

Summary
Amyotrophic lateral sclerosis (ALS) is a progressive, fatal neurodegenerative disease characterized by motor neuron cell death. However, not all motor neurons are equally susceptible. Most of what we know about the surviving motor neurons comes from gene expression profiling; less is known about their functional traits. We found that resistant motor neurons cultured from SOD1 ALS mouse models have enhanced axonal outgrowth and dendritic branching. They also have an increase in the number and size of actin-based structures like growth cones and filopodia. These phenotypes occur in cells cultured from presymptomatic mice and mutant SOD1 models that do not develop ALS but not in embryonic motor neurons. Enhanced outgrowth and upregulation of filopodia can be induced in wild-type adult cells by expressing mutant SOD1. These results demonstrate that mutant SOD1 can enhance the regenerative capability of ALS-resistant motor neurons. Capitalizing on this mechanism could lead to new therapeutic strategies.
Subject Areas: Biological Sciences, Genetics, Neuroscience, Cell Biology
Graphical Abstract
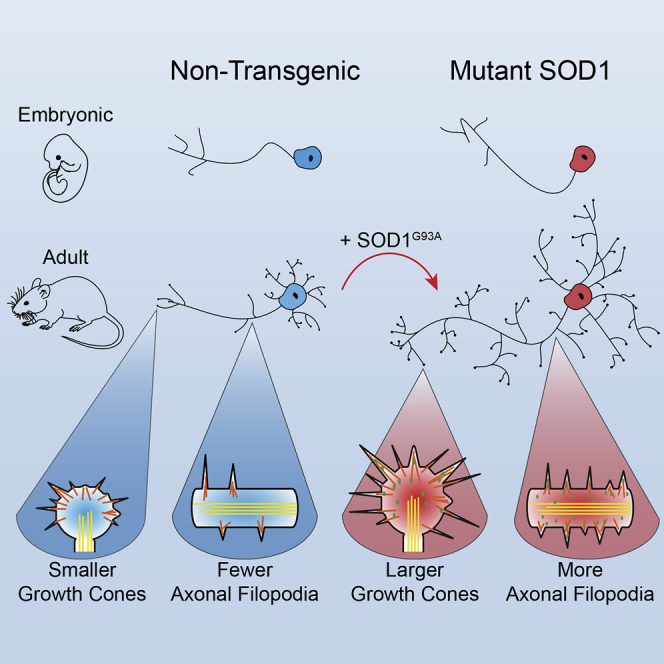
Highlights
-
•
Motor neurons from end-stage SOD1 ALS mice have enhanced neurite outgrowth/branching
-
•
Increased outgrowth occurs only in adult neurons and is independent of ALS symptoms
-
•
SOD1G93A adult motor neurons have larger growth cones and more axonal filopodia
-
•
Acute SOD1G93A expression upregulates outgrowth in wild-type adult motor neurons
Biological Sciences; Genetics; Neuroscience; Cell Biology
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal, adult-onset neurodegenerative disorder in which there is selective loss of motor neurons in the cerebral cortex, brainstem, and spinal cord (Ince et al., 1998). Approximately 90% of ALS cases are sporadic with unknown etiology; the remaining 10% are inherited and known as familial ALS (fALS), of which over 20% have mutations in the gene encoding Cu/Zn superoxide dismutase 1 (SOD1) (Brown and Al-Chalabi, 2017). To date, over 155 different mutations have been identified in SOD1 either in isolated cases of ALS or more commonly in patients from families showing autosomal dominant patterns of inheritance (Andersen and Al-Chalabi, 2011, Pasinelli and Brown, 2006). ALS-linked SOD1 mutations are thought to induce a toxic gain of function in the protein, which becomes prone to misfolding and subsequent aggregation (Karch et al., 2009, Saccon et al., 2013). However, expression of mutant SOD1 can affect a number of cellular processes, causing ER distress, mitochondrial dysfunction, excitotoxicity, defects in axonal transport, and inhibition of the proteasome (Ilieva et al., 2009). Despite being the first gene identified with mutations that cause fALS (Rosen et al., 1993) and providing the basis of the first ALS animal model (Gurney, 1994), there is still no consensus about how mutant SOD1 specifically alters motor neuron physiology.
Although most studies have focused on the cellular mechanisms and genes that induce motor neuron death in ALS, less is known about the neurons that do survive, including their ability to resist stress-induced cell death and to compensate for dying motor neurons. Not all motor neurons are equally susceptible to cell death during ALS disease progression. ALS mostly targets motor neurons required for voluntary movement, whereas motor neurons of the autonomic system are less sensitive (Piccione et al., 2015). There is also a gradient of vulnerability among spinal motor neurons, whereby faster motor units become affected before slower muscle types (Pun et al., 2006). Motor neurons that are less ALS susceptible can compensate for the cells that initially die by establishing new connections with the motor endplate, although many of these will eventually succumb to the disease (Schaefer et al., 2005). This selective neuronal vulnerability is present in both sporadic ALS and familial ALS and is also recapitulated in rodent models, such as the SOD1G93A mouse (Gurney, 1994, Nimchinsky et al., 2000).
Most of our current knowledge about surviving spinal motor neurons in ALS mouse models has largely been generated by gene expression profiling of tissue and cells (Bandyopadhyay et al., 2013, Brockington et al., 2013, de Oliveira et al., 2013, Ferraiuolo et al., 2007, Lobsiger et al., 2007, Saxena et al., 2009). However, these studies provide just a single snapshot of the motor neuron's biology and only allow for inferences to be made about how changes in gene expression alter motor neuron physiology, allow them to resist degeneration, or compensate for dying neurons by forming new motor endplate attachments. In the current study, we sought to functionally characterize ALS-resistant motor neurons by culturing them in vitro, where we would be able to directly assess dynamic cellular properties such as outgrowth, branching, and regulation of the cytoskeleton.
Results
Axon Outgrowth and Branching Are Increased in Adult Motor Neurons from Symptomatic SOD1-ALS Mice
To functionally characterize ALS-resistant motor neurons, we isolated them from adult mice expressing human SOD1G93A at low copy number (referred to as G93A-DL) (Acevedo-Arozena et al., 2011). This model expresses between six and eight copies of the human SOD1G93A transgene, resulting in the onset of ALS symptoms around 9 months of age. We corroborated these findings with the more extensively studied SOD1G93A high copy number mouse model (referred to as G93A), which expresses SOD1G93A at around 3-fold that of the G93A-DL model. These mice develop symptoms more rapidly, with hindlimb paralysis seen as early as 5 months of age (Gurney, 1994). Using a well-characterized protocol for the high yield extraction of spinal motor neurons from adult mice (Beaudet et al., 2015), we established cultures of adult motor neurons (Figure S1) from mutant SOD1 and non-transgenic mice (referred to as NTg). We then performed a large-scale quantitative analysis of these cells' ability to extend new processes. Since the isolation protocol severs all established neuronal projections, this assay is a direct measure of in vitro neurite regeneration.
Motor neurons from G93A-DL mice displayed significantly increased outgrowth in comparison with age- and sex-matched NTg controls, both in axon length (∼55% longer) and in overall neurite branching complexity (approximately three times as many intersections 60 μm from the soma center) (Figures 1A–1D and S2). Motor neurons isolated from late-stage G93A mice also demonstrated increased neurite branching and axonal outgrowth relative to NTg mice (Figures 1C and 1D). In contrast, motor neurons from adult mice overexpressing wild-type SOD1 (WT SOD1) exhibited a slight decrease in outgrowth and branching (Figures 1C and 1D). This was an important control since the SOD1G93A mutant maintains its enzymatic ability to remove superoxide radicals (Nishida et al., 1994). The reduction in axon extension and branching in motor neurons from WT SOD1 mice is consistent with previous findings where ROS depletion results in negative effects on neurite outgrowth (Munnamalai and Suter, 2009). Thus, the enhanced regeneration seen in late-stage motor neurons is specific to the SOD1G93A ALS mice and occurs with both high and low expression levels of the mutant gene.
Figure 1.

Adult Motor Neurons Cultured from Symptomatic SOD1G93A ALS Mouse Models Have Enhanced Axonal Outgrowth and Neurite Branching
(A) Confocal images of representative motor neurons harvested from non-transgenic (NTg), low-copy-number SOD1G93A (G93A-DL), SOD1G93A (G93A), and wild-type SOD1 (WT) transgenic mice. Motor neurons from both strains of SOD1G93A mice were harvested at their respective symptomatic stages (approximately 9 months for G93A-DL and approximately 6 months for G93A), and motor neurons from WT mice were harvested at 6 months of age. Scale bar represents 30 μm.
(B) Representative images of motor neurons harvested from NTg and ALS symptomatic G93A-DL mice. These are the traced, segmented neurites that were used for Sholl analysis. The original images can be found in Figure S3. The pseudocolor image of G93A-DL motor neuron demonstrates the number of intersections measured at progressive distances from the soma. Scale bar represents 50 μm. The line graph represents the average number of intersections measured at a given distance from the soma for all NTg or G93A-DL neurons.
(C) Box-and-whisker plots of the axonal outgrowth (measured as the length of the longest neurite branch) of motor neurons cultured from G93A-DL, G93A, and WT mice relative to their age-matched non-transgenic (NTg) controls.
(D) Branching of motor neurons was quantified by measuring the average number of intersections found 60 μm from the center of the soma (noted as dotted line in [B]). Bar graphs depict motor neuron arborization seen in G93A-DL, G93A, and WT motor neurons compared with their respective NTg controls. For (C) and (D): G93A-DL dataset: n = 56 cells from three NTg mice and n = 62 cells from three G93A-DL mice; G93A dataset: n = 88 cells from four NTg mice and n = 120 cells from five G93A mice; WT dataset: n = 82 cells from two NTg mice and n = 89 cells from three WT mice.
(E) Representative images of G93A-DL and G93A growth cones of motor neurons harvested from symptomatic mice with representative NTg growth cone. Scale bar represents 5 μm.
(F–H) Plots depicting growth cone area, average filopodia length filopodia per growth cone, and number of filopodia per growth cone demonstrated in G93A and G93A-DL motor neurons from symptomatic mice relative to their NTg controls. For (F–H) n = 25 for NTg and n = 25 for G93A-DL, harvested from two mice for each for G93A-DL dataset; n = 38 cells for NTg and n = 28 cells for G93A, harvested from two mice each for G93A dataset; and n = 30 from two mice for NTg and n = 45 from three mice for WT dataset.
(I) Representative images of NTg, G93A-DL, and G93A axonal filopodia. Scale bar represents 10 μm.
(J) Bar graph of axonal filopodia density of G93A-DL, G93A, and WT-SOD1 motor neurons compared with age- and sex-matched control cells. n = 46 cells from four mice for NTg and n = 49 cells from four mice for G93A-DL, and n = 30 cells from two mice for both NTg and G93A; n = 30 from two mice for NTg and n = 45 from three mice for WT dataset.
Box-and-whisker plots denote the 95th (top whisker), 75th (top edge of box), 25th (bottom edge of box), and 5th (bottom whisker) percentiles and the median (bold line in box). Data in bar graphs are represented as mean ±95% confidence intervals. p Values were obtained from a two-tailed Student's t test.
Actin-Based Structures Are Increased in Adult Motor Neurons from Symptomatic SOD1-ALS Mice
Growth cones are the motile organelles found at the tip of axonal and dendritic projections that play a pivotal role in outgrowth and pathfinding, including during in vivo adult motor neuron regeneration (Kang and Lichtman, 2013). The peripheral region of the growth cone contains actin-based lamellipodia and filopodia, two types of membrane protrusions that function in growth cone movement and environment sensing (Vitriol and Zheng, 2012). We observed a marked increase in the size of growth cones and filopodia in spinal motor neurons isolated from symptomatic G93A-DL and G93A mice. G93A-DL growth cones were on average greater than twice the size of those from NTg controls; G93A growth cones exhibited a similar increase in size (Figures 1E and 1F). Growth cone filopodia from both ALS mouse lines were also significantly longer than those from NTg controls, with G93A-DL cells exhibiting the largest size difference (Figure 1G). G93A-DL growth cones also contained more filopodia (Figure 1H). Growth cones from WT-SOD1 motor neurons were not significantly larger than those from NTg controls, nor did they have an increased number of filopodia (Figure 1F). However, the filopodia that were present were significantly longer (Figure 1G).
Axonal filopodia are actin-based structures extending off of the main axon terminal that serve as precursors for collateral branches, which are involved in building complex neural circuits (Gallo, 2013). In ALS, the formation of new collateral branches occurs in the resistant motor units as they attempt to expand their synaptic connections to compensate for early denervation events (Clark et al., 2016, Schaefer et al., 2005). In motor neurons isolated from both G93A-DL and G93A mice, we observed a marked increase in axonal filopodia density relative to NTg controls (Figures 1I and 1J). There was no difference in axonal filopodia of motor neurons from WT SOD1 mice compared with the NTg controls (Figure 1J). Thus, the surviving motor neurons isolated from symptomatic ALS mice exhibit an upregulation of multiple actin-based structures associated with outgrowth and regeneration.
Enhanced Regeneration of Mutant SOD1 Motor Neurons Occurs Only in Adult Cells and Is Independent of ALS Onset
Our results were surprising since previous studies have shown that the expression of G93A is either inhibitory or has no effect on outgrowth and regeneration in motor neurons. However, these studies were conducted using either embryonic cells (Nagai et al., 2007) or iPSC-derived motor neurons (Isobe et al., 2015, Karumbayaram et al., 2009), which more closely resemble the embryonic state (Ho et al., 2016) and may respond differently to the mutant SOD1expression. When we cultured motor neurons from G93A-DL and NTg pups at E14, there was no significant difference observed in outgrowth or branching after 3 days in vitro (DIV) (Figures 2A and 2B). We then cultured motor neurons from adult G93A-DL mice at different time points prior to the onset of ALS symptoms (1, 2, and 6 months of age). Increased axonal outgrowth and neurite branching relative to NTg controls were observed at the 2- and 6-month time points, with a more significant difference at 6 months (Figures 2C and 2D). These data reveal a trend whereby regeneration is enhanced relative to NTg controls as the mice age and become closer to developing ALS. However, if the actual size of the processes is plotted instead of their relative size (to NTg controls), G93A-DL motor neurons maintain the same level of outgrowth (axon is ∼120 μm) throughout their lifespan, whereas NTg motor neurons actually become progressively smaller. The same trend exists for branching (Figure 2D). This could be interpreted as G93A-DL motor neurons having a preserved, rather than an enhanced, ability to regenerate.
Figure 2.

Enhanced Outgrowth and Branching of Adult Motor Neurons from Mutant SOD1 Mice Is Specific to Adult Cells and Independent of ALS Onset
(A) Representative images and Sholl graph of E14 motor neurons harvested from NTg and G93A-DL mice and imaged after 2 DIV. Scale bar represents 100 μm.
(B) Box-and-whisker plot and bar graph showing axonal outgrowth (neuron radius) and neurite branching (number of intersections found 60 μm from the center of the soma, noted as dotted line in [A]) measurements for embryonic NTg and G93A-DL motor neurons.
(C) Representative images of adult motor neurons harvested at 2, 6, and 9 months from NTg and G93A-DL mice. Scale bar represents 50 μm.
(D) Line graphs depicting axonal outgrowth and neurite branching as a function of age. For the dataset taken at 1 month, n = 49 for NTg and n = 47 for G93A-DL, taken from three mice each; for 2 months n = 95 for NTg and n = 80 for G93A-DL, harvested from three mice each. Data representing 6 months was taken from three mice each for NTg and G93A-DL, with n = 88 and n = 80, respectively. Data representing symptomatic stage (indicated with red rectangle) was taken from three NTg mice and three G93A-DL mice, with n = 56 and n = 62, respectively. Asterisk indicates p value of less than 0.001.
(E) Representative images and Sholl graph of motor neurons harvested from NTg and G85R mice at 5 months of age. Scale bar represents 50 μm.
(F) Box-and-whisker plot and bar graph showing axonal outgrowth (neuron radius) and neurite branching (number of intersections found 60 μm from the center of the soma, noted as dotted line in [E]) measurements for G85R and NTg motor neurons. n = 23 from two mice for NTg and n = 51 from three mice for G85R.
Box-and-whisker plots denote the 95th (top whisker), 75th (top edge of box), 25th (bottom edge of box), and 5th (bottom whisker) percentiles and the median (bold line in box). Data in bar graphs are represented as mean ±95% confidence intervals. p Values were obtained from a two-tailed Student's t test. The images of motor neurons in A, C, and E are the traced, segmented neurites that were used for Sholl analysis. The original images can be found in Figure S3.
To verify that enhanced outgrowth of adult motor neurons from mutant SOD1 mouse models occurs independently of developing ALS, we isolated cells from transgenic mice overexpressing YFP-SOD1G85R. YFP-SOD1G85R homozygous mice develop ALS, whereas the heterozygous mice (referred to as G85R-het) do not develop symptoms (Bruijn et al., 1997, Wang et al., 2009a). Thus, the heterozygous model is a useful tool for studying the effects of mutant SOD1 overexpression independently of the effects of ALS progression. Motor neurons were isolated from G85R-het mice at 5 months of age, which is when the G93A mice typically start to show symptoms of ALS and also an age where pre-symptomatic G93A-DL mice still have substantial increases in outgrowth and branching relative to controls (Figures 1C, 1D, and 2D). G85R-het motor neurons exhibited an increase in axon length and neurite branching comparable with that seen in end-stage G93A mice (Figures 2E and 2F). Thus, expression of mutant SOD1 can enhance regeneration independently of ALS symptoms even in a model in which there is no selection for surviving cells or stress from motor neuron death that signals the remaining neurons to reinnervate lost connections (Höke et al., 2006). This strongly suggests that it is the expression of mutant SOD1, not external factors caused by ALS, which increases outgrowth and regeneration of adult motor neurons.
Expression of SOD1G93A Enhances Outgrowth and Branching of Wild-Type Adult Motor Neurons
All of the experiments described earlier are performed with animal models where the cells express a mutant transgene for months in vivo. In fact, the enhanced outgrowth and branching phenotypes becomes apparent only after the mouse is 2 months old (Figure 2D). Therefore, it could be argued that enhanced regeneration is the result of an accumulated effect caused by long-term expression of the mutant gene, thus explaining the differences seen between adult (Figures 1 and 2) and embryonic motor neurons (Figures 2A and 2B). To determine if acute expression of SOD1G93A was sufficient to increase outgrowth and branching in adult motor neurons, we cultured wild-type cells from 9- to 12-month-old NTg mice and transduced them with adeno-associated virus (AAV) to express wild-type SOD1 (WT-YFP), SOD1G93A (G93A-YFP), or a GFP control. Interestingly, acute expression of G93A-YFP was sufficient to increase axonal outgrowth relative to NTg motor neurons (Figures 3A and 3B). Although branching was not significantly increased, there was a significant positive correlation between G93A-YFP expression levels and both outgrowth parameters measured (Figure 3C). The difference between cells expressing G93A-YFP and WT-YFP was even more significant for outgrowth and branching (Figure 3B). WT-YFP-positive cells did not have statistically significant differences in outgrowth or branching relative to GFP controls, but expression of WT-YFP was negatively correlated with outgrowth, mimicking the trend observed in the SOD1 overexpressing transgenic mouse models (Figure 1). Thus, acute expression of mutant SOD1 was sufficient to increase regeneration of wild-type adult motor neurons.
Figure 3.

Acute Expression of SOD1G93A Increases Outgrowth and Branching in Non-transgenic Adult Motor Neurons
(A) Representative images of NTg motor neurons from 9- to 12-month-old mice transduced with AAV to overexpress GFP, wild-type SOD1-YFP (WT-YFP), or SOD1G93A-YFP (G93A-YFP). Scale bar represents 50 μm.
(B) Box-and-whisker plot depicting axonal outgrowth (neuron radius) and bar graph showing branching (number of intersections found 100 μm from the center of the soma) measurements of transduced NTg motor neurons. n = 38 cells from six mice, n = 37 cells from six mice, and n = 48 cells from six mice for AAV-GFP, AAV-WT-YFP, and AAV-G93A-YFP, respectively.
(C) Scatterplot depicting the relationship between the mean cellular fluorescence intensity of G93A-YFP axon outgrowth and neurite branching. Pearson's correlation coefficient (Pearson) R values are depicted under the respective bars in (B). R values were reported only if the correlation was statistically significant (p < 0.05); otherwise, they are listed as not significant (n.s.). n = 46, n = 45, and n = 57 for NTg motor neurons infected with AAV-GFP, AAV-WT-YFP, and AAV-G93A-YFP, respectively; cells harvested from nine mice (three trials, three mice/trial).
Box-and-whisker plots denote the 95th (top whisker), 75th (top edge of box), 25th (bottom edge of box), and 5th (bottom whisker) percentiles and the median (bold line in box). Data in bar graphs are represented as mean ±95% confidence intervals. p Values were obtained by ANOVA followed by Tukey's post hoc test.
SOD1G93A Increases Axonal Filopodia and Localizes to Actin-Based Structures
Since actin-based structures were upregulated in motor neurons from G93A mouse models (Figures 1E–1J), we wanted to determine if expression of SOD1G93A was sufficient to increase such structures in wild-type motor neurons. Cells from NTg mice were infected with AAV expressing GFP, WT-SOD1-YFP (WT-YFP), and G93A-YFP, after which axonal filopodia were measured. Quantification of growth cone parameters was not possible because of the length of time the cells were cultured to achieve robust transgene expression (10 DIV compared with 3 DIV for our analysis of end-stage G93A/G93A-DL motor neurons). Growth cones are most prevalent at 3 DIV. By 10 DIV few cells still had growth cones, but filopodia were still ubiquitously present on neurite projections, so we quantified axonal filopodia density. Both WT-YFP- and G93A-YFP-expressing cells had significantly higher axonal filopodia densities compared with GFP-expressing cells, with G93A-YFP-expressing cells having the largest increase in filopodia (Figure 4A). G93A-YFP expression resulted in a 33% increase in filopodia density over SOD1-YFP and a 100% increase over GFP (Figure 4A). Furthermore, we observed that G93A-YFP was localized to actin-based structures such as growth cones (the few that were present) and filopodia more than SOD1-YFP (Figure 4B).
Figure 4.

SOD1G93A Localizes to and Increases Filopodia
(A) Representative images and quantification of axonal filopodia density (measured on the longest projection) from AAV-GFP-, AAV-WT-YFP-, or AAV-G93A-YFP-transduced NTg motor neurons. n = 38 cells from six mice, n = 37 cells from six mice, and n = 48 cells from six mice for AAV-GFP, AAV-SOD1-YFP, or AAV-G93A-YFP, respectively. Scale bar represents 50 μm.
(B) Confocal images of neurites from NTg adult motor neurons transduced with AAV-WT-YFP and AAV-G93A-YFP. Arrows indicate localization of G93A-YFP to filopodia. Scale bar represents 5 μm.
(C) Representative images of neurite-like projections in Cath-a-differentiated (CAD) cells that have been transfected with YFP, wild-type SOD1-YFP (WT-YFP), or SOD1G93A-YFP (G93A-YFP). Scale bar represents 5 μm.
(D) Bar graph measuring filopodia density in CAD cells transfected with YFP, WT-YFP, or G93A-YFP. n = 71 cells for YFP, n = 48 cells for SOD1-YFP, and n = 72 for G93A-YFP, three separate transfections per condition.
(E) Enlarged view of YFP fluorescence from dashed boxes from (C) showing the increased localization of G93A-YFP into filopodia compared with WT-YFP. Scale bar represents 1 μm.
(F) Box-and-whisker plot quantifying YFP fluorescence intensity in filopodia in CAD cells. n = 1,359 filopodia from 38 cells for YFP, n = 1,367 filopodia from 40 cells for WT-YFP, and n = 1,524 from 39 cells for G93A-YFP, three separate transfections per condition.
Box-and-whisker plots denote the 95th (top whisker), 75th (top edge of box), 25th (bottom edge of box), and 5th (bottom whisker) percentiles and the median (bold line in box). Data in bar graphs are represented as mean ±95% confidence intervals. p Values were obtained by ANOVA followed by Tukey's post hoc test.
To determine if the localization to, and enhancement of, filopodia by SOD1G93A is a general phenomenon, we overexpressed G93A-YFP, WT-SOD1-YFP (WT-YFP), or YFP in Cath-a-differentiated (CAD) cells. CAD cells are a CNS-derived cell line that extend neurite-like projections that are highly similar to actual neurites upon serum withdrawal (Qi et al., 1997) that contain numerous filopodia (Kapustina et al., 2016). Transfected CAD cells were differentiated for 18 hr, labeled with phalloidin, and then imaged using deconvolution-based super-resolution confocal microscopy (Wilson, 2011). Filopodia were segmented and analyzed with the ImageJ plugin Filopodyan (Urbančič et al., 2017), which allowed us to measure the hundreds of individual filopodia per condition. We observed a significant increase in the localization of WT-YFP and G93A-YFP into filopodia over the YFP control, with G93A having the most robust filopodia localization (Figures 4C, 4E, and 4F). This is consistent with previous work showing that both WT and G93A SOD1 bind actin, but the interaction is increased with the G93A mutant (Takamiya et al., 2005). Additionally, we observed an increase in filopodia density when G93A-YFP was expressed (Figure 4D) that was similar to experiments performed with adult motor neurons (Figure 4A). Thus, SOD1G93A localizes to and increases filopodia in multiple cell types, indicating it is a general mechanism of actin regulation.
Discussion
This study demonstrates that SOD1G93A expression can have pro-regenerative effects on adult motor neurons. Not only was enhanced neurite regeneration observed in motor neurons isolated from mutant SOD1 transgenic mice (Figure 2) but also the expression of SOD1G93A alone was sufficient to increase outgrowth in non-transgenic primary motor neurons (Figure 3). Furthermore, SOD1G93A localized to actin-based cellular structures and increased their size and number (Figure 4). Finally, an important take-home message from this study is the importance of working with the right cell type, as mutant SOD1 expression had no effect on embryonic motor neuron regeneration (Figures 2A and 2B) but had a substantial effect on adult motor neuron outgrowth and branching (Figures 1, 2, and 3). Although it has been speculated that upregulation of regenerative/injury pathways is merely a compensatory response to mutant-SOD1-induced toxicity (Lobsiger et al., 2007, Pun et al., 2006), our work suggests a novel gain of function for mutant SOD1, where it can help preserve motor neuron plasticity.
There are two mechanisms through which this could occur. The first is a direct regulation of actin by mutant SOD1. Although the relationship between mutant SOD1 and the actin cytoskeleton is not well characterized, it has been shown that SOD1G93A directly interacts with actin (Takamiya et al., 2005, Zetterstrom et al., 2011). Interestingly, mutant SOD1 has a significantly higher affinity for actin than the wild-type protein (Takamiya et al., 2005). That SOD1 is found in filopodia is also of interest. Since filopodia are extremely thin (∼200 nm) extensions of the cellular membrane that are tightly packed with rearward-flowing actin filaments (Mattila and Lappalainen, 2008), localization there strongly indicates a specific interaction (Bird et al., 2017). It also might suggest that SOD1 preferentially binds or bundles linear arrays of filaments since the most prevalent structures we found to be upregulated by expression of mutant SOD1 were axonal filopodia. Axonal filopodia are precursor membrane protrusions to collateral branches (Gallo, 2013). New collateral branches form during the early stages of ALS, when the resistant neurons try to establish new synaptic connections to compensate for the denervation caused by the loss of the most susceptible neurons (Clark et al., 2016, Schaefer et al., 2005). Axonal filopodia must recruit microtubules to become collateral branches (Ketschek et al., 2015). Interestingly, in addition to actin, mutant SOD1 can also interact with the microtubule cytoskeleton (Kabuta et al., 2009). Thus, mutant SOD1 may have a dual role in the formation of new branches by increasing axonal filopodia (Figures 4A and 4D) and then helping microtubules to enter them. However, future studies will be required to determine if mutant SOD1 is directly involved in cytoskeletal regulation.
The second way that mutant SOD1 expression could enhance outgrowth and branching would be through upregulation of pro-regenerative signaling and cytoskeletal pathways. There are several published studies characterizing genetic changes in motor neurons from G93A mice at various stages of disease progression (D'Arrigo et al., 2010, de Oliveira et al., 2013, Ferraiuolo et al., 2007, Guipponi et al., 2010, Offen et al., 2009, Perrin et al., 2005, Saris et al., 2013, Yu et al., 2013). However, these studies have not reached consensus regarding the underlying genetics promoting ALS resistance, probably because of the variation in experimental design and tissue sampling. For example, using G93A mice, one study found a massive upregulation of genes involved in cell growth and/or maintenance in micro-dissected motor neurons from the lumbar spinal cord (Perrin et al., 2005), whereas another study found Wnt signaling to be significantly activated when homogenized whole spinal cord was used for RNA extraction (Yu et al., 2013). It has also been shown that upregulation of axonal guidance genes and actin cytoskeletal genes (including α-actin and β-actin) occurs from the lumbar spinal cord of pre-symptomatic G93A mouse (de Oliveira et al., 2013). Thus, SOD1G93A may prime adult motor neurons for outgrowth and regeneration through a positive activation of genes that regulate the actin cytoskeleton. However, it should be noted that many of the initially resistant motor neurons eventually succumb to ALS (Schaefer et al., 2005). The positive influence of mutant SOD1 expression on adult motor neuron regeneration most likely reflects an intermediate state occurring before cytotoxicity overwhelms the cells. However, instead of trying to completely silence mutant SOD1 expression, as with antisense oligonucleotide therapy (Smith et al., 2006), capitalizing on mutant SOD1's pro-regenerative effects while combating its toxicity could be a useful strategy for future therapeutics.
Limitations of the Study
The results of this study were not corroborated with in vivo studies of motor neuron regeneration, so the direct relevance of this work to ALS disease progression remains to be determined.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Dr. Benoit Giasson (University of Florida) for providing antibodies used in this study. This project was supported by a grant from the National Institutes of Health (NIH) (R01 NS092788) (D.R.B.), an NIH Pathway to Independence Award (R00 NS087104) (E.A.V.), and a Starter Grant from the ALS Association (E.A.V. and T.-A.R).
Author Contributions
Conceptualization, T.-A.R. and E.A.V.; Methodology, T.-A.R. and E.A.V.; Formal Analysis, Z.O., R.H., K.S., A.R.E., T.-A.R., E.A.V.; Investigation, Z.O., J.I.A., T.-A.R.; Resources, J.I.A., H.B., D.R., T.E.G., D.R.B.; Writing – Original Draft, Z.O., J.I.A., T.-A.R., E.A.V.; Writing – Review & Editing, Z.O., T.-A.R., E.A.V.; Visualization, Z.O., E.A.V., Supervision, T.-A.R., E.A.V.; Project Administration, E.A.V., Funding Acquisition, D.R.B., T.-A.R., E.A.V.
Declaration of Interests
The authors declare no competing interests.
Published: January 25, 2019
Footnotes
Supplemental Information includes Transparent Methods and three figures and can be found with this article online at https://doi.org/10.1016/j.isci.2018.12.026.
Contributor Information
Tracy-Ann Read, Email: taread@ufl.edu.
Eric A. Vitriol, Email: evitriol@ufl.edu.
Supplemental Information
References
- Acevedo-Arozena A., Kalmar B., Essa S., Ricketts T., Joyce P., Kent R., Rowe C., Parker A., Gray A., Hafezparast M. A comprehensive assessment of the SOD1G93A low-copy transgenic mouse, which models human amyotrophic lateral sclerosis. Dis. Model. Mech. 2011;4:686–700. doi: 10.1242/dmm.007237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P.M., Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 2011;7:603–615. doi: 10.1038/nrneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay U., Cotney J., Nagy M., Oh S., Leng J., Mahajan M., Mane S., Fenton W.A., Noonan J.P., Horwich A.L. RNA-Seq profiling of spinal cord motor neurons from a presymptomatic SOD1 ALS mouse. PLoS One. 2013;8:e53575. doi: 10.1371/journal.pone.0053575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudet M.J., Yang Q., Cadau S., Blais M., Bellenfant S., Gros-Louis F., Berthod F. High yield extraction of pure spinal motor neurons, astrocytes and microglia from single embryo and adult mouse spinal cord. Sci. Rep. 2015;5:16763. doi: 10.1038/srep16763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird J.E., Barzik M., Drummond M.C., Sutton D.C., Goodman S.M., Morozko E.L., Cole S.M., Boukhvalova A.K., Skidmore J., Syam D. Harnessing molecular motors for nanoscale pulldown in live cells. Mol. Biol. Cell. 2017;28:463–475. doi: 10.1091/mbc.E16-08-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockington A., Ning K., Heath P.R., Wood E., Kirby J., Fusi N., Lawrence N., Wharton S.B., Ince P.G., Shaw P.J. Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 2013;125:95–109. doi: 10.1007/s00401-012-1058-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R.H., Al-Chalabi A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017;10:162–172. doi: 10.1056/NEJMra1603471. [DOI] [PubMed] [Google Scholar]
- Bruijn L.I., Becher M.W., Lee M.K., Anderson K.L., Jenkins N.A., Copeland N.G., Sisodia S.S., Rothstein J.D., Borchelt D.R., Price D.L. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Clark J.A., Southam K.A., Blizzard C.A., King A.E., Dickson T.C. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016;76:35–47. doi: 10.1016/j.jchemneu.2016.03.003. [DOI] [PubMed] [Google Scholar]
- D'Arrigo A., Colavito D., Peña-Altamira E., Fabris M., Dam M., Contestabile A., Leon A. Transcriptional profiling in the lumbar spinal cord of a mouse model of amyotrophic lateral sclerosis: a role for wild-type superoxide dismutase 1 in sporadic disease? J. Mol. Neurosci. 2010;41:404–415. doi: 10.1007/s12031-010-9332-2. [DOI] [PubMed] [Google Scholar]
- de Oliveira G.P., Alves C.J., Chadi G. Early gene expression changes in spinal cord from SOD1(G93A) Amyotrophic Lateral Sclerosis animal model. Front. Cell. Neurosci. 2013;7:216. doi: 10.3389/fncel.2013.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo L., Heath P.R., Holden H., Kasher P., Kirby J., Shaw P.J. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J. Neurosci. 2007;27:9201–9219. doi: 10.1523/JNEUROSCI.1470-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo G. Mechanisms underlying the initiation and dynamics of neuronal filopodia: from neurite formation to synaptogenesis. Int. Rev. Cell Mol. Biol. 2013;301:95–156. doi: 10.1016/B978-0-12-407704-1.00003-8. [DOI] [PubMed] [Google Scholar]
- Guipponi M., Li Q.X., Hyde L., Beissbarth T., Smyth G.K., Masters C.L., Scott H.S. SAGE analysis of genes differentially expressed in presymptomatic TgSOD1G93A transgenic mice identified cellular processes involved in early stage of ALS pathology. J. Mol. Neurosci. 2010;41:172–182. doi: 10.1007/s12031-009-9317-1. [DOI] [PubMed] [Google Scholar]
- Gurney M.E. Transgenic-mouse model of amyotrophic lateral sclerosis. N. Engl. J. Med. 1994;331:1721–1722. doi: 10.1056/NEJM199412223312516. [DOI] [PubMed] [Google Scholar]
- Ho R., Sances S., Gowing G., Amoroso M.W., O'Rourke J.G., Sahabian A., Wichterle H., Baloh R.H., Sareen D., Svendsen C.N. ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat. Neurosci. 2016;19:1256–1267. doi: 10.1038/nn.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höke A., Redett R., Hameed H., Jari R., Zhou C., Li Z.B., Griffin J.W., Brushart T.M. Schwann cells express motor and sensory phenotypes that regulate axon regeneration. J. Neurosci. 2006;26:9646–9655. doi: 10.1523/JNEUROSCI.1620-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H., Polymenidou M., Cleveland D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince P.G., Lowe J., Shaw P.J. Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology. Neuropathol. Appl. Neurobiol. 1998;24:104–117. doi: 10.1046/j.1365-2990.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Isobe T., Tooi N., Nakatsuji N., Aiba K. Amyotrophic lateral sclerosis models derived from human embryonic stem cells with different superoxide dismutase 1 mutations exhibit differential drug responses. Stem Cell Res. 2015;15:459–468. doi: 10.1016/j.scr.2015.09.006. [DOI] [PubMed] [Google Scholar]
- Kabuta T., Kinugawa A., Tsuchiya Y., Kabuta C., Setsuie R., Tateno M., Araki T., Wada K. Familial amyotrophic lateral sclerosis-linked mutant SOD1 aberrantly interacts with tubulin. Biochem. Biophys. Res. Commun. 2009;387:121–126. doi: 10.1016/j.bbrc.2009.06.138. [DOI] [PubMed] [Google Scholar]
- Kang H., Lichtman J.W. Motor axon regeneration and muscle reinnervation in young adult and aged animals. J. Neurosci. 2013;33:19480–19491. doi: 10.1523/JNEUROSCI.4067-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapustina M., Read T.A., Vitriol E.A. Simultaneous quantification of actin monomer and filament dynamics with modeling-assisted analysis of photoactivation. J. Cell Sci. 2016;129:4633–4643. doi: 10.1242/jcs.194670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch C.M., Prudencio M., Winkler D.D., Hart P.J., Borchelt D.R. Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc. Natl. Acad. Sci. U S A. 2009;106:7774–7779. doi: 10.1073/pnas.0902505106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karumbayaram S., Kelly T.K., Paucar A.A., Roe A.J., Umbach J.A., Charles A., Goldman S.A., Kornblum H.I., Wiedau-Pazos M. Human embryonic stem cell-derived motor neurons expressing SOD1 mutants exhibit typical signs of motor neuron degeneration linked to ALS. Dis. Model. Mech. 2009;2:189–195. doi: 10.1242/dmm.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketschek A., Jones S., Spillane M., Korobova F., Svitkina T., Gallo G. Nerve growth factor promotes reorganization of the axonal microtubule array at sites of axon collateral branching. Dev. Neurobiol. 2015;75:1441–1461. doi: 10.1002/dneu.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobsiger C.S., Boillée S., Cleveland D.W. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc. Natl. Acad. Sci. U S A. 2007;104:7319–7326. doi: 10.1073/pnas.0702230104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila P.K., Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 2008;9:446–454. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- Munnamalai V., Suter D.M. Reactive oxygen species regulate F-actin dynamics in neuronal growth cones and neurite outgrowth. J. Neurochem. 2009;108:644–661. doi: 10.1111/j.1471-4159.2008.05787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M., Re D.B., Nagata T., Chalazonitis A., Jessell T.M., Wichterle H., Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky E.A., Young W.G., Yeung G., Shah R.A., Gordon J.W., Bloom F.E., Morrison J.H., Hof P.R. Differential vulnerability of oculomotor, facial, and hypoglossal nuclei in G86R superoxide dismutase transgenic mice. J. Comp. Neurol. 2000;416:112–125. doi: 10.1002/(sici)1096-9861(20000103)416:1<112::aid-cne9>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Nishida C.R., Gralla E.B., Valentine J.S. Characterization of three yeast copper-zinc superoxide dismutase mutants analogous to those coded for in familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U S A. 1994;91:9906–9910. doi: 10.1073/pnas.91.21.9906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offen D., Barhum Y., Melamed E., Embacher N., Schindler C., Ransmayr G. Spinal cord mRNA profile in patients with ALS: comparison with transgenic mice expressing the human SOD-1 mutant. J. Mol. Neurosci. 2009;38:85–93. doi: 10.1007/s12031-007-9004-z. [DOI] [PubMed] [Google Scholar]
- Pasinelli P., Brown R.H. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Perrin F.E., Boisset G., Docquier M., Schaad O., Descombes P., Kato A.C. No widespread induction of cell death genes occurs in pure motoneurons in an amyotrophic lateral sclerosis mouse model. Hum. Mol. Genet. 2005;14:3309–3320. doi: 10.1093/hmg/ddi357. [DOI] [PubMed] [Google Scholar]
- Piccione E.A., Sletten D.M., Staff N.P., Low P.A. Autonomic system and amyotrophic lateral sclerosis. Muscle Nerve. 2015;51:676–679. doi: 10.1002/mus.24457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun S., Santos A.F., Saxena S., Xu L., Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Qi Y., Wang J.K., McMillian M., Chikaraishi D.M. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J. Neurosci. 1997;17:1217–1225. doi: 10.1523/JNEUROSCI.17-04-01217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen D.R., Siddique T., Patterson D., Figlewicz D.A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J.P., Deng H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Saccon R.A., Bunton-Stasyshyn R.K., Fisher E.M., Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain. 2013;136:2342–2358. doi: 10.1093/brain/awt097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saris C.G., Groen E.J., van Vught P.W., van Es M.A., Blauw H.M., Veldink J.H., van den Berg L.H. Gene expression profile of SOD1-G93A mouse spinal cord, blood and muscle. Amyotroph. Lateral Scler. Frontotemporal Degener. 2013;14:190–198. doi: 10.3109/21678421.2012.749914. [DOI] [PubMed] [Google Scholar]
- Saxena S., Cabuy E., Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Schaefer A.M., Sanes J.R., Lichtman J.W. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J. Comp. Neurol. 2005;490:209–219. doi: 10.1002/cne.20620. [DOI] [PubMed] [Google Scholar]
- Smith R.A., Miller T.M., Yamanaka K., Monia B.P., Condon T.P., Hung G., Lobsiger C.S., Ward C.M., McAlonis-Downes M., Wei H. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 2006;116:2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamiya R., Takahashi M., Park Y.S., Tawara Y., Fujiwara N., Miyamoto Y., Gu J., Suzuki K., Taniguchi N. Overexpression of mutated Cu,Zn-SOD in neuroblastoma cells results in cytoskeletal change. Am. J. Physiol. Cell Physiol. 2005;288:C253–C259. doi: 10.1152/ajpcell.00014.2004. [DOI] [PubMed] [Google Scholar]
- Urbančič V., Butler R., Richier B., Peter M., Mason J., Livesey F.J., Holt C.E., Gallop J.L. Filopodyan: an open-source pipeline for the analysis of filopodia. J. Cell Biol. 2017;216:3405–3422. doi: 10.1083/jcb.201705113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitriol E.A., Zheng J.Q. Growth cone travel in space and time: the cellular ensemble of cytoskeleton, adhesion, and membrane. Neuron. 2012;73:1068–1081. doi: 10.1016/j.neuron.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Farr G.W., Zeiss C.J., Rodriguez-Gil D.J., Wilson J.H., Furtak K., Rutkowski D.T., Kaufman R.J., Ruse C.I., Yates J.R. Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc. Natl. Acad. Sci. U S A. 2009;106:1392–1397. doi: 10.1073/pnas.0813045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson T. Resolution and optical sectioning in the confocal microscope. J. Microsc. 2011;244:113–121. doi: 10.1111/j.1365-2818.2011.03549.x. [DOI] [PubMed] [Google Scholar]
- Yu L., Guan Y., Wu X., Chen Y., Liu Z., Du H., Wang X. Wnt Signaling is altered by spinal cord neuronal dysfunction in amyotrophic lateral sclerosis transgenic mice. Neurochem. Res. 2013;38:1904–1913. doi: 10.1007/s11064-013-1096-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterstrom P., Graffmo K.S., Andersen P.M., Brannstrom T., Marklund S.L. Proteins that bind to misfolded mutant superoxide dismutase-1 in spinal cords from transgenic amyotrophic lateral sclerosis (ALS) model mice. J. Biol. Chem. 2011;286:20130–20136. doi: 10.1074/jbc.M111.218842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.