

Abstract
Adaptive radiations provide an opportunity to examine complex evolutionary processes such as ecological specialization and speciation. While a well-resolved phylogenetic hypothesis is critical to completing such studies, the rapid rates of evolution in these groups can impede phylogenetic studies. Here we study the quinaria and testacea species groups of the immigrans-tripunctata radiation of Drosophila, which represent a recent adaptive radiation and are a developing model system for ecological genetics. We were especially interested in understanding host use evolution in these species. In order to infer a phylogenetic hypothesis for this group we sampled loci from both the nuclear genome and the mitochondrial DNA to develop a dataset of 43 protein-coding loci for these two groups along with their close relatives in the immigrans-tripunctata radiation. We used this dataset to examine their evolutionary relationships along with the evolution of feeding behavior. Our analysis recovers strong support for the monophyly of the testacea but not the quinaria group. Results from our ancestral state reconstruction analysis suggests that the ancestor of the testacea and quinaria groups exhibited mushroom-feeding. Within the quinaria group, we infer that transition to vegetative feeding occurred twice, and that this transition did not coincide with a genome-wide change in the rate of protein evolution.
Keywords: ancestral state reconstruction, mycophagy, host specialization, phylogeny, Drosophila
Graphical Abstract
1. Introduction
Recent adaptive radiations are a fertile ground for understanding evolutionary processes such as how speciation occurs and how adaptive traits evolve (Givnish and Sytsma, 1997; Grant and Grant, 2008; Schluter, 2000). Critical to these inferences is a well-resolved and accurate phylogenetic history of the group being studied (Hahn and Nakhleh, 2016; O’Meara, 2012). In this study, we investigate the phylogenetic history of the quinaria and testacea groups of the genus Drosophila, which are a developing model system for ecological genetic studies. Both of these groups occur within the immigrans-tripunctata radiation of the subgenus Drosophila and are found in temperate and boreal forests of the Northern hemisphere. The quinaria group is a young adaptive radiation (~19.5MYA; Izumitani et al., 2016) composed of 26 species, and the testacea group is smaller, comprising four species.
Within the quinaria and testacea groups, there is a remarkable amount of morphological and life history variation. Most species in the quinaria group and all members of the testacea group exhibit mushroom-feeding, though within the quinaria group several species have switched to utilizing decaying vegetation. In addition to variation in food substrate preference, within and among these species there is well characterized variation in body and wing pigmentation (Dombeck and Jaenike, 2004; Werner et al., 2010), circadian rhythms (Simunovic and Jaenike, 2006), parasite prevalence and resistance (Haselkorn et al., 2009; Perlman and Jaenike, 2003; Perlman et al., 2003), mycotoxin tolerance (Jaenike et al., 1983; Spicer and Jaenike, 1996; Stump et al., 2011), endosymbiont infection and resistance (Dyer and Jaenike, 2004; Haselkorn et al., 2013; Jaenike, 2007; Stahlhut et al., 2010; Unckless and Jaenike, 2012; Werren and Jaenike, 1995), cold and desiccation tolerance (Gibbs and Matzkin, 2001; Kimura, 2004), behavioral isolation (Arthur and Dyer, 2015; Humphreys et al., 2016; Jaenike et al., 2006), sexual signals used in mate discrimination (Dyer et al., 2014; Giglio and Dyer, 2013), and meiotic drivers and suppressors (Dyer, 2012; Dyer et al., 2007; Jaenike, 1999).
Several aspects promote the use of these species groups as a model for evolutionary ecology studies. The mushroom-feeders in both groups are composite generalists on fleshy basidiomycete mushrooms, and all life stages revolve around mushrooms: the adults are attracted to mushrooms to mate, and then females lay their eggs on mushrooms that serve as a substrate for larval development. Species that consume decaying vegetation are specialists on water plants such as skunk cabbage (Grimaldi and Jaenike, 1983). These species are easy to collect from baits or naturally occurring hosts, and large numbers of flies can be reared in the lab, where they have a 2–3 week generation time. In addition, many can be hybridized in the lab (e.g., Bray et al., 2014; Humphreys et al., 2016; Shoemaker et al., 1999), which facilitates forward genetic studies. Genetic transformation has been performed in D. guttifera, enabling reverse genetics (Werner et al., 2010), and genomic resources are being developed that will aide in downstream genetic studies.
Here we use a multi-locus genome-wide approach to resolve the phylogeny of the quinaria and testacea group species, which will allow ecological and genetic questions to be addressed in an evolutionary framework. When branch lengths are short as in recent radiations, processes such as incomplete lineage sorting and hybridization can alter the evolutionary history of molecular characters (Edwards, 2009; Mallet et al., 2016). Thus, when attempting to reconstruct a species tree for a recently diverging group, it is important to consider the type of data, such as the genomic region being sampled, as well as the analytical method(s), to avoid being misled by non-representative phylogenetic signal. To date, the phylogenetic inference of these groups (Dyer et al., 2011b; Perlman et al., 2003; Spicer and Jaenike, 1996), as well as of their placement in the immigrans-tripunctata radiation (Hatadani et al., 2009; Izumitani et al., 2016; Morales-Hojas and Vieira, 2012; O’Grady and DeSalle, 2018), has used datasets primarily composed of mitochondrial (mtDNA) markers, while some datasets have also included the Y-chromosome and a few nuclear loci. The results across loci and studies are largely incongruent.
Several aspects about the biology of these flies may complicate phylogenetic inferences. First, based on previous molecular evolutionary results, divergence times are low and thus the speciation rates appear to be high (Perlman et al., 2003; Spicer and Jaenike, 1996), and based on population genetic work the effective population sizes of these species are large (e.g. Dyer et al., 2013; Dyer et al., 2007; Dyer and Jaenike, 2005; Dyer et al., 2011b); both of these factors can increase the amount of incomplete lineage sorting. In lineages where incomplete lineage sorting has occurred, phylogenetic analyses based on concatenated multi-gene datasets can recover strong support for an incorrect topology (Kubatko and Degnan, 2007; Mendes and Hahn, 2018; Roch and Steel, 2014). An alternative approach is to recover species trees using a multispecies coalescent model, treating each locus as a distinct unit and then inferring the species tree from individual gene trees (Edwards et al., 2016). Second, many species within each group can hybridize and some show evidence of recent hybridization (e.g. Bray et al., 2014; Dyer et al., 2011b; Humphreys et al., 2016; Jaenike et al., 2006; Patterson and Stone, 1952). Third, several species are infected with Wolbachia and other maternally-inherited endosymbionts, which can cause the linked mtDNA to show non-neutral or incongruent evolutionary patterns (Cariou et al., 2017; Hurst and Jiggins, 2005; Shoemaker et al., 2004).
In this study, we sampled all of the testacea group species, most of the quinaria group species, and several closely related outgroups in the immigrans-tripunctata radiation. We constructed a dataset composed of sequence data for 43 protein-coding loci that are located throughout the genome (i.e., on the autosomes, sex chromosomes, and mtDNA). To account for the natural history of these species, we reconstructed phylogenetic hypotheses for the quinaria and testacea groups using both analyses of a concatenated molecular dataset as well as a coalescent-based model approach that recovers a species tree from individual gene trees. We then compared the resulting topologies and examined phylogenetic incongruence in the context of specific genomic regions. Finally, we used these results to infer the evolutionary history of feeding behavior within the group using comparative approaches. Specifically, we examined the transitions between mushroom- and vegetation-feeding life histories and the associated rates of protein evolution.
2. Materials and Methods
2.1. Taxon sampling
Fly stocks used in this study were either derived from wild-caught strains or obtained from the Drosophila Species Stock Center. In Table 1, we list the 27 species used, along with the origin of the sequenced strain and reported feeding behavior of the species. We used one strain per species, and our species sampling includes all four of the known testacea group species and 18 of the 26 known quinaria group species. As there is strong behavioral isolation among populations of D. subquinaria (Jaenike et al., 2006), we included two strains of this species, originating from inland (Hinton, Alberta) and coastal (Portland, Oregon) regions of its geographic range. Several quinaria group species were not represented in this study because fresh genomic DNA was not available, and they include the North American species D. macroptera, D. rellima, and D. palustris, the European species D. limbata, and the Asian species D. curvispina, D. kuntzei, and D. unispina. We also included representative members of other species groups known to occur in the immigrans-tripunctata radiation, including D. immigrans, D. tripunctata, and D. bizonata from the immigrans, tripunctata and bizonata species groups respectively, and D. cardini and D. acutilabella from the cardini group.
Table 1.
Species included in this study.
| Species Group | Species | Feeding Behavior | Wolbachia | Range | Hybridization |
|---|---|---|---|---|---|
| bizonata | bizonata | Ma | A | ||
| cardini | cardini | M,Fb,c | NA | ||
| acutilabella | M,Fb,c | NA | |||
| immigrans | immigrans | Fd | NA, E | ||
| quinaria | angularis | Mc | A | ||
| brachynephros | Mc | A | |||
| deflecta | Vd | NA | |||
| falleni | Md | NA | |||
| guttifera | Md | NA | |||
| innubila | M | Y | NA | ||
| magnaquinaria | V | NA | |||
| munda | M | NA | |||
| nigromaculata | M,V,Fe | A | |||
| occidentalis | M | NA | suboccidentalis | ||
| phalerata | Mf | E | |||
| quinaria | Vd | Y | NA | ||
| recens | Md | Y | NA | subquinaria, | |
| transversa, | |||||
| tenebrosa | |||||
| suboccidentalis | M | NA | occidentalis | ||
| tenebrosa, | |||||
| subpalustris | |||||
| subpalustris | Vd | NA | tenebrosa, | ||
| subquinaria | Md | NA | recens, transversa | ||
| (Inland) | |||||
| subquinaria | Md | NA | recens, transversa | ||
| (Coastal) | |||||
| tenebrosa | M | NA | suboccidentalis, | ||
| subpalustris, recens | |||||
| transversa | M | A, E | recens, subquinaria | ||
| testacea | neotestacea | Me | Y | NA | |
| orientacea | Mg | Y | A | testacea | |
| putrida | Me | Y | NA | ||
| testacea | Mg | Y | E | orientacea | |
| tripunctata | tripunctata | M,Fe | NA |
NOTE – The species representing each of the species groups included in the analysis are listed. In addition, the collection location, Wolbachia infection status, and potential to hybridize are provided for each species. M: Mushrooms, V: Decaying vegetation, F: Fermenting fruit; Geographic Range: Asian (A), North America (NA), Europe (E).
2.2. DNA sequencing
We sequenced portions of 43 protein-coding loci from each species (Table S1). The names of the loci, their genomic location using the corresponding Müller Element in Drosophila melanogaster, and the primers for PCR and sequencing are in Tables S2 and S3. These loci are spread throughout the genome and based on gene location in D. melanogaster this includes ten loci on Müller element A (which is the X-chromosome in these species), five on element B, eight on element C, three on element D, ten on element E, two on the ‘dot’ element F, three on the mtDNA, and two on the Y chromosome. There is general conservation across Drosophila of genes to Müller elements, but substantial scrambling within chromosomes due to rearrangements (Bhutkar et al., 2008). Some of the sequences for the Y-chromosome and mtDNA loci were obtained from previous studies (Dyer et al., 2011b; Perlman et al., 2003).
Genomic DNA was extracted from single flies using the Qiagen Puregene Kit, and PCR used standard methods. PCR amplicons were purified using Exosap-IT (Thermo Fisher Scientific), sequenced in both directions using Big Dye 3.1 chemistry (Applied Biosystems), and run on an ABI 3730xl DNA Analyzer at the Georgia Genomics Facility at the University of Georgia. Chromatograms were analyzed using Sequencher (Gene Codes). Sites that were heterozygous (as evident by double peaks) were left as heterozygous in the analyses. All sequences have been deposited in Genbank (Accession numbers MK016667-MK017684).
The sequences were aligned in Geneious v10 (Kearse et al., 2012). Open reading frames were assigned using annotated D. melanogaster orthologs as a guide (Flybase.org). Initial alignments of coding sequences were completed using the protein translation implementing a BLOSUM weight matrix, and then the sequences were converted back to DNA sequences and manually adjusted. Noncoding sequences (introns and untranslated regions) were removed; these contained many gaps among species and thus were of limited use at this phylogenetic scale. For one locus (period) a highly repetitive region of the coding sequence that we could not align with confidence was also excluded from the analyses. The final dataset included 21,486 base pairs per species. We used MEGA v7 (Kumar et al., 2016) to assess the number of informative sites in each alignment (Table S3).
2.3. Phylogenetic methods
To reconstruct species trees, we completed phylogenetic analyses from concatenated datasets using maximum likelihood and Bayesian inference methods as well as from individual gene trees using a coalescent-based approach. For the analyses of the concatenated datasets and individual gene sequences, data partitions and models of evolution were determined using a ‘greedy’ search and the Bayesian Information Criterion (BIC) implemented in PartitionFinder v2.1.1 (Guindon et al., 2010; Lanfear et al., 2012; Lanfear et al., 2017). See Appendix S1 for the optimal partitioning scheme and models of evolution used in the phylogenetic analyses.
For the phylogenetic analysis of the concatenated datasets, we used maximum likelihood (ML) and Bayesian inference (BI) analyses on three datasets: one that contained all 43 loci (21,486 bp), another that excluded the three mtDNA loci (40 loci, 19,973 bp), and one composed only of the three mtDNA loci. The ML analysis was completed with RAxML-HPC2 v8.2.8 (Stamatakis, 2006, 2014). To obtain branch support for the most likely topology we completed 1,000 bootstrap replications, and only support values greater than 50% are provided in the results. We conducted the BI analysis using MrBayes v3.2.6 (Ronquist et al., 2012). When the best-fit model of evolution identified by PartitionFinder could not be implemented due to software limitations, we used the next most complex model that could be implemented. The BI analyses were performed with four chains, one cold and three hot, using the default temperature settings. The analysis consisted of two simultaneous, independent runs of 20,000,000 generations with samples drawn every 1,000 generations. The first 5,000,000 generations were discarded as ‘burn-in’. To confirm that the two runs had converged, the potential scale reduction factor (PSRF; Gelman and Rubin 1992) was calculated. The PSRF should approach 1.000 as independent runs converge. Posterior probability percent values provided the branch support values. Both the ML and BI analyses were completed on the CIPRES scientific gateway (Miller et al., 2010), and the resulting phylogenies were rooted using D. immigrans.
We inferred species trees using the coalescent-based model implemented in ASTRAL-III v5.6.1 (Zhang et al., 2017). First, using RAxML v8.2.4 we obtained the most likely unrooted topology (i.e., gene tree) along with 1,000 bootstrap replicates for each individual locus from the nuclear DNA. We concatenated the three mtDNA loci and analyzed them as a single locus. Then, we reconstructed a species tree using the unrooted gene trees of the 40 nuclear loci plus the mtDNA topology. From these gene trees, we also estimated species trees using several subsets of the data, including the loci from each Müller element separately, the Y-chromosome, and all 40 nuclear loci excluding the mtDNA. We used the ‘multi-locus bootstrapping’ option in ASTRAL-III to infer the species tree, and we used the ‘-q’ option in ASTRAL-III to score the species trees produced by the analyses of the concatenated datasets. The ML analyses of the individual loci and the ASTRAL-III analyses were completed using the Georgia Advanced Computing Resource Cluster. The species trees resulting from the summary coalescent model analysis were rooted using D. immigrans.
2.4. Ancestral state reconstruction
To reconstruct the evolutionary history of the feeding behaviors in this group, the feeding behaviors of the 27 species included in the phylogenetic analyses were divided into the following categories: (A) mushroom, (B) vegetative, and (C) fruit (Table 1; Appendix S2). Species that exhibit multiple feeding behaviors were coded as possessing multiple states. For instance, D. tripunctata feeds on both mushrooms and fruit and thus was coded as AC rather than a unique state. We reconstructed the evolution of feeding behavior using a Bayesian approach, implementing the Bayesian Binary MCMC (BBM) ancestral state reconstruction method in RASP (Yu et al., 2015). The analysis was run for 5,000,000 generations using four chains that were sampled every 100 generations. A fixed Jukes-Cantor model with null root distribution was used for the analysis, and the first 1,000 samples were discarded as burn-in.
2.5. Rates of molecular evolution
The vegetative species in the quinaria group are thought to feed only on a limited range of decaying water plants, whereas the mushroom-feeding species are generalists on fleshy basidiomycete mushrooms and thus may have a broader host range. Host specialization may decrease the effective population size (e.g., Li et al., 2014), which in turn may render purifying selection less efficient and result in a higher rate of protein evolution due to the fixation of slightly deleterious mutations. We tested for relaxation of purifying selection in the vegetative species by analyzing the rates of protein evolution (ω, or dN/dS) of each locus. We implemented branch models in codeml of PAML v4.8a (Yang, 2007), and for each locus we compared a model with a single ω value across the entire phylogeny (ω-all) versus a model with two ω values, one for the clade(s) with the vegetative species (ω-veg) and another for the rest of the phylogeny (ω-nonveg). For each locus, we accounted for gene and species tree differences by completing the analyses using the individual gene tree from the RAxML analyses, the pruned summary species tree from the coalescent model analysis run in ASTRAL-III, and the pruned ML tree from the concatenated dataset. We used likelihood ratio tests to ask whether the model with two ω values was a better fit than the model with one ω value. We determined each P-value using a χ2-distribution with one degree of freedom; for each phylogeny we implemented a Bonferroni correction and thus the adjusted significance threshold is P = 0.05/40 loci = 0.0013. We used nonparametric statistics to compare the ω values across loci from each type of analysis.
3. Results
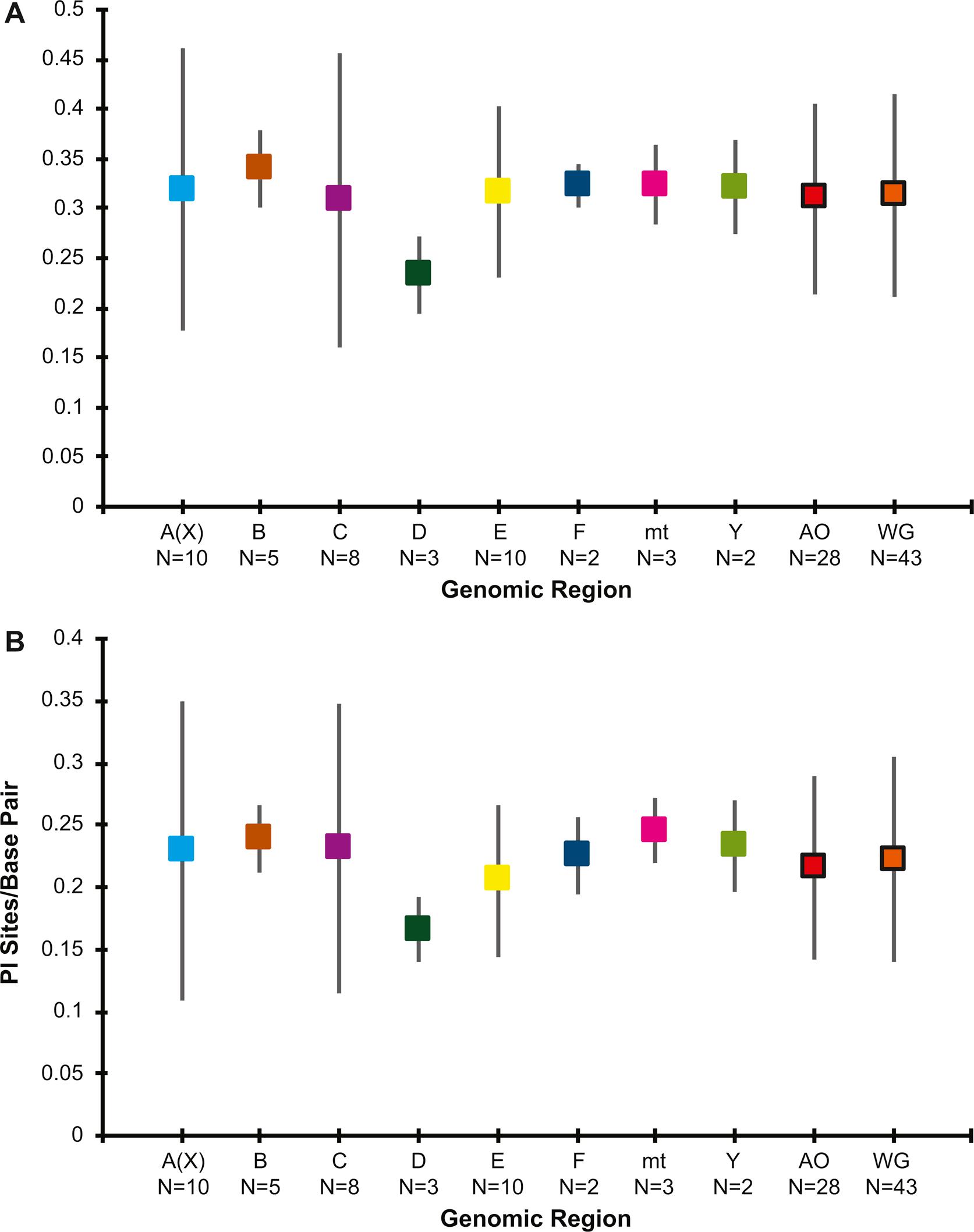
To examine the evolutionary relationships within the quinaria and testacea groups, we constructed a dataset of 43 protein-coding loci that represents every autosome, both sex chromosomes, and the mtDNA from 27 species (Table S1). The final dataset contained 21,486 bp per species, of which 7,051 were variable sites (VS) and 5,022 were parsimony informative sites (PI) (Table S3). Not every locus could be sequenced in every species (Table S1). The average number of loci sequenced per species was 39 of 43 loci (std dev = 2.7) and each locus was sequenced in an average of 26 of 27 species (std dev = 2.8). The proportion of VS and PI sites did not differ across the chromosomes (p = 0.96 and p = 0.94 respectively; Fig. S1).
3.1. Phylogenetic Analyses
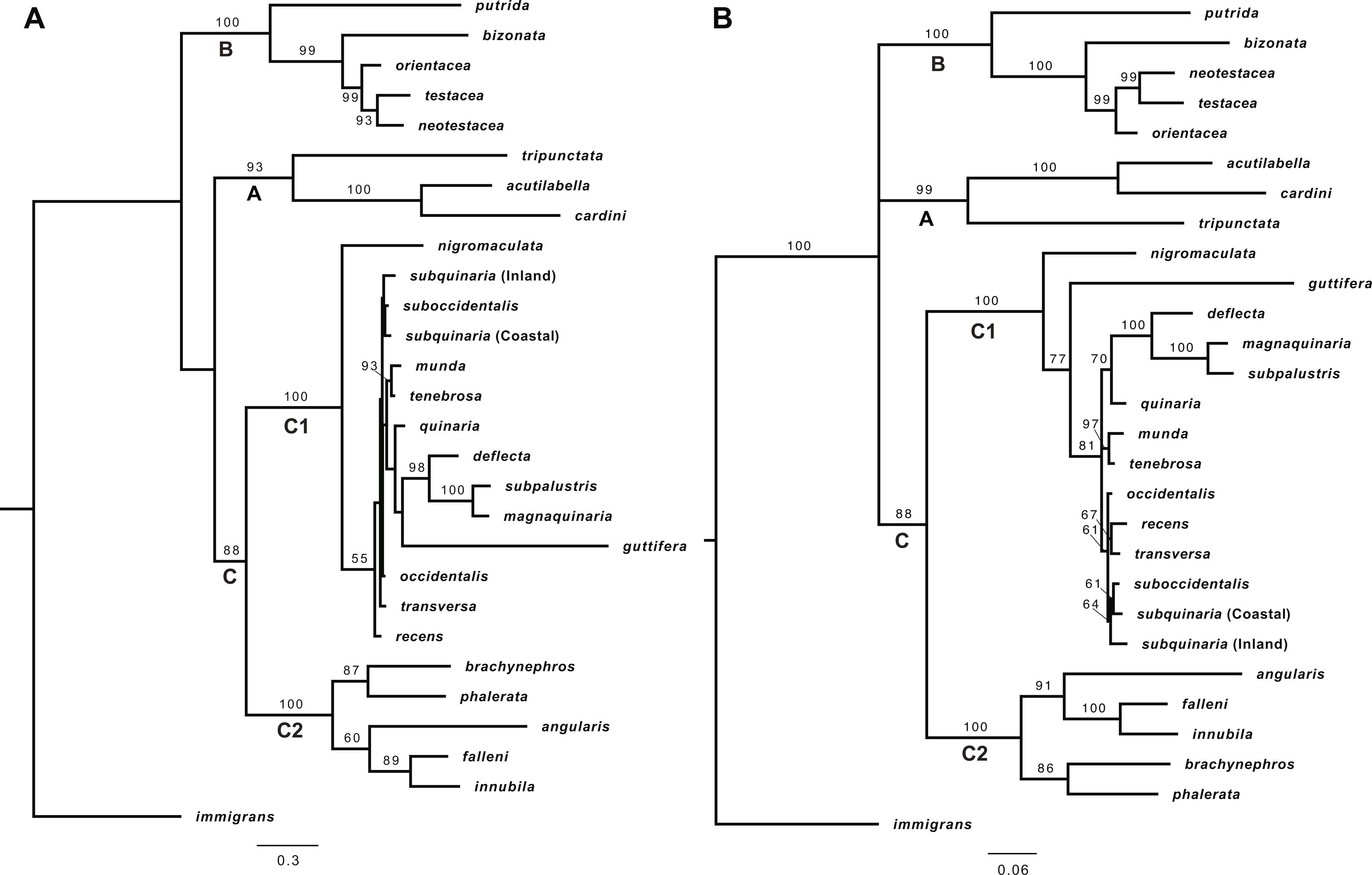
The ML and BI analyses of the concatenated dataset that included all of the nuclear and mtDNA loci recovered well-resolved and strongly supported species trees (Fig. 1 A–B). The topologies were largely congruent, and both contained three clades composed of the same species, which we refer to as clades A, B, and C. Clade A (Bootstrap (BS) = 100; Posterior Probability (PP) = 100), which contained the species representing the tripunctata and cardini groups, is sister to Clades B + C. Clade B is composed of the bizonata and testacea groups (BS = 100; PP = 100) and is sister to a monophyletic quinaria group (BS = 100; PP = 100). We refer to the entire quinaria group as Clade C, and within the quinaria group, the analyses recovered two divergent subgroups, which we refer to as Clades C1 and C2 (BS = 100; PP = 100 for both). Clade C2 is comprised of D. angularis, D. falleni, D. innubila, D. brachynephros, and D. phalerata, while Clade C1 is comprised of all other quinaria group species (Fig. 1). In both the ML and BI species trees, the two subgroups were comprised of the same species, and the relationships among the species were also identical.
Figure 1.
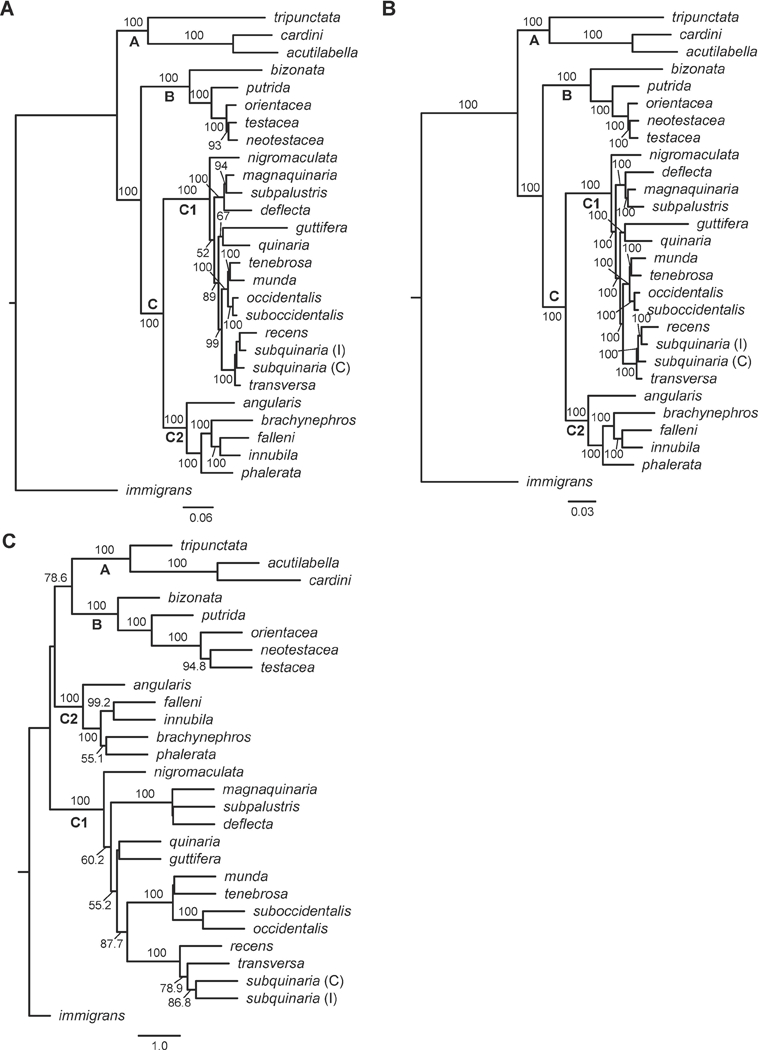
Comparison of phylogenetic relationships within the quinaria and testacea groups using the entire dataset of 43 loci. (A) Analyses of the concatenated dataset using maximum likelihood. Shown is the most likely cladogram, with a LnL = −111850.75. BS ≥ 50 is given above branches; (B) Analyses of the concatenated dataset using Bayesian inference. Shown is the most probable topology, with a Harmonic Mean = −108525.50. PP is given above the branches; (C) The species tree resulting from the ASTRAL analysis of all 43 gene trees, with BS ≥ 50 given above branches. All analyses were rooted with D. immigrans. Clade A represents the tripunctata and cardini group species; Clade B represents the testacea and bizonata group species, and Clade C represents the quinaria group species, with C1 and C2 representing the two divergent clades within this group. Within D. subquinaria, the Coastal and Inland samples are designated by C and I, respectively.
The coalescent-based model analysis in ASTRAL-III using all the loci also recovered a well-resolved, strongly supported species tree (Fig. 1C). While this phylogeny contained two of the same main clades from the ML and BI analyses (Clades A and B; BS = 100 for both), it did not support the monophyly of the quinaria group (Clade C). Instead, the quinaria group was paraphyletic, though the two quinaria subgroups were recovered (Clade C1, BS = 100; Clade C2, BS = 100). The C2 quinaria subgroup (BS < 50) was sister to the lineage (BS > 78.6) containing the tripunctata + cardini (Clade A; BS = 100) and bizonata + testacea clades (Clade B; BS = 100). The C1 subgroup was sister to the other three clades (A, B, and C2).
In addition to differences among the species relationships recovered in the analyses of the concatenated dataset and the coalescent-based model analysis, we scored the species trees from each analysis of the 43 locus dataset in ASTRAL-III to determine the fraction of the induced quartet trees resulting from the input set of gene trees that are in the given species tree (i.e., normalized quartet score (NQS)). The NQS can serve as a measure of the presence of incomplete lineage sorting (Mai et al., 2017; Sayyari and Mirarab, 2016). For each of the three analytical methods, there was only moderate congruence of the gene trees and the species trees. Specifically, the discordance between the gene trees and species trees was approximately the same in the coalescent-based (NQS = 0.7895), the BI analysis (NQS = 0.7858), and the ML analysis (NQS = 0.7858). These findings indicate that 79% of the quartet trees generated from the gene trees could be found in the species trees of each of the three analytical methods.
As previous examinations of the evolutionary relationships of these species utilized datasets composed primarily of mtDNA markers (Dyer et al., 2011b; Hatadani et al., 2009; Izumitani et al., 2016; Morales-Hojas and Vieira, 2012; Perlman et al., 2003; Spicer and Jaenike, 1996), we examined the phylogenetic signal in the three mtDNA loci separately from the nuclear loci using ML and BI analyses (Fig. S2 A, B). Both analyses recovered topologies that contained the three main clades (Clades A-C) and the two quinaria subgroups (Clades C1 and C2) found in the dataset containing all loci (Fig. 1 A, B). However, the relationships among the three clades differed from what was observed in the analysis of all the loci and neither analysis was able to resolve the relationships of these groups (Fig S2 A, B). Both analyses recovered moderate to weak support for the monophyly of the quinaria group (BS > 88; PP > 88), but the evolutionary relationships among the species within each subgroup differed from those found in the all gene analyses.
Given these findings, we completed a ML, BI, and coalescent-based analysis using only the 40 nuclear loci (Fig. 2 A–C). In each of the three analyses, the main clades and the relationships among them did not change with the exclusion of the mtDNA relative to those same analyses with the full dataset. With the ML analysis (Figs. 1A, 2A), the only difference between the topologies occurred among the species relationships recovered for D. magnaquinaria, D. deflecta, and D. subpalustris in subgroup C1. Overall the support values for some relationships showed minor decreases, and the resulting ML species tree exhibited slightly more discordance (NQS = 0.7847) with the gene trees. For the BI analysis (Figs. 1B, 2B), the only topological change occurred among the same three species as in the ML analysis, but the support values for all relationships remained at PP = 100. As with the species tree from the ML analysis, the BI species tree obtained exhibits greater discordance with the gene trees (NQS = 0.7847). The species tree obtained from the coalescent-based analysis (Fig. 2C) was also identical to that recovered by the analysis of the 43 loci (Fig. 1C). Exclusion of the mtDNA increased support in the sister relationship between the C2 clade and Clades A + B (BS > 60.5). In addition, the amount of discordance between the species tree and gene trees did not change for the coalescence-based analysis (NQS = 0.7894). Because of the incongruence of the mtDNA vs. nuclear loci and the well-known issues with using mtDNA in phylogenetic analyses of insects (Cariou et al., 2017; Hurst and Jiggins, 2005), we used the ML and coalescent-based species tree topologies obtained from the nuclear loci for all downstream analyses.
Figure 2.
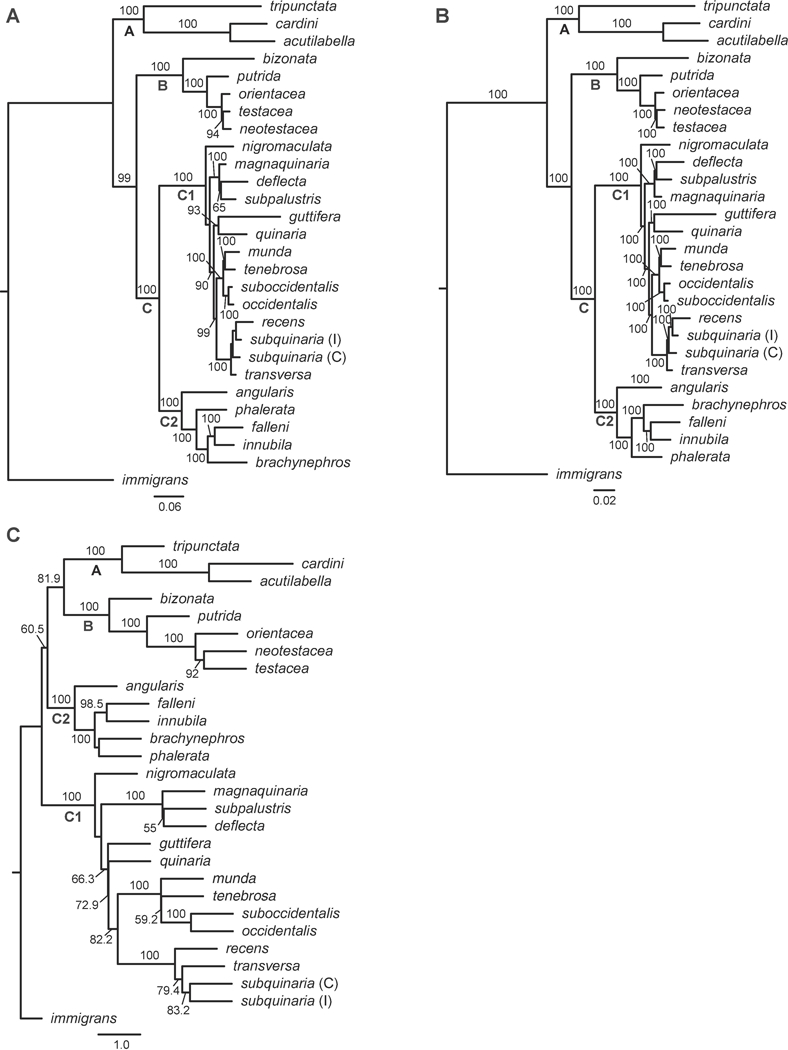
Phylogenetic relationships within the quinaria and testacea groups resulting from the analyses of 40 nuclear loci. BS ≥ 50 are given above branches. (A) Analyses of the concatenated dataset using maximum likelihood. Shown is the most likely cladogram, with a LnL = −102720.99. BS ≥ 50 is given above branches; (B) Analyses of the concatenated dataset using Bayesian inference. Shown is the most probable topology, with a Harmonic Mean = −99861.12. PP is given above the branches; (C) The species tree resulting from the ASTRAL analysis of all 40 gene trees, with BS ≥ 50 given above branches. As in Fig. 1, Clade A represents the tripunctata and cardini group species; Clade B represents the testacea and bizonata group species, and Clade C represents the quinaria group, with C1 and C2 representing the two divergent clades within this group. Within D. subquinaria, the Coastal and Inland samples are designated by C and I, respectively.
3.2. Genomic Incongruence
To identify incongruence among the loci in the different genomic regions, we used a coalescent-based model analysis to assemble species trees from subsets of the molecular dataset corresponding to different genomic regions. To assess the incongruence among the nuclear loci, we recovered species trees for each Müller element and the Y chromosome (Fig. S3A-G) and compared them to species tree recovered by the ASTRAL analysis of the 40 nuclear loci dataset (Fig. 2C). Every genome region except Müller D recovered a phylogeny that contained the A, B, C1, and C2 clades each as a monophyletic group. In the Müller D species tree (Fig. S3D), both the C1 and C2 clades were found to be paraphyletic. The trees recovered from the Müller B, C, and F supported a monophyletic quinaria group (i.e., Clade C), whereas Müller elements A, D, E, and the Y-chromosome did not support a monophyletic quinaria group. When the quinaria group was not monophyletic, it was always the C2 clade that was more closely related to either the testacea + bizonata groups (Clade B) or both this and the tripunctata + cardini groups (Clades A and B). In general, the different genomic regions did not recover consistent sister relationships among the A, B, and C clades. Non-recombining and/or regions involved in reproductive isolation (i.e., Müller F and the sex chromosomes) may show reduced incomplete lineage sorting and/or reduced introgression, but even comparing these three regions they all differed in the sister relationships of the main clades and the monophyly of the quinaria group.
3.3. Feeding behavior evolution
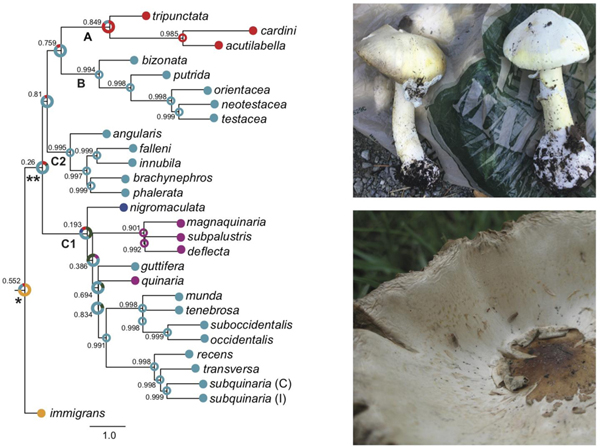
To infer the instances of host switching in the quinaria group, we reconstructed the most likely ancestral feeding behavior states on the ML and ASTRAL species trees recovered from the analysis of all loci except the mtDNA (Fig. 3A,B). Despite the differences in the evolutionary relationships identified by the two phylogenetic analyses, they each suggest similar patterns of feeding behavior evolution. First, within the quinaria group there were two independent instances of switching from mushrooms to vegetation, both within the C1 clade. Three of the vegetative species, D. deflecta, D. magnaquinaria, and D. subpalustris, form a monophyletic group whose common ancestor was likely vegetative feeding (ML: 89.18%, Probability (P)=0.885; ASTRAL: 90.76%, P=0.901). The fourth vegetative species in our analysis, D. quinaria, is a distinct lineage in the group and a separate evolutionary instance of host switching, and we recovered moderate to strong support that its ancestor most likely exhibited mushroom feeding (ML: 87.26%, P=0.873; ASTRAL: 82.81%, P=0.694). Second, the common ancestor of the testacea and quinaria groups was likely mushroom feeding. This is more strongly supported in the ML species tree, where these groups are monophyletic (60.74%, P=0.536). This node on the ASTRAL tree also contains the tripunctata and cardini groups that consume fruit and mushrooms, thus the support for this finding is substantially weaker (75.57%, P=0.26). Third, we recovered moderate to strong support (ML: P=0.511; ASTRAL: P=0.552) that the most likely feeding behavior of the ancestor of the immigrans-tripunctata radiation (Node 1) was fruit feeding (ML: 84.11%; ASTRAL: 73.08%), and weak to moderate support that the ancestor of the ingroup taxa (Node 2) exhibited mushroom feeding behavior (ML: 60.74%, P=0.536; ASTRAL: 75.57%, P=0.26).
Figure 3.
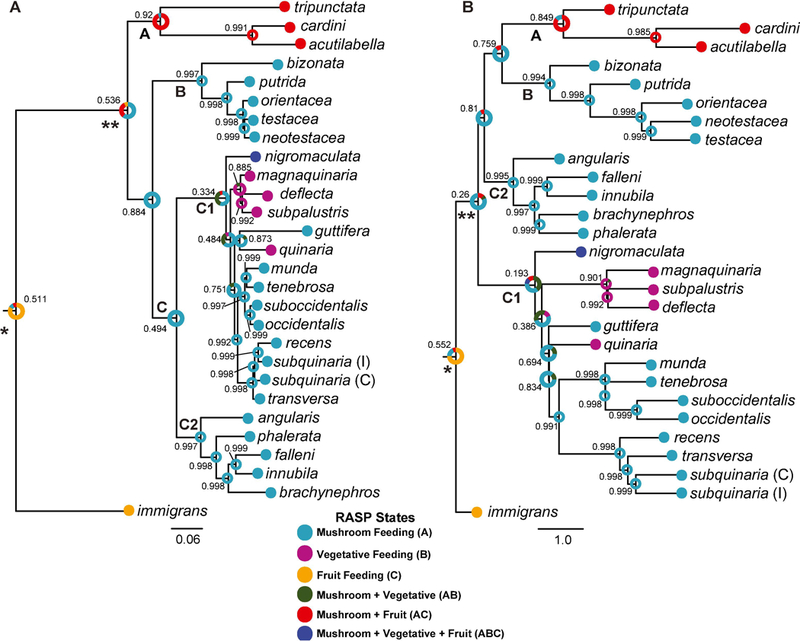
Evolutionary hypothesis of feeding behavior using species trees reconstructed from the 40 nuclear loci. (A) Feeding behavior reconstruction based on ML topology; (B) Feeding behavior reconstruction based on the ASTRAL species tree. The most likely ancestral feeding behavior reconstruction and alternative reconstructions with a likelihood > 5% is indicated on each branch. The probability (P) of the most likely ancestral state is provided for each node. Clade A represents the tripunctata and cardini group species; Clade B represents the testacea and bizonata group species, and Clade C represents a monophyletic quinaria group, with C1 and C2 representing the two divergent clades within this group; * represents Node 1, the ancestor of the immigrans-tripunctata radiation; ** represents Node 2, the ancestor of the ingroup taxa. Within D. subquinaria, the Coastal and Inland samples are designated by C and I, respectively.
3.4. Rates of protein evolution
Within the quinaria group some species are generalists on fleshy basidiomycete mushrooms while others are specialists on decaying water plants. A narrower host range of the vegetative species may result in a decrease in the effective population size and thus a relaxation of purifying selection. However, we do not find compelling evidence for relaxation of purifying selection in these lineages (Fig. S4). For each locus, we compared a model with two ω (or dN/dS) values, one for the clade(s) of the vegetative species (ω-veg; see Table 1) and another for the rest of the phylogeny (ω-nonveg), with a model with one ω for the entire tree (ω-all). We found a two ω model to be a better fit for one locus using the ML species tree (per), for four loci using the ASTRAL species tree (ebony, mof, per, skp), and for two loci using the gene trees (per, skp) (Table S4). The per locus was the only locus with consistent evidence of an elevated dN/dS value in the vegetative lineages (Table S4). Most of the loci appear to be under strong purifying selection (Fig. S4), and across all 40 nuclear loci overall we found no significant differences between the ω values calculated from any of the three topologies (e.g., ML species tree: median ω-all = 0.031, median ω-veg = 0.031, median ω-nonveg = 0.033; Steel-Dwass pairwise tests, all P > 0.85).
4. Discussion
Previous phylogenetic studies of the quinaria and testacea groups and their placement within the immigrans-tripunctata radiation have produced results that are largely incongruent due, in part, to limited sampling of genomic compartments (i.e., just mtDNA, Y-chromosome, and/or a few nuclear loci) (Dyer et al., 2011a; Dyer et al., 2011b; Hatadani et al., 2009; Izumitani et al., 2016; Morales-Hojas and Vieira, 2012; Perlman et al., 2003; Spicer and Jaenike, 1996). In addition, hybridization and incomplete lineage sorting can obscure phylogenetic signal in recent adaptive radiations, and both may be occurring in species groups. Our study represents the first attempt to analyze evolutionary relationships within and among the quinaria and testacea groups and several other closely species groups in the immigrans-tripunctata radiation using many loci that are distributed across all of the chromosomes of the genome.
4.1. Congruence among loci and approaches
Given our knowledge of the natural history and genetics of the quinaria group, there is a strong possibility for the phylogenetic history of the individual loci in our dataset to differ from that of the “true” species tree. Thus, we reconstructed species trees using ML and BI analyses of a concatenated dataset and also using a coalescent-based model analysis of the individual gene trees. We found that the species tree from each of three analyses contained the same four clades (A, B, C1, C2), though the evolutionary relationships among these clades varied depending on the type of analysis. Most significantly for our species of interest, the C1 and C2 clades that comprise the quinaria group species were not always monophyletic, though the same species were always found within each clade. While the analyses of the concatenated dataset recovered strong support for the monophyly of the quinaria group, the coalescent-based species analysis found the quinaria group to be paraphyletic with respect to the other ingroup taxa. Given this incongruence across analysis methods, we examined subsets of the data to understand the factors that may be contributing to topology differences both within the quinaria group as well as among the other species groups we sampled.
mtDNA is especially prone to incongruent phylogenetic patterns due to transfer between species through hybridization and linkage with maternally-transmitted endosymbionts such as Wolbachia and Spiroplasma. We know that both of these phenomena occur in the quinaria group – for instance, some D. quinaria harbor a highly divergent mtDNA haplotype at low frequency that originates from an unknown and/or extinct species (Dyer et al., 2011a) and D. subquinaria harbors a low frequency of D. recens mtDNA (Bewick and Dyer, 2014; Jaenike et al., 2006). Both of these events are likely due to past hybridization events. Indeed, in our study, the phylogenies constructed from the mtDNA are incongruent with the nuclear loci. For instance, the mtDNA trees did not support the monophyly of the testacea group, nor did they support the sister relationship between D. occidentalis and D. suboccidentalis (Fig. S3). This is in contrast to all of our species trees, no matter the analytical method, as well as the previous parsimony-based analyses of these species that used only these mtDNA loci (e.g. Perlman et al. 2003). Thus, our findings support many previous suggestions that the mtDNA can bias species-level phylogenies, and we suggest that the species tree excludes this genomic region. Given the extensive sampling of nuclear loci in this study, exclusion of the mtDNA loci from the analysis did not significantly alter the evolutionary relationships recovered for any of the three species trees (Figs. 1, 2). The relationships of the four clades (A, B, C1, and C2) were all the same, and the only difference in the species relationships within the groups was in the relationships of some of the species in the C1 clade of the ML and BI tree.
We also assessed concordance of the different chromosomes of the nuclear genome. While the major groupings (A, B, C1, and C2 clades) were present in the species tree from each chromosome, the relationships among and within them varied. This incongruence among the different genomic regions is likely to be in part responsible for the low branch support values along the backbone of the phylogeny in the coalescent analysis. Our findings of the major species groups (Clades A, B, C1, and C2) and the species found within each group are consistent with other recent studies that used a smaller number of loci (Dyer et al., 2011b; Hatadani et al., 2009; Izumitani et al., 2016; Morales-Hojas and Vieira, 2012; Perlman et al., 2003). Thus, these clades likely represent natural groups. To recover the relationships among these groups it will be necessary to sample additional taxa from other species groups within the immigrans-tripunctata radiation (e.g., funebris, guarani, and others) and employ a larger number of markers.
Other studies have also found that the quinaria group is comprised of two divergent subgroups, and we cannot resolve whether these subgroups form a monophyletic group. The species trees from Müller B, C, and F supported the monophyly of the quinaria group (C1 and C2 clades), whereas for the remaining four genomic regions (Müller A(X), D, and E and the Y-Chromosome), the quinaria group is paraphyletic. While regions of the genome that have reduced recombination (i.e. Müller F and the Y-chromosome) or are generally involved in reproductive isolation (i.e. the sex chromosomes) may show reduced incomplete lineage sorting due to a smaller effective population size and/or reduced introgression in other species, these regions do not show any consistent pattern in our dataset. Given the large effective population sizes and potential for incomplete lineage sorting and hybridization, the resolution of the monophyly of the quinaria group will require larger-scale approaches than used here.
4.2. Patterns of reproductive isolation and divergence
Within the quinaria group, both of the C1 and C2 clades contain New and Old-World species. While Patterson and Stone (1952) lists instances of potential hybridization between C1 and C2 species, all of the cases we have been able to confirm in the laboratory are between species pairs in the C1 clade. Furthermore, within the C1 clade, species that hybridize generally do not co-occur, even if they are closely related (Coyne and Orr, 1989). For instance, D. tenebrosa is found only at high elevation in the mountains of the southeast United States and Mexico, and can hybridize with D. suboccidentalis, D. recens, and D. palustris, all of which occur only in northern North America (Dyer, unpublished). Likewise, D. suboccidentalis and D. occidentalis can produce fertile F1 hybrids but do not co-occur (Arthur and Dyer, 2015). The exception is D. recens and D. subquinaria, which co-occur in central Canada (Jaenike et al. 2006). These species along with D. transversa show incomplete reproductive isolation: hybrid sons of D. recens and either D. subquinaria or D. transversa are sterile, while hybrid sons and daughters between D. subquinaria and D. transversa are fully fertile (Humphreys et al., 2016; Shoemaker et al., 1999). The three species tree analyses all result in a different relationship of these species, which is likely due to recent hybridization and incomplete lineage sorting. Even the two geographic populations of D. subquinaria (coastal and inland) are not consistently monophyletic among analyses. Given the complex demography and incomplete isolation among these species, studies are underway that use more samples from each species, which may sort out some of these incongruences.
Within the testacea group, our results are consistent with previous studies in identifying that the North American species D. putrida is sister to the other three species in the group (i.e., Perlman et al. 2003, Dyer et al. 2011b, Izumitani et al. 2016). This is also consistent with patterns of reproductive isolation, as D. putrida will not form hybrids with any of the other testacea group species and it is both ecologically and morphologically distinct from the others. Considering the other three species in this group, most of our analyses suggest that D. neotestacea and D. testacea are most closely related, with D. orientacea as the outgroup. However, this is contrary to patterns of reproductive isolation: D. orientacea and D. testacea can form fertile F1 female hybrids, whereas neither of these species form hybrids with D. neotestacea (Dyer et al., 2011b; Grimaldi et al., 1992). A further puzzle is that in the analyses that do not replicate the pattern of D. orientacea being the outgroup of this species trio, D. orientacea is found to be sister to D. neotestacea and not D. testacea (Müller C and ML of the full dataset, also Perlman et al. 2003), or the three species form a polytomy (Müller D and Y-chromosome). All three of these species are morphologically identical; D. neotestacea is found in the New World and the other two species occur in the Old World, where it is unknown whether their ranges overlap. Finally, little is known about the Asian species D. bizonata, which is also now found in Hawaii (Leblanc et al., 2009), but it is morphologically very similar to the testacea group species. It is found to be sister to the testacea group in most of our analysis as well as in another recent analysis (Izumitani et al., 2016).
4.3. Evolution of feeding strategies
Species within the immigrans-tripunctata radiation feed on mushrooms, decaying vegetation, sap fluxes, fermenting fruits, flowers, or combinations of multiple host sources (Markow and O’Grady, 2008). We reconstructed the feeding history of the species we sampled, focusing on the quinaria and testacea groups. First, within the quinaria group, species are primarily mushroom feeders, but several have switched to utilizing decaying vegetation (Markow and O’Grady, 2008; Morales-Hojas and Vieira, 2012; Spicer and Jaenike, 1996). We found that there were likely two independent host-switching events within the C1 clade of the quinaria group from mushroom-feeding to vegetation-feeding (Fig. 3). It will be interesting to determine whether the preference for vegetation has the same or different genetic basis in these two lineages. We do not find a difference in the rate of protein evolution in the vegetative versus non-vegetative lineages (Fig. S4). However, it is interesting to note that period is both the fastest evolving locus in our dataset and the only locus to have a consistently higher rate of protein evolution in vegetative versus non-vegetative species (Table S4), though it does not display evidence of positive selection (i.e. dN/dS is not greater than 1). This gene is involved in circadian rhythms and courtship behaviors (reviewed in Nitabach and Taghert (2008) and Tataroglu and Emery (2015)). Previous work in the quinaria group found that the vegetative species are active throughout the day, whereas the mushroom species tended to restrict their activity to the morning and evening (Simunovic and Jaenike, 2006), thus an intriguing possibility is that a shift in the selective pressures of circadian feeding behaviors is reflected in the molecular evolution of the per locus.
At a deeper phylogenetic scale, we found that the common ancestor of the quinaria and testacea groups was probably mushroom-feeding, though additional species group sampling within this radiation will be needed to confirm this finding. It will be interesting to determine if traits present in these groups associated with mushroom-feeding such as the ability to tolerate mushroom toxins also had a single origin. Finally, at the base of our phylogeny we found that the ancestral state of the immigrans-tripunctata radiation was probably fruit feeding. This seems likely given that species within the immigrans group and closely related groups outside of the radiation are primarily fruit feeders (Kimura, 1980; Mitsui et al., 2010; Morales-Hojas and Vieira, 2012). Within the cardini, immigrans, and tripunctata groups a variety of feeding strategies occur, many of which were not included in our study. For instance, some species only exhibit only the fruit feeding strategy, others are fruit or flower feeders (e.g., guarani and pallidipennis), and others (e.g., funebris group) exhibit mushroom feeding but only consume polypore rather than basidiomycete mushrooms (Lacy, 1984; Markow and O’Grady, 2008; Morales-Hojas and Vieira, 2012). In general, the inclusion of additional taxa from these groups in future studies will help resolve the ancestral feeding strategy along the backbone of the phylogeny.
5. Conclusions
Our results represent the first well-sampled phylogeny of the quinaria and testacea groups of Drosophila. The findings from our different phylogenetic analyses highlight the importance of careful selection of molecular markers and types of analyses employed, particularly in recent radiations. This study also provides a framework phylogeny for the immigrans-tripunctata radiation that can be used to guide the taxon selection for future studies. The inclusion of representatives of additional species groups and additional species from the immigrans, cardini, and tripunctata groups could help to resolve the deeper relationships within the radiation as well as whether the quinaria group should be split into two species groups. Given that we sampled almost all of the species currently placed within the testacea and quinaria groups, it is unlikely that additional taxon sampling will help to resolve relationships, particularly in subgroup C1. However, employing phylogenomic approaches may resolve some of the more recent nodes where reproductive isolation among species is not complete and they have a complex demographic history (e.g., exhibit incomplete lineage sorting, hybridization, and/or a combination of both factors). Even with these uncertainties, we are able to infer that the evolution of mushroom-feeding in the quinaria and testacea groups likely occurred once, and that within the quinaria group there were probably two independent instances of host-switching from mushroom-feeding to vegetation feeding. Future studies will disentangle whether these host switches occurred via a similar genetic mechanism.
Supplementary Material
Sequence variation by genomic region. (A) Mean number of variable sites per base pair and standard deviation for each region; (B) Mean number of parsimony informative sites per base pair and standard deviation for each region. B – F = Müller elements B – F; A(X) and Y = Sex chromosomes; mt = mtDNA; AO = autosomes only; WG = whole genome. The number of loci sampled from each genomic region is indicated below the label on the X-axis.
{kind=link}
Phylogenetic relationships resulting from analyses of the three mtDNA loci. (A) Analyses of the concatenated mtDNA dataset using maximum likelihood. Shown is the most likely cladogram, with a LnL = −8181.07. BS ≥ 50 is given above branches; (B) Analyses of the concatenated mtDNA dataset using Bayesian inference. Shown is the most probable topology, with a Harmonic Mean = −8103.53. PP is given above the branches. Clade A represents the tripunctata and cardini group species; Clade B represents the testacea and bizonata group species, and Clade C represents the quinaria group species, with C1 and C2 representing the two divergent clades within this group.
{kind=link}
Visualization of incongruence in different genomic regions sampled. A comparison between the ASTRAL species tree recovered from the 40 gene dataset (on the left) and the tree recovered from each individual autosome and sex chromosome (on the right) included in the dataset. (A) Müller Element A (X-chromosome); (B) Müller Element B; (C) Müller Element C; (D) Müller Element D; (E) Müller Element E; (F) Müller Element F; (G) Y-chromosome. BS ≥ 50 given above branches. All analyses were rooted with D. immigrans. Clade A represents the tripunctata and cardini group species; Clade B represents the testacea and bizonata group species, and Clade C represents the quinaria group species, with C1 and C2 representing the two divergent clades within this group.
Results from the PAML analysis. Shown is a boxplot created from the individual locus values of ω (dN/dS). Results are shown by phylogenetic tree and by branch model (one ω for the entire tree or two ω, including one value for the vegetation-consuming clade and another for the rest of the tree).
Tabular data (.xlsx); The optimal partitioning scheme identified with PartitionFinder and used in the ML and BI analyses.
Tabular data (.csv); The codings for feeding state(s) used in the RASP analysis.
Tabular data (.xlsx); Species used in the study and the sequence coverage in each. For each species included in the analysis, the species group, feeding behavior, stock name, and strain source are included. Successfully amplified loci are indicated with +.
Tabular data (.xlsx); Loci used in this study. Includes the full locus name, abbreviation, Flybase ID, genome location (Müller element), presence of introns, and primers used to amplify and sequence each.
Tabular data (.xlsx); Phylogenetic information and genetic variation found in each locus. Includes the Müller Element of the locus, the length of the alignment, the number of variable sites (VS), the number of parsimony informative (PI) sites in each locus, the variance of each locus in VS/BP and PI/BP, and the total values for the genomic location.
Tabular data (.xlsx); Results from codon models of molecular evolution. Each locus was analyzed using each of the pruned species tree and the locus-specific gene tree. Shown is the ω value for each model (1ω and 2ω) as well as the significance of each comparison using a likelihood ratio test. Includes the genomic location (autosome, dot, or X and Y-chromosome).
Highlights.
Phylogenetic analyses support monophyly of testacea group but not quinaria group
Phylogeny offers framework to guide future study of immigrans-tripunctata radiation
Ancestor of the tripunctata and quinaria groups exhibited mushroom-feeding
Two transitions to vegetative feeding occurred in the quinaria group
No change in rate of protein evolution associated with switch to vegetative feeding
Acknowledgements
This work was funded by National Science Foundation grants DEB-1149350 to KAD, DEB-1457707 to Corbin Jones and KAD, DEB-1737869 to LKR and CHSC, DEB-1737824 to KAD, a National Institute of Health grant R01 GM098856 to LKR, a University of Alabama – RSP Postdoctoral Fellowship to CHSC. We are grateful to John Jaenike for sharing fly stocks and his natural history knowledge about these flies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arthur NJ, Dyer KA, 2015. Asymmetrical sexual isolation but no postmating isolation between the closely related species of Drosophila suboccidentalis and D. occidentalis. BMC Evolutionary Biology 15, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewick ER, Dyer KA, 2014. Reinforcement shapes clines in mate discrimination in Drosophila subquinaria. Evolution 68, 3082–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutkar A, Schaeffer SW, Russo SM, Xu M, Smith TF, Gelbart WM, 2008. Chromosomal rearrangement inferred from comparisons of 12 Drosophila genomes. Genetics 179, 1657–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray MJ, Werner T, Dyer KA, 2014. Two genomic regions together cause dark abdominal pigmentation in Drosophila tenebrosa. Heredity 112, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariou M, Duret L, Charlat S, 2017. The global impact of Wolbachia on mitochondrial diversity and evolution. Journal of Evolutionary Biology 30, 2204–2210. [DOI] [PubMed] [Google Scholar]
- Colon-Parrilla WV, Perez-Chiesa I, 1999. Ethanol tolerance and alcohol dehydrogenase (ADH; EC1.1.1.1) activity in species of the cardini group of Drosophila. Biochemical Genetics 37, 95–107. [DOI] [PubMed] [Google Scholar]
- Coyne JA, Orr HA, 1989. Patterns of speciation in Drosophila. Evolution 43, 362–381. [DOI] [PubMed] [Google Scholar]
- Dombeck I, Jaenike J, 2004. Ecological genetics of abdominal pigmentation in Drosophila falleni. Evolution 58, 587–596. [PubMed] [Google Scholar]
- Dyer KA, 2012. Local selection underlies the geographic distribution of sex-ratio drive in Drosophila neotestacea. Evolution 66, 974–984. [DOI] [PubMed] [Google Scholar]
- Dyer KA, Bray MJ, Lopez SJ, 2013. Genomic conflict drives pattern of X-linked population structure in Drosophila neotestacea. Molecular Ecology 22, 157–169. [DOI] [PubMed] [Google Scholar]
- Dyer KA, Burke C, Jaenike J, 2011a. Wolbachia-mediated persistence of mtDNA from a potentially extinct species. Molecular Ecology 20, 2805–2817. [DOI] [PubMed] [Google Scholar]
- Dyer KA, Charlesworth B, Jaenike J, 2007. Chromosome-wide linkage disequilibrium as a consequence of meiotic drive. Proceedings of the National Academy of Sciences of the United States of America 104, 1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer KA, Jaenike J, 2004. Evolutionary stable infection by a male-killing endosymbiont in Drosophila innubila: Molecular evidence from the host and parasite genomes. Genetics 168, 1443–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer KA, Jaenike J, 2005. Evolutionary dynamics of a spatially structured host-parasite association: Drosophila innubila and male-killing Wolbachia. Evolution 59, 1518–1528. [PubMed] [Google Scholar]
- Dyer KA, White BE, Bray MJ, Piqué DG, Betancourt AJ, 2011b. Molecular evolution of a Y chromosome to autosome gene duplication in Drosophila. Molecular Biology and Evolution 28, 1293–1306. [DOI] [PubMed] [Google Scholar]
- Dyer KA, White BE, Sztepanacz J, Bewick ER, Rundle HD, 2014. Reproductive character displacement of epicuticular compounds and their contribution to mate choice in Drosophila subquinaria and D. recens. Evolution 68, 1163–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SV, 2009. Is a new and general theory of molecular systematics emerging? Evolution 63, 1–19. [DOI] [PubMed] [Google Scholar]
- Edwards SV, Xi Z, Janke A, Faircloth BC, McCormack JE, Glenn TC, Zhong B, Wu S, Lemmon EM, Lemmon AR, Leache AD, Liu L, Davis CC, 2016. Implementing and testing the multispecies coalescent model: A valuable paradigm for phylogenomics. Molecular Phylogenetics and Evolution 94, 447–462. [DOI] [PubMed] [Google Scholar]
- Gelman A, Rubin DB, 1992. Inference from iterative simulation using multiple sequences. Statistical Science 7, 457–511. [Google Scholar]
- Gibbs AG, Matzkin LM, 2001. Evolution of water balance in the genus Drosophila. Journal of Experimental Biology 204, 2331–2338. [DOI] [PubMed] [Google Scholar]
- Giglio EM, Dyer KA, 2013. Divergence of premating behaviors in the closely related species Drosophila subquinaria and D. recens. Ecology and Evolution 3, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givnish TJ, Sytsma KJ (Eds.), 1997. Molecular evolution and adaptive radiation Cambridge University Press, Cambridge, UK. [Google Scholar]
- Grant PR, Grant BR, 2008. How and why species multiply: The radiation of Darwin’s finches Princeton University Press, Princeton, New Jersey, USA. [Google Scholar]
- Grimaldi D, Jaenike J, 1983. The Diptera breeding on skunk cabbage, Symplocarpus foetidus (Araceae). Journal of the New York Entomological Society 91, 83–89. [Google Scholar]
- Grimaldi D, James AC, Jaenike J, 1992. Systematics and modes of reproductive isolation in the Holarctic Drosophila testacea species group (Diptera: Drosophilidae). Annals of the Entomological Society of America 85, 671–685. [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O, 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology 59, 307–321. [DOI] [PubMed] [Google Scholar]
- Hahn MW, Nakhleh L, 2016. Irrational exuberance for resolved species trees. Evolution 70, 7–17. [DOI] [PubMed] [Google Scholar]
- Haselkorn TS, Cockburn SN, Hamilton PT, Perlman SJ, Jaenike J, 2013. Infectious adaptation: potential host range of a defensive endosymbiont in Drosophila. Evolution 67, 934–945. [DOI] [PubMed] [Google Scholar]
- Haselkorn TS, Markow TA, Moran NA, 2009. Multiple introductions of the Spiroplasma bacterial endosymbiont into Drosophila. Molecular Ecology 18, 1294–1305. [DOI] [PubMed] [Google Scholar]
- Hatadani LM, McInerney JO, de Medeiros HF, Junqueira AC, de Azeredo-Espin AM, Klaczko LB, 2009. Molecular phylogeny of the Drosophila tripunctata and closely related species groups (Diptera: Drosophilidae). Molecular Phylogenetics and Evolution 51, 595–600. [DOI] [PubMed] [Google Scholar]
- Humphreys DP, Rundle HD, Dyer KA, 2016. Patterns of reproductive isolation in the Drosophila subquinaria complex: Can reinforced premating isolation cascade to other species? Current Zoology 62, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst GDD, Jiggins FM, 2005. Problems with mitochondrial DNA as a marker in population phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proceedings: Biological Sciences 272, 1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumitani HF, Kusaka Y, Koshikawa S, Toda MJ, Katoh T, 2016. Phylogeography of the subgenus Drosophila (Diptera: Drosophilidae): evolutionary history of faunal divergence between the Old and the New Worlds. PLoS One 11, e0160051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenike J, 1999. Suppression of sex-ratio meiotic drive and the maintenance of Y-chromosome polymorphism in Drosophila. Evolution 53, 164–174. [DOI] [PubMed] [Google Scholar]
- Jaenike J, 2007. Spontaneous emergence of a new Wolbachia phenotype. Evolution 61, 2244–2252. [DOI] [PubMed] [Google Scholar]
- Jaenike J, Dyer KA, Cornish C, Minhas MS, 2006. Asymmetrical reinforcement and Wolbachia infection in Drosophila. PLoS Biology 4, e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenike J, Grimaldi DA, Sluder AE, Greenleaf AL, 1983. α-Amanitin tolerance in mycophagous Drosophila. Science 221, 165–167. [DOI] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stone-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A, 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequences data. Bioinformatics 28, 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura MT, 1980. Evolution of food preferences in fungus-feeding Drosophila: an ecological study. Evolution 34, 1009–1018. [DOI] [PubMed] [Google Scholar]
- Kimura MT, 2004. Cold and heat tolerance of drosophilid flies with reference to their latitudinal distributions. Oecologia 140, 442–449. [DOI] [PubMed] [Google Scholar]
- Kimura M, Toda M, Beppu K, Watabe H, 1977. Breeding sites of Drosophilid flies in and near Sapporo, northern Japan, with supplementary notes on adult feeding habits. Japanese Journal of Entomology 45, 571–582 [Google Scholar]
- Kubatko LS, Degnan JH, 2007. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Systematic Biology 56, 17–24. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K, 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy RC, 1984. Predictability, toxicity, and trophic niche breadth in fungus-feeding Drosophilidae (Diptera). Ecological Entomology 9, 43–54. [Google Scholar]
- Lanfear R, Calcott B, Ho SYW, Guindon S, 2012. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Molecular Biology and Evolution 29, 1695–1701. [DOI] [PubMed] [Google Scholar]
- Lanfear R, Frandsen PB, Wright AP, Senfeld T, Calcott B, 2017. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Molecular Biology and Evolution 34, 772–773. [DOI] [PubMed] [Google Scholar]
- Leblanc L, O’Grady PM, Rubinoff D, Montgomery SL, 2009. New immigrant Drosophilidae in Hawaii and a checklist of the established immigrant species. Proceedings of the Hawaiian Entomological Society 41, 121–127. [Google Scholar]
- Li S, Jovelin R, Yoshiga T, Tanaka R Cutter AD, 2014. Specialist versus generalist life histories and nucleotide diversity in Caenorhabditis nematodes. Proceedings of the Royal Society B 281, 20132858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai U, Sayyari E, Mirarab S, 2017. Minimum variance rooting of phylogenetic trees and implications for species tree reconstruction. PLoS One 12, e0182238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J, Besansky N, Hahn MW, 2016. How reticulated are species? Bioessays 38, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markow TA, O’Grady P, 2008. Reproductive ecology of Drosophila. Functional Ecology 22, 747–759. [Google Scholar]
- Mendes FK, Hahn MW, 2018. Why concatenation fails near the anomaly zone. Systematic Biology 67, 158–169. [DOI] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T, 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, pp. 1–8. [Google Scholar]
- Mitsui H, Beppu K, Kimura MT, 2010. Seasonal life cycles and resource uses of flower- and fruit-feeding drosophilid flies (Diptera: Drosophilidae) in central Japan. Entomological Science 13, 60–67. [Google Scholar]
- Morales-Hojas R, Vieira J, 2012. Phylogenetic patterns of geographical and ecological diversification in the subgenus Drosophila. PLoS One 7, e49552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitabach MN, Taghert PH, 2008. Organization of the Drosophila circadian control circuit. Current Biology 18, R84–R93. [DOI] [PubMed] [Google Scholar]
- O’Meara BC, 2012. Evolutionary inferences from phylogenies: a review of methods. Annual Review of Ecology, Evolution, and Systematics 43, 267–285. [Google Scholar]
- O’Grady PM, DeSalle R, 2018. Phylogeny of the genus Drosophila. Genetics 209, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson JT, Stone WS, 1952. Evolution in the genus Drosophila The Macmillan Company, New York, USA. [Google Scholar]
- Perlman SJ, Jaenike J, 2003. Infection success in novel hosts: an experimental and phylogenetic study of Drosophila-parasitic nematodes. Evolution 57, 544–557. [DOI] [PubMed] [Google Scholar]
- Perlman SJ, Spicer GS, Shoemaker DD, Jaenike J, 2003. Associations between mycophagous Drosophila and their Howardula nematode parasites: a worldwide phylogenetic shuffle. Molecular Ecology 12, 237–249. [DOI] [PubMed] [Google Scholar]
- Roch S, Steel M, 2014. Likelihood-based tree reconstruction on a concatenation of aligned sequence data sets can be statistically inconsistent. Theoretical population biology 100C, 56–62. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP, 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyari E, Mirarab S, 2016. Fast coalescent-based computation of local branch support from quartet frequencies. Molecular Biology and Evolution 33, 1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter D, 2000. The ecology of adaptive radiation Oxford University Press Inc., Oxford, New York. [Google Scholar]
- Shoemaker DD, Dyer KA, Ahrens M, McAbee K, Jaenike J, 2004. Decreased diversity but increased substitution rate in host mtDNA as a consequence of Wolbachia endosymbiont infection. Genetics 168, 2049–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker DD, Katju V, Jaenike J, 1999. Wolbachia and the evolution of reproductive isolation between Drosophila recens and Drosophila subquinaria. Evolution 53, 1157–1164. [DOI] [PubMed] [Google Scholar]
- Simunovic A, Jaenike J, 2006. Adaptive variation among Drosophila species in their circadian rhythms. Evolutionary Ecology Research 8, 803–811. [Google Scholar]
- Spicer GS, Jaenike J, 1996. Phylogenetic analysis of breeding site use and α-Amanitin tolerance within the Drosophila quinaria species group. Evolution 50, 2328–2337. [DOI] [PubMed] [Google Scholar]
- Stahlhut JK, Desjardins CA, Clark ME, Baldo L, Russell JA, Werren JH, Jaenike J, 2010. The mushroom habitat as an ecological arena for global exchange of Wolbachia. Molecular Ecology 19, 1940–1952. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, 2006. RAxML-VI-HPC: Maximum Likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump AD, Jablonski SE, Bouton L, Wilder JA, 2011. Distribution and mechanism of alpha-amanitin tolerance in mycophagous Drosophila (Diptera: Drosophilidae). Environmental Entomology 40, 1604–1612. [DOI] [PubMed] [Google Scholar]
- Tataroglu O, Emery P, 2015. The molecular ticks of the Drosophila circadian clock. Current Opinion in Insect Science 7, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuno N, Takahashi KH, Yamashita H, Osawa N, Tanaka C, 2007Tolerance of Drosophila flies to ibotenic acid poisons in mushrooms. Journal of Chemical Ecology 33, 311–317. [DOI] [PubMed] [Google Scholar]
- Unckless RL, Jaenike J, 2012. Maintenance of a male-killing Wolbachia in Drosophila innubila by male-killing dependent and male-killing independent mechanisms. Evolution 66, 678–689. [DOI] [PubMed] [Google Scholar]
- Werner T, Jaenike J, 2017. Drosophilids of the Midwest and Northeast River Campus Libraries, University of Rochester, Rochester, NY. [Google Scholar]
- Werner T, Koshikawa S, Williams TM, Carroll SB, 2010. Generation of a novel wing color pattern by the Wingless morphogen. Nature 464, 1143–1148. [DOI] [PubMed] [Google Scholar]
- Werren JH, Jaenike J, 1995. Wolbachia and cytoplasmic incompatibility in mycophagous Drosophila and their relatives. Heredity 75, 320–326. [DOI] [PubMed] [Google Scholar]
- Yang Z, 2007. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yu Y, Harris AJ, Blair C, He X, 2015. RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Molecular Phylogenetics and Evolution 87, 46–49. [DOI] [PubMed] [Google Scholar]
- Zhang C, Sayyari E, Mirarab S, 2017. ASTRAL-III: increased scalability and impacts of contracting low support branches. In: Meidanis J, Nakhleh L (Eds.), Comparative Genomics: 15th International Workshop Springer International Publishing, Barcelona, Spain, pp. 53–75. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence variation by genomic region. (A) Mean number of variable sites per base pair and standard deviation for each region; (B) Mean number of parsimony informative sites per base pair and standard deviation for each region. B – F = Müller elements B – F; A(X) and Y = Sex chromosomes; mt = mtDNA; AO = autosomes only; WG = whole genome. The number of loci sampled from each genomic region is indicated below the label on the X-axis.
Phylogenetic relationships resulting from analyses of the three mtDNA loci. (A) Analyses of the concatenated mtDNA dataset using maximum likelihood. Shown is the most likely cladogram, with a LnL = −8181.07. BS ≥ 50 is given above branches; (B) Analyses of the concatenated mtDNA dataset using Bayesian inference. Shown is the most probable topology, with a Harmonic Mean = −8103.53. PP is given above the branches. Clade A represents the tripunctata and cardini group species; Clade B represents the testacea and bizonata group species, and Clade C represents the quinaria group species, with C1 and C2 representing the two divergent clades within this group.
Visualization of incongruence in different genomic regions sampled. A comparison between the ASTRAL species tree recovered from the 40 gene dataset (on the left) and the tree recovered from each individual autosome and sex chromosome (on the right) included in the dataset. (A) Müller Element A (X-chromosome); (B) Müller Element B; (C) Müller Element C; (D) Müller Element D; (E) Müller Element E; (F) Müller Element F; (G) Y-chromosome. BS ≥ 50 given above branches. All analyses were rooted with D. immigrans. Clade A represents the tripunctata and cardini group species; Clade B represents the testacea and bizonata group species, and Clade C represents the quinaria group species, with C1 and C2 representing the two divergent clades within this group.
Results from the PAML analysis. Shown is a boxplot created from the individual locus values of ω (dN/dS). Results are shown by phylogenetic tree and by branch model (one ω for the entire tree or two ω, including one value for the vegetation-consuming clade and another for the rest of the tree).
Tabular data (.xlsx); The optimal partitioning scheme identified with PartitionFinder and used in the ML and BI analyses.
Tabular data (.csv); The codings for feeding state(s) used in the RASP analysis.
Tabular data (.xlsx); Species used in the study and the sequence coverage in each. For each species included in the analysis, the species group, feeding behavior, stock name, and strain source are included. Successfully amplified loci are indicated with +.
Tabular data (.xlsx); Loci used in this study. Includes the full locus name, abbreviation, Flybase ID, genome location (Müller element), presence of introns, and primers used to amplify and sequence each.
Tabular data (.xlsx); Phylogenetic information and genetic variation found in each locus. Includes the Müller Element of the locus, the length of the alignment, the number of variable sites (VS), the number of parsimony informative (PI) sites in each locus, the variance of each locus in VS/BP and PI/BP, and the total values for the genomic location.
Tabular data (.xlsx); Results from codon models of molecular evolution. Each locus was analyzed using each of the pruned species tree and the locus-specific gene tree. Shown is the ω value for each model (1ω and 2ω) as well as the significance of each comparison using a likelihood ratio test. Includes the genomic location (autosome, dot, or X and Y-chromosome).