

Abstract
Tyrosine kinase (TK), vascular endothelial growth factor (VEGF), and matrix metalloproteinases (MMP) are important cancer therapeutic target proteins. Based on reported anti-cancer and cytotoxic activities of Caesalpinia bonduc, this study isolated phytochemicals from young twigs and leaves of C bonduc and identified the interaction between them and cancer target proteins (TK, VEGF, and MMP) in silico. AutoDock Vina, iGEMDOCK, and analysis of pharmacokinetic and pharmacodynamic properties of the isolated bioactives as therapeutic molecules were performed. Seven phytochemicals (7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone, 4,4′-dihydroxy-2’-methoxy-chalcone, 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone, luteolin, quercetin-3-methyl, kaempferol-3-O-β-d-xylopyranoside and kaempferol-3-O-α-l-rhamnopyranosyl-(1 → 2)-β-D-xylopyranoside) were isolated. Molecular docking analysis showed that the phytochemicals displayed strong interactions with the proteins compared with their respective drug inhibitors. Pharmacokinetic and pharmacodynamic properties of the compounds were promising suggesting that they can be developed as putative lead compounds for developing new anti-cancer drugs.
Keywords: Caesalpinia bonduc, anti-cancer, molecular docking, tyrosine kinase, vascular endothelial growth factor, matrix metalloproteinases, in silico
Introduction
Cancer is one of the non-communicable diseases that pose a great risk to public health and tough challenge to modern medicine. It is a major cause of reported human deaths worldwide with approximately 9 million deaths and more than 14 million new cases reported each year.1 Deep understanding of the mechanisms of formation and spread of tumour cells is important in the development of new and effective therapeutic agents to induce apoptosis. Apoptosis is a physiological process of cell death, which is well-regulated; thus, cellular inflammatory responses are not induced, making it a safer and better feature of a therapeutic candidate.2 Some techniques such as chemotherapy, radiation, checkpoint inhibitors,3 anti-cancer antibodies,4 and adoptive cell therapies5 have been developed to induce apoptosis. Inhibition of angiogenesis, tumour vascularization, and tyrosine kinase (TK) activity are key therapeutic points that prevent metastasis and cause apoptosis.1 Thus, TK, vascular endothelial growth factor (VEGF), and matrix metalloproteinases (MMP) are important therapeutic targets. Natural products, medicinal plants, and plant-based food have played an important role in management, treatment, and prevention of cancer.6 Plants are exceptional and dependable sources of novel anti-cancer therapeutics responsible for more than 60% of the various anti-cancer agents available.7 Caesalpinia bonduc (Linn) Roxb., commonly known as Gray Nicker nut and Ayo by the Yoruba tribe in south-west Nigeria, is a prickly shrub with a hard, grey, globular-shaped, and smooth shining surface seeds found in tropical and subtropical Africa, Asia, and the Caribbean.8 Glycosides, alkaloids, and cassane/voucapane diterpenoids in seed kernel; cassane diterpene, caesaldekarins (-F, -G, and -C), caesalpinin, and bonducellpins (-A, -B, -C, and –D) in roots; homoisoflavonoids, 6-o-methylcaesalpinianone, and caesalpinianone in bark; and pinitol, brazillin, and bonducin in leaves are various bioactive phytoconstituents that have been identified.9,10 Isolated flavonoids from C bonduc have been reported to have exhibit antiplasmodial and anti-cancer activities against chloroquine sensitive strain of Plasmodium falciparum (FCR-3)8and HeLa11 cells, respectively. Anti-cancer and cytotoxic activities of various parts of C bonduc7,10,11,12 as well as its isolated phytoconstituents13,14 have been reported, with upregulation of Bax and activation of poly (ADP-ribose) polymerase (PARP) as a possible mechanism of apoptosis induction. Despite these broad studies, there is a dearth of information on the molecular interaction between the phytoconstituents and proteins involved in angiogenesis, metastasis, and apoptosis. This study, therefore, investigates the molecular interaction of identified phytochemicals present in young twigs and leaves of C bonduc with human VEGF, TK, and MMP as well as their absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties in silico.
Materials and Methods
Plant material
Young twigs and leaves of C bonduc (Linn) Roxb. were collected from Forestry Research Institute of Nigeria (FRIN), Ibadan, Oyo state, Nigeria. Plant identification was done by Prof. Conrad Omonhinmi, Department of Biological Sciences, Covenant University, Ota, Ogun state, Nigeria. Authentication and voucher referencing were carried out at FRIN with voucher specimen SHI108408 deposited in their herbarium.
Extraction and solvent fractionation of plant material
The leaves and young twigs of the plant collected were air dried at room temperature and powdered. Powdered plant (8.8 kg) was extracted with 75% v/v ethanol (50 L) at normal room temperature (25oC) by maceration for 72 hours using 3 consecutive extractions. The total filtrate was concentrated to dryness with rotary evaporator at 50oC. The dried ethanolic extract of the plant (1120 g) was re-suspended in distilled water (H2O) and partitioned in sequence with petroleum ether, ethyl acetate, and n-butanol. The different solvent fractions were concentrated with rotary evaporator to yield petroleum ether – soluble fraction (150 g), ethyl acetate – soluble fraction (120 g), n-butanol – soluble fraction (170 g), and H2O – soluble fraction (630 g).
Fractionation, isolation, and identification of compounds
Ethyl acetate and petroleum ether soluble fractions were combined (270 g) and separated by column chromatography (CC; 3 kg silica gel, mesh 100–200; solvent: chloroform-methanol [100:1]). Fractions were collected in gradient and this afforded 20 different fractions (C1-C20). Successive column separations of C6 in (silica gel, mesh 200-300; solvent: petroleum ether-acetone [15:1]) and (silica gel, mesh 10-40; solvent: chloroform-ethyl acetate [30:1]) afforded compound 1. Successive CC separation of C10 in (silica gel, mesh 200-300; solvent: chloroform-acetone [30:1]), (silica gel, mesh 200-300; solvent: chloroform-methanol [120:1]), and (silica gel, mesh 10-40; solvent: chloroform-methanol [60:1]) and further separation by high-performance liquid chromatography (HPLC column: YMC-Pack ODS-A, 10 mm × 15 cm, flow rate of 2 mL/min; solvent: methanol-water [60:40] and [58:42]) led to the separation of compounds 2 and 3, respectively. Series of CC separation of C12 in (silica gel, mesh 200-300; solvent: chloroform-methanol [200:1]), (silica gel, mesh 10-40; solvent: chloroform-methanol [40:1]), and (Sephadex LH-20, solvent: chloroform-methanol [1:1]) and further separation by HPLC (YMC-Pack ODS-A, 10 mm × 15 cm, flow rate of 2 mL/min; MeOH: H2O [50:50]) led to the separation of compounds 4 and 5. Sequential separation of C16 by CC in (Sephadex LH-20, solvent: chloroform-methanol [1:1]) and (silica gel, mesh 10-40; chloroform-methanol [20:1]) and separation by HPLC (YMC-Pack ODS-A, 10 mm × 15 cm, flow rate of 2 mL/min; solvent: methanol-water [47:53]) afforded compound 6. Sequential CC separation of C20 in (Sephadex LH-20; solvent: chloroform-methanol [1:1]) and further separation by medium-pressure liquid chromatography (MPLC; RP-18; MeOH: solvent: water [10%-90%]) and HPLC (YMC-Pack ODS-A, 10 mm × 15 cm, flow rate of 2 mL/min; solvent: methanol-water (60:40)) afforded compound 7. Structural identification was carried out by spectroscopic methods.
Ligand modeling
The structures (.sd) of the identified compounds were sketched using ChemDraw and used as ligands.15 The SD files were converted to their corresponding three-dimensional (3D) structures and saved as .pdb format using Open Babel.16 Gasteiger charges were added and non-polar hydrogens were merged using AutoDock4.2.17
In silico analysis of drug-likeness
The drug-likeness of the screened compounds was calculated using SwissADME.18 The ligands were subjected to Lipinski,19 Ghose,20 Veber,21 Egan,22 and Muegge23 screening. Only ligands that were able to satisfy these variants without default were used for docking simulation.
Protein preparation
The 3D crystal structures of human VEGF, human TK, and human MMP were retrieved from Research Collaboratory for Structural Bioinformatics (RCSB) protein data bank (PDB) as 1vpf, 1fku, and 2oxu as their respective PDB codes without any complexed ligands. The protein structures were cleaned, water molecules removed, Gasteiger charges computed, polar hydrogens added, and non-polar hydrogens merged using AutoDock4.2.17
Active site prediction
The possible active binding sites of the proteins were obtained using DoGSiteScorer.24 The binding sites with the best volume, surface area, and druggability score were selected for this study. DoGSiteScorer is a grid-based method, which uses a difference of Gaussian filter to detect potential binding pockets, solely based on the 3D structure of the protein, and splits them into subpockets.
Docking simulations
AutoDock4.2 was used to transform both receptor and ligand structures to pdbqt file format, which includes atomic charges, atom-type definitions and, for ligands, topological information (rotatable bonds) with a grid centred to ensure coverage of the binding site of the structure. AutoDock Vina was used to perform docking simulations, generating 9 conformations of ligand in complex with the receptor, which were finally ranked on the basis of binding energy.25 Target proteins and ligands were docked with iGEMDOCK (version 2.1). The genetic algorithm (GA) parameters, which guided the docking procedure, were set as 200 (population size), 70 (generations), and 3 (number of solution). Bond energies, such as hydrogen bond (Hb), van der Waals (VdW), and electrostatic interaction that occurred between the proteins and ligands were identified. The resulting conformations were visualized in the Discovery Studio Visualizer26 and PyMol.27
ADMET properties
The in silico ADMET properties of the selected compounds were calculated as an alternative approach to the expensive experimental evaluation of ADMET profiles.28 For ADMET assessment, we used admetSAR32 to examine the different pharmacokinetic parameters of the docked ligands.
Results and Discussion
Fractionation, isolation, and identification results
Figure 1 illustrates the 7 flavonoids (7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone; 4,4′-dihydroxy-2’-methoxy-chalcone; 7,4’-dihydroxy-3,11-dehydrohomoisoflavanone; luteolin; quercetin-3-methyl; kaempferol-3-O-β-D-xylopyrano-side; and kaempferol-3-O-α-L-rhamnopyranosyl-(1→2)-β-D-xylopyranoside) that were isolated via bioassay-guided fractionation of both the petroleum ether and ethyl acetate fractions of C bonduc ethanolic extract. These compounds have previously been isolated from different parts of Caesalpinia spp., Draba nemorosa and Bauhinia racemosa.29,30,31,32
Figure 1.
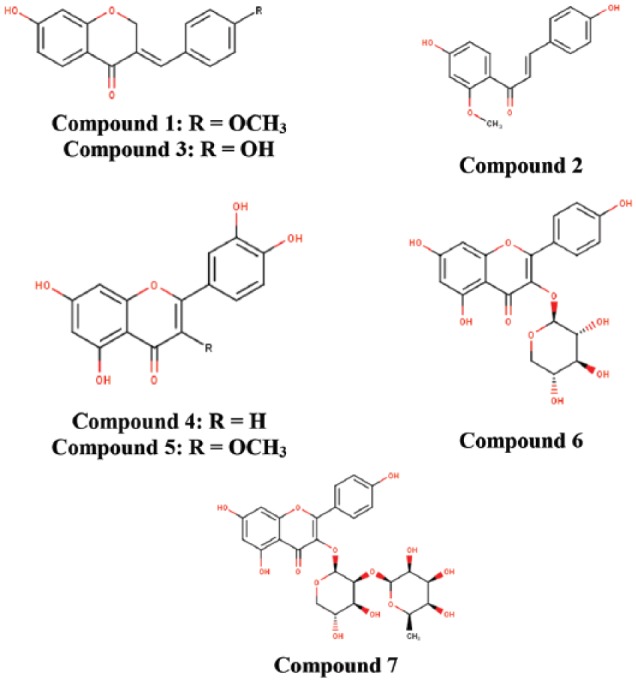
Chemical structures of isolated flavonoids of Caesalpinia bonduc.
Drug-likeness results
In estimating drug-likeness of a compound, the compliance of the compounds’ physicochemical properties with filter variants such as Lipinski rule of 5 (RO5), Ghose filter, Veber rule, Egan rule, and Muegge rule is paid close attention. Properties such as the number of hydrogen bond donors (HBD), hydrogen bond acceptors (HBA), molecular mass, logP, molar refractivity, rotatable bonds, topological polar surface area (TPSA), and number of rotatable bonds are taken into consideration.20,21,22,23,33 Among these isolated phytochemicals, only 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone; 4,4′-dihydroxy-2′-methoxy-chalcone; 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone; luteolin; and quercetin-3-methyl passed all the various drug-likeness tests they were subjected to without any default. Kaempferol-3-O-β-D-xylopyranoside and kaempferol-3-O-α-L-rhamnopyranosyl-(1 → 2)-β-D-xylopyranoside had defaults in all variants except for Ghose test of drug-likeness which kaempferol-3-O-β-D-xylopyranoside passed (Table 1). Lead compounds whose properties successfully pass these variants are considered to possess good pharmacokinetic properties and thus are further subjected to various in silico techniques such as molecular docking.34 Out of the 7 isolated compounds, 5 passed all the filter variants without any default and thus could be considered as lead compounds with good pharmacokinetic properties.
Table 1.
Druglikeness violation of isolated compounds.
| Compound | Name | Number of violations | ||||
|---|---|---|---|---|---|---|
| Lipinski | Ghose | Veber | Egan | Muegge | ||
| 1. | 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone | – | – | – | – | – |
| 2. | 4,4′-dihydroxy-2′-methoxy-chalcone | – | – | – | – | – |
| 3. | 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone | – | – | – | – | – |
| 4. | Luteolin | – | – | – | – | – |
| 5. | Quercetin-3-methyl | – | – | – | – | – |
| 6. | Kaempferol-3-O-β-D-xylopyranoside | 1 | – | 1 | 1 | 2 |
| 7. | Kaempferol-3-O-α-L-rhamnopyranosyl-(1 → 2)-b-D-xylopyranoside | 3 | 3 | 1 | 1 | 3 |
Docking simulations results
Molecular docking is an important in silico technique, which predicts the mode of interaction between a small ligand and target protein for an established binding site.35 Binding energy informs us on the strength and how much affinity a compound binds to the pocket of a target protein (as shown in Figure 2). A compound with a lower binding energy is preferred as a possible drug candidate and vice versa.36,37 Vina uses a sophisticated gradient optimization method in its local optimization procedure25 while iGEMDOCK uses an empirical scoring function and an evolutionary approach.38 Table 2 shows the binding affinity of the 5 isolated compounds as simulated by AutoDock Vina ranging from −5.9 to −7.0, −7.7 to −8.8, and −7.8 to −10.7 kcal/mol for VEGF, TK, and MMP, respectively. These values are comparable with the inhibitors (pazopanib [−7.7 kcal/mol] and imatinib [−10.3 kcal/mol]) of VEGF and TK, respectively. However, these isolated compounds showed a stronger binding affinity compared with batimastat (−6.7 kcal/mol), which is a major drug inhibitor of MMP. In spite of hydrogen, van der Waal, and pi bonds stabilizing interactions between the ligands and amino acids residues present in the active site of the proteins, hydrogen bond was absent in the interaction between 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone and 4,4′-dihydroxy-2′-methoxy-chalcone in the binding pockets of VEGF and TK. Gly 59, Met 341, Thr 338, Lys 295, Asp 404, Leu 273, and Leu 393; Pro 238, Leu 181, Leu 214, Phe 237, Lys 241, His 218, Glu 219, Thr 215, and Thr 239 are the amino acid residues in the binding pockets of VEGF, TK, and MMP that are responsible for the various interactions between these proteins and the various ligands docked (Figures 3, 4 and 5). The docking energies of the 5 isolated compounds as simulated by iGEMDOCK ranged from −81.58 to −96.80, −88.59 to −100.35, and −105.47 to −115.72 kcal/mol for VEGF, TK, and MMP respectively. These values are comparable with the inhibitors (pazopanib [−95.60 kcal/mol] and imatinib [−111.55 kcal/mol]) of VEGF and TK, respectively. The same result was obtained for MMP, whereby all isolated compounds showed a stronger binding affinity than batimastat (−88.10 kcal/mol; Table 3). Hydrogen, van der Waal, and various pi bonds stabilizing interactions were present between the ligands and amino acid residues present in the active site of the proteins. Arg 160 and Asn 397 were amino acid residues in the pockets responsible for the binding of all ligands to TK, while Ala182, Lys 233, and 241 were responsible for ligand binding to MMP (Figures 6, 7 and 8). Despite the differences in the scoring algorithm of both AutoDock Vina and iGEMDOCK, the predicted stronger binding affinity of the isolated compounds to MMP binding pocket than batimastat is predictive of a better Ki value qualifying them as hit inhibitors of MMP and potential leads against cancer.39,40 Several amino acid residues present in the binding pocket of proteins are involved in the various interactions that occur between ligand and their targets. These residues are ligand-dependent as the isolated phytochemicals bind to different residues within the binding pocket. However, Lys 241 was involved in stabilizing MMP-ligand interactions in both AutoDock Vina and iGEMDOCK. This amino acid residue is somewhat amphipathic with a long hydrophobic carbon tail close to the backbone and a positively charged amino group on the side chain. It is frequently found in the binding sites of proteins where they form hydrogen bonds with ligands and also function in acid-base catalysis,41,42 making it a viable drug target.43
Figure 2.

The 3D crystal structures of human (A) VEGF (1vpf), (B) TK (1fmk), and (C) MMP (2oxu) with their binding pockets highlighted in purple and golden brown. MMP indicates matrix metalloproteinases; TK, tyrosine kinase; VEGF, vascular endothelial growth factor.
Table 2.
Molecular docking analysis using AutoDock Vina for isolated compounds showing estimated binding free energy and interacting residues in the binding site of VEGF, TK, and MMP.
| Protein (PDB code) | Ligand | Binding affinity (kcal/mol) | Hb-ir (Å) | VdW-ir | Pi-ir |
|---|---|---|---|---|---|
| VEGF (1vpf) | 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone | −6.7 | – | Ile 29, Gly 58, Gly 59, Leu 32, Thr 31, Glu 30, Cys 57 | Ile 29, Gly 58, Gly 59, Leu 32, Thr 31 |
| 4,4′-dihydroxy-2′-methoxy-chalcone | −5.9 | – | Ile 29, Gly 58, Gly 59, Leu 32, Thr 31, Glu 30, Cys 57, Arg 56 | Ile 29, Gly 58, Gly 59, Leu 32, Thr 31 | |
| 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone | −6.6 | Lys 107 (3.34) | Gly 59, Cys 60, Cys 61, Glu 64, Glu 67, Ser 50, Asp 63, Phe 47, Phe 36, Leu 66 | Ile 46, Cys 68 | |
| Luteolin | −7.0 | Cys 61 (3.33), Cys 68 (3.38) | Phe 36, Phe 47, Lys 107, Glu 67, Leu 66, Cys 60, Gly 59 Asn 62, Asp 63 | Ser 50,Glu 64, Ile 46 | |
| Quercetin-3-methyl | −6.3 | Asp 63 (3.26) | Gly 59, Cys 68, Cys 61, Cys 51, Glu 67, Lys 107, Leu 66, Phe 36, Ile 46, Asn 62, Asp 34 | Cys 60, Glu 65, Ser 50 | |
| Pazopanib | −7.7 | Gly 59 (2.52), Asp 34 (2.40), Ser 50 (2.89) | Thr 31, Glu 64, Glu 67, Glu 30, Leu 66, Leu 32, Asp 63, Asp 34, Gly 58, Cys 61, Cys 68, Cys 51, Lys 107, Phe 36, Val 33 | Leu 32, Ile 29, Cys 60, Thr 31 | |
| TK (1fmk) | 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone | −8.6 | – | Gly 344, Tyr 340, Met 341, Val 323, Phe 405, Ile 336, Ser 345, Asp 348 | Asp 404, Ala 403, Ala 293, Lys 295, Leu 273, Leu 393, Thr 338, Val 281 |
| 4,4′-dihydroxy-2′-methoxy-chalcone | −7.7 | – | Phe 405, Ile 336,Val 323, Tyr 340, Leu 393, Ser 345, Ser 342, Met 341, Gly 344, Gly 274, Asp 348 | Asp 404, Ala 404, Ala 293, Lys 295, Leu 273, Thr 338 | |
| 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone | −8.8 | Asp 348 (3.38) | Gly 344, Tyr 340, Met 341, Val 323, Val 281, Ile 336, Ser 345 | Leu 273, Leu 393, Lys 295, Ala 293, Ala 403 Asp 404, Thr 338 | |
| Luteolin | −8.7 | Asp 404 (3.03), Met 341 (3.11, 3.47). | Tyr 340, Val 323, Ile 336 | Lys 295, Ala 403, Ala 293, Leu 273, Leu 393, Val 281,Gly 344, Ser 345, Thr 338, Asp 404 | |
| Quercetin-3-methyl | −7.7 | Asn 391 (3.05) | Met 341, Ala 293, Thr 338, Asp 404,Arg 388, Cys 277, Gln 275, Ser 345, Gly 274, Gly 344 | Leu 273, Ala 390, Leu 393, Val 281, Gly 276 | |
| Imatinib | −10.3 | – | Leu 325, Ile 336, Gln 275, Gly 274, Gly 279, Gly 276, Gly 406, Cys 277, Asp 386, Arg 388, Ala 390, Ser 345, Met 341, Tyr 340, Glu 339, | Val 323, Val 281, Ala 293, Ala 403, Lys 295, Leu 273, Leu 393, Phe 405, Phe 278, Asp 404, Thr 338, Asn 391 | |
| MMP (2oxu) | 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone | −10.7 | Lys 233 (3.00), Lys 241(3.31) | Pro 232, Ala 234, Phe 237, Tyr 240, Thr 215, Leu 181, Phe 248, Leu 214, Pro 238, Zn 264 | Val 243, Val 235, His 218, Thr 239, Glu 219 |
| 4,4′-dihydroxy-2′-methoxy-chalcone | −9.3 | Lys 241 (3.19), Lys 233 (2.89) | Pro 232, Ala 234, Phe 237, Tyr 240, Thr 215, Leu 181, Phe 248, Leu 214, Pro 238, Glu 219, | Val 243, Val 235, His 218, Thr 239 | |
| 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone | −10.4 | Lys 233 (3.06) | Pro 232, Ala 234, Phe 237, Tyr 240, Thr 215, Leu 181, Phe 248, Leu 214, Pro 238, Lys 241 | Val 243, Val 235, His 218, Thr 239, Glu 219 | |
| Luteolin | −8.8 | Pro 232 (2.46), Pro 238 (2.88) | Phe 248, Val 243, Ala 234, Phe 237, Ala 182, Thr 215, Leu 214, Zn 264 | Tyr 240, His 218, Lys 241, Thr 239, Leu 181, Thr 239, Glu 219 | |
| Quercetin-3-methyl | −7.8 | Phe 237 (3.37), Thr 239 (3.36) | His 183, Ala 184, His 222, Ala 182, Glu 219, Val 235, Lys 241, Leu 214, Tyr 240, Leu 181, Pro 238, Zn 264 | Ile 180, His 218, Thr 215, Gly 179 | |
| Batimastat | −6.5 | Ala 182 (3.00, 2.77), Glu 219 (2.19,2.37,2.45), Leu 181 (3.12) | Thr 239, Thr 215, Leu 214, Ala 234, Ala 184, His 218, His 222, His 183, His 172, His 228, Phe 237, Pro 238, Gly 179, Ile 180, Zn 264 | Leu 181, Tyr 240, Val 235, Lys 241 |
Abbreviations: Hb-ir, hydrogen bonds interacting residues; MMP, matrix metalloproteinases; Pi-ir, Pi bonds interacting residues; TK, tyrosine kinase; VdW-ir, van der Waals interacting residues; VEGF, vascular endothelial growth factor.
Figure 3.

The 2D and 3D views of (A) 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone, (B) 4,4′-dihydroxy-2′-methoxy-chalcone, (C) 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone, (D) luteolin, (E) quercetin-3-methyl, and (F) pazopanib interactions with VEGF using AutoDock Vina. In each case, the hydrogen, pi-donor hydrogen, pi-alkyl, pi-sigma, and pi-anion bond interactions are shown as green, light blue, magenta, purple, and orange broken lines, respectively, while ligand backbones are green. 2D indicates two-dimensional; 3D, three-dimensional; VEGF, vascular endothelial growth factor.
Figure 4.

The 2D and 3D views of (A) 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone, (B) 4,4′-dihydroxy-2′-methoxy-chalcone, (C) 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone, (D) luteolin, (E) quercetin-3-methyl, and (F) imatinib interactions with TK using AutoDock Vina. In each case, the hydrogen, pi-donor hydrogen, pi-alkyl, pi-sigma, pi-pi, and pi-anion bond interactions are shown as green, light blue, magenta, purple, indigo, and orange broken lines, respectively, while ligand backbones are represented in green. 2D indicates two-dimensional; 3D, three-dimensional; TK, tyrosine kinase.
Figure 5.

The 2D and 3D views of (A) 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone, (B) 4,4′-dihydroxy-2′-methoxy-chalcone, (C) 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone, (D) luteolin, (E) quercetin-3-methyl, and (F) batimastat interactions with MMP using AutoDock Vina. In each case, the hydrogen, pi-donor hydrogen, pi-alkyl, pi-sigma, pi-pi, pi-sulphur, and pi-anion bond interactions are shown as green, light blue, magenta, purple, indigo, golden brown, and orange broken lines, respectively, while ligand backbones are represented in green. MMP indicates matrix metalloproteinases; 2D, two-dimensional; 3D, three-dimensional.
Table 3.
Molecular docking energies for isolated compounds and standards in the binding site of VEGF, TK, and MMP using iGEMDOCK.
| Protein (PDB code) | Ligand | TE (kcal/mol) | VdW (kcal/mol) | Hb (kcal/mol) | EI (kcal/mol) |
|---|---|---|---|---|---|
| 1vpf | 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone | −87.6998 | −81.1329 | −6.56699 | 0 |
| 4,4′-dihydroxy-2′-methoxy-chalcone | −81.5765 | −77.2059 | −4.37062 | 0 | |
| 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone | −88.408 | −73.3137 | −15.0943 | 0 | |
| Luteolin | −87.7781 | −79.8252 | −7.95291 | 0 | |
| Quercetin-3-methyl | −96.7964 | −88.7148 | −8.0816 | 0 | |
| Pazopanib | −95.5916 | −75.2912 | −20.3724 | 0 | |
| 1fmk | 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone | −96.0817 | −90.777 | −5.30469 | 0 |
| 4,4′-dihydroxy-2′-methoxy-chalcone | −88.5854 | −73.2362 | −15.3492 | 0 | |
| 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone | −89.4101 | −65.0273 | −24.3828 | 0 | |
| Luteolin | −102.786 | −77.3195 | −25.4669 | 0 | |
| Quercetin-3-methyl | −100.347 | −79.3807 | −20.9667 | 0 | |
| Imatinib | −111.552 | −104.552 | −7 | 0 | |
| 2oxu | 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone | −114.791 | −106.307 | −8.48373 | 0 |
| 4,4′-dihydroxy-2′-methoxy-chalcone | −115.724 | −101.233 | −14.4907 | 0 | |
| 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone | −111.687 | −99.0438 | −12.6431 | 0 | |
| Luteolin | −105.468 | −91.6619 | −13.8058 | 0 | |
| Quercetin-3-methyl | −110.402 | −88.9414 | −21.4609 | 0 | |
| Batimastat | −88.1012 | −78.874 | −9.22726 | 0 |
Abbreviation: EI, electrostatic interaction; HB, hydrogen bond; TE, total energy; VdW, van der Waals interaction.
Figure 6.

The 2D and 3D views of (A) 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone, (B) 4,4′-dihydroxy-2′-methoxy-chalcone, (C) 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone, (D) luteolin, (E) quercetin-3-methyl, and (F) pazopanib interactions with VEGF using iGEMDOCK. In each case, the hydrogen, pi-donor hydrogen, pi-alkyl, amide pi-stacked, pi-sulphur, pi-lone pair, and pi-cation bond interactions are shown as green, light blue, magenta, purple, indigo, golden brown, light green, and orange broken lines, respectively, while ligand backbones are represented in green. 2D indicates two-dimensional; 3D, three-dimensional; VEGF, vascular endothelial growth factor.
Figure 7.

The 2D and 3D views of (A) 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone, (B) 4,4′-dihydroxy-2′-methoxy-chalcone, (C) 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone, (D) luteolin, (E) quercetin-3-methyl, and (F) imatinib interactions with TK using iGEMDOCK. In each case, the hydrogen, pi-donor hydrogen, pi-alkyl, pi-sigma, pi-pi stacked, and pi-cation bond interactions are shown as green, light blue, magenta, purple, indigo, and orange broken lines, respectively, while ligand backbones are represented in green. 2D indicates two-dimensional; 3D, three-dimensional; TK, tyrosine kinase.
Figure 8.

The 2D and 3D views of (A) 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone, (B) 4,4′-dihydroxy-2′-methoxy-chalcone, (C) 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone, (D) luteolin, (E) quercetin-3-methyl, and (F) Batimastat interactions with MMP using iGEMDOCK. In each case, the hydrogen, pi-donor hydrogen, pi-alkyl, pi-sigma, pi-pi stacked, pi-cation, and unfavourable bump bond interactions are shown as green, light blue, magenta, purple, indigo, orange, and red broken lines, respectively, while ligand backbones are represented in green. MMP indicates matrix metalloproteinases; 2D, two-dimensional; 3D, three-dimensional.
ADMET results
In the preliminary stages of the drug discovery process, it is important to measure various indices of absorption, distribution, metabolism, excretion, and toxicology (ADMET).44 It is a key and considerable step, which eliminates lead compounds with the ability to elicit hazardous side effects.43 Laboratory experimentation of ADMET is very expensive and time consuming, making in silico ADMET evaluation a much viable option, thereby preventing potential drug failures during clinical trials.45 The docked ligands as illustrated in Table 4, showed no inhibitory side effects on renal organic cation transport. Positive results were observed for human intestinal absorption (HIA), Caco-2 permeability (CP), and blood-brain barrier (BBB) penetrability. However, 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone, quercetin-3-methyl, and 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone exhibited negative results for BBB. When a drug is administered orally, absorption occurs primarily in the intestine. Transporter proteins, efflux proteins, and phase II conjugation enzymes are highly expressed by Caco-2 cells, which make them useful models of transcellular pathways and metabolic biotransformation of test.34,46 The compounds exhibited positive results to both HIA and CP, indicating that these compounds could be absorbed or assimilated through human intestine. Compounds that exhibited positive results for BBB might also have pharmacological brain function. Luteolin being the exception, all other compounds were P-glycoprotein (P-gp) substrates. Nevertheless, none of the ligands were identified as P-gp non inhibitors (Table 4). In metabolism, no ligand was a substrate for all cytochrome isoforms (2C9, 2D6, and 3A4). On the contrary, they all exhibited high cytochrome P450 inhibitory promiscuity (CYP IP). Despite inhibiting isoforms 1A2, 2C9, and 2C19, the isolated compounds are not inhibitors of isoform 2D6 (Table 5). Cytochrome P450 enzymes are responsible for the metabolism and clearance of drugs and xenobiotics from biological systems. Five isoforms (1A2, 2C9, 2C19, 2D6, and 3A4) predominantly expressed in the liver and also in the small intestine, lungs, placenta, and kidneys important in xenobiotic metabolism.47 When any isoform of CYP is inhibited, malfunctioning of drug metabolism and elevation of toxicity is always a usual occurrence.48 The inhibition of 1A2, 2C9, and 2C19 CYP isoforms by the compounds suggests that the phytochemicals may affect drug metabolism; thus, the dosage concentration should be a critical factor to consider during drug design of these lead compounds. The safety of the compounds is absolutely crucial for a successful drug. A drug like a candidate should meet certain ADMET parameters, which is as critical as therapeutic properties.34 The isolated phytochemicals were weak inhibitors of human ether-a-go-go-related genes and exhibit no properties that exert significant toxicity in humans. Only 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone and 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone were positive for Ames toxicity. In contrast, all of the selected compounds were found to present high toxicity for fish, Tetrahymena pyriformis, and honey bees (Table 6). The toxicity profiles of the docked compounds revealed that they were non-mutagenic and carcinogenic.
Table 4.
Predicted molecular adsorption profile of isolated compounds from Caesalpinia bonduc.
| Compound | BBB | HIA | Caco-2 permeability | Caco-2 permeability (Log Papp, cm/s) | Aqueous solubility (LogS) | P-gp substrate | P-gp inhibitor 1 | P-gp inhibitor 2 | ROCT |
|---|---|---|---|---|---|---|---|---|---|
| 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone | − | + | + | 1.6006 | −3.3153 | + | + | − | − |
| 4,4′-dihydroxy-2′-methoxy-chalcone | + | + | + | 1.2136 | −3.3655 | + | − | − | − |
| 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone | − | + | + | 1.4709 | −2.9877 | + | − | − | − |
| Luteolin | + | + | + | 0.9536 | −4.1810 | − | − | − | − |
| Quercetin-3-methyl | − | + | + | 0.3244 | −3.0097 | + | − | + | − |
Abbreviation: BBB, blood-brain barrier penetrability; HIA, human intestinal absorption; P-gp, P-glycoprotein; ROCT, renal organic cation transporter.
Table 5.
Predicted molecular metabolism profile of isolated compounds from Caesalpinia bonduc.
| Compound | CYP450 2C9 substrate | CYP450 2D6 substrate | CYP450 3A4 substrate | CYP450 1A2 inhibitor | CYP450 2C9 inhibitor | CYP450 2D6 inhibitor | CYP450 2C19 inhibitor | CYP450 3A4 inhibitor | CYP IP |
|---|---|---|---|---|---|---|---|---|---|
| 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone | − | − | − | + | + | − | + | + | High |
| 4,4′-dihydroxy-2′-methoxy-chalcone | − | − | − | + | + | − | + | − | High |
| 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone | − | − | − | + | + | − | + | − | High |
| Luteolin | − | − | − | + | + | − | + | − | High |
| Quercetin-3-methyl | − | − | − | + | − | − | − | + | High |
Abbreviation: CYP450, cytochrome P450; CYP IP, cytochrome P450 inhibitory promiscuity.
Table 6.
Predicted molecular toxicity profile of isolated compounds from Caesalpinia bonduc.
| Compound | HEaggRGI 1 | HEaggRGI 2 | AMES toxicity | Carcinogens (Carcinogenicity [three classes]) | FT (pLC50, mg/L) | TPT (pIGC50, μg/L) | HBT | Biodegradation | AOT | RAT (LD50, mol/kg) |
|---|---|---|---|---|---|---|---|---|---|---|
| 7-hydroxy-4′-methoxy-3,11-dehydrohomoisoflavanone | Weak | − | + | − (Not required) | High (−0.9293) | High (1.2201) | High | + | III | 3.0399 |
| 4,4′-dihydroxy-2′-methoxy-chalcone | Weak | − | − | − (Not required) | High (−0.2645) | High (2.5521) | High | − | III | 2.0019 |
| 7,4′-dihydroxy-3,11-dehydrohomoisoflavanone | Weak | − | + | − (Not required) | High (−0.6181) | High (1.0444) | High | + | II | 3.2798 |
| Luteolin | Weak | − | − | − (Not required) | High (0.1014) | High (0.3577) | High | − | III | 2.5046 |
| Quercetin-3-methyl | Weak | − | − | − (Not required) | High (0.5863) | High (1.1489) | High | − | III | 2.6388 |
Abbreviations: AOT, acute oral toxicity; FT, fish toxicity; HBT, honey bee toxicity; HEaggRGI, human ether-a-go-go-related gene inhibition; RAT, rat acute toxicity; TPT, Tetrahymena pyriformis toxicity.
Conclusions
This study focused on the discovery of potent inhibitors of VEGF, TK, and MMP as a potential therapy for cancer using in silico methods. When compared with already established anti-cancer drugs, the binding energy was highly comparable as well as interactions with the proteins. They also possess good ADMET properties, indicating these phytochemical isolates can also be considered safe and thus be further developed into active commercial anti-cancer drugs. Nevertheless, during lead optimization process, the CYP inhibitory promiscuity of the compounds needs to be improved and Lys 241 amino acid residue can be targeted as a promising mode of action to modulate the activity of MMP. Molecular dynamics simulation as well as molecular mechanics energies combined with the Poisson-Boltzmann or generalized Born and surface area continuum solvation (MM-PBSA/GBSA) analyses can be performed to further validate docking results. Further in vitro and in vivo investigations can be performed to further authenticate the physiological relevance of these results.
Footnotes
Funding:The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: FNI and OOO Put up the general concepts and design of the study. FNI and OOO Carried out the implementation of these concepts and design of the work. FNI, OOO, OEO, II and JO Carried out the analysis of the work. FNI Drafted the first manuscript. All authors read and approved the final manuscript.
ORCID iDs: Franklyn Nonso Iheagwam  https://orcid.org/0000-0001-8487-4052
https://orcid.org/0000-0001-8487-4052
Jelili Oyelade
https://orcid.org/0000-0002-5476-4992
References
- 1. Hegedűs C, Kovács K, Polgár Z, et al. Redox control of cancer cell destruction. Redox Biol. 2018;16:59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chakravarti B, Maurya R, Siddiqui JA, et al. In vitro anti-breast cancer activity of ethanolic extract of Wrightia tomentosa: role of pro-apoptotic effects of oleanolic acid and urosolic acid. J Ethnopharmacol. 2012;142:72–79. [DOI] [PubMed] [Google Scholar]
- 3. Simeone E, Ascierto PA. Immunomodulating antibodies in the treatment of metastatic melanoma: the experience with anti-CTLA-4, anti-CD137, and anti-PD1. J Immunotoxicol. 2012;9:241–247. [DOI] [PubMed] [Google Scholar]
- 4. Sun J, Chia S. Adjuvant chemotherapy and HER-2-directed therapy for early-stage breast cancer in the elderly. Br J Cancer. 2017;116:4–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ramos CA, Heslop HE, Brenner MK. CAR-T cell therapy for lymphoma. Annu Rev Med. 2016;67:165–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iheagwam FN, Nsedu EI, Kayode KO, Emiloju OC, Ogunlana OO, Chinedu SN. Bioactive screening and in vitro antioxidant assessment of Nauclea latifolia leaf decoction. AIP Conf Proc. 2018;1954:030015. [Google Scholar]
- 7. Ubhenin A, Uwakwe A, Falodun A, Engel N, Onwuka F, Langer P. Anti-proliferative and pro-apoptotic effects of Caesalpinia bonduc extract and its fractions in estrogen-sensitive human breast adenocarcinoma cell line. J Herbs Spices Med Plants. 2013;19:159–167. [Google Scholar]
- 8. Ogunlana OO, Kim HS, Wataya Y, Olagunju JO, Akindahunsi AA, Tan NH. Antiplasmodial flavonoid from young twigs and leaves of Caesalpinia bonduc (Linn) Roxb. J Chem Pharm Res. 2015;7:931–937. [Google Scholar]
- 9. Shivaprakash P, Balaji KS, Lakshmi GM, et al. Methanol extract of Caesalpinia bonducella induces apoptosis via up-regulation of Bax and activation of PARP in Ehrlich Ascites tumor cells. Med Aromat Plants. 2016;5:2167–0412. [Google Scholar]
- 10. Sandhia KG, Bindu AR. Phytochemical, anti-inflammatory and in vitro anticancer activities of Caesalpinia bonduc stem bark. Int J Pharm Sci Res. 2015;6:50–56. [Google Scholar]
- 11. Ogunlana OO, He WJ, Fan JT, et al. Cytotoxic flavonoids from the young twigs and leaves of Caesalpinia bonduc (Linn) Roxb. Pak J Pharm Sci. 2015;28:2191–2198. [PubMed] [Google Scholar]
- 12. Pillai PG, Suresh P, Mishra G. Induction of apoptosis and inhibitory potential of the methanol extract of Caesalpinia bonduc (L.) Roxb against breast cancer. Ann Plant Sci. 2013;2:284–291. [Google Scholar]
- 13. Yadav PP, Maurya R, Sarkar J, et al. Cassane diterpenes from Caesalpinia bonduc. Phytochemistry. 2009;70:256–261. [DOI] [PubMed] [Google Scholar]
- 14. Wu L, Luo J, Zhang Y, Wang X, Yang L, Kong L. Cassane-type diterpenoids from the seed kernels of Caesalpinia bonduc. Fitoterapia. 2014;93:201–208. [DOI] [PubMed] [Google Scholar]
- 15. Mills N. ChemDraw ultra 10.0. J Am Chem Soc. 2006;128:13649–13650. [Google Scholar]
- 16. O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28:1145–1152. [DOI] [PubMed] [Google Scholar]
- 18. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. [DOI] [PubMed] [Google Scholar]
- 20. Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999;1:55–68. [DOI] [PubMed] [Google Scholar]
- 21. Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. [DOI] [PubMed] [Google Scholar]
- 22. Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43:3867–3877. [DOI] [PubMed] [Google Scholar]
- 23. Muegge I, Heald SL, Brittelli D. Simple selection criteria for drug-like chemical matter. J Med Chem. 2001;44:1841–1846. [DOI] [PubMed] [Google Scholar]
- 24. Volkamer A, Kuhn D, Grombacher T, Rippmann F, Rarey M. Combining global and local measures for structure-based druggability predictions. J Chem Inf Model. 2012;52:360–372. [DOI] [PubMed] [Google Scholar]
- 25. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Accelrys. Discovery Studio Visualizer Software. San Diego, CA: Accelrys; 2014. [Google Scholar]
- 27. DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific LLC; 2013. http://www.pymol.org. [Google Scholar]
- 28. Cheng F, Li W, Zhou Y, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52:3099–3105. [DOI] [PubMed] [Google Scholar]
- 29. Liu AL, Shu SH, Qin HL, Lee SM, Wang YT, Du GH. In vitro anti-influenza viral activities of constituents from Caesalpinia sappan. Planta Med. 2009;75:337–339. [DOI] [PubMed] [Google Scholar]
- 30. Purushothaman KK, Kalyani K, Subramaniam K, Shanmughanathan SP. Structure of bonducellin. A new homoisoflavone from Caesalpinia bonducella. Indian J Chem. 1982;21:383–386. [Google Scholar]
- 31. Namikoshi M, Nakata H, Nuno M, Ozawa T, Saitoh T. Homoisoflavonoids and related compounds. III. Phenolic constituents of Caesalpinia japonica SIEB. et ZUCC. Chem Pharm Bull. 1987;35:3568–3575. [Google Scholar]
- 32. Moon SS, Rahman AA, Manir M, Ahamed VJ. Kaempferol glycosides and cardenolide glycosides, cytotoxic constituents from the seeds of Draba nemorosa (Brassicaceae). Arch Pharm Res. 2010;33:1169–1173. [DOI] [PubMed] [Google Scholar]
- 33. Lipinski CA. Rule of five in 2015 and beyond: target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv Drug Deliv Rev. 2016;101:34–41. [DOI] [PubMed] [Google Scholar]
- 34. Azad I, Nasibullah M, Khan T, Hassan F, Akhter Y. Exploring the novel heterocyclic derivatives as lead molecules for design and development of potent anticancer agents. J Mol Graph Model. 2018;81:211–228. [DOI] [PubMed] [Google Scholar]
- 35. Ripphausen P, Stumpfe D, Bajorath J. Analysis of structure-based virtual screening studies and characterization of identified active compounds. Future Med Chem. 2012;4:603–613. [DOI] [PubMed] [Google Scholar]
- 36. Mirza MU, Noor-Ul-Huda Ghori NI, Adil AR, Manzoor S. Pharmacoinformatics approach for investigation of alternative potential hepatitis C virus nonstructural protein 5B inhibitors. Drug Des Devel Ther. 2015;9:1825–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nisha CM, Kumar A, Nair P, et al. Molecular docking and in silico ADMET study reveals acylguanidine 7a as a potential inhibitor of β-secretase. Adv Bioinformatics. 2016;2016:9258578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang JM, Chen CC. GEMDOCK: a generic evolutionary method for molecular docking. Proteins. 2004;55:288–304. [DOI] [PubMed] [Google Scholar]
- 39. Hughes JP, Rees S, Kalindjian SB, Philpott KL. Principles of early drug discovery. Br J Pharmacol. 2011;162:1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chinthala Y, Thakur S, Tirunagari S, et al. Synthesis, docking and ADMET studies of novel chalcone triazoles for anti-cancer and anti-diabetic activity. Eur J Med Chem. 2015;93:564–573. [DOI] [PubMed] [Google Scholar]
- 41. Betts MJ, Russell RB. Amino acid properties and consequences of substitutions. In: Barnes MR, Gray IC. eds. Bioinformatics for Geneticists. 1st ed Hoboken, NJ: John Wiley & Sons; 2003:289–316. doi: 10.1002/0470867302.ch14. 2003. [DOI] [Google Scholar]
- 42. Sokalingam S, Raghunathan G, Soundrarajan N, Lee SG. A study on the effect of surface lysine to arginine mutagenesis on protein stability and structure using green fluorescent protein. PLoS One. 2012;7:e40410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Encinar JA, Fernández-Ballester G, Galiano-Ibarra V, Micol V. In silico approach for the discovery of new PPARγ modulators among plant-derived polyphenols. Drug Des Devel Ther. 2015;9:5877–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Onawole AT, Kolapo TU, Sulaiman KO, Adegoke RO. Structure based virtual screening of the Ebola virus trimeric glycoprotein using consensus scoring. Comput Biol Chem. 2018;72:170–180. [DOI] [PubMed] [Google Scholar]
- 45. Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nature Biotechnol. 2014;32:40–51. [DOI] [PubMed] [Google Scholar]
- 46. Raj U, Kumar H, Gupta S, Varadwaj PK. Novel DOT1L receptor natural inhibitors involved in mixed lineage leukemia: a virtual screening, molecular docking and dynamics simulation study. Asian Pac J Cancer Prev. 2015;16:3817–3825. [DOI] [PubMed] [Google Scholar]
- 47. Olsen L, Oostenbrink C, Jørgensen FS. Prediction of cytochrome P450 mediated metabolism. Adv Drug Deliv Rev. 2015;86:61–71. [DOI] [PubMed] [Google Scholar]
- 48. Malik A, Manan A, Mirza MU. Molecular docking and in silico ADMET studies of silibinin and glycyrrhetic acid anti-inflammatory activity. Trop J Pharm Res. 2017;16:67–74. [Google Scholar]