

Abstract
Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized by the death of dopaminergic (DA) neurons in the substantia nigra. To develop therapeutic strategies to halt or slow the neurodegenerative process, it is imperative that we understand the pathogenesis of PD. With the current state of knowledge, multiple pathological pathways such as oxidative stress, inflammation due to microglial activation, apoptotic pathway activation via Abelson (c-Abl)tyrosine kinase enzyme, and DA toxins have been incriminated in causing DA neuronal death in PD. In the recent times, there is growing evidence of the role of c-Abl nonreceptor tyrosine kinase in the pathogenesis of PD. We give a short account of the potential of c-Abl inhibitors, the currently used anticancer drugs such as nilotinib in preventing the neurodegenerative process in PD.
Keywords: C-Abl, Nilotinib, Parkinson's disease
INTRODUCTION
Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized by the death of dopaminergic (DA) neurons in the substantia nigra (SN) and formation of inclusions known as Lewy bodies (LBs), which primarily contain aggregated alpha-synuclein.[1] The cardinal symptoms of PD are the result of depletion of striatal DA due to neuronal loss in SN. The current therapeutic options are all targeted toward symptomatic relief, and no therapeutic modality can halt the neurodegenerative process.
To develop therapeutic strategies to halt or slow the neurodegenerative process, it is imperative that we understand the pathogenesis of PD. With the current state of knowledge, multiple pathological pathways such as oxidative stress, inflammation due to microglial activation, apoptotic pathway activation via Abelson (c-Abl), and DA toxins have been incriminated in causing DA neuronal death in PD.[2]
ROLE OF C-ABL IN PARKINSON'S DISEASE PATHOGENESIS
One of the important hypotheses for pathogenesis of PD is the role of increased oxidative stress in DA neurons of the SN which leads to progressive neuronal loss. This hypothesis is backed by animal studies and also studies on postmortem PD patient's brain. c-Abl nonreceptor tyrosine kinase regulates the proteolytic cleavage of protein kinase C. The activation of protein kinase C by phosphorylation at tyrosine residue leads to the mitochondrial apoptotic pathway cascade activation and further leads to mitochondrial dysfunction and cell death.[3]
In genetically inherited PD, mutation in parkin ligase expression gene accounts for 50% of autosomal recessive form of PD and 18% of early-onset PD. Parkin has cytoprotective function by preventing the accumulation of toxic parkin substrates as Aminoacyl-tRNA synthetase-interacting fructose-1,6-bisphosphatase1, multifunctional protein type 2 (AIMP2), and PARkin-interacting substrate (PARIS).[4] Parkin mutation has also been linked to sporadic forms of PD. In animal studies, the knockdown of c-Abl is neuroprotective and prevents neuronal loss in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD. By knockdown of c-Abl, there is a suppression of (a) tyrosine phosphorylation of parkin, (b) upregulation of PARIS, and (c) suppression of aminoacyl-tRNA synthetase complex-interacting multifunctional protein 2 (AIMP-2). In brain samples from patients with PD, animal models of α-synucleinopathies, and animal models of MPTP-induced PD, it has been demonstrated that the higher levels of phosphorylated c-Abl are found.[5]
Alpha-synuclein protein misfolding and aggregation leads to the formation of LBs which are characteristically seen in PD, DLBD, and multiple system atrophy. In mice studies, injecting lentiviral c-Abl into the SN significantly increases both monomeric and aggregated SNCA levels and this conversely leads to c-Abl activation.[6] This association between c-Abl activation and SNCA accumulation has been demonstrated in human postmortem PD striatal specimens.[6] All these suggest that preventing c-Abl activation may be a promising disease-modifying therapy.
TYROSINE KINASE C-ABL INHIBITOR
Tyrosine kinase inhibitors were first used for the treatment of Philadelphia chromosome-positive patients of chronic myeloid leukemia (CML). Imatinib is considered the prototype drug of this group and is considered as the first-line drug for treatment of CML.[7] Nilotinib compared to imatinib has higher potency and increased brain bioavailability and was approved for use in CML by the Food and Drug Administration in 2007.
PRECLINICAL STUDIES OF C-ABL INHIBITOR IN PARKINSON'S DISEASE
In many preclinical studies [Table 1], c-abl inhibitors have been studied to investigate the neuroprotective effects in PD animal models. Studies in transgenic and lentiviral gene transfer models have found that nilotinib accelerates autophagic clearance of SNCA and decreases c-Abl activity.[6] These studies have proved that c-Abl inhibitors have good penetration into brain and have neuroprotective effects which have been demonstrated in animal models, MPTP neurotoxin model, and model induced by lentiviral overexpression of α-synuclein in the midbrain. Imam et al. reported that STI-571, a selective c-Abl inhibitor, prevented tyrosine phosphorylation of parkin and restored its E3 ligase activity and cytoprotective function both in vitro and in vivo.[5] In MPTP-induced model of PD, nilotinib reduces c-Abl activation and the levels of PARIS, resulting in prevention of DA neuron loss and behavioral deficits following MPTP intoxication. Surprisingly, there was no reduction in the tyrosine phosphorylation of parkin and the parkin substrate, AIMP2, suggesting that the protective effect of nilotinib may, in part, be parkin independent.[8] Imam et al. demonstrated that second-generation irreversible Abl kinase inhibitor, INNO-406, is capable of preventing the progression of DA neuronal damage in a toxin-induced C57 mouse model of PD INNO-406 given to C57 mice for 1 week before MPTP treatment and then for 1 week after MPTP treatment decreased the loss of dopamine in the striatum by 45% and the loss of TH + neurons in SN pars compacta by 40%.[9] Hebron et al. evaluated the effects of Abl inhibition via nilotinib treatment on clearance of α-synuclein in transgenic α-synuclein mice models of synucleinopathies and reported enhanced autophagic clearance.[6]
Table 1.
Preclinical studies of tyrosine kinase inhibitors
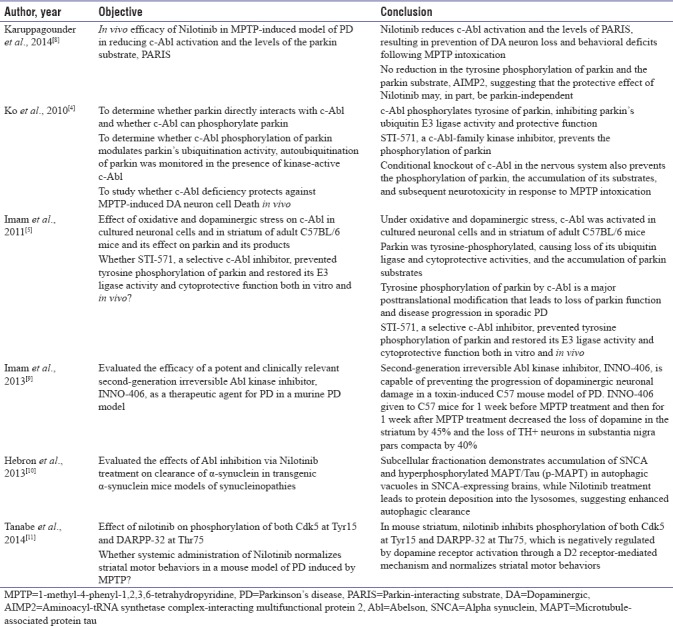
All the above preclinical studies have suggested that inhibition of c-Abl may have the potential to modify the clinical course of PD.
CLINICAL STUDIES OF C-ABL INHIBITOR IN PARKINSON'S DISEASE
After getting these findings from the preclinical data, the international committee of PD experts came together twice in 2012 and 2013 to discuss if nilotinib should be taken for a drug-repurposing candidate in PD.[12]
Pagan et al. published the results of the clinical trial of nilotinib on PD patients as an abstract at the Society of Neuroscience in Chicago, October 2015. The study enrolled 12 patients, of whom 1 had PD, 5 had DLBD, and 6 had parkinsonian disorder with cognitive deficit, and were randomized into two groups: Group A – nilotinib 300 mg (n = 7) and Group B – nilotinib 150 mg (n = 5). After 24 weeks, compared to baseline, unified Parkinson's disease rating scale (scores decrease by 3.4 points (nilotinib 300 mg) and 3.6 points (nilotinib 150 mg) and the study concluded that nilotinib was safe and tolerated. These benefits disappeared after 12 weeks of cessation of drug treatment, thus raising doubts whether the effects were in first place true to reduced neurodegeneration or not. However, most of the patients developed psychiatric manifestations, such as hallucination, agitation, and paranoia. One patient had myocardial infarction. When cerebrospinal fluid synuclein was measured at 2 and 6 months, the levels were slightly reduced in the group on 150 mg dose and unchanged in the 300 mg dose group. The study had no placebo group and the baseline characteristics of patients were inhomogeneous to begin with in both groups.[13]
The drug was all over the newspapers and was termed as the most promising new treatment in PD in decades. There was a media frenzy regarding the results of this small study and the scientific community had concerns regarding the limited data that had not been scrutinized by peer-reviewed research. Many experts raised concerns about the hyperbolic media exposure about results of non-placebo-controlled trial. Nilotinib has a black box warning for cardiac conduction side effects such as QT prolongation and also sudden cardiac death has been reported in patients taking nilotinib.
While c-Abl inhibition is promising in preclinical models, many challenges remain in translating this into human studies. First is the limited blood–brain barrier penetrance of the commonly used inhibitors such as imatinib and nilotinib. The second and most important is to ensure the specificity of kinase inhibition. The nonspecific kinase inhibition may lead to off-target deleterious adverse effects.
Not all hope is lost for patients with PD, as Michael J Fox Foundation for Parkinson's Research started a placebo-controlled trail of nilotinib in 2017 “Randomized, Double-Blind, Placebo-Controlled, Phase IIa, Parallel Group, Two Cohort Study to Define the Safety, Tolerability, Clinical and Exploratory Biological Activity of the Chronic Administration of Nilotinib in Participants With Parkinson's Disease,” which would give answers to the safety and efficacy profile of nilotinib in patients with PD (ClinicalTrials.gov Identifier: NCT03205488).[14]
To sum up, the preclinical and clinical trials definitely give a new direction, to target a molecular pathway that has not been assessed previously for neuroprotection in PD. Large-sized, well-designed trials, vigorously assessing the safety and possible neuroprotective action of nilotinib or newer molecules with similar property, are required to see if the hope can match the hype surrounding this molecule. Researchers worldwide should continue to strive to develop drugs, which are selective and safe, and can arrest the neurodegenerative process ongoing in PD. Only time will tell if this group of molecule will have long-term clinical use in a chronic disease like PD.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Dauer W, Przedborski S. Parkinson's disease: Mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 2.Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302:819–22. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 3.Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- 4.Ko HS, Lee Y, Shin JH, Karuppagounder SS, Gadad BS, Koleske AJ, et al. Phosphorylation by the c-Abl protein tyrosine kinase inhibits Parkin's ubiquitination and protective function. Proc Natl Acad Sci U S A. 2010;107:16691–6. doi: 10.1073/pnas.1006083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imam SZ, Zhou Q, Yamamoto A, Valente AJ, Ali SF, Bains M, et al. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: Implications for Parkinson's disease. J Neurosci. 2011;31:157–63. doi: 10.1523/JNEUROSCI.1833-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hebron ML, Lonskaya I, Moussa CE. Tyrosine kinase inhibition facilitates autophagic SNCA/α-synuclein clearance. Autophagy. 2013;9:1249–50. doi: 10.4161/auto.25368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J, et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–42. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 8.Karuppagounder SS, Brahmachari S, Lee Y, Dawson VL, Dawson TM, Ko HS, et al. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson's disease. Sci Rep. 2014;4:4874. doi: 10.1038/srep04874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Imam SZ, Trickler W, Kimura S, Binienda ZK, Paule MG, Slikker W, Jr, et al. Neuroprotective efficacy of a new brain-penetrating C-Abl inhibitor in a murine Parkinson's disease model. PLoS One. 2013;8:e65129. doi: 10.1371/journal.pone.0065129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hebron ML, Lonskaya I, Moussa CE. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson's disease models. Hum Mol Genet. 2013;22:3315–28. doi: 10.1093/hmg/ddt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanabe A, Yamamura Y, Kasahara J, Morigaki R, Kaji R, Goto S, et al. A novel tyrosine kinase inhibitor AMN107 (nilotinib) normalizes striatal motor behaviors in a mouse model of Parkinson's disease. Front Cell Neurosci. 2014;8:50. doi: 10.3389/fncel.2014.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brundin P, Barker RA, Conn PJ, Dawson TM, Kieburtz K, Lees AJ, et al. Linked clinical trials – the development of new clinical learning studies in Parkinson's disease using screening of multiple prospective new treatments. J Parkinsons Dis. 2013;3:231–9. doi: 10.3233/JPD-139000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pagan F, Hebron M, Valadez EH, Torres-Yaghi Y, Huang X, Mills RR, et al. Nilotinib effects in Parkinson's disease and dementia with lewy bodies. J Parkinsons Dis. 2016;6:503–17. doi: 10.3233/JPD-160867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Available from: https://www.clinicaltrials.gov/ct2/show/NCT03205488 .