

Abstract
The induction of mitochondrial reactive oxygen species (mtROS) by hyperglycemia is a key event responsible for endothelial activation and injury. Heat shock protein 22 (HSP22) is a stress-inducible protein associated with cytoprotection and apoptosis inhibition. However, whether HSP22 prevents hyperglycemia-induced vascular endothelial injury remains unclear. Here, we investigated whether HSP22 protects the vascular endothelium from hyperglycemia-induced injury by reducing mtROS production. We used a high-fat diet and streptozotocin injection model to induce type 2 diabetes mellitus (T2DM, metabolic syndrome) and exposed human umbilical vein endothelial cells (HUVECs) to high glucose following overexpression or silencing of HSP22 to explore the role of HSP22. We found that HSP22 markedly inhibited endothelial cell activation and vascular lesions by inhibiting endothelial adhesion and decreasing cytokine secretion. We performed confocal microscopy and flow cytometry assays using HUVECs and showed that HSP22 attenuated mtROS and mitochondrial dysfunction in hyperglycemia-stimulated endothelial cells. Mechanistically, using the mtROS inhibitor MitoTEMPO, we demonstrated that HSP22 suppressed endothelial activation and injury by eliminating hyperglycemia-mediated increases in mtROS. Furthermore, we found that HSP22 maintained the balance of mitochondrial fusion and fission by mitigating mtROS in vitro. HSP22 attenuated the development of vascular lesions by suppressing mtROS-mediated endothelial activation in a T2DM mouse model. This study provides evidence that HSP22 may be a promising therapeutic target for vascular complications in T2DM.
Keywords: Hyperglycemia, HSP22, Inflammation, mtROS, Oxidative stress, T2DM
Graphical abstract
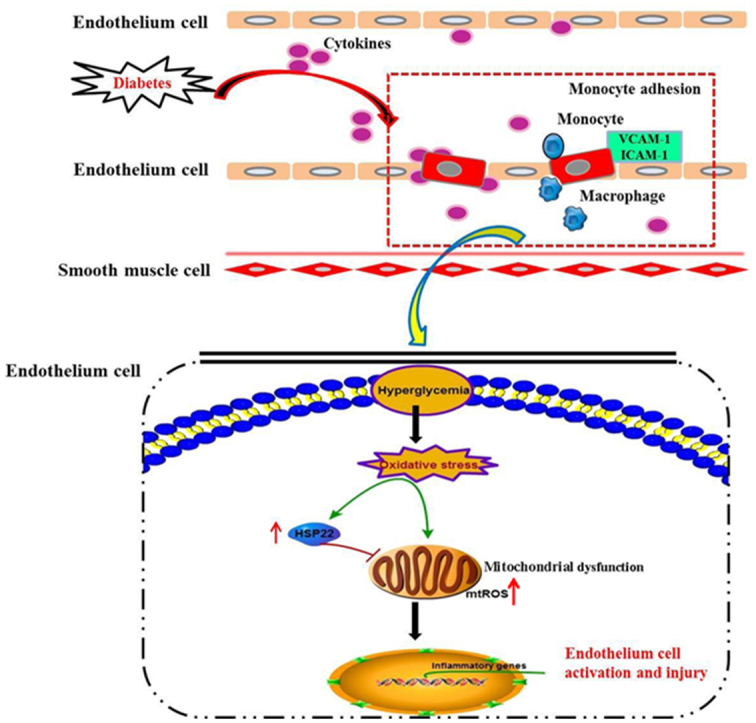
The role of HSP22 in hyperglycemia-stimulated endothelial cell activation and injury. Diabetes induces oxidative stress, mtROS production and mitochondrial injury, which promote endothelial activation and impair endothelial function. Under diabetic conditions, the upregulation of HSP22 protein expression decreases endothelial activation and improves endothelial function by inhibiting mtROS production and mitochondrial dysfunction.
Highlights
-
•
HSP22 reduces endothelial inflammation under diabetic conditions.
-
•
HSP22 restrains hyperglycemia-induced oxidative stress in the vascular endothelium.
-
•
HSP22 reduces hyperglycemia-induced mtROS and endothelial mitochondrial dysfunction.
-
•
HSP22 maintains the balance of mitochondrial fusion and fission by mitigating mtROS.
1. Introduction
Human lifestyle changes have recently led to a rapid increase in type 2 diabetes mellitus (T2DM) [1], [2]. T2DM is a chronic inflammatory and progressive disease that induces multiple complications, such as atherosclerosis, diabetic nephropathy, diabetic retinopathy, and cardiovascular disease, and can be life-threatening [3], [4], [5], [6]. Endothelial cell injury plays a critical role in the development and progression of diabetic-induced complications [5], [7], [8]. However, the molecular mechanisms underlying T2DM-induced endothelial dysfunction remain incompletely elucidated.
Mitochondria are highly dynamic organelles that are involved in multiple cellular processes, including energy metabolism, reactive oxygen species (ROS) production, inflammation and cell death [9]. To date, accumulating evidence suggests that increased mitochondrial reactive oxygen species (mtROS) represent the major intracellular source of superoxide anions in diabetes and that mtROS disrupt mitochondrial homeostasis and ultimately lead to cell damage [9], [10]. In addition, excessive mtROS and mitochondrial damage are critical for diabetes-induced endothelial dysfunction and complications [9], [11]. MtROS are signaling mediators that drive the activation of proinflammatory transcription factors, such as activator protein-1 (AP-1) and nuclear factor-κB (NF-κB), and promote inflammatory cytokine production [10], [12]. Therefore, we focused on the molecular mechanism by which mtROS mediate endothelial injury to further understand the pathologic process of diabetes-induced vascular injury and to identify a potential therapeutic target for T2DM.
Small heat shock proteins (sHSPs) are induced upon stress and pathological processes and protect cells against various stress stimuli [9], [13]. To date, 10 members of the sHSP family have been identified in the human genome, and only four sHSPs, including heat shock protein 27 (HSP27), αB-crystallin, heat shock protein 20 (HSP20) and heat shock protein 22 (HSP22), are ubiquitously expressed in various tissues [9]. Thus far, information regarding the protective effects of sHSPs in diabetes has been generally limited to HSP27, HSP20 and αB-crystallin. For example, HSP20 is downregulated in the heart and retina of diabetic rats, and overexpression of HSP20 reduces cell injury [6], [13]. HSP27 and αB-crystallin contribute to the protection of the endothelium against apoptosis under high-glucose conditions [14], [15], [16], [17], [18]. Although HSP22 has also been reported to be upregulated in the diabetic retina and heart, whether HSP22 plays a role in diabetes remains largely unknown [6], [13]. An increasing number of studies have confirmed that HSP22 is involved in resistance to oxidative stress and cytoprotection. Qiu et al. showed that HSP22 deletion accelerated heart failure by impairing both nuclear and mitochondrial functions [19]. Laure found that overexpression of HSP22 provided cardioprotection by modulating mtROS production [20]. HSP22 also inhibited oxidative stress-induced hippocampal neuronal cell death [21]. Interestingly, HSP22 interacts with phosphoglucomutase to promote the synthesis of glycogen and plays roles in cell growth, cytoprotection and metabolic adaptation to stress conditions [22]. Hence, investigating whether HSP22 is expressed in endothelial cells and reduces mtROS-mediated endothelial injury under hyperglycemia is warranted.
In the current study, we used a mouse model of T2DM that was developed in three types of mice, HSP22 transgenic (TG) mice, HSP22 knockout (KO) mice and matched wild-type (WT) littermates, and human umbilical vein endothelial cells (HUVECs) treated with high glucose (HG) to investigate whether HSP22 is involved in hyperglycemia-stimulated endothelial injury. Our results show that overexpression of HSP22 alleviated hyperglycemia-induced endothelial dysfunction and mitochondrial damage. Silencing HSP22 had the opposite effect in vitro and in vivo. Furthermore, we found that silencing HSP22 eliminated the effects of the mtROS inhibitor MitoTEMPO, which suppressed hyperglycemia-induced endothelial cell activation and mitochondrial damage in vitro. In summary, our study presents the first evidence that HSP22 can inhibit mtROS-mediated endothelial injury during diabetes.
2. Research design and methods
2.1. Animal studies
Male C57BL/6N mice obtained from Charles River (Wilmington, MA) were housed conventionally at 22 °C under a 12-h light-dark cycle with free access to water and food [23]. C57BL/6 mice were chosen for the animal model because these mice develop the typical characteristics of human metabolic syndrome, including hyperglycemia, when fed a high-fat diet (HFD) [23], [24]. Generation of both the TG mouse model with the overexpression of HSP22 and the KO mouse model with silencing of HSP22 has been previously described [19], [20], [25], [26], [27], [28]. All animal experiments were conducted in compliance with the National Institutes of Health (NIH) policies in the Guide for the Care and Use of Laboratory Animals and were approved by the Nanchang University Animal Care and Use Committee (SYXK(G) 2015-0001).
Male HSP22-TG mice, HSP22-KO mice and WT control mice (aged 8–12 weeks) were prepared for induction of T2DM. A group of 30 WT mice was randomly divided into three experimental groups of 10 mice each, and two groups of 20 HSP22-TG mice and 20 HSP22-KO mice were both randomly divided into two experimental groups of 10 mice each. We used a HFD and streptozotocin injection model to induce T2DM as previously described [29], [30], [31]. All animals were fed either a HFD (60% of calories from fat, Research Diets, #D12492, USA) or normal chow. After 4 weeks, the HFD mice who exhibited insulin resistance received an intraperitoneal (i.p.) injection of streptozotocin (STZ, Sigma, USA) at a single dose of 100 mg/kg dissolved in citrate buffer (0.1 mol/l Na citrate, pH 4.5) to partly destroy islet function and raise glucose levels. The normal chow mice received citrate buffer alone and were processed in parallel with the diabetic mice. All mice were maintained on their respective diets until the end of the study. The fasting blood glucose (FBG) level and body weight were measured using a blood glucose monitor (OneTouch Ultra2; LifeScan, USA.), and their weights were monitored once per month. Mice with blood glucose levels > 200 mg/dl were considered diabetic [5]. At the end of the experiments, the mice were anesthetized intraperitoneally using sodium pentobarbital (50 mg/kg), and the adequacy of anesthesia was confirmed by the absence of a reflex response to a foot squeeze [13]. Subsequently, the aortas were perfused and isolated for collection of frozen sections and paraffin sections for immunohistochemistry, ROS production, PCR and Western blot analyses.
2.2. Tissue pathological morphology
The aortic pathological morphology was observed using hematoxylin and eosin staining (H&E). The fixed paraffin embedded aortic rings (5-µm sections) were stained with H&E and examined under an optical microscope (Olympus, Japan).
2.3. Immunohistochemistry and immunofluorescence
Immunohistochemical staining of the aortas to detect HSP22, intracellular adhesion molecule 1 (ICAM-1), and vascular cell adhesion molecule 1 (VCAM-1) was performed as previously described [32]. Briefly, the aortas were embedded in paraffin, and 5-µm slices were cut from the embedded blocks. After heat-induced antigen retrieval, slides were incubated with primary antibodies diluted in phosphate-buffered saline (PBS). Primary antibodies for the immunohistochemistry study (HSP22 (ab151552, rabbit monoclonal, at 1:50), ICAM-1 (ab119871, rat monoclonal, at 1:200), and VCAM-1 (ab134047, rabbit monoclonal, at 1:200)) were acquired from Abcam (MA, USA). After incubation with primary antibodies overnight, the sections were washed in PBS and incubated with biotinylated IgG (1:250) for 1 h and then with streptavidin-HRP for 30 min at room temperature. Next, 50 μl of DAB was added to each section for 1–5 min for staining. After washing, the slides were counterstained with hematoxylin for 2 min. The slides were then mounted and observed under a microscope. Semiquantitative analysis of the tissue staining was performed using the Image-Pro Plus 6.0 System (Media cybernetics, USA).
Immunofluorescence staining was performed to detect the expression of HSP22 and CD31 proteins in HUVECs. After treatments, the cells were fixed in cold 4% paraformaldehyde for 15 min and soaked in PBS with 0.1% Triton, 1% bovine serum albumin, 10% normal goat serum (Sigma) and 0.3 M glycine for 1 h at room temperature. Then, the cells were incubated with primary antibodies overnight at 4 °C. Primary antibodies for the immunofluorescence assay (HSP22 (ab151552, rabbit monoclonal, at 1:50) and CD31 (ab24590, mouse monoclonal, at 1:50)) were acquired from Abcam (MA, USA). The cells were washed repeatedly with PBS and incubated with Alexa Fluor® 488-conjugated goat anti-rabbit IgG secondary antibody and Alexa Fluor™ 594-conjugated goat anti-mouse IgG secondary antibody (Molecular Probes, Invitrogen, USA) for 1 h. Nuclear DNA was labeled with DAPI (Invitrogen, USA). The fluorescence intensity was examined using a confocal laser scanning microscope (Leica, Germany), and the images were analyzed using ImageJ (Bethesda, MD, USA).
2.4. Cytokine antibody array
Serum was obtained from each group of mice and analyzed using a mouse cytokine array kit (R&D Systems; QAH-TH17-1, RayBiotech, USA) according to the manufacturer's specifications [33], [34]. The fluorescence intensities were measured by an axon scanner 4000B using GenePix software. Then, the results were analyzed using a RayBio Analysis Tool Excel sheet.
2.5. Cell culture and transfections
HUVECs were obtained from American Type Culture Collection (Manassas, VA) and grown in endothelial basal medium (EBM) with 10% FBS and growth supplement (Gibco, USA). The cells were incubated at 37 °C in a humidified atmosphere of 5% CO2 and 95% air and grown to 70–80% confluence. Cells at passages three to eight were used in the experiments. HUVECs were treated with normal glucose (NG, 5 mM D-Glucose), osmotic control (OC, 30 mM D-mannitol) or HG (30 mM D-Glucose).
The cells were transfected with plasmids containing the HSP22 gene or a dominant-negative form and HSP22-specific siRNA oligos or dominant-negative siRNA oligos using Lipofectamine 3000 (Invitrogen, USA) according to the manufacturer's instructions. Forty-eight hours after transfection, the cells were treated as indicated, and changes in cell morphology and function were assessed; the molecular mechanisms were also investigated.
2.6. Cell viability detection
Viability was determined by a Cell Counting Kit-8 (CCK-8, Dojindo, Japan) as previously described [35], [36]. Briefly, HUVECs in 96-well plates (2 × 104 cells/well) were cocultured with CCK-8 for 1 h. CCK-8 was then reduced to formazan by the activity of mitochondrial dehydrogenases, and the absorbance, which is directly proportional to the number of viable cells was measured using a microplate reader (Bio-Rad, USA) at 450 nm. Cell viability was calculated based on the relative optical density compared with that of untreated controls.
2.7. Toxicology assay
Cytotoxicity was measured in vitro using a Lactic Dehydrogenase-based Toxicology Assay Kit (Sigma, USA) as previously described [36], [37]. Briefly, cytoplasmic lactate dehydrogenase (LDH) released into the medium was measured based on the reduction in NAD by LDH. The resulting reduced NADH was converted to a colored compound and measured by a microplate reader (Bio-Rad, USA). The LDH activity was calculated by subtracting the 630 nm absorbance value (background signal from instrument) from the 490 nm absorbance value.
2.8. Adhesion assays
Adhesion assays were performed as previously described [38]. Briefly, primary human peripheral mononuclear cells (PBMCs) were grown in RPMI 1640 containing 10% FBS (Gibco, USA) at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Then, PBMCs were stained with 2 μM calcein green AM for 30 min at 37 °C. After the HUVECs were seeded in 96-well plates and treated, the labeled PBMCs (1 × 106 cells/ml) were added to the HUVECs in each well. After 1 h of coincubation at 37 °C, the cells were washed with PBS to remove the unattached PBMCs, and the plates were read using a microplate reader (Bio-Rad, USA).
2.9. Assessment of reactive oxygen species production
Dihydroethidium (DHE) is a lipophilic cell-permeable dye that can be oxidized by O2•- to form ethidium bromide. The intracellular ROS in the freshly isolated aortas were measured using DHE (Sigma, USA) staining. Upon reacting with the superoxide anion, DHE forms a red fluorescent product, 2-hydroxyethidium, with maximum excitation and emission peaks at 500 and 580 nm, respectively. Briefly, DHE was reconstituted in anhydrous DMSO and diluted with Krebs buffer (containing 20 mM HEPES) to a concentration of 5 μmol/l before use. The freshly isolated aortic rings (10-µm sections) were incubated with dihydroethidium in Krebs buffer at 37 °C for 20 min in the dark. After washing, the sections were incubated with DAPI. The fluorescence intensity was examined under a fluorescence microscope (Olympus, Japan), and all images were analyzed using ImageJ software.
MtROS production in the HUVECs was assessed using the mtROS-specific probe MitoSOX (Thermo Fisher Scientific, USA) according to the manufacturer's specifications and previously described [38], [39]. The cells were incubated with Hank's balanced salt solution containing 5 μmol/l MitoSOX at 37 °C for 15 min. Then, the cells were collected, washed, and resuspended in 200 μl Hank's balanced salt solution. The data were analyzed using flow cytometry (FCM, BD, USA) with excitation and emission at 510 and 580 nm, respectively.
2.10. ELISA of 8-hydroxy-desoxyguanosine (8-OHdG)
Serum was obtained from each group of mice and analyzed using a mouse 8-hydroxy-desoxyguanosine ELISA kit (CUSABIO, Wuhan, China) according to the manufacturer's instructions. In brief, 50 μl of serum was added to a microplate strip well and,50 μl of HRP-conjugate (1×) was added to each well; the plates were incubated for 30 min at 37 °C. Then, 90 μl of TMB Substrate solution was applied to each well and incubated for 20 min at 37 °C. After 50 μl of Stop Solution was added to each well, chemiluminescent absorbance was determined using a microplate reader (Bio-Rad, USA). The 8-OHdG level was quantified by relating the sample readings to the generated standard curve.
2.11. Detection and quantification of mitochondrial morphology
Mitochondrial morphology was measured using MitoTracker Red (Invitrogen, USA) according to the manufacturer's instructions. Briefly, the cells were incubated with MitoTracker Red at 0.1 μM in EBM for 20 min. Fluorescence was detected (490 nm excitation/525 nm emission) at 63× by confocal laser scanning microscopy (Leica, Germany), and the images were analyzed using ImageJ (Bethesda, MD, USA). Briefly, the images were contrast enhanced at 0.5% saturation levels, treated with a smoothing linear filter and convoluted with a 5 × 5 Laplacian kernel with nonzero diagonal terms to define the structures based on abrupt spatial changes in fluorescence. Following the convolution, the images were translated into the frequency domain by ImageJ's Fast Fourier Transform algorithm, and a bandpass filter was applied (to filter the structures below the optical resolution limit of 0.2 µm). After translation into the spatial domain, the images were segmented by thresholding, binarized and skeletonized according to the detected thresholds. The morphology of each mitochondrial object in the final image was described by scatter plots of the aspect ratio vs. form factor that were generated for each image.
2.12. Mitochondrial membrane potential measurement
The mitochondrial membrane potential was measured using JC-1 immunochemistry staining as previously described [37]. JC-1 is a lipophilic, cationic dye that can selectively enter mitochondria. In healthy cells with a normal mitochondrial membrane potential, JC-1 forms J-aggregates showing red fluorescence at the 585-nm/590-nm wavelengths. However, in cells with a collapsed mitochondrial membrane potential, JC-1 remains in the monomeric form and shows green fluorescence at the 514-nm/529-nm wavelengths. Increased green fluorescence levels and decreased red fluorescence levels indicate mitochondrial membrane potential collapse. Briefly, the cells were incubated with JC-1 at 10 μg/ml for 15 min at 37 °C and then observed by confocal laser scanning microscopy (Leica, Germany). All images were analyzed using ImageJ.
2.13. ATP measurement
ATP in the cell homogenates was measured using an ATP measurement kit (Thermo Fisher Scientific, USA) according to the manufacturer's instructions. Briefly, 1 × 106 cells grown in 6-well plates were washed with cold PBS, homogenized and sonicated in isolation buffer. The supernatants were retrieved by centrifugation at 12,000 × g for 5 min. ATP content was determined by measuring the luminescence of supernatants mixed with luciferase assay buffer using a microplate reader (Bio-Rad, USA). The ATP luminescence was normalized by the protein concentration.
2.14. Quantitative real-time PCR
Total RNA was extracted from the aorta tissue and cells using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA) according to the manufacturer's protocol and as described in our previous study [13]. cDNA synthesis was performed using random hexamer primers and a TaqMan reverse transcription kit (TaKaRa, Japan). Then, quantitative Real-Time PCR was performed using a SYBR® PremixEx Taq II Kit (TaKaRa, Japan) on a 7500 Fast Real Time PCR system from Applied Biosystems (Bio-Rad, USA). The relative gene expression levels were determined by the ΔΔCt method using GAPDH as a reference gene. All primer sequences are listed in Table 1.
Table 1.
Primers used for real-time RT-PCR analysis.
| Gene | Primers (5′→3′) | |
|---|---|---|
| Mouse primers | ||
| HSP22 Fwd | 5′-ATGAAGAGAAGCAGCAGGAAGGTG-3′ | |
| Rev | 5′-TGGTTGTCTTGAGGAAGCTCGTTG-3′ | |
| IL5 Fwd | 5′-GGCCACTGCCATGGAGATTCC-3′ | |
| Rev | 5′-AGCCTCATCGTCTCATTGCTTGTC-3′ | |
| IL6 Fwd | 5′-AGTTGCCTTCTTGGGACTG-3′ | |
| Rev | 5′-AGGTCTGTTGGGAGTGGTATC-3′ | |
| IL13 Fwd | 5'-CCTGGCTCTTGCTTGCCTT-3' | |
| Rev | 5'-GGTCTTGTGTGATGTTGCTCA-3' | |
| TGFβ1 Fwd | 5′-CACTGGAGTTGTACGGCAGT-3′ | |
| Rev | 5′-AGAGCAGTGAGCGCTGAATC-3′ | |
| MIP3αFwd | 5′-GTGGCAAGCGTCTGCTCTTCC-3′ | |
| Rev | 5′-AGAGGCGTCTGTATGTACGAGAGG-3′ | |
| GAPDH Fwd | 5′-AAGAAGGTGGTGAAGCAGGCATC-3′ | |
| Rev | 5′-CGGCATCGAAGGTGGAAGAGTG-3′ | |
| Human primers | ||
| HSP22 Fwd | 5′-GCCAGAGGAGTTGATGGTGA-3′ | |
| Rev | 5′-ACAATGCCACCTTCTTGCTG-3′ | |
| IL5 Fwd | 5′-TACGTGTATGCCATCCCCAC-3′ | |
| Rev | 5′-CCCCCTTGCACAGTTTGACT-3′ | |
| IL6 Fwd | 5'-ATGAACTCCTTCTCCACAAGC-3' | |
| Rev | 5'-GTTTTCTGCCAGTGCCTCTTTG-3' | |
| IL13 Fwd | 5′-TGGAGCATCAACCTGACAGC-3′ | |
| Rev | 5′-AGAATCCGCTCAGCATCCTC-3′ | |
| TGFβ1 Fwd | 5′-AAGTGGACATCAACGGGTTC-3′ | |
| Rev | 5′-CTCCGTGGAGCTGAAGCAATA-3′ | |
| MIP3αFwd | 5′-CGGCGAATCAGAAGCAAGC-3′ | |
| Rev | 5′-TTGGATTTGCGCACACAGAC-3′ | |
| ICAM1 Fwd | 5′-GTCACCTATGGCAACGACTCCTTC-3′ | |
| Rev | 5′-AGTGTCTCCTGGCTCTGGTTCC-3′ | |
| VCAM1 Fwd | 5′-GATGAGTGGTGGCCTCGTGAATG-3′ | |
| Rev | 5′-AATTCAATCTCCAGCCGGTCAAGG-3′ | |
| GAPDH Fwd | 5′-CCACTAGGCGCTCACTGTTC-3′ | |
| Rev | 5′-CCCCATACGACTGCAAAGAC-3′ | |
2.15. Western blot analysis
The proteins were extracted using RIPA lysis buffer (Cell Signaling Technology, Danvers, MA, USA) supplemented with a protease inhibitor (Sigma, USA). The total protein concentrations were measured using a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, USA). Equal amounts of total protein (50 μg) were separated by SDS-PAGE electrophoresis and electrotransferred to a PVDF membrane. The membranes were blocked in TBST buffer and then further incubated with antibodies. Primary antibodies for Western blot analysis (HSP22 (ab151552, rabbit monoclonal, at 1:200), GAPDH (ab8245, mouse monoclonal, at 1:2000), Drp1 (#8570, rabbit monoclonal, at 1:1000), and phospho-DRP1 (Ser637, #6319, rabbit monoclonal, at 1:500)) were acquired from Abcam (MA, USA) and Cell Signaling Technology (MA, USA). The antibody complexes were visualized with a chemiluminescent substrate (Thermo Scientific) using a ChemiDoc XRS device (Bio-Rad, American). For quantification, the electrochemiluminescence (ECL) signals were digitized using Quantity One software.
2.16. Statistical analyses
The data are presented as the means ± S.E.M. Two independent samples with normal distributions and homogeneity of variance were analyzed using t-tests. For comparisons among multiple groups, one-way ANOVA was used with Dunnett or Bonferroni tests as appropriate. P < 0.05 was considered statistically significant. The statistical analyses were performed using Statistical Product and Service Solutions 19.0 (SPSS, Systat Software, San Jose, CA, USA).
3. Results
3.1. Hyperglycemia induces HSP22 upregulation and endothelial injury
We subjected HFD mice to STZ treatment to induce T2DM using protocols established in previous animal models of diabetes [23], [29]. The body weights and 4–6 h FBG levels were monitored monthly. The diabetic mice exhibited significantly higher blood glucose levels and lower body weights than the nondiabetic control mice (Fig. 1A–B). Hyperglycemia has been shown to induce endothelial injury [5], [7], [9]. Therefore, the aortas from different groups of mice were evaluated by H&E staining to estimate the degree of diabetes-induced endothelial injury. Endothelial activation initiates endothelial injury [11], [40]. We further detected the levels of ICAM-1 and VCAM-1 proteins in the aortas from each group. Compared with the basic diet group and the HFD group, the diabetes group showed significant changes in aortic pathological morphology (Fig. 1C). The protein expression levels of ICAM-1 and VCAM-1 in the aortic endothelium from the diabetic mice were higher than those in the nondiabetic control mice by immunohistochemistry (Fig. 1D–E). Using a cytokine antibody array containing 18 cytokines [33], [34], we found that 5 serum cytokines—IL5, IL6, IL13, Mip3a and TGFβ1— were clearly elevated in diabetic group (Fig. 1F–G). To further determine diabetes-induced endothelial activation, we investigated whether these five cytokines changed in the aortic endothelium of the diabetic mice. Using RT-PCR, we confirmed that hyperglycemia increased the expression of IL5, IL6, IL13, Mip3α and TGFβ1 (Fig. 1H). HSP22 has been reported to be upregulated in the diabetic retina and heart [6], [13]. Therefore, we evaluated whether HSP22 is expressed in endothelial cells under hyperglycemic conditions. We examined the upregulation of HSP22 in the aortas from the diabetic mice using immunohistochemistry with an HSP22 antibody and RT-PCR (Fig. 1D, I–J).
Fig. 1.

Hyperglycemia induces HSP22 upregulation and endothelial injuryin vivo. Type 2 diabetes mellitus (T2DM) mice were induced with a high-fat diet (HFD; 60% of calories from fat) and injected with streptozotocin (STZ; 100 mg/kg) or buffer for 12 weeks (n = 8 per group). A) Blood glucose concentrations in control, HFD-control and diabetic mice were measured at baseline and 4, 8, and 12 weeks after diabetes induction. B) The body weights of the control, HFD-control and diabetic mice were measured at baseline and 4, 8, and 12 weeks after diabetes induction. C) Representative images of hematoxylin and eosin staining (H&E, original magnification ×40). D) Representative immunohistochemical staining images of ICAM-1, VCAM-1 and HSP22 in aortic sections (original magnification ×40). E) Quantification of positive staining for ICAM-1 and VCAM-1 in aortic rings. n = 5. F) Representative images of a gene heat map from a cytokine array. Serum was obtained from each group of mice for a murine cytokine array analysis. Unsupervised hierarchical clustering of genes was performed to generate heat maps showing the genes that were regulated by hyperglycemia (n = 3 per group). G) Quantification of the fluorescence intensities of cytokine levels in serum. H) mRNA expression of IL5, IL6, IL13, Mip3α and TGFβ1 in the aorta. I) Quantification of positive staining for HSP22 in aortic sections. n = 5. J) mRNA expression of HSP22 in the aorta. *P < 0.05 vs. Control; #P < 0.05 vs. HFD-control.
Cell viability was also measured in hyperglycemia-treated HUVECs by CCK8 and LDH. Compared with NG and OC, HG significantly decreased cell viability and increased cytotoxicity (Supplemental Fig. 1A–B). The monocyte adhesion assay showed that compared with the control treatment groups, the HG-treated HUVECs exhibited increased adhesion to untreated monocytes (Supplemental Fig. 1C). Using RT-PCR, we observed that the levels of ICAM-1 and VCAM-1 were increased in the HG-induced HUVECs (Supplemental Fig. 1D). Furthermore, endothelial cell activation-related cytokines were examined in HUVECs in response to HG (Supplemental Fig. 1E). Taken together, these results suggest that hyperglycemia induces endothelial activation and injury. Subsequently, we examined the expression level of HSP22 in HUVECs. Compared with the NG and OC treatment groups, a significant increase in HSP22 expression was found in the HG group by immunofluorescence, RT-PCR and Western blotting, but the vascular endothelial specific marker CD31 was significantly decreased (Supplemental Fig. 1F–J).
3.2. Hyperglycemia induces oxidative stress, mtROS production and mitochondrial impairment
Accumulating evidence has confirmed that diabetes-induced mtROS contributes to mitochondrial damage, resulting in endothelial dysfunction [9], [10], [11]. We evaluated the ROS levels in the aortas by DHE staining. Compared with the nondiabetic mice, the diabetic mice showed higher levels of ROS in the aortas (Fig. 2A–B). Accumulation of oxidative mitochondrial DNA (mtDNA) lesions is associated with mtROS and mitochondrial dysfunction. 8-OHdG has been used as a marker for common DNA modifications resulting from ROS. We then examined 8-OHdG levels in the aortas by ELISA, and the results showed that 8-OHdG accumulation in the diabetic mice was substantially greater than that in the other groups (Fig. 2C). We further examined the mitochondrial superoxide anion production in vitro by incubating HUVECs with the fluorophore MitoSOX Red, which selectively targets mitochondria. As expected, HG-induced increased mtROS was found by MitoSOX and flow cytometry (Supplemental Fig. 2A). The mitochondrial morphology and function were observed to determine the effects of HG on HUVECs. Using confocal microscopy to visualize MitoTracker Red and JC-1 staining, the mitochondrial fluorescence density and the membrane potential, we found that the mitochondrial fluorescence density and membrane potential in the HG group were significantly lower than those in the NG and OC groups (Supplemental Fig. 2B–E).
Fig. 2.

Hyperglycemia induces aortic oxidative stressin vivo. A) Frozen aortic sections were incubated with 5 μmol/l DHE for 20 min and analyzed using fluorescence microscopy (original magnification ×40). B) Quantification of the fluorescence intensity of ROS levels in the aorta. n = 4. C) Quantification of 8-hydroxy-desoxyguanosine (8-OHdG) levels in the aorta. n = 4. *P < 0.05 vs. Control; #P < 0.05 vs. HFD-control.
3.3. HSP22 alleviates hyperglycemia-induced endothelial injury
HSP22 has been reported to play roles in resistance to oxidative stress and cytoprotection [19], [20], [21], [22]. However, whether HSP22 plays a role in hyperglycemia-induced endothelial dysfunction remains unclear. We generated HSP22 TG mice to explore whether HSP22 is involved in diabetes-induced endothelial dysfunction. Compared with the WT-diabetic mice, HSP22 overexpression had no effect on the FBG level in the TG-diabetic mice, and their body weights were not significantly lower than those of the WT-diabetic mice (Fig. 3A–B). The aortic pathological morphology in the TG-diabetic mice was found to be significantly improved compared with that in the WT-diabetic mice (Fig. 3C). We then detected the expression of HSP22 in the aorta by immunohistochemistry and RT-PCR (Fig. 3D–F). The expression levels of the ICAM-1 and VCAM-1 proteins in the aortas of the TG mice were found to be lower than those in the WT mice (Fig. 3C, G). Similarly, the levels of cytokines were decreased in the aortas from the TG mice compared with those in the WT mice (Fig. 3H).
Fig. 3.

HSP22 reduces diabetes-induced endothelial injuryin vivo. Diabetes was induced in wild-type (WT) and transgenic (TG) mice by HFD adjuvanted with STZ injection for 12 weeks (n = 8 per group). A) Blood glucose concentrations in the WT-control, TG-control, WT-diabetic and TG-diabetic mice were measured at baseline and 4, 8, and 12 weeks after diabetes induction. B) The body weights of the WT-control, TG-control, WT-diabetes and TG-diabetes mice were measured at baseline and 4, 8, and 12 weeks after diabetes induction. C) Representative images of H&E staining (original magnification ×40). D) Representative immunohistochemical staining images of HSP22, ICAM-1 and VCAM-1 in aortic sections (original magnification ×40). E) Quantification of positive staining for HSP22 in aortic sections. n = 5. F) mRNA expression of HSP22 in the aorta. G) Quantification of positive staining for ICAM-1 and VCAM-1 in aortic rings. n = 5. H) mRNA expression of IL5, IL6, IL13, Mip3α and TGFβ1 in the aorta. *P < 0.05 vs. WT-control; #P < 0.05 vs. WT-diabetes;&P < 0.05 vs. TG-control.
Next, we examined the effects of the overexpression of HSP22 on HUVECs. Compared with the negative control group, a clear increase in cell viability and a decrease in cytotoxicity were observed in the HG-treated endothelial cells after overexpression of HSP22 (Supplemental Fig. 3A–C). Compared with the HG group with an NC form of HSP22, the levels of ICAM-1 and VCAM-1 and the monocyte adhesion of endothelial cells were decreased in the HSP22-higher expression group (Supplemental Fig. 3D–E). Furthermore, HG-induced increases in endothelial cell activation-related cytokines were reduced by overexpression of HSP22 (Supplemental Fig. 3F). Taken together, these results suggest that HSP22 reduces endothelial dysfunction.
3.4. HSP22 inhibits hyperglycemia-induced oxidative stress, mtROS production and mitochondrial impairment
Subsequently, we sought to determine whether HSP22 is related to endothelial mitochondrial oxidative stress. Our results showed that the HSP22 TG aortas had lower levels of ROS than the WT aortas (Fig. 4A–B). The 8-OHdG levels in the WT-diabetic group were obviously higher than those in the HSP22 TG group (Fig. 4C).
Fig. 4.

HSP22 inhibits diabetes-induced endothelial oxidative stressin vivo. A) Frozen aortic sections were incubated with 5 μmol/L DHE for 20 min and analyzed using fluorescence microscopy (original magnification ×40). B) Quantification of the fluorescence intensity of ROS levels in the aorta. n = 4. C) Quantification of 8-OHdG levels in the aorta. n = 4. *P < 0.05 vs. WT-control; #P < 0.05 vs. WT-diabetes.
We also demonstrated that high HSP22 expression reduced mtROS production and improved mitochondrial function by using flow cytometry and confocal microscopy (Supplemental Fig. 4A–E). These data suggest that HSP22 inhibits hyperglycemia-induced mtROS and mitochondrial impairment in endothelial cells.
3.5. HSP22 deficiency aggravates hyperglycemia-induced endothelial injury
We further elucidated the role of HSP22 in hyperglycemia-induced endothelial injury using HSP22 KO mice and an HSP22-silenced endothelial cell line. There was no difference between the KO-diabetic mice and the WT mice in the FBG level or body weight (Fig. 5A–B). HSP22 expression in the aorta was tested by immunohistochemistry and RT-PCR (Fig. 5D–F). Endothelial morphology and activation were detected by H&E staining and immunohistochemistry, and the results suggested that HSP22 deficiency changes the endothelial morphology and upregulates the expression of ICAM-1 and VCAM-1 (Fig. 5C–D, G). The levels of cytokines in the aortas from the KO mice were significantly increased compared with those in the WT mice (Fig. 5H).
Fig. 5.

HSP22 deficiency exacerbates diabetes-induced endothelial injuryin vivo. Diabetes was induced in wild-type (WT) and knockout (KO) mice by HFD adjuvanted with STZ injection for 12 weeks (n = 8 per group). A) Blood glucose concentrations in the WT-control, KO-control, WT-diabetes and KO-diabetes mice were measured at baseline and 4, 8, and 12 weeks after diabetes induction. B) The body weights of the WT-control, KO-control, WT-diabetes and KO-diabetes mice were measured at baseline and 4, 8, and 12 weeks after diabetes induction. C) Representative images of H&E (original magnification ×40). D) Representative immunohistochemical staining images of HSP22, ICAM-1 and VCAM-1 in aortic sections (original magnification ×40). E) Quantification of positive staining for HSP22 in aortic sections. n = 5. F) mRNA expression of HSP22 in the aorta. G) Quantification of positive staining for ICAM-1 and VCAM-1 in aortic sections. n = 5. H) mRNA expression of IL5, IL6, IL13, Mip3α and TGFβ1 in the aorta. *P < 0.05 vs. WT-control; #P < 0.05 vs. WT-diabetes; &P < 0.05 vs KO-control.
The effects of HSP22 deficiency on HUVECs were the same as those observed in the aortas. Compared with the NC group, a decreased cell viability and higher cytotoxicity were observed following the silencing of HSP22 (Supplemental Fig. 5A–C). Monocyte adhesion of HG-treated endothelial cells was significantly increased following the silencing of HSP22 compared with that in the NC group (Supplemental Fig. 5D). Similar trends were observed in the expression of ICAM-1, VCAM-1 and cytokines in the HSP22-deficient group (Supplemental Fig. 5E–F).
3.6. HSP22 deficiency exacerbates oxidative stress, endothelial mtROS production and mitochondrial impairment under hyperglycemic conditions
HSP22 deficiency increases oxidative stress, endothelial mtROS production and mitochondrial impairment under hyperglycemic conditions. HSP22 deficiency clearly increased mitochondrial oxidative stress in vivo and in vitro, which was opposite to the effects of HSP22 overexpression. Compared with the WT group, HSP22 deficiency significantly promoted ROS production and 8-OHdG accumulation in the aortas of the diabetic mice (Fig. 6A–C).
Fig. 6.

HSP22 deficiency increases diabetes-induced endothelial oxidative stressin vivo. A)Frozen aortic sections were incubated with 5 μmol/l DHE for 20 min and analyzed using fluorescence microscopy (original magnification ×40). B) Quantification of the fluorescence intensity of ROS levels in the aorta. n = 4. C) Quantification of 8-OHdG levels in the aorta. n = 4. *P < 0.05 vs. WT-control; #P < 0.05 vs. WT-diabetes.
Silencing HSP22 consistently increased mtROS in the HG-treated HUVECs (Supplemental Fig. 6A). Next, we performed confocal microscopy with MitoTracker and JC-1 staining to further examine the mitochondrial density and membrane potential. As expected, the mitochondrial immunofluorescence intensity and membrane potential were largely decreased in the HSP22 siRNA group compared with in the NC group (Supplemental Fig. 6B–E).
3.7. HSP22 deficiency exacerbates oxidative stress, endothelial mtROS production and mitochondrial impairment under hyperglycemic conditions
A compelling body of evidence has demonstrated that hyperglycemia-mediated mtROS is critical for endothelial dysfunction [9], [11]. However, whether HSP22 attenuates endothelial dysfunction by inhibiting mtROS remains unclear. To gain insight into the mechanism by which HSP22 prevents endothelial dysfunction, we assessed the antioxidative stress effect on HUVECs using MitoTEMPO, which is a mitochondrial-targeted potent antioxidant, and by silencing HSP22. The effects of MitoTEMPO observed on HUVECs were consistent with those observed in a previous report [38]. Endothelial cells treated with MitoTEMPO were rescued under high-glucose conditions; however, HSP22 deficiency impeded this process. As a result, compared with the HG group, increased cell viability and decreased cytotoxicity were observed following MitoTEMPO treatment, but silencing HSP22 reduced this trend (Fig. 7A–B). The same trend was observed for monocyte adhesion and the levels of adhesion molecules and cytokines (Fig. 7C–E).
Fig. 7.

HSP22 deficiency offsets MitoTEMPO-inhibited endothelial injury under hyperglycemic conditionsin vitro. HUVECs were transfected with negative control siRNA (NC siRNA) or HSP22 siRNA for 48 h and then treated with high glucose with/without MitoTEMPO (1 μM) for 24 h. A) Cells in 96-well plates were stained with Cell Counting Kit-8 (CCK8) to evaluate cell viability. n = 3. B) Cytotoxicity was measured by LDH release in the cell media. n = 3. C) Adhesion of nonstimulated PBMCs to stimulated HUVECs was quantified. n = 3. D) mRNA expression of ICAM-1 and VCAM-1 was determined by RT-PCR. n = 3. E) Endothelial cell activation-related cytokines were examined by RT-PCR. n = 3. *P < 0.05 vs. NC-NG; #P < 0.05 vs. HG-MitoTEMPO.
The same trend was also observed for mitochondrial oxidative stress in endothelial cells. Compared with the NC form in the HSP22 group, there was an apparent increase in the level of mtROS following the silencing of HSP22 in the MitoTEMPO treatment group (Fig. 8A). Following MitoTEMPO treatment, we also observed an injured mitochondrial morphology and a decline in the membrane potential in the HSP22-deficient group (Fig. 8B–E). Mitochondrial injury disrupts respiratory enzyme activity and energy production in diabetic mice; therefore, we evaluated mitochondrial function by measuring ATP levels [41], [42]. The results showed that MitoTEMPO treatment recovered the ATP levels following HG-mediated mitochondrial injury, but the silencing of HSP22 reduced this trend (Supplemental Fig. 7A). Imbalances in mitochondrial dynamics contribute to oxidative stress and hyperglycemia-induced mitochondrial injury, resulting in endothelial dysfunction [9], [41], [42], [43]. Finally, we investigated whether there were changes in mitochondrial fusion and fission. Western blotting results showed that the HG-induced increases in DRP1 and p-DRP1 protein expression were downregulated by MitoTEMPO treatment in the HUVECs, but these changes were blocked after further silencing HSP22 (Supplemental Fig. 7B–C). Overall, these results suggest that HSP22 rescues endothelial dysfunction by inhibiting hyperglycemia-mediated mtROS.
Fig. 8.

HSP22 deficiency offsets MitoTEMPO-inhibited hyperglycemia-mediated mtROSin vitro. A) Mitochondrial superoxide was measured by flow cytometry. HUVECs were incubated in Hank's balanced salt solution containing 5 μmol/L MitoSOX for 15 min. n = 3. B) Representative images of MitoTracker Red staining showing the mitochondrial morphology in each group of cells (original magnification ×63). C) Mitochondrial density was quantified. n = 3. D) Representative images of JC-1 immunochemistry staining showing the mitochondrial membrane potential in each group of cells (original magnification ×63). E) Mitochondrial membrane potential was quantitated as a percentage in the gate. n = 3. *P < 0.05 vs. NC-NG; #P < 0.05 vs. HG-MitoTEMPO.
4. Discussion
In the present study, we found that HSP22 inhibits hyperglycemia-induced endothelial cell activation and injury by suppressing cell adhesion and cytokines. These experiments provide evidence that HSP22 restrains hyperglycemia-induced mtROS and mitochondrial dysfunction in endothelial cells. Therefore, HSP22 reduces hyperglycemia-induced endothelial injury by mediating mtROS.
This study focused on the expression of HSP22, and we found that it was upregulated in diabetic-stimulated endothelial cells, which is consistent with the findings reported in former studies showing that HSP22 is increased in response to stress [6], [13]. The upregulation of HSP22 protects against hyperglycemia-induced endothelial injury. This protective effect is similar to other reported sHSPs, including HSP27, HSP20 and αB-crystallin [6], [13], [14], [15], [16], [17], [18]. Previous reports have suggested that HSP22, which is increased in response to oxidative stress, is mainly regulated by heat shock factor (HSF) signaling; however, HSP22 was prevented from further increasing due to a negative feedback loop between HSF and HSP22 [44]. That is, HSP22 may not constantly increase and fully exert its effects under stress. In our study, HSP22 overexpression increased resistance to oxidative damage in diabetic mice and hyperglycemia-induced endothelial cell injury, which was consistent with the results reported by Sui et al. [45]. Three reasons may explain these results. First, HSP22 is a molecular chaperone that functions in helping to clear aggregated proteins [6]. Next, HSP22 has been reported to protect against mitochondrial oxidative stress by mediating the activity of transcription factors, such as NF-κB and STATE [19], [46]. Moreover, HSP22 interacts with other sHSPs, such as HSP27 and B-crystallin, to promote cell survival and decrease apoptosis [6], [47], [48]. And all above effects need HSP22 overexpression to induce. Multiple studies have suggested that the heat shock response is an intrinsic defense mechanism that protects cells against various stress stimuli and pathological conditions [49], [50]. Our results and other studies offer strong evidence that sHSPs are involved in resistance to diabetes-induced cell injury.
Endothelial activation is considered an initial event responsible for endothelial injury [11], [40]. The increased adhesion and secretion of endothelial cell adhesion molecules, such as ICAM-1 and VCAM-1, and the elevated cytokines are additional related and specific features of endothelial activation [51]. In our study, overexpression of HSP22 reduced endothelial cell adhesion and downregulated adhesion molecules and inflammation-related cytokines, leading to decreased endothelial cell injury. When HSP22 was silenced, the opposite results were observed. We provide compelling evidence that HSP22 suppresses diabetic-induced endothelial activation by decreasing endothelial adhesion and cytokine secretion, thus reducing endothelial dysfunction. Our findings are consistent with those reported by Li et al. and Shao et al., who found that the inhibition of hyperglycemia-induced inflammation contributed to decreased endothelial activation, which is a key initial stage of endothelial dysfunction [11], [52]. Our study is the first to provide evidence supporting the hypothesis that HSP22 is a key factor that suppresses hyperglycemia-induced endothelial activation and injury.
In our study, HSP22 reduced oxidative stress and mtROS production in vivo and in vitro, leading to attenuated mitochondrial damage in endothelial cells. These results are consistent with those of previous studies reporting that HSP22 protects cells from damage by conferring resistance to oxidative stress and modulating mtROS production. Accumulating evidence has indicated that excessive mtROS disrupts mitochondrial function and contributes to cell damage in diabetes [9], [10]. In addition, the mechanism of excessive mtROS-mediated endothelial activation has been supported by previous findings concerning hyperglycemia [53]. Taken together, these results indicate that HSP22 may attenuate hyperglycemia-induced endothelial activation by restraining the release of mtROS.
Consistent with other reports using MitoTEMPO, which is an mtROS scavenger, showing that the inhibition of mtROS in the hyperglycemia-stimulated endothelium rescues cell injury, we demonstrated that eliminating mtROS lowered endothelial cell adhesion and the release of inflammatory cytokines [5], [38]. However, after silencing the expression of HSP22, the protective effect of MitoTEMPO was decreased, leading to a change in mitochondrial morphology, decreased membrane potential, and increased adhesion and endothelial activation-related cytokine release. These results further indicate that HSP22, which is an upstream event, attenuates hyperglycemia-induced endothelial activation by regulating the levels of mtROS, which helps reduce damage to endothelial cells. Mitochondrial homeostasis is crucial for the maintenance of mitochondrial morphology and function [9], [43], [54]. Previous studies have reported that hyperglycemia disrupts the balance between mitochondrial fusion and fission by upregulating DRP1, which is a regulator of mitochondrial fission events, leading to excessive mtROS production and mitochondrial dysfunction [9], [43], [54]. In our study, we found that the hyperglycemia-stimulated upregulation of DRP1 and pDRP1 expression further increased when HSP22 was silenced, but MitoTEMPO reversed the situation. In addition, MitoTEMPO increased ATP levels, which were reduced by HSP22 deficiency. Briefly, HSP22 maintained mitochondrial balance and function by suppressing mtROS in hyperglycemia-stimulated endothelial cells. Based on these results, we propose a new mechanism by which HSP22 protects the endothelium, which may provide a new treatment strategy for T2DM vascular complications.
In summary, our findings offer new insight into the protection conferred by HSP22 in diabetic endothelial injury, indicating that HSP22 might serve as a biomarker of mitochondrial oxidative stress and a potential therapeutic target. We provide evidence that overexpression of HSP22 rescues mitochondrial dysfunction by reducing the expression of mtROS and endothelial inflammation under diabetic conditions. The elimination of mtROS and inflammation restrains diabetes-stimulated endothelial cell activation by decreasing endothelial cell adhesion and cytokine release, leading to decreased endothelial injury. Although considerable knowledge regarding vascular endothelial cell protection has accumulated in recent years, insight into the protection mechanisms is inadequate and requires further investigation.
Author contributions
L.Y. designed the study, researched and analyzed the data, and wrote the manuscript. Q.L and W.Z. conceived and designed the study, analyzed the data, and drafted the manuscript. L.Y., M.W., Q.L. and M.B. performed research and analyzed and interpreted the data. H.B. performed research and reviewed and revised the manuscript. H.B. and X.C. designed the study and critically revised the manuscript. X.C. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81560051, 81260023, 81460045 and 1460010), the Science and Technology Planning Project of Jiangxi Province (No. 2016YNQN12034), a China Postdoctoral Science Foundation funded project (2017M622107) and the Postdoctoral Science Foundation of Jiangxi Province (No. 2016KY51).
Competing interest
The authors declare that there is no competing interest associated with the manuscript.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.101095.
Contributor Information
Huihui Bao, Email: huihui_bao77@126.com.
Xiaoshu Cheng, Email: xiaoshumenfan@126.com.
Appendix A. Supplementary material
Fig. S1.

Hyperglycemia induces HSP22 upregulation and endothelial activationin vitro. Human umbilical vein endothelial cells (HUVECs) were cultured in endothelial basal medium (EBM) containing normal glucose (NG, 5 mM D-Glucose), osmotic control (OC, 30 mM D-mannitol), or high glucose (HG, 30 mM D-Glucose) for 24 h. A) Cell viability was evaluated in cells in 96-well plates by a Cell Counting Kit-8 (CCK8) after treatment with NG, OC or HG for 24 h. n = 3. B) HUVECs were cultured in EBM containing NG, OC or HG for 24 h, and the cell media were collected for the measurement of lactate dehydrogenase (LDH) release as an indicator of cytotoxicity. n = 3. C) HUVECs were cultured in EBM containing NG, OC or HG for 24 h, and the adhesion of nonstimulated primary human peripheral mononuclear cells (PBMCs) to stimulated HUVECs was quantified. n = 3. D) mRNA expression of ICAM-1 and VCAM-1 was determined by RT-PCR. n = 3. E) Endothelial cell activation-related cytokines were examined by RT-PCR. n = 3. F) Representative immunofluorescence staining images of HSP22 (green), the endothelial cell marker CD31 (red), and nuclei (blue) (original magnification ×63). G) Quantification of the fluorescence intensity of HSP22 and CD31 in HUVECs. n = 3. H) mRNA expression of HSP22 was determined by RT-PCR. n = 3. I) HSP22 protein expression was determined by Western blotting. J) Quantification of HSP22 protein expression. GAPDH was used as a loading control. n = 3. *P < 0.05 vs. NG; #P < 0.05 vs. OC.
Fig. S2.

Hyperglycemia induces mtROS production and mitochondrial impairment in endothelial cellsin vitro. A) Mitochondrial superoxide was measured by flow cytometry. HUVECs were incubated in Hanks' balanced salt solution containing 5 μmol/l MitoSOX for 15 min. n = 3. B) Representative images of MitoTracker Red staining showing the mitochondrial morphology in each group of cells (original magnification ×63). C) Mitochondrial density was quantified. n = 3. D) Representative images of JC-1 immunochemistry staining showing the mitochondrial membrane potential in each group of cells (original magnification ×63). E) Mitochondrial membrane potential was quantitated as a percentage in the gate. n = 3. *P < 0.05 vs. NG; #P < 0.05 vs. OC.
Fig. S3.

HSP22 reduces hyperglycemia-induced endothelial injuryin vitro. HUVECs were transfected with plasmids containing HSP22 or a dominant-negative form for 48 h and then stimulated with high glucose for 24 h. A) HSP22 protein expression was determined by Western blotting. GAPDH was used as a loading control. n = 3. B) Cells in 96-well plates were stained with a Cell Counting Kit-8 (CCK8) to evaluate cell viability. n = 3. C) Cytotoxicity was measured by LDH release in the cell media. n = 3. D) Adhesion of nonstimulated primary human peripheral mononuclear cells (PBMCs) to stimulated HUVECs was quantified. n = 3. E) mRNA expression of ICAM-1 and VCAM-1 was determined by RT-PCR. n = 3. F) Endothelial cell activation-related cytokines were examined by RT-PCR. n = 3. *P < 0.05 vs. NC-NG; #P < 0.05 vs. NC-HG.
Fig. S4.

HSP22 inhibits hyperglycemia-induced mtROS production and mitochondrial impairment in endothelial cellsin vitro. A) Mitochondrial superoxide was measured by flow cytometry. HUVECs were incubated in Hanks' balanced salt solution containing 5 μmol/l MitoSOX for 15 min. n = 3. B) Representative images of MitoTracker Red staining showing the mitochondrial morphology in each group of cells (original magnification ×63). C) Mitochondrial density was quantified. n = 3. D) Representative images of JC-1 immunochemistry staining showing the mitochondrial membrane potential in each group of cells (original magnification ×63). E) Mitochondrial membrane potential was quantitated as a percentage in the gate. n = 3. *P < 0.05 vs. NC-NG; #P < 0.05 vs. NC-HG.
Fig. S5.

HSP22 deficiency exacerbates hyperglycemia-induced endothelial injuryin vitro. HUVECs were transfected with negative control siRNA (NC siRNA) or HSP22 siRNA for 48 h and then cultured with high-glucose medium for 24 h. A) HSP22 protein expression was determined by Western blotting. GAPDH was used as a loading control. n = 3. B) Cells in 96-well plates were stained with a Cell Counting Kit-8 (CCK8) to evaluate cell viability. n = 3. C) Cytotoxicity was measured by LDH release in the cell media. n = 3. D) Adhesion of nonstimulated PBMCs to stimulated HUVECs was quantified. n = 3. E) mRNA expression of ICAM-1 and VCAM-1 was determined by RT-PCR. n = 3. F) Endothelial cell activation-related cytokines were examined by RT-PCR. n = 3. *P < 0.05 vs. NC-NG; #P < 0.05 vs. NC-HG.
Fig. S6.

HSP22 deficiency increases endothelial mtROS production and mitochondrial impairment under hyperglycemic conditionsin vitro. A) Mitochondrial superoxide was measured by flow cytometry. HUVECs were incubated in Hank's balanced salt solution containing 5 μmol/l MitoSOX for 15 min. n = 3. B) Representative images of MitoTracker Red staining showing the mitochondrial morphology in each group of cells (original magnification ×63). C) Mitochondrial density was quantified. n = 3. D) Representative images of JC-1 immunochemistry staining showing the mitochondrial membrane potential in each group of cells (original magnification ×63). E) The mitochondrial membrane potential was quantitated as a percentage of the gate. n = 3. *P < 0.05 vs. NC-NG; #P < 0.05 vs. NC-HG.
Fig. S7.

HSP22 deficiency offsets MitoTEMPO-inhibited hyperglycemia-induced mitochondrial fission in endothelium. ATP was measured in cell homogenates. n = 3. B) Representative immunoreactive bands of DRP1, Ser637 phosphorylated DRP1 (p-DPR1) and GAPDH. C) Quantification of the densitometry of the immunoreactive bands of DRP1/p-DPR1. GAPDH was used as a loading control. n = 3. *P < 0.05 vs. NC-NG; #P < 0.05 vs. HG-MitoTEMPO.
References
- 1.Viswambharan H., Yuldasheva N.Y., Sengupta A., Imrie H., Gage M.C., Haywood N., Walker A.M., Skromna A., Makova N., Galloway S., Shah P., Sukumar P., Porter K.E., Grant P.J., Shah A.M., Santos C.X., Li J., Beech D.J., Wheatcroft S.B., Cubbon R.M., Kearney M.T. Selective enhancement of insulin sensitivity in the endothelium in vivo reveals a novel proatherosclerotic signaling loop. Circ. Res. 2017;120:784–798. doi: 10.1161/CIRCRESAHA.116.309678. [DOI] [PubMed] [Google Scholar]
- 2.Kubota T., Kubota N., Kadowaki T. Imbalanced insulin actions in obesity and type 2 diabetes: key mouse models of insulin signaling pathway. Cell Metab. 2017;25:797–810. doi: 10.1016/j.cmet.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Balkau B., Hu G., Qiao Q., Tuomilehto J., Borch-Johnsen K., Pyorala K. Prediction of the risk of cardiovascular mortality using a score that includes glucose as a risk factor. The DECODE study. Diabetologia. 2004;47:2118–2128. doi: 10.1007/s00125-004-1574-5. [DOI] [PubMed] [Google Scholar]
- 4.Mazzone T., Chait A., Plutzky J. Cardiovascular disease risk in type 2 diabetes mellitus: insights from mechanistic studies. Lancet. 2008;371:1800–1809. doi: 10.1016/S0140-6736(08)60768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qi H., Casalena G., Shi S., Yu L., Ebefors K., Sun Y., Zhang W., D'Agati V., Schlondorff D., Haraldsson B., Bottinger E., Daehn I. Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic kidney disease susceptibility. Diabetes. 2017;66:763–778. doi: 10.2337/db16-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reddy V.S., Raghu G., Reddy S.S., Pasupulati A.K., Suryanarayana P., Reddy G.B. Response of small heat shock proteins in diabetic rat retina. Invest. Ophthalmol. Vis. Sci. 2013;54:7674–7682. doi: 10.1167/iovs.13-12715. [DOI] [PubMed] [Google Scholar]
- 7.Liu J., Wu J., Sun A., Sun Y., Yu X., Liu N., Dong S., Yang F., Zhang L., Zhong X., Xu C., Lu F., Zhang W. Hydrogen sulfide decreases high glucose/palmitate-induced autophagy in endothelial cells by the Nrf2-ROS-AMPK signaling pathway. Cell Biosci. 2016;6:33. doi: 10.1186/s13578-016-0099-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lv Q., Xue Y., Li G., Zou L., Zhang X., Ying M., Wang S., Guo L., Gao Y., Li G., Xu H., Liu S., Xie J., Liang S. Beneficial effects of evodiamine on P2X(4)-mediated inflammatory injury of human umbilical vein endothelial cells due to high glucose. Int. Immunopharmacol. 2015;28:1044–1049. doi: 10.1016/j.intimp.2015.08.020. [DOI] [PubMed] [Google Scholar]
- 9.Wang Q., Zhang M., Torres G., Wu S., Ouyang C., Xie Z., Zou M.H. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. 2017;66:193–205. doi: 10.2337/db16-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang H., Hu N., Zhao Q., Wang B., Zhou H., Fu Q., Shen L., Chen X., Shen F., Lyu J. mtDNA haplogroup N9a increases the risk of T2DM by altering mitochondrial function and intracellular mitochondrial signals. Diabetes. 2018 doi: 10.2337/db17-0974. [DOI] [PubMed] [Google Scholar]
- 11.Shao Y., Cheng Z., Li X., Chernaya V., Wang H., Yang X.F. Immunosuppressive/anti-inflammatory cytokines directly and indirectly inhibit endothelial dysfunction – a novel mechanism for maintaining vascular function. J. Hematol. Oncol. 2014;7:80. doi: 10.1186/s13045-014-0080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X., Fang P., Yang W.Y., Chan K., Lavallee M., Xu K., Gao T., Wang H., Yang X. Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells. Can. J. Physiol. Pharmacol. 2017;95:247–252. doi: 10.1139/cjpp-2016-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X., Gu H., Huang W., Peng J., Li Y., Yang L., Qin D., Essandoh K., Wang Y., Peng T., Fan G.C. Hsp20-mediated activation of exosome biogenesis in cardiomyocytes improves cardiac function and angiogenesis in diabetic mice. Diabetes. 2016;65:3111–3128. doi: 10.2337/db15-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu B., Bhat M., Nagaraj R.H. AlphaB-crystallin inhibits glucose-induced apoptosis in vascular endothelial cells. Biochem. Biophys. Res. Commun. 2004;321:254–258. doi: 10.1016/j.bbrc.2004.06.151. [DOI] [PubMed] [Google Scholar]
- 15.Chen W., Lu Q., Lu L., Guan H. Increased levels of alphaB-crystallin in vitreous fluid of patients with proliferative diabetic retinopathy and correlation with vascular endothelial growth factor. Clin. Exp. Ophthalmol. 2017;45:379–384. doi: 10.1111/ceo.12891. [DOI] [PubMed] [Google Scholar]
- 16.Schalkwijk C.G., van Bezu J., van der Schors R.C., Uchida K., Stehouwer C.D., van Hinsbergh V.W. Heat-shock protein 27 is a major methylglyoxal-modified protein in endothelial cells. Febs Lett. 2006;580:1565–1570. doi: 10.1016/j.febslet.2006.01.086. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez-Nino M.D., Sanz A.B., Sanchez-Lopez E., Ruiz-Ortega M., Benito-Martin A., Saleem M.A., Mathieson P.W., Mezzano S., Egido J., Ortiz A. HSP27/HSPB1 as an adaptive podocyte antiapoptotic protein activated by high glucose and angiotensin II. Lab. Invest. 2012;92:32–45. doi: 10.1038/labinvest.2011.138. [DOI] [PubMed] [Google Scholar]
- 18.Dai T., Patel-Chamberlin M., Natarajan R., Todorov I., Ma J., LaPage J., Phillips L., Nast C.C., Becerra D., Chuang P., Tong L., de Belleroche J., Wells D.J., Wang Y., Adler S.G. Heat shock protein 27 overexpression mitigates cytokine-induced islet apoptosis and streptozotocin-induced diabetes. Endocrinology. 2009;150:3031–3039. doi: 10.1210/en.2008-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiu H., Lizano P., Laure L., Sui X., Rashed E., Park J.Y., Hong C., Gao S., Holle E., Morin D., Dhar S.K., Wagner T., Berdeaux A., Tian B., Vatner S.F., Depre C. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation. 2011;124:406–415. doi: 10.1161/CIRCULATIONAHA.110.013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laure L., Long R., Lizano P., Zini R., Berdeaux A., Depre C., Morin D. Cardiac H11 kinase/Hsp22 stimulates oxidative phosphorylation and modulates mitochondrial reactive oxygen species production: involvement of a nitric oxide-dependent mechanism. Free Radic. Biol. Med. 2012;52:2168–2176. doi: 10.1016/j.freeradbiomed.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 21.Jo H.S., Kim D.W., Shin M.J., Cho S.B., Park J.H., Lee C.H., Yeo E.J., Choi Y.J., Yeo H.J., Sohn E.J., Son O., Cho S.W., Kim D.S., Yu Y.H., Lee K.W., Park J., Eum W.S., Choi S.Y. Tat-HSP22 inhibits oxidative stress-induced hippocampal neuronal cell death by regulation of the mitochondrial pathway. Mol. Brain. 2017;10:1. doi: 10.1186/s13041-016-0281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L., Zajac A., Hedhli N., Depre C. Increased expression of H11 kinase stimulates glycogen synthesis in the heart. Mol. Cell. Biochem. 2004;265:71–78. doi: 10.1023/b:mcbi.0000044311.58653.54. [DOI] [PubMed] [Google Scholar]
- 23.Rendina-Ruedy E., Hembree K.D., Sasaki A., Davis M.R., Lightfoot S.A., Clarke S.L., Lucas E.A., Smith B.J. A comparative study of the metabolic and skeletal response of C57BL/6J and C57BL/6N mice in a diet-induced model of type 2 diabetes. J. Nutr. Metab. 2015;2015:758080. doi: 10.1155/2015/758080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulze C., Bangert A., Kottra G., Geillinger K.E., Schwanck B., Vollert H., Blaschek W., Daniel H. Inhibition of the intestinal sodium-coupled glucose transporter 1 (SGLT1) by extracts and polyphenols from apple reduces postprandial blood glucose levels in mice and humans. Mol. Nutr. Food Res. 2014;58:1795–1808. doi: 10.1002/mnfr.201400016. [DOI] [PubMed] [Google Scholar]
- 25.Lizano P., Rashed E., Kang H., Dai H., Sui X., Yan L., Qiu H., Depre C. The valosin-containing protein promotes cardiac survival through the inducible isoform of nitric oxide synthase. Cardiovasc Res. 2013;99:685–693. doi: 10.1093/cvr/cvt136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Depre C., Hase M., Gaussin V., Zajac A., Wang L., Hittinger L., Ghaleh B., Yu X., Kudej R.K., Wagner T., Sadoshima J., Vatner S.F. H11 kinase is a novel mediator of myocardial hypertrophy in vivo. Circ. Res. 2002;91:1007–1014. doi: 10.1161/01.res.0000044380.54893.4b. [DOI] [PubMed] [Google Scholar]
- 27.Depre C., Wang L., Sui X., Qiu H., Hong C., Hedhli N., Ginion A., Shah A., Pelat M., Bertrand L., Wagner T., Gaussin V., Vatner S.F. H11 kinase prevents myocardial infarction by preemptive preconditioning of the heart. Circ. Res. 2006;98:280–288. doi: 10.1161/01.RES.0000201284.45482.e8. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto N., Tokuda H., Kuroyanagi G., Kainuma S., Matsushima-Nishiwaki R., Fujita K., Kozawa O., Otsuka T. Heat shock protein 22 (HSPB8) limits TGF-beta-stimulated migration of osteoblasts. Mol. Cell. Endocrinol. 2016;436:1–9. doi: 10.1016/j.mce.2016.07.011. [DOI] [PubMed] [Google Scholar]
- 29.Li H., Li Y., Xiang L., Zhang J., Zhu B., Xiang L., Dong J., Liu M., Xiang G. GDF11 attenuates development of type 2 diabetes via Improvement of Islet beta-cell function and survival. Diabetes. 2017;66:1914–1927. doi: 10.2337/db17-0086. [DOI] [PubMed] [Google Scholar]
- 30.Lee J.Y., Jeong E.A., Kim K.E., Yi C.O., Jin Z., Lee J.E., Lee D.H., Kim H.J., Kang S.S., Cho G.J., Choi W.S., Choi S.Y., Kwon H.M., Roh G.S. TonEBP/NFAT5 haploinsufficiency attenuates hippocampal inflammation in high-fat diet/streptozotocin-induced diabetic mice. Sci. Rep. 2017;7:7837. doi: 10.1038/s41598-017-08319-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kusakabe T., Tanioka H., Ebihara K., Hirata M., Miyamoto L., Miyanaga F., Hige H., Aotani D., Fujisawa T., Masuzaki H., Hosoda K., Nakao K. Beneficial effects of leptin on glycaemic and lipid control in a mouse model of type 2 diabetes with increased adiposity induced by streptozotocin and a high-fat diet. Diabetologia. 2009;52:675–683. doi: 10.1007/s00125-009-1258-2. [DOI] [PubMed] [Google Scholar]
- 32.Wang S., Xu J., Song P., Viollet B., Zou M.H. In vivo activation of amp-activated protein kinase attenuates diabetes-enhanced degradation of GTP cyclohydrolase I. Diabetes. 2009;58:1893–1901. doi: 10.2337/db09-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Velsko I.M., Chukkapalli S.S., Rivera-Kweh M.F., Zheng D., Aukhil I., Lucas A.R., Larjava H., Kesavalu L. Periodontal pathogens invade gingiva and aortic adventitia and elicit inflammasome activation in alphavbeta6 integrin-deficient mice. Infect. Immun. 2015;83:4582–4593. doi: 10.1128/IAI.01077-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.By Reduced CCL2 Expression.
- 35.Fang H., Zhang F., Li F., Shi H., Ma L., Du M., You Y., Qiu R., Nie H., Shen L., Bai Y., Lyu J. Mitochondrial DNA haplogroups modify the risk of osteoarthritis by altering mitochondrial function and intracellular mitochondrial signals. Biochim. Biophys. Acta. 2016;1862:829–836. doi: 10.1016/j.bbadis.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 36.MitoTEMPO prevents oxalate induced injury in NRK-52E cells via inhibiting mitochondrial dysfunction and modulating oxidative stress, Oxid. Med. Cell. Longev., 2017, 2017, 7528090. [DOI] [PMC free article] [PubMed]
- 37.Xi H., Zhang Y., Xu Y., Yang W.Y., Jiang X., Sha X., Cheng X., Wang J., Qin X., Yu J., Ji Y., Yang X., Wang H. Caspase-1 inflammasome activation mediates homocysteine-induced pyrop-apoptosis in endothelial cells. Circ. Res. 2016;118:1525–1539. doi: 10.1161/CIRCRESAHA.116.308501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X., Fang P., Li Y., Kuo Y.M., Andrews A.J., Nanayakkara G., Johnson C., Fu H., Shan H., Du F., Hoffman N.E., Yu D., Eguchi S., Madesh M., Koch W.J., Sun J., Jiang X., Wang H., Yang X. Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arterioscler. Thromb. Vasc. Biol. 2016;36:1090–1100. doi: 10.1161/ATVBAHA.115.306964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Batandier C., Fontaine E., Keriel C., Leverve X.M. Determination of mitochondrial reactive oxygen species: methodological aspects. J. Cell. Mol. Med. 2002;6:175–187. doi: 10.1111/j.1582-4934.2002.tb00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deanfield J., Donald A., Ferri C., Giannattasio C., Halcox J., Halligan S., Lerman A., Mancia G., Oliver J.J., Pessina A.C., Rizzoni D., Rossi G.P., Salvetti A., Schiffrin E.L., Taddei S., Webb D.J. endothelial function and dysfunction. Part I: methodological issues for assessment in the different vascular beds: a statement by the Working Group on endothelin and endothelial factors of the European Society of hypertension. J. Hypertens. 2005;23:7–17. doi: 10.1097/00004872-200501000-00004. [DOI] [PubMed] [Google Scholar]
- 41.Shenouda S.M., Widlansky M.E., Chen K., Xu G., Holbrook M., Tabit C.E., Hamburg N.M., Frame A.A., Caiano T.L., Kluge M.A., Duess M.A., Levit A., Kim B., Hartman M.L., Joseph L., Shirihai O.S., Vita J.A. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444–453. doi: 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edwards J.L., Quattrini A., Lentz S.I., Figueroa-Romero C., Cerri F., Backus C., Hong Y., Feldman E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia. 2010;53:160–169. doi: 10.1007/s00125-009-1553-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang S., Wang Y., Gan X., Fang D., Zhong C., Wu L., Hu G., Sosunov A.A., McKhann G.M., Yu H., Yan S.S. Drp1-mediated mitochondrial abnormalities link to synaptic injury in diabetes model. Diabetes. 2015;64:1728–1742. doi: 10.2337/db14-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tower J. Hsps and aging. Trends Endocrinol. Metab. 2009;20:216–222. doi: 10.1016/j.tem.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sui X., Li D., Qiu H., Gaussin V., Depre C. Activation of the bone morphogenetic protein receptor by H11kinase/Hsp22 promotes cardiac cell growth and survival. Circ. Res. 2009;104:887–895. doi: 10.1161/CIRCRESAHA.108.192328. [DOI] [PubMed] [Google Scholar]
- 46.Lizano P., Rashed E., Kang H., Dai H., Sui X., Yan L., Qiu H., Depre C. The valosin-containing protein promotes cardiac survival through the inducible isoform of nitric oxide synthase. Cardiovasc. Res. 2013;99:685–693. doi: 10.1093/cvr/cvt136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun X., Fontaine J.M., Rest J.S., Shelden E.A., Welsh M.J., Benndorf R. Interaction of human HSP22 (HSPB8) with other small heat shock proteins. J. Biol. Chem. 2004;279:2394–2402. doi: 10.1074/jbc.M311324200. [DOI] [PubMed] [Google Scholar]
- 48.Benndorf R., Sun X., Gilmont R.R., Biederman K.J., Molloy M.P., Goodmurphy C.W., Cheng H., Andrews P.C., Welsh M.J. HSP22, a new member of the small heat shock protein superfamily, interacts with mimic of phosphorylated HSP27 ((3D)HSP27) J. Biol. Chem. 2001;276:26753–26761. doi: 10.1074/jbc.M103001200. [DOI] [PubMed] [Google Scholar]
- 49.Richter K., Haslbeck M., Buchner J. The heat shock response: life on the verge of death. Mol. Cell. 2010;40:253–266. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 50.Booth L.N., Brunet A. Shockingly early: chromatin-mediated loss of the heat shock response. Mol. Cell. 2015;59:515–516. doi: 10.1016/j.molcel.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 51.Yang X.F., Yin Y., Wang H. Vascular inflammation and atherogenesis are activated via receptors for PAMPs and suppressed by regulatory T cells. Drug Discov. Today Ther. Strateg. 2008;5:125–142. doi: 10.1016/j.ddstr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lenin R., Nagy P.G., Alli S., Rao V.R., Clauss M.A., Kompella U.B., Gangaraju R. Critical role of endoplasmic reticulum stress in chronic endothelial activation-induced visual deficits in tie2-tumor necrosis factor mice. J. Cell. Biochem. 2018 doi: 10.1002/jcb.27072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li X., Fang P., Mai J., Choi E.T., Wang H., Yang X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013;6:19. doi: 10.1186/1756-8722-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu N., Wu J., Zhang L., Gao Z., Sun Y., Yu M., Zhao Y., Dong S., Lu F., Zhang W. Hydrogen sulphide modulating mitochondrial morphology to promote mitophagy in endothelial cells under high-glucose and high-palmitate. J. Cell. Mol. Med. 2017;21:3190–3203. doi: 10.1111/jcmm.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]