

Abstract
Background
Co-colonization by Pseudomonas aeruginosa and Staphylococcus aureus is frequent in cystic fibrosis patients. Polymicrobial infections involve both detrimental and beneficial interactions between different bacterial species. Such interactions potentially indirectly impact the human host through virulence, antibiosis and immunomodulation.
Results
Here we explored the responses triggered by the encounter of these two pathogens to identify early processes that are important for survival when facing a potential competitor. Transcriptional profiles of both bacteria were obtained after 3 h co-culture and compared to the respective mono-culture using RNAseq. Global responses in both bacteria included competition for nitrogen sources, amino acids and increased tRNA levels. Both organisms also induced lysogenic mechanisms related to prophage induction (S. aureus) and R- and F- pyocin synthesis (P. aeruginosa), possibly as a response to stress resulting from nutrient limitation or cell damage. Specific responses in S. aureus included increased expression of de novo and salvation pathways for purine and pyrimidine synthesis, a switch to glucose fermentation, and decreased expression of major virulence factors and global regulators.
Conclusions
Taken together, transcriptomic data indicate that early responses between P. aeruginosa and S. aureus involve competition for resources and metabolic adaptations, rather than the expression of bacteria- or host-directed virulence factors.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-5398-y) contains supplementary material, which is available to authorized users.
Keywords: Pseudomonas aeruginosa, Staphylococcus aureus, Transcriptome, RNAseq, Bacterial competition
Background
Since the recent advent of culture-independent analyses of complete microbial populations, microbe-host-microbiota interactions have opened new avenues for the study of infectious diseases. In this respect, detrimental and beneficial interactions between co-colonizing bacteria may influence the development and persistence of co-infections. Co-infection by different pathogenic bacterial species often increases the severity of the underlying condition, and are more difficult to treat [1]. For instance, Pseudomonas aeruginosa and Staphylococcus aureus are frequently co-isolated in leg ulcers [2], rhino-sinusitis [3], burn wounds and cystic fibrosis (CF) [4, 5]. Studies on animal models have shown that co-infection with these two pathogens leads to delayed wound repair and increased antibiotic resistance in mice [6], and increased mortality in flies [7].
While S. aureus is the dominant bacterial species in the lungs of young CF-patients, P. aeruginosa is dominant during adulthood [8]. Co-infection by P. aeruginosa and S. aureus frequently occurs and is associated with poor patient prognosis [5, 9]. However, data from in vitro and in vivo studies on the interaction between these pathogens remain scarce. Some reports show evidence for increased virulence of co-colonizing strains and decreased antibiotic treatment efficiencies [10, 11], while other studies describe commensal interactions between P. aeruginosa and S. aureus [12, 13]. These studies compared well-characterized laboratory strains of P. aeruginosa to clinical isolates of P. aeruginosa that have evolved within the CF-lung where they might interact with the co-colonizing S. aureus. They showed that laboratory strains of P. aeruginosa such as PA14 and early isolates from CF-patients outcompete S. aureus, while isolates after years of colonization have lost their ability to outcompete S. aureus in co-cultures [13]. This suggests that host-microbe or microbe-microbe adaptations occur during chronic colonization, which may subsequently influence infection progression and outcome.
Microbes can engage in mutualistic, neutral or antagonistic interactions, when sharing the same environment. During the infection, bacteria encounter nutrient poor environments leading to competition for resources and to antibiosis, although cooperative behaviors have also been described [14]. Carbon and nitrogen sources as well as iron are essential nutrients, which are often limited in the host. Thus nutrient scavenging is a widely used strategy among bacteria [15–17]. Antibiosis, which is defined by detrimental interactions between at least two species, usually involves the synthesis of antimicrobial molecules that inhibit growth or kill other bacterial species [18]. Finally, cooperation between bacterial species also occurs, but rather governs the interactions within the microbiota [19]. Interactions between P. aeruginosa and S. aureus have been reported from both in vitro and in vivo conditions. One prominent example is the inhibition of the cytochrome b mediated e-transport, mediated by 2-heptyl-4-quinolone N-oxide (HQNO) produced by P. aeruginosa [20], resulting in formation of small colony variants by S. aureus [21]. Furthermore, P. aeruginosa can lyse S. aureus through the action of the secreted LasA protease [22], allowing P. aeruginosa to get access to iron [23, 24]. Studies have also reported cooperative behaviors, in which P. aeruginosa protects S. aureus from phagocytosis by Dictyostelium discoideum through formation of mixed biofilms [25], and commensal interactions with adapted P. aeruginosa CF-isolates [12, 13, 26].
In this study, we present data from dual transcriptome experiments, which examine the early responses occurring after 3 h co-incubation in both P. aeruginosa and S. aureus. Our data reveal metabolic changes mainly in nucleotide and nitrogen metabolism, decreased expression of virulence factors and induction of phage-related genes in both bacteria. Our results are in agreement with an initial competition for nutrient resources rather than a direct interspecies competition response.
Results
Transcriptional responses in P. aeruginosa and S. aureus
To study the respective responses of P. aeruginosa and S. aureus at the very early stage of interaction, we compared the gene expression profile of both bacteria after 3 h of co-culture versus mono-culture. At 3 h the CFU ratio of P. aeruginosa and S. aureus was approximately 50% and comparable to the initial ratio at t = 0 (Fig. 1) suggesting that effects of potential competition had not translated into changes in population dynamics. S. aureus CFU started to decrease after 4 h (data not shown). Despite the conserved population structure after 3 h of co-culture, the transcriptome in each bacterium was significantly affected by the presence of the other one, when compared to the mono-culture transcriptome. Table 1 shows the significantly differentially expressed genes (false discovery rate (FDR) 5%). 1726 and 1718 genes were down and up-regulated, respectively, in P. aeruginosa co-culture versus mono-culture, while 761 and 730 were down and up-regulated, respectively, when S. aureus was growing in co-culture compared to the mono-culture. The proportion of genes that showed a more than 2-fold change in expression (log2 ratio ≥ 1 or ≤ − 1), accounted for 9.4% of the P. aeruginosa genome (545 genes) and 18.6% of the S. aureus genome (450 genes). Among these, 181 genes (33.2%) and 189 genes (42.1%) were up-regulated, whereas 364 genes (66.8%) and 261 genes (47.9%) were down-regulated in P. aeruginosa and S. aureus, respectively (Table 1, Fig. 2, Additional file 1: Figure S1). The functional classification of the differentially regulated genes showed that metabolism and transport were the most affected classes. Interestingly the proportions of these categories were similar in both organisms with 28.8% (P. aeruginosa) and 27.8% (S. aureus) for metabolism and 12.5% (P. aeruginosa) and 13.3% (S. aureus) for transport functions (Fig. 3). RNAseq data were validated by quantitative real-time PCR for six genes per bacterium (3 up- and 3 down-regulated genes), showing a linear correlation with the transcriptome data (Additional file 2: Figure S2).
Fig. 1.

Experimental setup for culture preparation and RNA extraction. S. aureus and P. aeruginosa strains were first grown separately in microtiter plates under static conditions and then mixed at the same proportion to prepare the co-cultures. Plate counts show no significant differences between replicates. Proportion of P. aeruginosa and S. aureus in co-cultures. Shown are average values of thre three replicates with standard deviations at the beginning of the co-culture (t = 3 h) and after 3 h of incubation (t = 6 h)
Table 1.
Differentially expressed genes in co-culture vs mono-culture (FDR < 5%)
| Condition | Up-regulated | Down-regulated | log2FC > 1 | log2FC < −1 |
|---|---|---|---|---|
| CC vs PM | 1726 | 1718 | 181 | 364 |
| CC vs SM | 761 | 730 | 189 | 261 |
CC co-culture, PM P. aeruginosa mono-culture, SM S. aureus mono-culture
FDR false discovery rate, FC fold-change
Fig. 2.
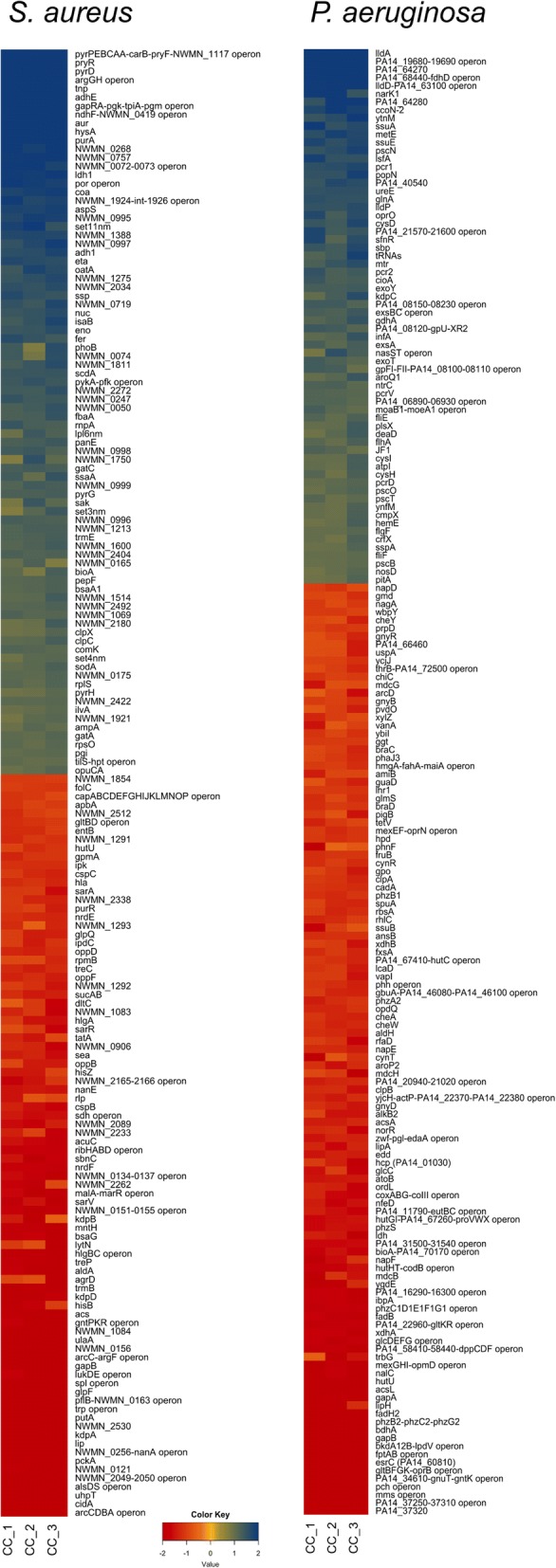
Heat map of differentially expressed genes in P. aeruginosa (left) and S. aureus (right) in co-cultures compared to mono-cultures expressed in log2 base. Only genes showing a log2 ratio ≤ −1 and ≥ 1, are shown. When applicable, genes were clustered in operons
Fig. 3.

Functional classification of genes and their change in expression (> 2-fold up or > 2-fold down) compared to the mono-culture. The x-axis shows the absolute number of genes differentially regulated in the presence of competitor versus the corresponding mono-culture
Oxygen limitation occurs early in co-culture
After 3 h of incubation, the transcriptome data was consistent with a switch from aerobic towards anoxic conditions. Reflected mainly by a 3 to 4-fold reduction in expression of the four cytochrome C oxidase genes (coxA-coxB-PA14_01310-coIII) and the apparent accumulation of nitrate in the cytosol that P. aeruginosa can use as an electron acceptor under anoxic conditions (Table 3). This is achieved by induction of narK1, encoding a cytoplasmic membrane antiporter that exports nitrite in exchange for nitrate [27], as well as nasS encoding a nitrate importer. In contrast, napF and napE, encoding subunits of the periplasmic nitrate reductase were down-regulated in P. aeruginosa. These observations indicate that nitrite production/import is inhibited thereby increasing the cytosolic nitrate concentration, which suggests a preferential use of the inner membrane-associated nitrate reductase (Nar), rather than the periplasmic nitrate reductase (Nap). To assess the oxygen status under the experimental setup, we added resazurin to mono and co-cultures, and followed fluorescence emission. Irreversible reduction of the non-fluorescent resazurin (blue) to the fluorescent resorufin (pink) occurred within less than 1 h for the S. aureus mono-culture and the co-culture (Fig. 4). Reduction was slower with the P. aeruginosa mono-culture reaching a maximal fluorescence after 2 h. Oxygen availability then decreased steadily in mono and co-cultures, but reached an almost undetectable fluorescence level after 14 h only in the co-culture. P. aeruginosa preferentially uses O2 as an electron acceptor and hence more rapidly depletes the available oxygen in the medium.
Table 3.
Selection of differentially expressed genes in P. aeruginosa in co-culture versus mono-culture
| gene | FC a | annotation | functional category |
|---|---|---|---|
| Up-regulated genes | |||
| lldA | 14.13 | L-lactate dehydrogenase | metabolism |
| PA14_19690 | 7.77 | CidB, LrgB anti-holin protein | cell envelope |
| PA14_19680 | 6.51 | CidA, LrgA holin like protein | cell envelope |
| fdnH | 5.55 | nitrate inducible formate DH accessory protein | metabolism |
| PA14_64270 | 5.08 | Leu/Ile/Val-binding protein family signature | transport |
| PA14_55631 | 5.05 | 23S ribosomal RNA | RNA synthesis/modif. |
| PA14_62060 | 5.05 | 23S ribosomal RNA | RNA synthesis/modif. |
| PA14_61830 | 4.88 | tRNA-Met | RNA synthesis/modif. |
| PA14_63100 | 4.38 | cytochrome type D-lactate DH (4Fe-4S) | metabolism |
| narK1 | 3.95 | nitrite extrusion protein I | transport |
| PA14_24780 | 3.87 | Amoonium transporter | transport |
| lldD | 3.64 | L-lactate dehydrogenase | metabolism |
| PA14_60150 | 3.26 | tRNA-Lys | RNA synthesis/modif. |
| popN | 3.13 | type III secretion system | virulence |
| PA14_08670 | 3.01 | tRNA-Thr | RNA synthesis/modif. |
| PA14_08660 | 2.96 | tRNA-Gly | RNA synthesis/modif. |
| lldP | 2.94 | L-lactate permease | transport |
| ureE | 2.93 | urease accessory protein UreE | metabolism |
| glnA | 2.93 | glutamine synthetase | N-metabolism |
| mtr | 2.74 | Tryptophan permease | transport |
| PA14_36220 | 2.61 | Amino acid permease | transport |
| exsB | 2.56 | TTSS regulator | virulence |
| PA14_08210 | 2.52 | F-pyocin | Phage/pyocin |
| gdhA | 2.49 | glutamate dehydrogenase | N-metabolism |
| PA14_06890 | 2.36 | pyruvate kinase pyridoxal phosphate | metabolism |
| PA14_06930 | 2.32 | glutamine amidotransferase | metabolism |
| PA14_08070 | 2.30 | R-pyocin, phage tail protein | Phage/pyocin |
| PA14_06920 | 2.04 | class III pyridoxal phosphate aminotransferase | metabolism |
| Down-regulated genes | |||
| PA14_37310 | −20.35 | allophanate hydrolase subunit II putative | metabolism |
| PA14_37290 | −18.80 | allophanate hydrolase subunit I putative | metabolism |
| PA14_37270 | −18.65 | LamB/YcsF, carbohydrate/lactam utilization | metabolism |
| mmsB | −16.48 | 3-hydroxyisobutyrate dehydrogenase | aa metabolism |
| mmsA | −15.68 | methylmalonate-semialdehyde dehydrogenase | aa metabolism |
| PA14_37260 | −15.13 | OpdO, lactam/pyroglutamate uptake | transport |
| pchB | −14.79 | pyochelin synthesis | virulence |
| PA14_37250 | −12.58 | MFS transporter | transport |
| bkdA1 | −11.87 | 2-oxoisovalerate dehydrogenase subunit alpha | aa metabolism |
| gnuT | −11.53 | gluconate permease | transport |
| pchC | −11.32 | pyochelin synthesis | virulence |
| pchG | −10.48 | pyochelin synthesis | virulence |
| pchA | −9.97 | pyochelin synthesis | virulence |
| pchF | −9.56 | pyochelin synthesis | virulence |
| PA14_23010 | −9.47 | GltK, ATP-binding component of ABC transporter | transport |
| PA14_23000 | −9.37 | permease of ABC sugar transporter | transport |
| PA14_22990 | −9.19 | permease of ABC sugar transporter | transport |
| PA14_23030 | −8.63 | OprB, Glucose/carbohydrate porin | transport |
| bkdA2 | −8.05 | 2-oxoisovalerate dehydrogenase subunit beta | aa metabolism |
| bdhA | −5.81 | 3-hydroxybutyrate dehydrogenase | metabolism |
| bkdB | −4.93 | branched-chain alpha-keto acid dehydrogenase | metabolism |
| lpdV | −3.58 | dihydrolipoamide dehydrogenase | metabolism |
| fptA | −4.85 | pyochelin receptor | virulence |
| mexG | −4.32 | RND efflux pump | transport |
| mexH | −4.12 | RND efflux pump | transport |
| opmD | −3.49 | RND efflux pump | transport |
| mexI | −3.43 | RND efflux pump | transport |
| coxA | −3.90 | Cytochrome c oxidase subunit I | energy metabolism |
| coxB | −2.98 | Cytochrome c oxidase subunit II | energy metabolism |
| PA14_01310 | −2.64 | Cytochrome c oxidase assembly protein | energy metabolism |
| coIII | −2.15 | Cytochrome c oxidase subunit III | energy metabolism |
| napF | −2.97 | periplasmic nitrate reducatse | N-metabolism |
| napE | −2.45 | periplasmic nitrate reducatse | N-metabolism |
aFC fold change; values are the average of three replicates after normalization
Fig. 4.
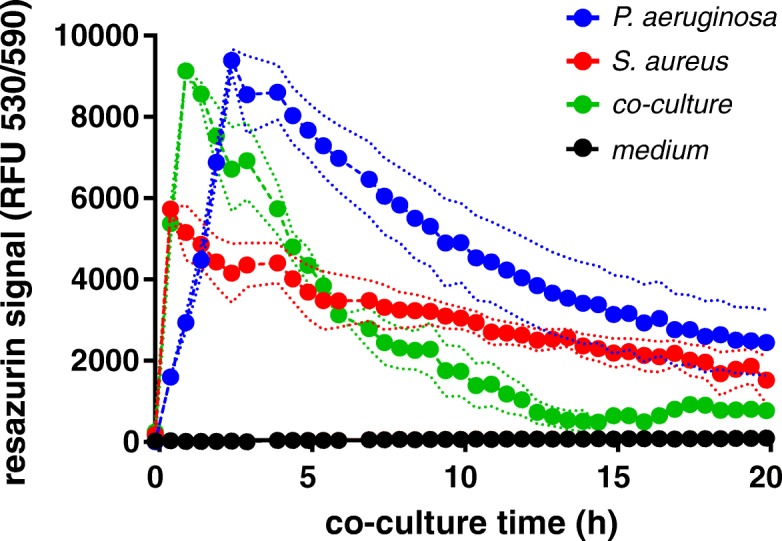
Measurement of oxygen status by resazurin. Oxygen is maximal 1 to 3 h post-inoculation. While in the S. aureus mono-culture, oxgen availability decreases slowly, oxygen depletion occurs faster in co-culture with P. aeruginosa and drops to microaerophilic conditions after 13 h. Shown is the average and the standard deviation (dotted lines) of three independently performed biological replicates. RFU, relative fluorescent units (excitation/emission wavelengths)
Increased expression of fermentation pathways in S. aureus and lactate utilization genes in P. aeruginosa
In response to the decreased oxygen availability, we noticed an increased expression of genes encoding enzymes for fermentation of glucose into pyruvate, encoded by the gap-pgk-tpi-pgm-eno operon in S. aureus (Table 2 and Additional file 3: Table S1). Genes for enzymes involved in conversion of pyruvate into formate (pflB) and acetoin (alsS, alsD) were repressed 6 and 10-fold, respectively. In contrast, the lactate dehydrogenase gene ldh1 and genes of the AdhE pathway (porAB, adhE, adh1) were upregulated 3 to 7-fold, indicating that S. aureus preferentially converted pyruvate into secreted lactate and ethanol. Hence, in our experimental setup, S. aureus mainly produced energy from glycolysis and fermentation of pyruvate into lactate (Table 2 and Additional file 3: Table S1). In line with these observations, the most upregulated gene of P. aeruginosa in response to S. aureus, was the membrane bound L-lactate dehydrogenase LldA (14-fold increase). Concomitant increase in expression (3 to 4-fold) of the second lactate dehydrogenase operon including lldD, the adjacent lldP lactate permease gene (3-fold) and PA14_63100 coding for a cytochrome type lactate dehydrogenase (Table 3 and Additional file 4: Table S2), strongly suggests that P. aeruginosa takes up lactate secreted by S. aureus to use it as a preferential carbon and energy source (Fig. 5).
Table 2.
Selection of differentially expressed genes in S. aureus in co-culture versus mono-culture
| gene | FCa | annotation | functional category |
|---|---|---|---|
| Up-regulated genes | |||
| pyrP | 255.62 | uracil permease | metabolism |
| pyrC | 126.35 | dihydroorotase | metabolism |
| pyrB | 94.35 | aspartate transcarbamoylase | metabolism |
| pyrA | 94.34 | Carbamoyl-phosphate synthase small chain | metabolism |
| carB | 56.66 | Carbamoyl-phosphate synthase large chain | metabolism |
| pyrR | 54.32 | Bifunctional protein PyrR | metabolism |
| pyrF | 37.64 | Orotidine 5′-phosphate decarboxylase | metabolism |
| pyrE | 29.69 | Orotate phosphoribosyltransferase | metabolism |
| pyrD | 24.95 | Dihydroorotate dehydrogenase (quinone) | metabolism |
| argG | 15.30 | Argininosuccinate synthase | aa metabolism |
| argH | 12.49 | Argininosuccinate lyase | aa metabolism |
| adhE | 7.64 | alcohol dehydrogenase | metabolism |
| NWMN_1896 | 6.34 | phage major capsid protein NM1 | phage |
| gapR | 6.00 | glycolytic gap-pgk-tpi-pgm-eno operon | metabolism |
| ndhF | 5.88 | NADH:menaquinone oxidoreductase (subunit 5) | metabolism |
| pgk | 5.81 | glycolytic gap-pgk-tpi-pgm-eno operon | metabolism |
| tpiA | 5.67 | glycolytic gap-pgk-tpi-pgm-eno operon | metabolism |
| gapA | 5.57 | glycolytic gap-pgk-tpi-pgm-eno operon | metabolism |
| pgm | 5.49 | glycolytic gap-pgk-tpi-pgm-eno operon | metabolism |
| porA | 5.18 | Pyruvate flavodoxin ferredoxin oxidoreductase | metabolism |
| purA | 4.42 | Adenylosuccinate synthetase | meatbolism |
| NWMN_0997 | 3.26 | phage NM2 | phage |
| adh1 | 3.17 | Alcohol dehydrogenase | metabolism |
| scdA | 2.72 | Iron-sulfur cluster repair protein ScdA | metabolism |
| NWMN_1213 | 2.37 | Glutathione peroxidase | metabolism |
| sodA | 2.18 | Superoxide dismutase | metabolism |
| hpt | 2.01 | Hypoxanthine phosphoribosyltransferase | metabolism |
| Down-regulated genes | |||
| arcB | −35.81 | Ornithine carbamoyltransferase | aa metabolism |
| arcA | −26.57 | Arginine deiminase | aa metabolism |
| arcD | −22.97 | Arginine/ornithine antiporter | aa metabolism |
| cidA | −16.67 | Holin-like protein CidA | cell envelope |
| alsD | −14.89 | Alpha-acetolactate decarboxylase | metabolism |
| alsS | −10.50 | Alpha-acetolactate synthase | metabolism |
| splD | −10.50 | Serine protease SplD | virulence |
| putA | −6.94 | Proline dehygrogenase | metabolism |
| pflB | −5.92 | Formate acetyltransferase | metabolism |
| capJ | −5.60 | Capsular polysaccharide biosynthesis | cell envelope |
| capI | −5.59 | Capsular polysaccharide biosynthesis | cell envelope |
| trpD | −5.57 | Anthranilate phosphoribosyltransferase | aa metabolism |
| lukE | −5.52 | leukotoxin LukE | virulence |
| glpF | −5.47 | glycerol uptake facilitator | transport |
| capN | −5.46 | Capsular polysaccharide biosynthesis | cell envelope |
| hisB | −5.39 | Imidazoleglycerol-phosphate dehydratas | metabolism |
| trpE | −5.31 | Anthranilate synthase component I | aa metabolism |
| capL | −5.25 | Capsular polysaccharide biosynthesis | cell envelope |
| capO | −4.94 | Capsular polysaccharide biosynthesis | cell envelope |
| splA | −3.91 | Serine protease SplA | virulence |
| splB | −3.44 | Serine protease SplB | virulence |
| sarV | −3.47 | Staphylococcal accessory regulator | signal transduction |
| murP | −3.04 | sucrose-specific PTS transporter IIBC component | transport |
| lytN | −2.35 | cell wall hydrolase | cell envelope |
| sarA | −2.30 | Global transcriptional regulator | signal transduction |
| sarR | −2.20 | Staphylococcal accessory regulator | signal transduction |
| purR | −2.18 | Pur operon repressor | metabolism |
| gltD | −2.11 | Glutamate synthase, small subunit | N-metabolism |
| gltB | −2.10 | Glutamate synthase, large subunit | N-metabolism |
aFC fold change; values are the average of three replicates after normalization
Fig. 5.

Schematic overview of the main reponses deduced by transcriptome analysis of P. aerugiosa and S. aureus after 3 h of co-culture. Vertical black arrows indicate genes up- or down-regulated in the presence of the competitor. Details are found in the text. Fold changes are reported in Tables 2 and 3 and in Additional file 3: Table S1 and Additional file 4: Table S2. Pathways involved in carbon and nitrogen metabolism affected in both organisms during the co-culture are shown in red and orange, respectively. Pathways affected in the co-culture and involved in the generation or in the response to oxidative stress are shown in green in S. aureus and in violet in P. aeruginosa. Induction of the holin/anti-holin like proteins (PA14_19680, PA14_19691) in P. aeruginosa are indicated in yellow (bottom right). After 3 h of co-culture, the synthesis of the siderophores pyoverdin and pyochelin as well as of the phenazines was strongly reduced in P. aeruginosa in response to S. aureus (box in the bottom left corner)
Co-culture induced starvation and competition for glutamine as nitrogen and energy source
After 3 h of co-culture, we detected signs of nutrient starvation and potential responses in both organisms. Nitrogen starvation by P. aeruginosa was highlighted by up-regulation of the master nitrogen regulator gene ntrC, which responds to limitation of N-sources, including urea, purines, amino acids and in particular glutamine [28]. In co-culture, increased expression of ntrC coincided with increased expression of gdhA, glnA and ureE, which respectively code for glutamate dehydrogenase, glutamine synthase, and a urease accessory factor, all involved in nitrogen assimilation (Table 3). In addition, the expression of the entire PA14_06890-PA14_06930 operon, which encodes putative glutamine amido/aminotransferases, was increased in the presence of S. aureus. Interestingly, we also observed a response compatible with nitrogen starvation in S. aureus, since both gltB and gltD, coding for the large and small subunit of glutamate synthase that converts glutamine to glutamate, were significantly decreased during co-culture. Moreover, both bacteria seemed to acquire extracellular N-sources as reflected by the increased expression of NWMN_1750 in S. aureus, encoding a glutamine binding protein, and PA14_24780 in P. aeruginosa, an ammonium transporter. Taken together, these findings indicate that co-culture rapidly induces conditions of nitrogen starvation in both bacteria, where glutamine seems to play a central role (Fig. 5).
Increased pyrimidine/purine synthesis and salvage pathways in S. aureus
Pyrimidines and purines are key components for DNA/RNA synthesis, cell-signaling ((p)ppGpp, c-di-GMP) and energy production (ATP, GTP). The most highly induced gene (255-fold) in S. aureus in response to P. aeruginosa, encodes the uracil transporter PyrP, followed by the pyrimidine de novo synthesis operon (pyrPEBCAA-carB-pyrF-NWMN_117) (126 to 25-fold) (Table 2, Fig. 2 and Additional file 3: Table S1), encoding all enzymes required for the conversion of glutamine to uridine mono-phosphate (UMP). Furthermore, we observed decreased expression of purR, encoding the pur operon repressor, with concomitant increased expression of purA of the de novo purine synthesis pathway. Together with the upregulation of hpt, encoding the hypoxanthine-guanine phosphoribosyl-transferase of the purine salvage pathway, this should result in increased intracellular purine levels. Taken together our observations indicate that activation of pyrimidine and purine synthesis via de novo and salvage pathways represent the most dramatic early response of S. aureus to the presence of P. aeruginosa (Fig. 5).
Amino acid starvation responses and decreased carbohydrate utilization
S. aureus strains are auxotrophic for arginine, proline, valine, cysteine and leucine [29], despite the fact that S. aureus possesses the corresponding biosynthetic pathways [30]. It was therefore surprising to find a very strong response in genes involved in arginine biosynthesis pathways. Genes arcA-arcB-arcC of the arginine catabolic operon were downregulated 33 to 7-fold, while argG and argH, encoding enzymes for conversion of citrulline to arginine were up-regulated (13 to 15-fold). This suggests a strong tendency to maintain or increase cytosolic arginine levels. Furthermore, the putA gene involved in an alternative arginine biosynthesis pathway was downregulated 7-fold (Table 2), probably to maintain sufficient proline concentrations [30]. In P. aeruginosa, a prototrophic organism, an increase in sspA, coding for the stringent starvation protein A, is indicative of amino acid shortage. In agreement with this, genes for branched chain amino acid (leucine, valine, isoleucine) transporters (PA14_64270, PA14_64280) and amino acid permeases (mtr, PA14_36220) were up-regulated 3 to 7-fold. Furthermore P. aeruginosa strongly repressed branched-chain amino acid degradation as illustrated by a 5 to 16-fold decrease in expression of the lpdV-bkdB-bdhA-bdkA1 and mmsA-mmsB operons (Table 3 and Additional file 4: Table S2). Since S. aureus requires leucine and valine, this response could further increase amino acid starvation in S. aureus.
Genes for sugar and carbohydrate metabolism in P. aeruginosa were among the most strongly downregulated genes in the presence of S. aureus. These include the operon containing genes for the glucose/carbohydrate specific OprB porin and the adjacent ABC-transporters (PA14_22990 and PA14_23000) (10-fold decrease), the operon containing the OpdO porin and the co-transcribed ABC-transporter PA14_37250 (15 to 20-fold decrease), involved in pyroglutamate/lactam uptake as well as the gluconate permease gene gnuT. The data clearly illustrate an effort in both organisms to maintain or increase intracellular amino acid pools, while downregulating sugar and carbohydrate uptake and metabolism.
Another indicator of amino acid starvation is the accumulation of probably uncharged tRNAs, observed in P. aeruginosa (Table 3 and Additional file 4: Table S2). Uncharged tRNAs bind the ribosome and stimulate the synthesis of the alarmones ppGpp and pppGppp by the RelA/SpoT enzymes, signaling nutrient starvation stress. In S. aureus we did not observe a similar increase in tRNA pools, but observed up-regulation of tRNA aminoacylation and rRNA genes (Additional file 3: Table S1).
Antioxidant response by S. aureus
The transcriptional profile of S. aureus indicated signs of oxidative stress as illustrated by the increased expression of several genes encoding antioxidant proteins and more strikingly, NWMN_0175, encoding an NO dioxygenase (2-fold) [31], catalyzing NO detoxification by conversion into nitrate as well as sodA (2-fold) encoding the superoxide dismutase, which detoxifies superoxide (O2−) into dioxygen (O2), hydrogen peroxide (H2O2) and H2O [32]. Expression of NWMN_1213, encoding a putative glutathione peroxidase, was also increased (2.4-fold), which would allow further detoxification of the produced hydrogen peroxide produced by turning it into H2O. Finally, expression of scdA, encoding a membrane-repairing protein induced by oxidative stress in cell membranes [33, 34] was increased (2.7-fold). Taken together, our results reveal strong evidence for increased oxidative stress sensed by S. aureus, which could result from acetate-induced ROS production under excess glucose concentrations or be induced by a metabolite [35, 36] (Fig. 5).
Co-culture decreases virulence in S. aureus and P. aeruginosa
P. aeruginosa possesses several multidrug efflux pumps to protect itself from harmful molecules including antibiotics [37]. The expression of one of the resistance-nodulation-division (RND) type efflux pumps, encoded by the mexGHI-opmD operon was decreased 4 to 5-fold in co-culture (Table 3). The MexGHI-OpmD pump has been linked to vanadium resistance [38] and has recently been shown to transport 5-methylphenazine-1-carboxylate, a toxic intermediate in phenazine synthesis [39]. This is in agreement with a 2 to 6-fold downregulation of genes belonging to the two phenazine biosynthesis operons. Unexpectedly, expression of the entire pyochelin synthesis operon (pchDCBA, pchEFG) and of the FptA pyochelin receptor gene was also downregulated 6 to 15-fold, which would indicate that iron was not limiting at this stage of growth. In contrast, expression of several type 3 secretion system (T3SS) genes (exsB-C, pscN, popN, pcr1 and pcr2) was increased 3-fold, despite the obvious absence of eukaryotic cells (Table 3 and Additional file 4: Table S2).
Similarly, S. aureus decreased expression of many virulence factors in response to P. aeruginosa. Among them, the global (sarA) and accessory (sarR, sarV) virulence regulator genes (2 to 3-fold) (Additional file 3: Table S1), the capsular polysaccharide operon capA-capP (2 to 8-fold), as well as leukocidine and the secreted serine protease genes (splA to splF) (Table 2 and Additional file 3: Table S1). Since many of these virulence factors are costly to produce and target host cells, their downregulation should save resources and energy required for metabolic processes. No change in the expression of antibiotic resistance genes was observed.
Opposite responses in cell wall lysis in S. aureus and P. aeruginosa
The CidA-CidB and LrgA/LrgB proteins constitute holin/anti-holin systems in S. aureus that regulate cell lysis in a programmed-cell-death manner and are required for biofilm formation through release of eDNA [40, 41]. Several genes that are involved in programmed cell lysis were significantly down-regulated in response to P. aeruginosa. In particular, the holin protein gene cidA was among the most affected genes (17-fold decrease) in co-culture. Furthermore, expression of the lytN cell wall hydrolase gene and the peptidoglycan degrading enzymes encoded in the NWMN_0134-murQ-murP-murR operon were also repressed in presence of P. aeruginosa (Table 2). In contrast, the so far uncharacterized cidA (PA14_16690, PA3431) and cidB (PA16680, PA3432) orthologues, present in P. aeruginosa were among the three most up-regulated genes in response to S. aureus (Table 3 and Additional file 4: Table S2). These holin/anti-holin systems regulate the access of endolysins to the periplasm and therefore control cell wall degradation and cell death.
Induction of phage and pyocin operons
Intriguingly and in contrast to the above observations, was the induction of all four prophage loci of the S. aureus Newman strain, including the anti-repressor genes of prophages NM2 and NM4, and of structural phage genes from the NM1 (3 to 9-fold induction) and NM3 prophage operons (Table 2 and Additional file 3: Table S1). Similarly, the R- and F-pyocin genes in P. aeruginosa were induced in response to S. aureus (Additional file 4: Table S2). R- and F- as well as S-pyocins are widely distributed in P. aeruginosa strains and are induced in response to mitomycin C, oxidatvive stress (H2O2), as well as ciprofloxacin [42]. Hence, the presence of the co-colonizing organism is recognized as a stress signal and likely causes lysis of a small fraction of the bacterial population.
Discussion
We attempted to analyze the early responses of P. aeruginosa and S. aureus when co-cultured for a short period (3 h) by transcriptomic analysis. Previous reports have focused on biofilm co-cultures in Brain Heart Infusion medium [43] and on growth on a CF-lung epithelium cell line performed in cell culture medium (MEM + 2 mM glutamine) [44]. Despite the very different culture conditions, we noticed some overlapping responses described in these two studies with our in vitro co-culture model but also identified some specific responses. A notable difference with the biofilm study was the very low number of genes (16) differentially regulated in P. aeruginosa [43], compared to the > 500 genes in our study using planktonic cells. This might be due to the different experimental setups, considering that metabolism is reduced during biofilm growth, while in our conditions both bacterial strains were in exponential growth phase requiring maximal metabolic activity.
It is therefore not surprising that the strongest responses measured by gene expression involved metabolic pathways in both bacteria. In particular, we found that S. aureus likely fermented glucose into lactate. As a logical consequence, we observed in P. aeruginosa, increased expression of lactate transporters, and pathways for conversion of lactate into pyruvate, which can subsequently fuel the TCA cycle. A similar response was observed by a previous transcriptome study on S. aureus-P. aeruginosa co-culture performed on a CF-respiratory epithelium, which showed increased lactate production in S. aureus in response to P. aeruginosa [44]. Moreover, the authors showed that P. aeruginosa preferentially uses lactate over glucose when both C-sources are available. Our data obtained after only 3 h of co-culture, and in the absence of epithelial cells, showed down-regulation of pflB, aldA and alsD-alsS genes (acetoin, formate synthesis), and upregulation of ldh (lactate) and adh (ethanol) genes in S. aureus, which is in agreement with the study of Filkins et al. showing similar changes after 16 h of co-culture [44]. This indicates that lactate production is an early co-culture response and is independent of the presence of host epithelial cells. Additionally, we observed upregulation of lactate utilization (lldA, lldD) and uptake (PA14_63100) genes in P. aeruginosa, corroborating their metabolic data.
After 3 h of co-culture the transcriptome showed signs of a transition from aerobic to anaerobic conditions. As shown by the resazurin experiments, oxygen availability decreased more rapidly in the co-culture compared to the mono-cultures. Also, the threshold levels for a switch from aerobic to microaerophilic conditions might differ between the two organisms. This might explain that some of the anaerobically induced pathways were detected in P. aeruginosa but not in S. aureus. Considering that the CF-lung represents an oxygen scarce environment [45], the observed metabolic changes observed between P. aeruginosa and S. aureus in vitro might be of particular importance also under these in vivo conditions [46].
Our data further suggest that the co-culture conditions lead to a state of nitrogen starvation in both bacteria. N-sources play a key role in all cellular anabolic processes such as protein and nucleotide biosynthesis. Purines and pyrimidines are constituents of both DNA and RNA, and purines are specifically important for energy metabolism (ATP, NADH, coenzyme A) and as secondary messengers (c-di-GMP, cAMP, (p)ppGpp). In this context, glutamine represents a high-yield nitrogen and carbon source for microorganisms when processed by aminotransferases [47]. Our data indicate that the strongest response in S. aureus included de novo synthesis and activation of the salvage pathway for both purines and pyrimidines. In addition, increased expression of tRNAs (Table 3 and Additional file 4: Table S2) coupled with signs of nitrogen starvation point to a state of increased protein synthesis in both bacteria in co-culture. We therefore believe that nitrogen sensing represents a specific early response during competition between these two organisms.
Detrimental interactions between different bacterial species may involve contact-independent inhibition, via synthesis of antimicrobial molecules, or through contact-dependent inhibition [14, 18]. In agreement with this hypothesis, we observed increased expression of numerous antioxidant responses in S. aureus and in particular the NO-dioxygenase, which detoxifies NO [31]. However, none of the known anti-staphylococcal products (LasA protease, HQNO, pyocyanin) were upregulated under our experimental conditions in P. aeruginosa. Since these products are quorum-sensing (QS)-controlled, their expression is likely to be induced during late exponential growth phase and independently of a competitor.
Interestingly, both organisms showed potential signs of cell lysis during co-culture, as suggested by induction of R- and F-pyocin synthesis genes in P. aeruginosa (Table 2) and several prophage genes (NM1 to NM4) in S. aureus (Table 3). Cell lysis may have occurred likely in a small sub-population of the culture in response to nutrient limitation or direct cell damage, and could allow the bacteria to detect the presence of a competitior, a mechanism known as competition sensing [48]. Prophage induction in S. aureus also occurs in the presence of hydrogen peroxide, produced for instance by Streptococcus pneumonia strains [49]. P. aeruginosa does not produce hydrogen peroxide but ROS or RNS species generated during the initial aerobic growth phase might induce this response in S. aureus.
Co-culture between P. aeruginosa and S. aureus also affected the expression of virulence factors in both bacteria. In order to colonize and survive within the host, bacteria express virulence factors, which allow them to adhere to host tissue and to get access to nutrients [50]. However, in this study S. aureus responded to the co-culture by decreasing the secretion of capsular polysaccharide, leukotoxins, general virulence regulators, serine proteases, none of which are known to have an effect on other bacteria. Similarly, P. aeruginosa showed decreased expression of biosynthesis pathways for the secreted virulence factors pyocyanin and pyochelin. The decreased expression of the RND multidrug efflux pump MexGHI-OpmD is consistent with these data [38]. Indeed, mutants in mexI and opmD are unable to produce the Pseudomonas Quinolone Signal (PQS, 2-heptyl-3-hydroxy-4-quinolone) and N-(3-oxododecanoyl)-L-homoserine lactone (3-oxo-C12-HSL), while showing a marked decreased production in N-butanoyl-L-homoserine lactone levels (C4-HSL). These defects have been reported to cause significant reduction in the production of QS-dependent virulence factors including elastase, rhamnolipids, pyocyanin, pyoverdin and significantly reduce swarming motility [51]. These observations suggest that in the absence of host cells, both organism decrease virulence factor production.
Conclusions
Our dual-transcriptome analysis on S. aureus and P. aeruginosa co-culture reveals an intricate pattern of genetic adaptations during the initial encounter between these bacterial pathogens (Fig. 5). The responses are dominated by metabolic changes, since the largest number of differentially expressed genes belong to the functional classes “metabolism” and “transport”. In particular, our data confirm the previous observation that P. aeruginosa drives S. aureus into fermentation, allowing the former organism to use lactate, produced by the latter organism, as a C-source [44]. Both organisms show signs of nitrogen starvation that likely induce purine and pyrimidine synthesis pathways in S. aureus. We found no evidence for induction of anti-staphylococcal pathways in P. aeruginosa, suggesting that resource competition rather than direct (interference) competition prevail during the early stages of co-culture. Both organisms seem to sense stress caused by nutrient deprivation, ROS-mediated attack or direct cell damage. These stressor may induce cell lysis in a subpopulation of the culture caused by induction of pyocins in P. aeruginosa and prophages in S. aureus.
Methods
Strains and culture conditions
Bacterial strains used in the study are P. aeruginosa strain PA14 [52] and S. aureus Newman [53]. Bacteria were inoculated from − 80 °C glycerol stocks and grown overnight on M14 agar plates. M14 medium is based on M9 salts (Na2HPO4 6 g/L; KH2PO4 3 g/L; NaCl 0.5 g/L; NH4Cl 1 g/L) [54] supplemented with 10 g/L casamino acids (BD™, United States), magnesium sulfate (MgSO4) 1 mM, 2 mg/L thiamine (vitamin B1), 2 mg/L niacin (vitamin B3), 2 mg/L calcium pantothenate (vitamin B5), 0.1 mg/L biotin (vitamin B9) and 2 g/L glucose (11 mM). In this medium S. aureus and P. aeruginosa displayed similar growth rates when incubated at 37 °C in a microtiter plate [55].
Short-term competition assay
The short-term competition assay was performed as illustrated in Fig. 1. P. aeruginosa PA14 and S. aureus Newman were grown overnight in M14 medium and diluted separately in this medium to an optical density (OD600) = 1.0. Ten μL from these bacterial suspensions were added to a microtiter plate (TPP®, Switzerland) containing 190 μL of M14 medium per well. One row was inoculated with P. aeruginosa and one row with S. aureus, only. The microtiter plate was incubated under static conditions in a Multi-Mode plate reader (Synergy H1, BioTek, USA) for 3 h. OD600 was measured at regular intervals following 1 min shaking. After the 3 h pre-incubation, co-cultures were prepared by mixing 100 μL of each mono-culture within an empty well obtaining a final volume of 200 μL. The remaining volume was discarded. The wells for the mono-culture were left unchanged (no addition of fresh medium). Rows A and H contained only medium to avoid evaporation at the plate edges and as a control for contamination. After 3 h of incubation, four identical wells of 200 μL were pooled together, representing one replicate. Three replicate samples were labelled as P. aeruginosa mono-cultures (PM1, PM2, and PM3), S. aureus mono-cultures (SM1, SM2, and SM3) and co-cultures (CC1, CC2, and CC3).
Plate countings were performed after the 3 h pre-incubation to determine the initial population size and after the following 3 h co-incubation period to assess the viability of both organisms (Fig. 1). Ten μL taken from four wells that constitute one sample were pooled and serially diluted in 0.9% NaCl. 100 μL aliquots of 10− 5, 10− 6 and 10− 7 dilutions were plated on LB agar plates supplemented with 12 μg/mL aztreonam (Merck, Switzerland) to eliminate P. aeruginosa, and on LB agar plates supplemented with 10 μg/mL vancomycin (Vancocin™, Sandoz Pharmaceuticals, Switzerland) to eliminate S. aureus. Colony-forming units (CFU) were counted after 18 h of incubation at 37 °C.
RNA extraction
The RNeasy Mini Kit (Qiagen, Switzerland) was used for RNA isolation according to the manufacturer’s protocol, which was modified to include combined lysozyme and lysostaphin (Sigma-Aldrich, USA) treatments. Once the short-term competition assay was completed, all samples were centrifuged at 6000 RPM for 5 min at 4 °C and the supernatant was discarded. Pellets were rinsed with 250 μL of an acetone-ethanol (1:1) solution. Tubes containing the cell pellets were stored at − 80 °C. The next day, samples were thawed on ice, centrifuged at 6000 RPM for 10 min at 4 °C. Remaining liquid was removed carefully by pipetting. The pellet was resuspended in 1 mL of TE (10 mM Tris, 1 mM EDTA) buffer at pH 8.0. All samples were again centrifuged at 6000 RPM for 10 min at 4 °C and the supernatant discarded. The pellet was resuspended in 100 μL of lysozyme solution prepared by dissolving 3 mg of lysozyme (Sigma-Aldrich, USA) in 1 ml of Tris-EDTA pH 8.0. Lysostaphin was added to the solution at a final concentration of 1 mg/mL and the samples were incubated at 37 °C for 5 min. From this step onward the manufacturer’s protocol (RNeasy Mini Kit RNA) was followed except that the final elution step was repeated once to increase RNA yield. DNase was removed by digestion with RQ1 RNase-Free DNase (Promega, USA) according to the manufacturer’s protocol. Another RNA cleanup was performed after the DNase treatment. The RNA concentration was then measured using a Nanodrop 1000 spectrophotometer (ThermoScientific, USA).
RNA sequencing and bioinformatics analysis
A total RNA amount of 1.3 μg was ribodepleted for each replicate sample using the Ribo-Zero rRNA removal Kit for bacteria (Epicentre, USA) following the manufacturer’s protocol. The TruSeq total RNA stranded kit (Illumina, USA) was used to prepare the libraries. The quantity of libraries was determined in a Qubit spectrophotometer and their quality assessed with a Tapestation on a DNA High sensitivity chip (Agilent Technologies, USA). The 18 libraries generated were pooled at equimolarity and loaded at 7 pM for clustering. The samples were sequenced in single reads of 100 bp using TruSeq SBS HS v3 chemistry on an Illumina HiSeq 2500 sequence (Illumina, USA). The sequencing quality control was done with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc), and all samples passed. Reads were mapped with the TopHat v.2 software [56] to the P. aeruginosa UCBP-PA14 RefSeq genome NC_008463.1 for samples PM1, PM2, PM3, and to the S. aureus Newman RefSeq genome NC_009641.1 for samples SM1, SM2, SM3, and a merge of the two for samples CC1, CC2, CC3. The tables of counts with the number of reads mapping to each gene feature were prepared with HTSeq v0.5.3 (https://htseq.readthedocs.io/en/release_0.11.1). The counts were normalized according to the library size and genes with less than one count per million reads (cpm) in all three samples were removed. The raw gene number of the set is 5977 for P. aeruginosa (analysis CC versus PM), and 2589 for S. aureus (analysis CC versus SM). Poorly or unexpressed genes were filtered out. The filtered data set consists of 5403 genes for P. aeruginosa PAO1 and 2418 genes for S. aureus. Additional file 5: Table S3 shows differentially expressed genes, using a False Discovery Rate of ≤5% (FDR).
Quantitative real-time PCR
Quantitative Real-Time PCR (qRT-PCR) was performed using the SYBR Green PCR Kit (Qiagen, Switzerland) in a Rotor-Gene 3000 Real-Time PCR machine (Corbette Research, Australia). Specific qRT-PCR primers are listed in Additional File 6: Table S4. The reaction mix was prepared in a final volume of 25 μL: 12.5 μL of Rotor-Gene SYBR Green PCR Master Mix™ 2X, 2.5 μL of forward primer, 2.5 μL of reverse primer, 1 μL of cDNA template diluted to 10 ng/μL in RNase-free water and 6.5 μL of RNase-free water. The program was as follows: 5 min at 95 °C, 5 s at 95 °C, 10 s at 60 °C (40 cycles). Melt curve analyses were performed at the end of the run by heating from 60 to 95 °C. Samples were run in triplicates. Standard curves were prepared by 10-fold dilutions of purified gDNA of P. aeruginosa and S. aureus. The obtained concentrations for each target gene were then normalized to the concentration of a housekeeping reference gene: rpsL for P. aeruginosa [57], NWMN_1382 for S. aureus [58]. Relative gene expression was calculated for each replicate by dividing the ratio of the target gene over reference gene in co-culture by the same ratio in mono-culture. Relative gene expression is expressed in base 2-logarithms.
Additional files
Figure S1. MA plots of the differentially expressed genes. (PDF 1068 kb)
Figure S2. Validation of RNAseq data. (PDF 269 kb)
Table S1. Differentially expressed genes in S. aureus in co-culture versus mono-culture (average of three replicates, after normalization). (XLSX 35 kb)
Table S2. Differentially expressed genes in P. aeruginosa in co-culture versus mono-culture (average of three replicates, after normalization). (XLSX 36 kb)
Table S3. RNAseq results and mapping statistics. (DOCX 15 kb)
Table S4. Primers used in this study. (DOCX 18 kb)
Acknowledgements
RNAseq was performed at the Genomics platform of the Medical Faculty of the University of Geneva. We thank Dr. M. Docquier and Dr. N. Civic for helpful discussions on the RNAseq data analyses and preparation of documents for submission of the RNAseq data to GEO.
Availablity of data and materials
The meta- and raw-data generated during this study are available at GEO under the following address: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122048
Funding
This research was conducted with the financial support of the Swiss National Science Foundation (grants n°140929 and n°159523).
Abbreviations
- CC
Co-culture
- CF
Cystic fibrosis
- FDR
False discovery rate
- HQNO
2-heptyl-4-quinolone N-oxide
- NO
Nitric oxide
- PM
Pseudomonas aeruginosa mono-culture
- PQS
Pseudomonas quinolone signal (2-heptyl-3-hydroxy-4-quinolone)
- qRT-PCR
Quantitiative RealTime-polymerase chain reaction
- QS
Quorum sensing
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- SM
S. aureus mono-culture
Authors’ contributions
MT and AL performed the experimental work and initial data analyses. TK and CvD conceived and supervised the study. MT and TK drafted the manuscript. CvD and TK obtained funding and finalized the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mikaël Tognon, Email: Mikael.Tognon@hcuge.ch.
Thilo Köhler, Email: thilo.kohler@unige.ch.
Alexandre Luscher, Email: Alexandre.Luscher@unige.ch.
Christian van Delden, Email: Christian.vanDelden@hcuge.ch.
References
- 1.Tay WH, Chong KK, Kline KA. Polymicrobial-host interactions during infection. J Mol Biol. 2016;428(17):3355–3371. doi: 10.1016/j.jmb.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Serra R, Grande R, Butrico L, Rossi A, Settimio UF, Caroleo B, et al. Chronic wound infections: the role of Pseudomonas aeruginosa and Staphylococcus aureus. Expert Rev Anti-Infect Ther. 2015;13(5):605–613. doi: 10.1586/14787210.2015.1023291. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Z, Adappa ND, Doghramji LJ, Chiu AG, Cohen NA, Palmer JN. Different clinical factors associated with Staphylococcus aureus and Pseudomonas aeruginosa in chronic rhinosinusitis. Int Forum Allergy Rhinol. 2015;5(8):724–733. doi: 10.1002/alr.21532. [DOI] [PubMed] [Google Scholar]
- 4.Ahlgren HG, Benedetti A, Landry JS, Bernier J, Matouk E, Radzioch D, et al. Clinical outcomes associated with Staphylococcus aureus and Pseudomonas aeruginosa airway infections in adult cystic fibrosis patients. BMC Pulm Med. 2015;15:67. doi: 10.1186/s12890-015-0062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maliniak ML, Stecenko AA, McCarty NA. A longitudinal analysis of chronic MRSA and Pseudomonas aeruginosa co-infection in cystic fibrosis: a single-center study. J Cyst Fibros. 2016;15(3):350–356. doi: 10.1016/j.jcf.2015.10.014. [DOI] [PubMed] [Google Scholar]
- 6.Dalton T, Dowd SE, Wolcott RD, Sun Y, Watters C, Griswold JA, et al. An in vivo polymicrobial biofilm wound infection model to study interspecies interactions. PLoS One. 2011;6(11):e27317. doi: 10.1371/journal.pone.0027317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sibley CD, Duan K, Fischer C, Parkins MD, Storey DG, Rabin HR, et al. Discerning the complexity of community interactions using a Drosophila model of polymicrobial infections. PLoS Pathog. 2008;4(10):e1000184. doi: 10.1371/journal.ppat.1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Hoiby N, et al. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol. 2012;10(12):841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- 9.Barnabie PM, Whiteley M. Iron-mediated control of Pseudomonas aeruginosa-Staphylococcus aureus interactions in the cystic fibrosis lung. J Bacteriol. 2015;197(14):2250–2251. doi: 10.1128/JB.00303-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeLeon S, Clinton A, Fowler H, Everett J, Horswill AR, Rumbaugh KP. Synergistic interactions of Pseudomonas aeruginosa and Staphylococcus aureus in an in vitro wound model. Infect Immun. 2014;82(11):4718–4728. doi: 10.1128/IAI.02198-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar A, Ting YP. Presence of Pseudomonas aeruginosa influences biofilm formation and surface protein expression of Staphylococcus aureus. Environ Microbiol. 2015;17(11):4459–4468. doi: 10.1111/1462-2920.12890. [DOI] [PubMed] [Google Scholar]
- 12.Michelsen CF, Christensen AM, Bojer MS, Hoiby N, Ingmer H, Jelsbak L. Staphylococcus aureus alters growth activity, autolysis, and antibiotic tolerance in a human host-adapted Pseudomonas aeruginosa lineage. J Bacteriol. 2014;196(22):3903–3911. doi: 10.1128/JB.02006-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldan R, Cigana C, Testa F, Bianconi I, De Simone M, Pellin D, et al. Adaptation of Pseudomonas aeruginosa in cystic fibrosis airways influences virulence of Staphylococcus aureus in vitro and murine models of co-infection. PLoS One. 2014;9(3):e89614. doi: 10.1371/journal.pone.0089614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.West SA, Diggle SP, Buckling A, Gardner A, Griffin AS. The social lives of microbes. Annu Rev Ecol Evol Syst. 2007;38(1):53–77. doi: 10.1146/annurev.ecolsys.38.091206.095740. [DOI] [Google Scholar]
- 15.West SA, Buckling A. Cooperation, virulence and siderophore production in bacterial parasites. Proc Biol Sci. 2003;270(1510):37–44. doi: 10.1098/rspb.2002.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arias AA, Lambert S, Martinet L, Adam D, Tenconi E, Hayette MP, et al. Growth of desferrioxamine-deficient Streptomyces mutants through xenosiderophore piracy of airborne fungal contaminations. FEMS Microbiol Ecol. 2015;91(7):1-9. [DOI] [PubMed]
- 17.Tanabe T, Funahashi T, Shiuchi K, Okajima N, Nakao H, Miyamoto K, et al. Characterization of Vibrio parahaemolyticus genes encoding the systems for utilization of enterobactin as a xenosiderophore. Microbiology. 2012;158(8):2039–2049. doi: 10.1099/mic.0.059568-0. [DOI] [PubMed] [Google Scholar]
- 18.Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8(1):15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickson RP, Erb-Downward JR, Huffnagle GB. Homeostasis and its disruption in the lung microbiome. Am J Physiol Lung Cell Mol Physiol. 2015;309(10):L1047–L1055. doi: 10.1152/ajplung.00279.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lightbown JW, Jackson FL. Inhibition of cytochrome system of heart muscle and of Staphylococcus aureus by 2-heptyl-4-hydroxyquinoline-N-oxide, an antagonist of dihydrostreptomycin. Biochem J. 1954;58(4):49. [PubMed]
- 21.Hoffman LR, Deziel E, D'Argenio DA, Lepine F, Emerson J, McNamara S, et al. Selection for Staphylococcus aureus small-colony variants due to growth in the presence of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2006;103:19890–19895. doi: 10.1073/pnas.0606756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kessler E, Safrin M, Olson JC, Ohman DE. Secreted LasA of Pseudomonas aeruginosa is a staphylolytic protease. J Biol Chem. 1993;268(10):7503–7508. [PubMed] [Google Scholar]
- 23.Mashburn LM, Jett AM, Akins DR, Whiteley M. Staphylococcus aureus serves as an iron source for Pseudomonas aeruginosa during in vivo coculture. J Bacteriol. 2005;187:554–566. doi: 10.1128/JB.187.2.554-566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fugere A, Lalonde Seguin D, Mitchell G, Deziel E, Dekimpe V, Cantin AM, et al. Interspecific small molecule interactions between clinical isolates of Pseudomonas aeruginosa and Staphylococcus aureus from adult cystic fibrosis patients. PLoS One. 2014;9(1):e86705. doi: 10.1371/journal.pone.0086705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang L, Liu Y, Wu H, Hoiby N, Molin S, Song ZJ. Current understanding of multi-species biofilms. Int J Oral Sci. 2011;3(2):74–81. doi: 10.4248/IJOS11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antonic V, Stojadinovic A, Zhang B, Izadjoo MJ, Alavi M. Pseudomonas aeruginosa induces pigment production and enhances virulence in a white phenotypic variant of Staphylococcus aureus. Infect Drug Resist. 2013;6:175–186. doi: 10.2147/IDR.S49039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma V, Noriega CE, Rowe JJ. Involvement of NarK1 and NarK2 proteins in transport of nitrate and nitrite in the denitrifying bacterium Pseudomonas aeruginosa PAO1. Appl Environ Microbiol. 2006;72(1):695–701. doi: 10.1128/AEM.72.1.695-701.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hervas AB, Canosa I, Santero E. Transcriptome analysis of Pseudomonas putida in response to nitrogen availability. J Bacteriol. 2008;190(1):416–420. doi: 10.1128/JB.01230-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emmett M, Kloos WE. Amino acid requirements of staphylococci isolated from human skin. Can J Microbiol. 1975;21(5):729–733. doi: 10.1139/m75-107. [DOI] [PubMed] [Google Scholar]
- 30.Nuxoll AS, Halouska SM, Sadykov MR, Hanke ML, Bayles KW, Kielian T, et al. CcpA regulates arginine biosynthesis in Staphylococcus aureus through repression of proline catabolism. PLoS Pathog. 2012;8(11):e1003033. doi: 10.1371/journal.ppat.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gardner PR, Gardner AM, Martin LA, Dou Y, Li T, Olson JS, et al. Nitric-oxide dioxygenase activity and function of flavohemoglobins. Sensitivity to nitric oxide and carbon monoxide inhibition. J Biol Chem. 2000;275(41):31581–31587. doi: 10.1074/jbc.M004141200. [DOI] [PubMed] [Google Scholar]
- 32.Fu H, Yuan J, Gao H. Microbial oxidative stress response: novel insights from environmental facultative anaerobic bacteria. Arch Biochem Biophys. 2015;584:28–35. doi: 10.1016/j.abb.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 33.Brunskill EW, de Jonge BL, Bayles KW. The Staphylococcus aureus scdA gene: a novel locus that affects cell division and morphogenesis. Microbiology. 1997;143(9):2877–2882. doi: 10.1099/00221287-143-9-2877. [DOI] [PubMed] [Google Scholar]
- 34.Kinkel TL, Roux CM, Dunman PM, Fang FC. The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. MBio. 2013;4(6):e00696–e00613. doi: 10.1128/mBio.00696-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas VC, Sadykov MR, Chaudhari SS, Jones J, Endres JL, Widhelm TJ, et al. A central role for carbon-overflow pathways in the modulation of bacterial cell death. PLoS Pathog. 2014;10(6):e1004205. doi: 10.1371/journal.ppat.1004205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patton TG, Rice KC, Foster MK, Bayles KW. The Staphylococcus aureus cidC gene encodes a pyruvate oxidase that affects acetate metabolism and cell death in stationary phase. Mol Microbiol. 2005;56(6):1664–1674. doi: 10.1111/j.1365-2958.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 37.Li XZ, Plesiat P, Nikaido H. The challenge of efflux-mediated antibiotic resistance in gram-negative bacteria. Clin Microbiol Rev. 2015;28(2):337–418. doi: 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aendekerk S, Ghysels B, Cornelis P, Baysse C. Characterization of a new efflux pump, MexGHI-OpmD, from Pseudomonas aeruginosa that confers resistance to vanadium. Microbiology. 2002;148(8):2371–2381. doi: 10.1099/00221287-148-8-2371. [DOI] [PubMed] [Google Scholar]
- 39.Sakhtah H, Koyama L, Zhang Y, Morales DK, Fields BL, Price-Whelan A, et al. The Pseudomonas aeruginosa efflux pump MexGHI-OpmD transports a natural phenazine that controls gene expression and biofilm development. Proc Natl Acad Sci U S A. 2016;113(25):E3538–E3547. doi: 10.1073/pnas.1600424113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ranjit DK, Endres JL, Bayles KW. Staphylococcus aureus CidA and LrgA proteins exhibit holin-like properties. J Bacteriol. 2011;193(10):2468–2476. doi: 10.1128/JB.01545-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rice KC, Mann EE, Endres JL, Weiss EC, Cassat JE, Smeltzer MS, et al. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc Natl Acad Sci U S A. 2007;104(19):8113–8118. doi: 10.1073/pnas.0610226104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brazas MD, Hancock RE. Ciprofloxacin induction of a susceptibility determinant in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2005;49:3222–3227. doi: 10.1128/AAC.49.8.3222-3227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller CL, Van Laar TA, Chen T, Karna SLR, Chen P, You T, et al. Global transcriptome responses including small RNAs during mixed-species interactions with methicillin-resistant Staphylococcus aureus and Pseudomonas aeruginosa. Microbiologyopen. 2017;6(3):e00427. doi: 10.1002/mbo3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Filkins LM, Graber JA, Olson DG, Dolben EL, Lynd LR, Bhuju S, et al. Coculture of Staphylococcus aureus with Pseudomonas aeruginosa Drives S. aureus towards fermentative metabolism and reduced viability in a cystic fibrosis model. J Bacteriol. 2015;197(14):2252–2264. doi: 10.1128/JB.00059-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alvarez-Ortega C, Harwood CS. Responses of Pseudomonas aeruginosa to low oxygen indicate that growth in the cystic fibrosis lung is by aerobic respiration. Mol Microbiol. 2007;65(1):153–165. doi: 10.1111/j.1365-2958.2007.05772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cowley ES, Kopf SH, LaRiviere A, Ziebis W, Newman DK. Pediatric cystic fibrosis sputum can be chemically dynamic, anoxic, and extremely reduced due to hydrogen sulfide formation. MBio. 2015;6(4):e00767. doi: 10.1128/mBio.00767-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aledo JC. Glutamine breakdown in rapidly dividing cells: waste or investment? BioEssays. 2004;26(7):778–785. doi: 10.1002/bies.20063. [DOI] [PubMed] [Google Scholar]
- 48.Cornforth DM, Foster KR. Competition sensing: the social side of bacterial stress responses. Nat Rev Microbiol. 2013;11(4):285–293. doi: 10.1038/nrmicro2977. [DOI] [PubMed] [Google Scholar]
- 49.Selva L, Viana D, Regev-Yochay G, Trzcinski K, Corpa JM, Lasa I, et al. Killing niche competitors by remote-control bacteriophage induction. Proc Natl Acad Sci U S A. 2009;106(4):1234–1238. doi: 10.1073/pnas.0809600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buckner MM, Finlay BB. Host-microbe interaction: innate immunity cues virulence. Nature. 2011;472(7342):179–180. doi: 10.1038/472179a. [DOI] [PubMed] [Google Scholar]
- 51.Aendekerk S, Diggle SP, Song Z, Hoiby N, Cornelis P, Williams P, et al. The MexGHI-OpmD multidrug efflux pump controls growth, antibiotic susceptibility and virulence in Pseudomonas aeruginosa via 4-quinolone-dependent cell-to-cell communication. Microbiology. 2005;151(4):1113–1125. doi: 10.1099/mic.0.27631-0. [DOI] [PubMed] [Google Scholar]
- 52.He J, Baldini RL, Deziel E, Saucier M, Zhang Q, Liberati NT, et al. The broad host range pathogen Pseudomonas aeruginosa strain PA14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc Natl Acad Sci U S A. 2004;101:2530–2535. doi: 10.1073/pnas.0304622101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J Bacteriol. 2008;190(1):300–310. doi: 10.1128/JB.01000-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rudin L, Sjostrom JE, Lindberg M, Philipson L. Factors affecting competence for transformation in Staphylococcus aureus. J Bacteriol. 1974;118(1):155–164. doi: 10.1128/jb.118.1.155-164.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tognon M, Kohler T, Gdaniec BG, Hao Y, Lam JS, Beaume M, et al. Co-evolution with Staphylococcus aureus leads to lipopolysaccharide alterations in Pseudomonas aeruginosa. ISME J. 2017;11(10):2233–2243. doi: 10.1038/ismej.2017.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dumas JL, van Delden C, Perron K, Kohler T. Analysis of antibiotic resistance gene expression in Pseudomonas aeruginosa by quantitative real-time-PCR. FEMS Microbiol Lett. 2006;254(2):217–225. doi: 10.1111/j.1574-6968.2005.00008.x. [DOI] [PubMed] [Google Scholar]
- 58.Khemici V, Prados J, Linder P, Redder P. Decay-initiating Endoribonucleolytic cleavage by RNase Y is kept under tight control via sequence preference and sub-cellular localisation. PLoS Genet. 2015;11(10):e1005577. doi: 10.1371/journal.pgen.1005577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. MA plots of the differentially expressed genes. (PDF 1068 kb)
Figure S2. Validation of RNAseq data. (PDF 269 kb)
Table S1. Differentially expressed genes in S. aureus in co-culture versus mono-culture (average of three replicates, after normalization). (XLSX 35 kb)
Table S2. Differentially expressed genes in P. aeruginosa in co-culture versus mono-culture (average of three replicates, after normalization). (XLSX 36 kb)
Table S3. RNAseq results and mapping statistics. (DOCX 15 kb)
Table S4. Primers used in this study. (DOCX 18 kb)