin controls body weight and mitochondrial biogenesis through negative regulation of PGC-1beta. EMBO J 34, 1042–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ji Y et al. (2016) The Sel1L-Hrd1 endoplasmic reticulum-associated degradation complex manages a key checkpoint in B cell development. Cell Rep 16, 2630–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shi G et al. (2017) ER-associated degradation is required for vasopressin prohormone processing and systemic water homeostasis. J. Clin. Invest 127, 3897–3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim GH et al. (2018) Hypothalamic ER-associated degradation regulates POMC maturation, feeding and age-associated obesity. J. Clin. Invest 128, 1125–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yamasaki S et al. (2007) Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. EMBO J 26, 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kong S et al. (2016) Endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 controls B-cell immunity through degradation of the death receptor CD95/Fas. Proc. Natl. Acad. Sci. U. S. A 113, 10394–10399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Francisco AB et al. (2010) Deficiency of suppressor enhancer lin12 1 like (SEL1L) in mice leads to systemic endoplasmic reticulum stress and embryonic lethality. J. Biol. Chem 285, 13694–13703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yagishita N et al. (2005) Essential role of synoviolin in embryogenesis. J. Biol. Chem 280, 7909–7916 [DOI] [PubMed] [Google Scholar]
- 113.Xu Y et al. (2016) The ER membrane-anchored ubiquitin ligase Hrd1 is a positive regulator of T-cell immunity. Nat. Commun 7, 12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Eura Y et al. (2012) Derlin-1 deficiency is embryonic lethal, Derlin-3 deficiency appears normal, and Herp deficiency is intolerant to glucose load and ischemia in mice. PLoS One 7, e34298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dougan SK et al. (2011) Derlin-2-deficient mice reveal an essential role for protein dislocation in chondrocytes. Mol. Cell. Biol 31, 1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Koenig PA et al. (2014) The E2 ubiquitin-conjugating enzyme UBE2J1 is required for spermiogenesis in mice. J. Biol. Chem 289, 34490–34502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Behnke J et al. (2016) Members of the Hsp70 family recognize distinct types of sequences to execute ER quality control. Mol. Cell 63, 739–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Buck TM et al. (2010) The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol. Biol. Cell 21, 1047–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dong M et al. (2008) ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol. Biol. Cell 19, 2620–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Travers KJ et al. (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258 [DOI] [PubMed] [Google Scholar]
- 121.Ghosh R et al. (2014) Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158, 534–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kyostila K et al. (2012) A SEL1L mutation links a canine progressive early-onset cerebellar ataxia to the endoplasmic reticulum-associated protein degradation (ERAD) machinery. PLoS Genet 8, e1002759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schulz J et al. (2017) Conserved cytoplasmic domains promote Hrd1 ubiquitin ligase complex formation for ER-associated degradation (ERAD). J. Cell Sci 130, 3322–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sanders SJ et al. (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sanches M et al. (2014) Structure and mechanism of action of the hydroxy-aryl-aldehyde class of IRE1 endoribonuclease inhibitors. Nat. Commun 5, 4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang L et al. (2014) IRE1 inhibition perturbs the unfolded protein response in a pancreatic beta-cell line expressing mutant proinsulin, but does not sensitize the cells to apoptosis. BMC Cell Biol 15, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Greenman C et al. (2007) Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xue Z et al. (2011) A conserved structural determinant located at the interdomain region of mammalian inositol-requiring enzyme 1alpha. J. Biol. Chem 286, 30859–30866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bujisic B et al. (2017) Impairment of both IRE1 expression and XBP1 activation is a hallmark of GCB DLBCL and contributes to tumor growth. Blood 129, 2420–2428 [DOI] [PubMed] [Google Scholar]
- 130.Horimoto S et al. (2013) The unfolded protein response transducer ATF6 represents a novel transmembrane-type endoplas-micreticulum-associated degradation substrate requiring both mannose trimming and SEL1L protein. J. Biol. Chem 288, 31517–31527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liou HC et al. (1988) Distinct cloned class II MHC DNA binding proteins recognize the X box transcription element. Science 242, 69–71 [DOI] [PubMed] [Google Scholar]
- 132.Kozutsumi Y et al. (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464 [DOI] [PubMed] [Google Scholar]
- 133.Shamu CE and Walter P (1996) Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J 15, 3028–3039 [PMC free article] [PubMed] [Google Scholar]
- 134.Bertolotti A et al. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol 2, 326–332 [DOI] [PubMed] [Google Scholar]
- 135.Urano F et al. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 [DOI] [PubMed] [Google Scholar]
- 136.Reimold AM et al. (2000) An essential role in liver development for transcription factor XBP-1. Genes Dev 14, 152–157 [PMC free article] [PubMed] [Google Scholar]
- 137.Meigs TE and Simoni RD (1992) Regulated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase in permeabilized cells. J. Biol. Chem 267, 13547–13552 [PubMed] [Google Scholar]
- 138.Fujita H et al. (2015) The E3 ligase synoviolin controls body weight and mitochondrial biogenesis through negative regulation of PGC-1beta. EMBO J 34, 1042–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hetz C and Glimcher LH (2009) Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol. Cell 35, 551–561 [DOI] [PMC free article] [PubMed] [Google Scholar]