

Abstract
Background
Poly(lactic-co-glycolic acid) (PLGA) has emerged as a promising anticancer drug delivery scaffold. Camptothecin (CPT) has been fabricated into a variety of nanosized formulations to improve drug action. We report an experimental study on the effect of CPT-encapsulated PLGA (PLGA-CPT) nanoparticles (NPs) on drug-metabolizing cytochrome P450 enzyme, CYP3A4.
Materials and methods
PLGA-CPT NPs were prepared by a single emulsion–solvent evaporation method.
Results
Transmission electron micrography showed that the NPs had a round and regular shape with a mean diameter of 94.6±5.7 nm. An in vitro drug release study showed that CPT was continuously released for 48 h. PLGA-CPT NPs showed greater cytotoxic effects on the HepG2 cell line compared with an equal dose of free CPT. Correlation with 4-h uptake data suggested that this was due to a higher cellular uptake amount of CPT from PLGA-CPT NPs than from free CPT. PLGA-CPT NPs tended to inhibit CYP3A4 activity isolated from HepG2 cells. However, PLGA-CPT NPs had no effect on the CYP3A4 mRNA levels. Furthermore, the interaction between PLGA-CPT NPs and CYP3A4 was investigated by ultraviolet–visible absorption spectroscopy and fluorescence spectroscopy.
Conclusion
Taken together, the results demonstrate that CYP3A4 may be inhibited by PLGA-CPT NPs and interference with biotransformation should be considered when using NPs as drug delivery vesicles.
Keywords: camptothecin, cytochrome P450, nanoparticle, drug delivery
Introduction
Camptothecin (CPT), a natural plant alkaloid extracted from Camptotheca acuminata, a plant native to China, shows significant antitumor activity against various tumors via inhibition of the activity of DNA topoisomerase I.1 However, CPT failed in clinical trials due to dose-limiting toxicities and, ultimately, poor efficacy. CPT is rapidly hydrolyzed at physiological pH from its active lactone form to a tenfold less active, more toxic carboxylate form, which is cleared rapidly once bound to plasma proteins.2 An effective drug delivery system for CPT is one strategy that could be used to improve drug action. CPT has been fabricated into nanoparticles (NPs),3 micelles,4 liposomes,5 and chitosan complexes6 to overcome its unstable characteristics.
Biotransformation of drugs, steroid hormones, and xenobiotics for excretion in the urine occurs by a two-stage process involving functional group oxidation (phase I enzymes) and subsequent coupling to, among others, glucuronic acid, glutathione, or sulfate groups (phase II enzymes).7 Phase I enzymes belong to the cytochrome P450 (CYP450) family of heme-based isoenzymes. Among the CYP isoforms, CYP1, CYP2, and CYP3 account for approximately 70% of human hepatic microsomes, with CYP1A2, CYP2C9, CYP2D6, and CYP3A4 being the major enzymes involved in drug metabolism.8 CYP3A4 represents 30%–60% of the liver CYP enzymes and CYP3A4 alone metabolizes more than 50% of all pharmaceuticals.9 Drugs, some food and beverages, and various chemicals such as those in cigarettes affect the activity of CYPs. Compared to the bulk materials, engineered NPs have attracted increasing attention because of their multiple applications and special physical–chemical properties.10–12 Based upon their structural characteristics and the fact that they could be taken up by hepatocytes, nanomaterials may be able to disrupt the function of CYPs.13 Nanomaterials have also been reported to affect CYP3A4 activity. Silver NPs dose-dependently decrease the amount of 6β-hydroxytestosterone, which is generated mainly by CYP3A4, in human liver microsomes.14 Nonmetallic carboxyl polystyrene NPs inhibit the activity of CYP3A4 in microsomes isolated from baculovirus-infected cells expressing wild-type CYP3A4.15 Seventy-nanometer silica NPs are localized in the cytoplasm, which contains many enzymes related to metabolism such as CYPs.16 As the understanding of malignant transformation has increased, it is clear that cancer involves changes in cell signaling and cell metabolism.17 Such studies have raised concerns and emphasize the importance of establishing the safety profile of the drug carriers and their effect on metabolic enzymes.
CYPs are a big class of terminal catalytically active components of monooxygenase systems where the substrate binds and is hydroxylated. NPs can interact with proteins through electrostatic, hydrophobic, and specific chemical interactions.18 Unraveling the interactions of NPs and CYPs is conductive to understanding the pharmacological actions of drugs, as well as designing and screening novel drugs with lower toxicities and side effects.19 Studies have shown that physical adsorption of bovine serum albumin and streptavidin on the carbon nanotube surface may lead to conformational changes in both secondary and tertiary structures.20
The structural changes of CYPs during metabolism need to be explored in order to understand the structure–function relationships and the underlying mechanism. However, little information is available about the effect of the formulation of CPT nanocarrier on CYP3A4 activity. Poly(lactic-co-glycolic acid) (PLGA) has been widely used in the delivery system for vaccines, peptides, proteins, and macromolecules over the last decades.21–23 The aim of this study was to evaluate whether the formulation of PLGA-CPT NPs would adversely affect its ability to modulate the expression and activity of CYP3A4. We examined CYP3A4 activity in microsomes isolated from human hepatocellular carcinoma cells (HepG2). Further, the structural properties of CYP3A4 were studied by spectroscopic techniques.
Materials and methods
Materials
CPT was bought from Sigma-Aldrich Co. (St Louis, MO, USA). PLGA 75/25 was bought from the Chinese Academy of Sciences (Sichuan, China). RPMI 1640, fetal bovine serum, glucose, penicillin, and streptomycin were obtained from Gibco (Thermo Fisher Scientific, Waltham, MA, USA).
Cell culture
Human liver HepG2 cells were purchased from the Institute of Hematology, Peking Union Medical College, China. Cells were maintained in RPMI 1640 medium supplemented with glucose, 10% fetal bovine serum, 100 units/mL penicillin, 100 µg/mL streptomycin at 37°C in 5% CO2 in a humidified incubator. Cells were incubated to 80%–90% confluency before they were subcultured for further experiments.
Preparation of NPs
NPs were prepared using a single emulsion–solvent evaporation method with slight modification.24 Briefly, 60 mg of PLGA and 6 mg of CPT were dissolved in 3 mL of dichloromethane. The dissolved mixture was added dropwise into 4 mL of polyvinyl alcohol solution (2 %, w/v) under sonication for 2 min in an ice bath in three 10-s bursts at 40% amplitude. The resulting oil-in-water emulsion was further processed by slowly dropping the mixture into 6 mL of 0.6% polyvinyl alcohol and stirring for 2 h at room temperature to allow the evaporation of dichloromethane. The NPs were then washed three times by centrifugation (2,000 rpm/10 min), lyophilized, and stored at −20°C until further use.
Characterization of NPs
The morphology of NPs was characterized using transmission electron microscopy (TEM). NPs were placed on a copper mesh coated with an amorphous carbon film at an acceleration voltage of 100 kV. Samples were imaged with a JEM-100CXII instrument (JEOL, Tokyo, Japan). The zeta-potential of the NPs was measured using dynamic light scattering (DLS) (ZEN 3600; Malvern Instruments, Malvern, UK).
Drug entrapment efficiency and drug loading determination
The entrapment efficiency (EE) and drug loading (DL) were determined by fluorescence. Briefly, freeze-dried NPs were dissolved in DMSO to a concentration of 1 mg/mL. The resulting solution was sonicated in a water bath for 1 h at 25°C. The intensity of the fluorescence of the CPT was determined using a fluorescence SpectraMax M5 microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA) with the excitation and emission wavelengths set at 365 and 430 nm, respectively. Control curves were constructed by dissolving blank NPs as described above and spiking with known amounts of CPT. The EE and DL of CPT were calculated using equations (1) and (2), respectively:25
| (1) |
| (2) |
In vitro release experiments
For the in vitro drug release study, drug-laden NPs were suspended in PBS containing Tween 80 (0.1%, w/v), which was placed in an air bath shaker at 150 rpm at 37°C±0.5°C. At scheduled time intervals, samples were collected and the same volume of fresh release medium was added. The collected samples were ultracentrifuged for 20 min at 80,000 rpm (Beckam Counter, Fullerton, CA, USA) and the supernatant was determined using the method described above. The experiments were run in triplicate.
Cell viability assay
The effects of prepared NPs on cell proliferation were examined by MTT assay as described previously.26 Briefly, HepG2 cell suspension (5×103 cells/well) was seeded into 96-well plates, incubated for 24 h, and subsequently treated with PLGA NPs, free CPT, or PLGA-CPT NPs at various CPT concentrations for an additional 48 or 72 h. Thereafter, MTT solution (20 µL, 5 mg/mL) was added and incubated at 37°C for 4 h, and any formazan crystals produced intracellularly were solubilized with the addition of DMSO. Cell viability was calculated as the ratio of the absorbance of treated and control wells. The absorbance was measured at 570 nm with a reference wavelength of 490 nm using a Spectra max PLUS 384 (Molecular Devices LLC).
Uptake of NPs by monolayer cells
HepG2 cells (5×104 cells) were cultured in a 24-well plate for 24 h, followed by treatment with free CPT or PLGA-CPT NPs at an equivalent CPT concentration for 4 h. The medium was removed and the cells were rinsed three times with ice-cold PBS to remove the extracellular CPT. An HCl/DMSO solution (0.04 M, 100 µL) was added to lyse the cells, and the intensity of fluorescence of CPT in the lysate was determined as mentioned above.
Preparation of microsomes from HepG2 cells
The preparation of microsomes from HepG2 cells was processed as reported.27 Briefly, 2×108 cells were harvested, centrifuged, and resuspended in 7 mL of HEPES buffer, followed by homogenizing in a Potter S (Sartorius, Staufen, Germany) homogenizer. The extract was then centrifuged at 6,000× g for 8 min to remove cell debris (Eppendorf 5702R, Eppendorf AG, Hamburg, Germany). The supernatant was centrifuged at 100,000× g for 1 h (Beckman) and the pellet was suspended in 200 µL buffer. Protein concentrations were determined using a BCA protein assay kit (Thermo Fisher, Waltham, MA, USA).
Functional study of CYP3A4 activity
The effect of PLGA-CPT NPs on CYP3A4 enzymatic activity was performed using CYP Vivid® kits (Life Technologies, Thermo Fisher Scientific) according to the manufacturer’s protocol. The experiment was measured in microsomes isolated from HepG2 cells. The assay was initiated by pipetting CPT, PLGA NPs, or PLGA-CPT NPs (40 µL) into a 96-well plate. Ketoconazole was chosen as the positive control, while solvent was the negative control. Samples were incubated with Master Pre-Mix (50 µL) at room temperature for 10 min. The Pre-Mix solution (10 µL) was added to initiate the enzyme reaction. The enzyme reaction was allowed to proceed and protected from light for 20 min at 37°C before it was quenched with the Stop Reagent (Tris base, 0.5 M, 10 µL). The fluorescence intensity was measured at excitation 415 nm and emission 460 nm. Results were calculated with a Kinetic Assay Mode using equation (3):
| (3) |
where X, A, and B represent the rates observed in the tested CPT, PLGA NPs, or CPT-PLGA NPs; the negative control; and the positive inhibition control, respectively.
CYP3A4 expression in the HepG2 cells
HepG2 cells were seeded onto a 6-cm dish at 2×105 cells per dish and allowed to attach overnight. Then, the medium was supplemented with PLGA NPs, free CPT, or PLGA-CPT NPs. Positive control cells were exposed to 10 µM ketoconazole. After 48 h of incubation, total RNA was isolated using TRIzol reagent (Life Technologies). Isolated RNA samples were quantified by a NanoDrop 3300 spectrophotometer (Thermo Scientific, NanoDrop Products, Wilmington, DE, USA). Synthesis of cDNA was done using a RevertAid First Strand cDNA Synthesis Kit following the manufacturer’s protocols (Thermo Scientific). CYP3A4 was amplified by a standard polymerase chain reaction protocol using 5′-TCAGCCTGGTGCTCCTCTAT-3′ as the forward primer, 5′-GGTAGGACAAAATATTTCCCAAAA-3′ as the reverse primer, and the prepared cDNA as a template. The internal control GAPDH adopted 5′-GAAGGTGAAGGTCGGAGTC-3′ as the forward primer and 5′-GAAGATGGTGATGGGATTTC-3′ as the reverse primer. The reaction mixtures were heated at 95°C for 10 min, followed by 30 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 20 s, and a final extension at 72°C for 5 min. Subsequently, polymerase chain reaction products were electrophoresed through 1.5% agarose gel and then subjected to a gel/fluorescence image analysis system for scanning. Each sample was analyzed in triplicate and the quantitative measurement of each gene was normalized to the amount of GAPDH cDNA.
Spectroscopy evaluation of the activity of CYP3A4
The ultraviolet–visible (UV–vis) absorption spectra of CYP3A4 were recorded using a spectrophotometer (Spectra Max Plus 384; Molecular Devices LLC). The absorption spectra of CYP3A4 before and after mixing with free CPT, PLGA NPs, or CPT-PLGA NPs were collected at 200–450 nm.
Fluorescence measurements were performed using a fluorescence spectrophotometer (F-4500; Hitachi Ltd., Tokyo, Japan), with 5/5 nm slit widths. The excitation wavelength of CYP3A4 was 280 nm, and the emission wavelengths were obtained at 290–600 nm. The concentration of CYP3A4 was maintained at 1 µM, and titrated with free CPT, PLGA NPs, or PLGA-CPT NPs. Mixed solutions rested for about 15 min before measurements. The synchronous fluorescence spectra of the above solutions were recorded at a temperature of 298 K from 200 to 350 nm at Δλ=60 and 15 nm, respectively.
Statistical analysis
Statistical analysis was performed using SPSS version 12.0 (SPSS Inc., Chicago, IL, USA). Results were expressed as mean±SD. Statistical significance was calculated using the t-test within groups or one-way analysis of variance between groups. A value of P<0.05 was considered statistically significant.
Results and discussion
Nanoparticle characterization
CPT has been formulated into a host of nanosized formulations in a bid to improve its in vivo bioavailability. In the present study, we constructed PLGA-CPT NPs and characterized their properties by morphology, particle size, size distribution, and surface charge. TEM analysis confirmed that the NPs possess a spherical morphology (Figure 1A). NPs were relatively monodisperse with a mean particle size of 94.6±5.7 nm, as measured by TEM. As depicted in Figure 1C, DLS measurement showed a hydrodynamic diameter of 128.4±8.6 nm and a zeta potential of −11.7±0.4 mV for PLGA-CPT NPs. The hydrodynamic size determined by DLS was slightly larger than that obtained by TEM because of the shrinkage of NPs during TEM sample preparation.28 In other words, the DLS determined the hydrodynamic diameter or the “equivalent sphere diameter” in solution, whereas the TEM images were obtained in the absence of a solvent.29
Figure 1.

Characterization of PLGA-CPT NPs.
Notes: (A) Representative transmission electron micrograph of NPs. (B) The in vitro release of CPT from PLGA-CPT NPs into buffer. (C) Physicochemical characterization of PLGA-CPT NPs. Data expressed as mean±SD from three independent experiments.
Abbreviations: DLS, dynamic light scattering; NP, nanoparticle; PLGA-CPT, camptothecin-encapsulated poly(lactic-co-glycolic acid); TEM, transmission electron microscopy.
Drug release behavior
In vitro release behavior of CPT was carried out under PBS containing Tween 80 at 37°C. The CPT release profile was obtained by measuring the percentage of drug released with respect to the total amount of CPT encapsulated in the NPs. As depicted in Figure 1B, CPT was released 54.7%±4.2% from PLGA-CPT NPs in the first 12 h, and reached a percentage of cumulative release of 87.3%±7.4% within 48 h. The EE of CPT was 64.8%±5.5% and the DL was 7.3%±0.6%.
In vitro cytotoxicity
The inhibitory effects of the prepared NPs on HepG2 cells were examined using the MTT assay after 48 or 72 h of incubation. As depicted in Figure 2, no significant difference was evident between PLGA NPs and the control group. PLGA-CPT NPs exerted a cytotoxic effect on HepG2 cells in a time- and concentration-dependent manner. Over the CPT concentration range of 25–100 µg/mL, PLGA-CPT NPs did not exhibit apparent toxic effects against the HepG2 cell viability after 48 h of exposure, compared with CPT treatment. PLGA-CPT NP treatment (150 µg/mL) for 48 h suppressed cell viability by 46.5%±4.8% (P<0.05 compared with CPT treatment), and this effect was exacerbated at higher concentration (200 µg/mL), with a maximal amount of suppression occurring at 72 h using a concentration of 200 µg/mL. The IC50 values of the nano-conjugates were much lower than that of the free CPT solution, which can be attributed to the increased solubility of CPT by nano-conjugation. The results were consistent with previous research that demonstrated CPT-conjugated NPs had a higher cell inhibition effect than free CPT against cervical cancer30 and colon cancer.31
Figure 2.

Cytotoxicity studies of different concentrations of CPT and PLGA-CPT NPs on HepG2 cells for 48 h (A) and 72 h (B) by the MTT method. Cell viability was calculated with respect to the PLGA NP-treated cells. Data expressed as mean±SD, n=6. #P<0.05.
Abbreviations: NP, nanoparticle; PLGA-CPT, camptothecin-encapsulated poly(lactic-co-glycolic acid).
Cellular uptake
It has been reported that the cellular uptake of NPs depends upon many factors, including particle size,32 the stage of the cell cycle,33 the sedimentation and diffusion velocity effects of large and dense particles,34 the surface charge,35 the composition of the protein corona on the NPs,36 and competition between receptors and ligands.37 HepG2 cells were treated with free CPT or PLGA-CPT NPs, and the uptake of CPT into the cells was measured. The cells were exposed to three concentrations (50, 100, and 150 µg/mL) of either CPT or PLGA-CPT NPs for 4 h (Figure 3). PLGA-CPT NPs (150 µg/mL) accumulated at higher intracellular concentrations after 4-h incubation compared with free CPT (P<0.05). However, no statistically significant differences were found in the amount of cellular uptake between the two groups at lower CPT concentrations (50 or 100 µg/mL). Our investigation of NP uptake by cells is similar to that observed with gold NPs or other PLGA NPs, and is a concentration-dependent endocytic process.32,38
Figure 3.
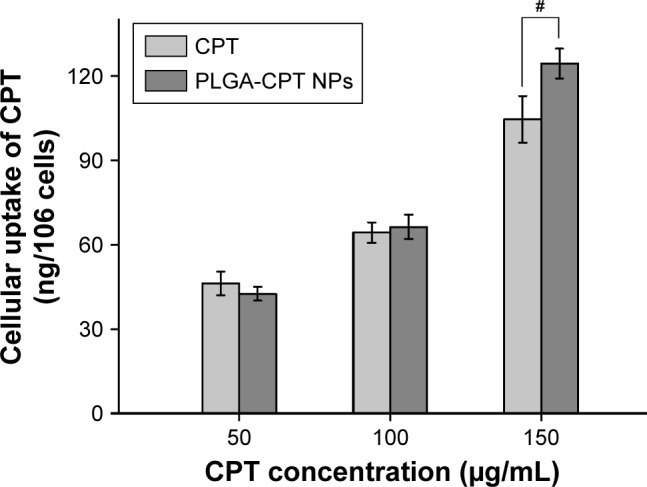
Uptake of CPT from PLGA-CPT NPs into HepG2 cells.
Notes: Data expressed as mean±SD, n.6. #P<0.05.
Abbreviations: NP, nanoparticle; PLGA-CPT, camptothecin-encapsulated poly(lactic-co-glycolic acid).
Effect of NPs on CYP3A4 activity and expression
NPs at various concentrations were incubated with microsomes isolated from HepG2 cells, and the metabolic activity of CYP3A4 was determined. As illustrated in Figure 4, the activity of CYP3A4 decreased dose-dependently upon incubation with CPT or PLGA-CPT NPs. PLGA NPs showed a much weaker CYP3A4 inhibitory effect, compared to PLGA-CPT NPs. Furthermore, there was no significant difference between CPT and PLGA-CPT NPs against the activity of CYP3A4, indicating that NP modification did not affect the inhibitory potential.
Figure 4.
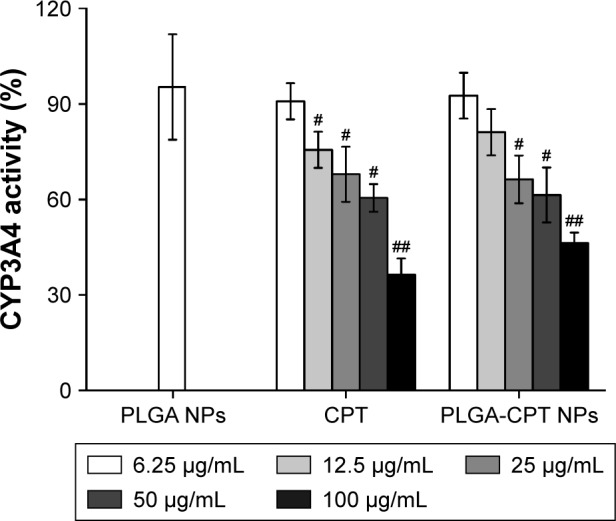
Effect of NPs on CYP3A4 activity in HepG2 cells.
Notes: Results representative of six separate experiments; data expressed as mean±SD. ##P<0.01 and #P<0.05 when compared with PLGA NP group, respectively.
Abbreviations: CYP3A4, cytochrome P450 enzyme; NP, nanoparticle; PLGA-CPT, camptothecin-encapsulated poly(lactic-co-glycolic acid).
The expression level of CYP3A4 in the HepG2 cells was investigated. As depicted in Figure 5, incubation of cells treated with CYP3A4 inhibitor ketoconazole upregulated the CYP3A4 mRNA level by 2.1-fold. However, cells pretreated with PLGA NPs, free CPT, or PLGA-CPT NPs did not have any significant effect on the CYP3A4 mRNA expression.
Figure 5.
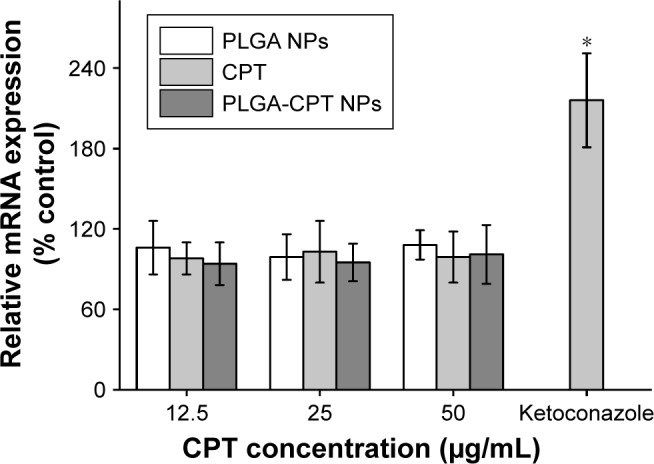
Effect of NPs on CYP3A4 mRNA expression.
Note: *P<0.05.
Abbreviations: CYP3A4, cytochrome P450 enzyme; NP, nanoparticle; PLGA-CPT, camptothecin-encapsulated poly(lactic-co-glycolic acid).
Spectroscopy assessment
The interactions between PLGA-CPT NPs and CYP3A4 were studied by UV–vis and fluorescence spectroscopy. A UV–vis absorption measurement is a simple and effective method to explore structural changes.39 Proteins exhibit UV absorptions owing to tryptophane (280 nm), tyrosine (280 nm), and phenylalanine (257 nm) as well as peptide bonds (225 nm).40 After adding PLGA-CPT NPs, the absorptions of protein chromophores are bound to change.41 As exhibited in Figure 6, the absorbance intensity of CYP3A4 decreased with the addition of PLGA-CPT NPs and the absorption at 280 nm shifted toward a longer wavelength region, which indicates the formulation of a complex between CYP3A4 and PLGA-CPT NPs.
Figure 6.
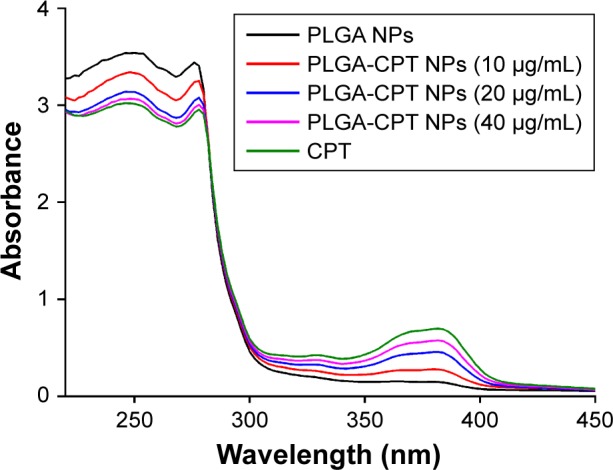
Ultraviolet–visible absorbance spectra of CYP3A4 in the presence of different concentrations of PLGA-CPT NPs.
Abbreviations: CYP3A4, cytochrome P450 enzyme; NP, nanoparticle; PLGA-CPT, camptothecin-encapsulated poly(lactic-co-glycolic acid).
It has been suggested that the fluorescence of proteins is due to their tyrosine, tryptophan, and phenylalanine residues. Synchronous fluorescence spectra can provide information about the molecular microenvironment in the vicinity of fluorophore functional groups.42 When the Δλ values between the excitation and emission wavelengths were stabilized at 15 and 60 nm, the synchronous fluorescence spectra exhibited the spectral characters of tyrosine and tryptophan residues, respectively.43 The fluorescence spectra of CYP3A4 in the absence and presence of PLGA-CPT NPs are shown in Figure 7. A significant decrease in fluorescence intensity was observed with the addition of PLGA-CPT NPs, suggesting that an interaction occurred between CYP3A4 and PLGA-CPT NPs. Meanwhile, the maximum emission wavelength of CYP3A4 slightly shifted toward the shorter wavelength region, which indicates that the addition of PLGA-CPT NPs decrease hydrophobicity. Furthermore, it is apparent from Figure 8A and B that the quenching of the intrinsic fluorescence of CYP3A4 is from tryptophan and tyrosine residues.
Figure 7.
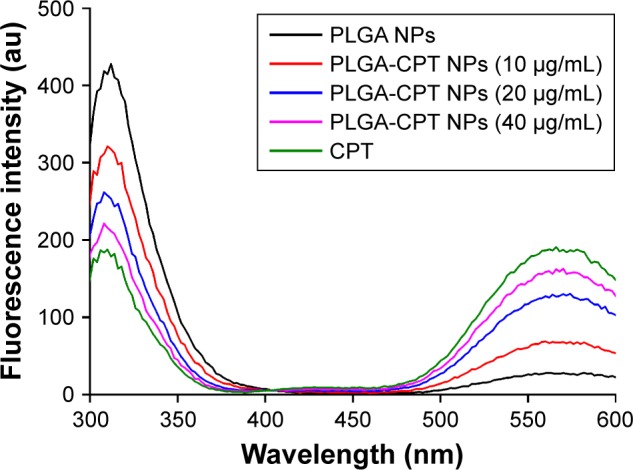
Fluorescence spectra of CYP3A4 in the presence of different concentrations of PLGA-CPT NPs.
Abbreviations: CYP3A4, cytochrome P450 enzyme; NP, nanoparticle; PLGA-CPT, camptothecin-encapsulated poly(lactic-co-glycolic acid).
Figure 8.
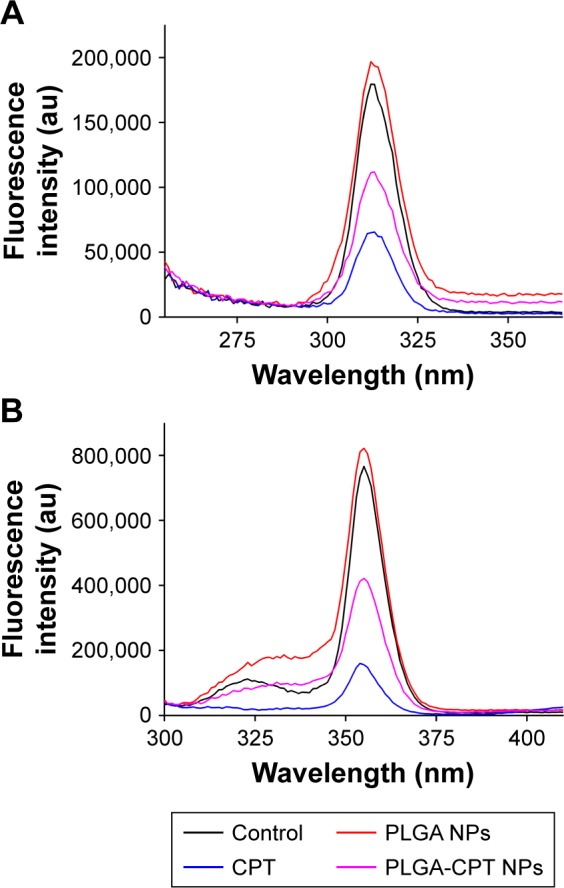
Synchronous fluorescence spectra of CYP3A4 with Δλ=15 nm (A) and Δλ=60 nm (B) in the presence of different concentrations of PLGA-CPT NPs.
Abbreviations: CYP3A4, cytochrome P450 enzyme; NP, nanoparticle; PLGA-CPT, camptothecin-encapsulated poly(lactic-co-glycolic acid).
The spectroscopy assessment is expected to provide important insights into the interaction mechanisms of PLGA-CPT NPs with CYP3A4, which could be a useful guideline for further investigations.
Conclusions
We reported for the first time about the interactions between PLGA-CPT NPs and CYP3A4. It can be concluded that PLGA-CPT NPs had the potential of inhibiting CYP3A4 activity. Our results suggest that the effects of the NP-based anticancer drug delivery system on CYP metabolic function should be considered.
Acknowledgments
This work was supported by Doctoral Foundation of Tianjin Medical University Cancer Institute and Hospital (B1205), the Tianjin Key Project in the Health Sector (14KG140), and the National Clinical Research Center for Cancer Cultivation Project.
Footnotes
Author contributions
Hanmei Bao and Zhao Yan designed the research. Hanmei Bao and Qing Zhang undertook the research. Hanmei Bao, Qing Zhang, and Zhao Yan analyzed the data. Hanmei Bao, Qing Zhang, and Zhao Yan wrote the paper. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Takimoto CH, Wright J, Arbuck SG. Clinical applications of the camptothecins. Biochimica et Biophysica Acta (BBA) – Gene Structure and Expression. 1998;1400(1–3):107–119. doi: 10.1016/s0167-4781(98)00130-4. [DOI] [PubMed] [Google Scholar]
- 2.Mross K, Richly H, Schleucher N, et al. A phase I clinical and pharmacokinetic study of the camptothecin glycoconjugate, BAY 38-3441, as a daily infusion in patients with advanced solid tumors. Ann Oncol. 2004;15(8):1284–1294. doi: 10.1093/annonc/mdh313. [DOI] [PubMed] [Google Scholar]
- 3.Householder KT, Diperna DM, Chung EP, et al. Intravenous delivery of camptothecin-loaded PLGA nanoparticles for the treatment of intra-cranial glioma. Int J Pharm. 2015;479(2):374–380. doi: 10.1016/j.ijpharm.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Yang Y, Ye J, Ma P, Xu X, Xia X, Liu Y. Folate and CPP mediated specific delivery of camptothecin to tumor pH and reduction dual-sensitive micelles. J Control Release. 2017;259:e164–e165. [Google Scholar]
- 5.Fang S, Hou Y, Ling L, et al. Dimeric camptothecin derived phospholipid assembled liposomes with high drug loading for cancer therapy. Colloids Surf B Biointerfaces. 2018;166:235–244. doi: 10.1016/j.colsurfb.2018.02.046. [DOI] [PubMed] [Google Scholar]
- 6.Dramou P, Fizir M, Taleb A, et al. Folic acid-conjugated chitosan oligosaccharide-magnetic halloysite nanotubes as a delivery system for camptothecin. Carbohydr Polym. 2018;197:117–127. doi: 10.1016/j.carbpol.2018.05.071. [DOI] [PubMed] [Google Scholar]
- 7.Fröhlich E, Kueznik T, Samberger C, Roblegg E, Wrighton C, Pieber TR. Size-dependent effects of nanoparticles on the activity of cytochrome P450 isoenzymes. Toxicol Appl Pharmacol. 2010;242(3):326–332. doi: 10.1016/j.taap.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Williams JA, Hyland R, Jones BC, et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32(11):1201–1208. doi: 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]
- 9.Keshava C, Mccanlies EC, Weston A. CYP3A4 polymorphisms – potential risk factors for breast and prostate cancer: a HuGE review. Am J Epidemiol. 2004;160(9):825–841. doi: 10.1093/aje/kwh294. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Z, Hu Y, Harmon T, Pentel PR, Ehrich M, Zhang C. Hybrid nanoparticle-based nicotine nanovaccines: boosting the immunological efficacy by conjugation of potent carrier proteins. Nanomedicine. 2018;14(5):1655–1665. doi: 10.1016/j.nano.2018.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ragelle H, Danhier F, Préat V, Langer R, Anderson DG. Nanoparticle-based drug delivery systems: a commercial and regulatory outlook as the field matures. Expert Opin Drug Deliv. 2017;14(7):851–864. doi: 10.1080/17425247.2016.1244187. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Z, Hu Y, Hoerle R, et al. A nanoparticle-based nicotine vaccine and the influence of particle size on its immunogenicity and efficacy. Nanomedicine. 2017;13(2):443–454. doi: 10.1016/j.nano.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scown TM, Goodhead RM, Johnston BD, et al. Assessment of cultured fish hepatocytes for studying cellular uptake and (eco)toxicity of nanoparticles. Environ Chem. 2010;7(1):36–49. [Google Scholar]
- 14.Lamb JG, Hathaway LB, Munger MA, Raucy JL, Franklin MR. Nanosilver particle effects on drug metabolism in vitro. Drug Metab Dispos. 2010;38(12):2246–2251. doi: 10.1124/dmd.110.035238. [DOI] [PubMed] [Google Scholar]
- 15.Fröhlich E, Kueznik T, Samberger C, Roblegg E, Wrighton C, Pieber TR. Size-dependent effects of nanoparticles on the activity of cytochrome P450 isoenzymes. Toxicol Appl Pharmacol. 2010;242(3):326–332. doi: 10.1016/j.taap.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Nabeshi H, Yoshikawa T, Matsuyama K, et al. Systemic distribution, nuclear entry and cytotoxicity of amorphous nanosilica following topical application. Biomaterials. 2011;32(11):2713–2724. doi: 10.1016/j.biomaterials.2010.12.042. [DOI] [PubMed] [Google Scholar]
- 17.Wilkes GM. Targeted therapy: attacking cancer with molecular and immunological targeted agents. Asia Pac J Oncol Nurs. 2018;5(2):137–155. doi: 10.4103/apjon.apjon_79_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung C. Fourier transform infrared spectroscopy as a tool to study structural properties of cytochromes P450 (CYPs) Anal Bioanal Chem. 2008;392(6):1031–1058. doi: 10.1007/s00216-008-2216-4. [DOI] [PubMed] [Google Scholar]
- 19.Shao J, Chen J, Li T, Zhao X. Spectroscopic and molecular docking studies of the in vitro interaction between puerarin and cytochrome P450. Molecules. 2014;19(4):4760–4769. doi: 10.3390/molecules19044760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong J, Song L, Meng J, et al. Bio–nano interaction of proteins adsorbed on single-walled carbon nanotubes. Carbon. 2009;47(4):967–973. [Google Scholar]
- 21.Bao H, Jin X, Li L, Lv F, Liu T. OX26 modified hyperbranched polyglycerol-conjugated poly(lactic-co-glycolic acid) nanoparticles: synthesis, characterization and evaluation of its brain delivery ability. J Mater Sci Mater Med. 2012;23(8):1891–1901. doi: 10.1007/s10856-012-4658-7. [DOI] [PubMed] [Google Scholar]
- 22.Ganipineni LP, Ucakar B, Joudiou N, et al. Magnetic targeting of paclitaxel-loaded poly(lactic-co-glycolic acid)-based nanoparticles for the treatment of glioblastoma. Int J Nanomedicine. 2018;13:4509–4521. doi: 10.2147/IJN.S165184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Z, Lou S, Hu Y, Zhu J, Zhang C. A nano-in-nano polymer-dendrimer nanoparticle-based nanosystem for controlled multidrug delivery. Mol Pharm. 2017;14(8):2697–2710. doi: 10.1021/acs.molpharmaceut.7b00219. [DOI] [PubMed] [Google Scholar]
- 24.Householder KT, Diperna DM, Chung EP, et al. Intravenous delivery of camptothecin-loaded PLGA nanoparticles for the treatment of intra-cranial glioma. Int J Pharm. 2015;479(2):374–380. doi: 10.1016/j.ijpharm.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Schafroth N, Arpagaus C, Jadhav UY, Makne S, Douroumis D. Nano and microparticle engineering of water insoluble drugs using a novel spray-drying process. Colloids Surf B Biointerfaces. 2012;90:8–15. doi: 10.1016/j.colsurfb.2011.09.038. [DOI] [PubMed] [Google Scholar]
- 26.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 27.Fröhlich E, Kueznik T, Samberger C, Roblegg E, Wrighton C, Pieber TR. Size-dependent effects of nanoparticles on the activity of cytochrome P450 isoenzymes. Toxicol Appl Pharmacol. 2010;242(3):326–332. doi: 10.1016/j.taap.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Yang M, Wang Q, et al. 10-Hydroxycamptothecin loaded nanoparticles: preparation and antitumor activity in mice. J Control Release. 2007;119(2):153–162. doi: 10.1016/j.jconrel.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 29.Morita T, Horikiri Y, Suzuki T, Yoshino H. Preparation of gelatin microparticles by co-lyophilization with poly(ethylene glycol): characterization and application to entrapment into biodegradable microspheres. Int J Pharm. 2001;219(1–2):127–137. doi: 10.1016/s0378-5173(01)00642-1. [DOI] [PubMed] [Google Scholar]
- 30.Rajan M, Krishnan P, Pradeepkumar P, et al. Magneto-chemotherapy for cervical cancer treatment with camptothecin loaded Fe3O4 functionalized β-cyclodextrin nanovehicle. RSC Adv. 2017;7(73):46271–46285. [Google Scholar]
- 31.Krishnan P, Rajan M, Kumari S, et al. Efficiency of newly formulated camptothecin with β-cyclodextrin-EDTA-Fe3O4 nanoparticle-conjugated nanocarriers as an anti-colon cancer (HT29) drug. Sci Rep. 2017;7(1):10962. doi: 10.1038/s41598-017-09140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bao H, Zhang Q, Xu H, Yan Z. Effects of nanoparticle size on anti-tumor activity of 10-hydroxycamptothecin-conjugated gold nanoparticles: in vitro and in vivo studies. Int J Nanomedicine. 2016;11:929–940. doi: 10.2147/IJN.S96422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JA, Åberg C, Salvati A, Dawson KA. Role of cell cycle on the cellular uptake and dilution of nanoparticles in a cell population. Nat Nanotechnol. 2012;7(1):62–68. doi: 10.1038/nnano.2011.191. [DOI] [PubMed] [Google Scholar]
- 34.Cho EC, Zhang Q, Xia Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nat Nanotechnol. 2011;6(6):385–391. doi: 10.1038/nnano.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim B, Han G, Toley BJ, Kim CK, Rotello VM, Forbes NS. Tuning payload delivery in tumour cylindroids using gold nanoparticles. Nat Nanotechnol. 2010;5(6):465–472. doi: 10.1038/nnano.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cedervall T, Lynch I, Lindman S, et al. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc Natl Acad Sci U S A. 2007;104(7):2050–2055. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar A, Ma H, Zhang X, et al. Gold nanoparticles functionalized with therapeutic and targeted peptides for cancer treatment. Biomaterials. 2012;33(4):1180–1189. doi: 10.1016/j.biomaterials.2011.10.058. [DOI] [PubMed] [Google Scholar]
- 38.Cartiera MS, Johnson KM, Rajendran V, Caplan MJ, Saltzman WM. The uptake and intracellular fate of PLGA nanoparticles in epithelial cells. Biomaterials. 2009;30(14):2790–2798. doi: 10.1016/j.biomaterials.2009.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashoka S, Seetharamappa J, Kandagal PB, Shaikh SMT. Investigation of the interaction between trazodone hydrochloride and bovine serum albumin. J Lumin. 2006;121(1):179–186. [Google Scholar]
- 40.Zhang F, Ni Y. A Comparison Study on the Interaction of Sunset Yellow and β-Carotene with Bovine Serum Albumin. Acta Chimica Sinica. 2012;70(12):1379–1384. [Google Scholar]
- 41.Channu BC, Kalpana HN, Eregowda GB, Dass C, Houghton PJ, Thimmaiah KN. Interaction of substituted phenoxazine chemosensitizers with bovine serum albumin. J Pharm Biomed Anal. 1999;21(4):775–785. doi: 10.1016/s0731-7085(99)00187-9. [DOI] [PubMed] [Google Scholar]
- 42.Yj H, Liu Y, Zb P, Ss Q. Interaction of cromolyn sodium with human serum albumin: a fluorescence quenching study. Bioorgan Med Chem. 2005;13:6609–6614. doi: 10.1016/j.bmc.2005.07.039. [DOI] [PubMed] [Google Scholar]
- 43.Congdon RW, Muth GW, Splittgerber AG. The binding interaction of Coomassie blue with proteins. Anal Biochem. 1993;213(2):407–413. doi: 10.1006/abio.1993.1439. [DOI] [PubMed] [Google Scholar]