

Abstract
Defective brain hormonal signaling has been associated with Alzheimer’s disease (AD), a disorder characterized by synapse and memory failure. Irisin is an exercise-induced myokine released upon cleavage of membrane-bound precursor protein FNDC5, also expressed in the hippocampus. Here we show that FNDC5/irisin levels are reduced in AD hippocampi and cerebrospinal fluid, and in experimental AD models. Knockdown of brain FNDC5/irisin impaired long-term potentiation and novel object recognition memory in mice. Conversely, boosting brain levels of FNDC5/irisin rescued synaptic plasticity and memory in AD mouse models. Peripheral overexpression of FNDC5/irisin rescued memory impairment, whereas blockade of either peripheral or brain FNDC5/irisin attenuated the neuroprotective actions of physical exercise on synaptic plasticity and memory in AD mice. By showing that FNDC5/irisin is an important mediator of the beneficial effects of exercise in AD models, our findings place FNDC5/irisin as a novel agent capable of opposing synapse failure and memory impairment in AD.
Keywords: Alzheimer’s disease, FNDC5, irisin, synaptic plasticity, memory, physical exercise
Introduction
The incidence of Alzheimer’s disease (AD), the most common form of dementia in the elderly, is increasing as the world population ages, with more than thirty-five million people now affected worldwide1. There is no current effective treatment for AD2, and significant efforts are aimed at developing strategies to counteract mechanisms leading to neuronal damage, synapse failure and memory impairment in AD.
Consolidated evidence indicates that the central nervous system (CNS) is an important target for the actions of peripheral hormones, including insulin, leptin, glucagon-like peptide 1 (GLP-1), glucocorticoids and others3-5. Insulin, leptin and GLP-1 stimulate neuronal survival, synaptic plasticity, and contribute to higher brain functions, including cognition6-9. Failure of hormone-initiated signaling pathways has been associated with brain disorders, including AD10. For example, brain insulin signaling is decreased in AD4,11-13, and strategies aimed at bolstering it are currently under clinical investigation14,15.
Irisin was recently identified as a myokine released into the circulation upon physical exercise, and capable of stimulating adipocyte browning and thermogenesis in mice and humans16,17. Irisin is cleaved from fibronectin type III domain containing 5 (FNDC5), a transmembrane precursor protein expressed in muscle under the control of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α). FNDC5/irisin stimulates the expression of brain-derived neurotrophic factor (BDNF) in the hippocampus18, a brain region centrally involved in learning and memory. This raises the possibility that FNDC5/irisin could play a neuroprotective role in brain disorders, such as AD. Here, we investigated FNDC5/irisin levels in brains and cerebrospinal fluid (CSF) of AD patients and in mouse models of AD, and tested the hypothesis that FNDC5/irisin could be a key mediator of the beneficial effects of exercise on synaptic plasticity and memory in AD models, thus holding promise as a potential target for therapeutic intervention in AD.
Results
Immunodetection of FNDC5/irisin in the brain
Since the original report16 describing irisin as a cleavage product derived from FNDC5 (Fig. 1a), the existence and possible physiological functions of irisin in rodents and humans have been a matter of controversy. Part of the debate has focused on the specificity and identity of proteins identified using anti-FNDC5/irisin antibodies. The existence and levels of irisin in human plasma have been settled using state-of-the-art mass spectrometry analysis17. Here, we first validated the rabbit polyclonal antibody we used (Abcam; anti-FNDC5, cat # ab131390) against recombinant irisin produced in CHO cells (Adipogen; cat # AG-40B-0136). In agreement with recent reports19,20, previous incubation of the antibody with antigen at increasing molar ratios effectively blocked immunodetection of recombinant irisin in Western blots (Extended Data Fig. 1a). We note, nonetheless, that because irisin is derived from FNDC5, immunological approaches for detection of irisin inherently detect its precursor FNDC5 in samples where the latter is also present. Moreover, irisin has been reported to exhibit an apparent molecular mass in the 22-32 kDa range resulting from dimerization and/or glycosylation15,18-21. This is similar to the molecular mass of FNDC516,21, making it difficult to discriminate between FNDC5 and irisin in immunoblots from tissue samples where both FNDC5 and irisin may be present. Thus, in the current study we refer to FNDC5/irisin when describing results based on immunological detection of irisin in brain tissue homogenates. On the other hand, because irisin is thought to comprise the majority of secreted FNDC5/irisin16, we refer to irisin when describing results obtained in CSF or plasma using an irisin ELISA kit.
Figure 1. CNS FNDC5/irisin is reduced in AD.
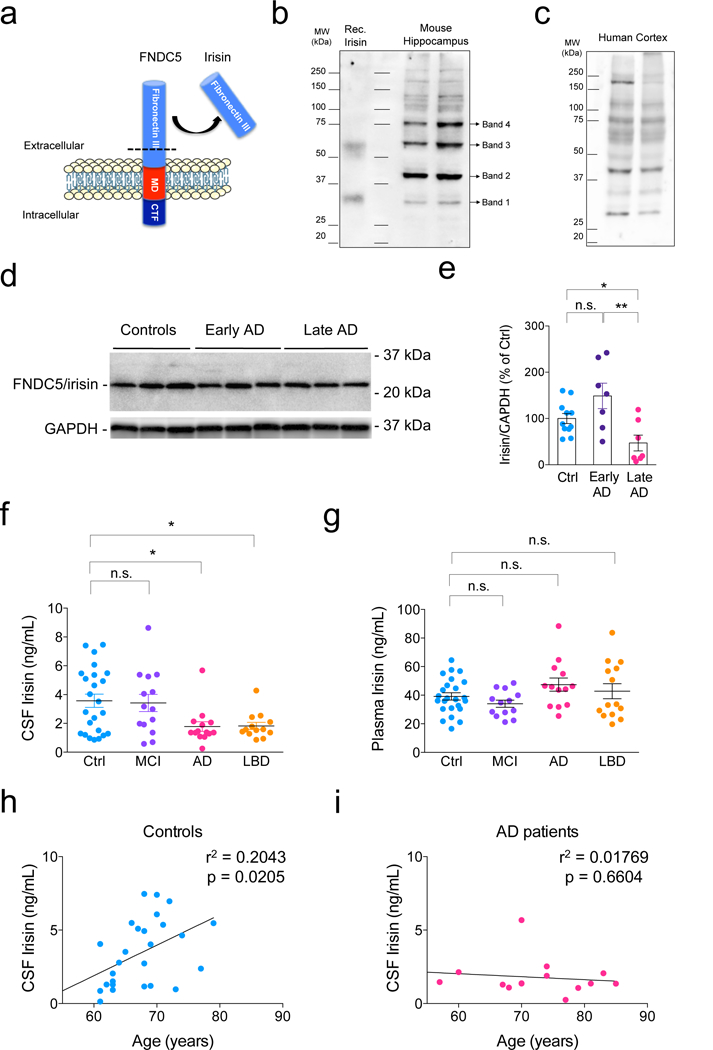
(a) Schematic representation of FNDC5 containing irisin, as part of the fibronectin III domain, which is cleaved by proteolysis and released to the extracellular medium. (b,c) Representative immunoblots of anti-FNDC5 (Abcam ab131390) in the mouse hippocampus (b) and human cortex (c). Bands analyzed by mass spectrometry (see Extended Data Figure 1 and Source Data 1) are indicated. These Western blots were repeated 3 times with similar results. (d,e) Summary quantification of human hippocampal irisin protein levels in late stage AD or early AD cases as compared to healthy controls (N = 11 controls, 7 early AD, 7 late AD). *p<0.05; **p<0.01, two-sided one-way ANOVA with Holm-Sidak post-test. Values are expressed as mean ± SEM. The experiments were repeated 2 times with similar results. See Source Data 2 for original data. (f) Summary quantification of irisin in the cerebrospinal fluid (CSF) of AD and Lewy body dementia (LBD) patients compared to healthy controls or mild cognitive impairment (MCI) patients. (N = 26 controls, 14 MCI, 14 AD, 13 LBD patients). *p<0.05, two-sided one-way ANOVA followed by Holm-Sidak post-test. Centre values are expressed as mean ± SEM. (g) Plasma levels of irisin in AD and LBD patients compared to healthy controls or MCI patients (N = 26 controls, 13 MCI, 13 AD, 14 LBD patients). (h, i) Correlation between age and CSF irisin in control (N = 31) and AD patients (N = 14; linear regression; r2 and p values as indicated).
We used the anti-FNDC5 antibody to identify FNDC5/irisin in mouse hippocampal (Fig. 1b) and human cortical homogenates (Fig. 1c). Immunoblots showed a band at 29 kDa, within the range reported for irisin, which migrated with similar electrophoretic mobility as recombinant irisin (Adipogen; cat # AG-40B-0136) (Fig. 1b). Mass spectrometry analysis in this band identified a peptide with amino acid sequence DNEPNNNK, which is contained within FNDC5 (Extended Data Fig. 1b,c; Source Data 1). Based on mass spectrometry results and because this band (which we termed band 1) was detected at the same apparent molecular mass as recombinant irisin, we used this band for quantification of FNDC5/irisin levels in subsequent experiments. In addition, we detected immunopositive bands at apparent molecular masses of ~ 40, 58 and 75 kDa (bands 2, 3 and 4, respectively; Fig. 1b). To rule out the possibility that bands 2-4 could reflect non-specific antibody labeling, we analyzed them by mass spectrometry. Results showed that bands 2 and 3 contained the same FNDC5 peptide as band 1. An additional peptide (DEVTMKEMGR) identified in band 4 was also comprised within FNDC5/irisin (Extended Data Fig. 1b,c; Source Data). Results indicate the presence of different forms (e.g., multimers and/or post-translationally modified species) of FNDC5/irisin in immunoblots from brain homogenates.
FNDC5/irisin is reduced in AD brains and CSF, and in AD experimental models
FNDC5/irisin was significantly reduced in the hippocampi of late-stage AD patients compared to age-matched early AD or cognitively normal subjects (Fig. 1d,e; Supplementary Table 1 for demographics). No changes were detected in βIII-tubulin (Tuj1), used as a neuron-specific control, indicating that the decrease in FNDC5/irisin was not caused by significant neuronal death in the samples analyzed (data not shown).
For CSF analysis of AD or control individuals, we employed an irisin ELISA kit (Phoenix Pharmaceuticals; cat # EK-067-29), which robustly recognizes recombinant irisin (Extended Data Fig. 1d). Irisin was decreased in the CSF of AD patients compared to mild cognitive impairment (MCI) or non-demented controls (Fig. 1f). Lewy body dementia (LBD) patients also presented reduced CSF levels of irisin (Fig. 1f). No significant alterations were found in plasma levels of irisin in AD or LBD patients compared to non-demented controls (Fig. 1g). CSF irisin levels showed a positive correlation with age in non-demented controls, but not in AD patients (Fig. 1h,i). While no correlation was found between CSF and plasma irisin levels in controls or AD patients (Extended Data Fig. 2a,b), we found a selective reduction in the CSF/plasma ratio of irisin in AD patients, but not in MCI or LBD (Extended Data Fig. 2c).
We next sought to determine whether Aβ oligomers (AβOs), soluble Aβ aggregates that accumulate in AD brains and are linked to synapse failure and memory loss2,22, impact FNDC5/irisin levels. Exposure of rat hippocampal cultures to AβOs (500 nM; 24 h), reduced FNDC5/irisin at both mRNA (Extended Data Fig. 3a) and protein levels (Extended Data Fig. 3b,c). FNDC5/irisin immunoreactivity was mostly associated with the surface of mature neurons (positively labeled for βIII-tubulin), with little immunoreactivity present in GFAP-positive astrocytes (Extended Data Fig. 3d,e). FNDC5/irisin immunoreactivity was reduced in AβO-exposed hippocampal cultures (Extended Data Fig. 3f,g). Specificity of surface staining by the anti-FNDC5 antibody was confirmed by our observation that lentiviral-mediated shRNA knockdown of FNDC5 markedly reduced its surface immunostaining (Extended Data Fig. 3h). We next exposed cultured human adult cortical slices23 to AβOs (500 nM) for 12 h, and observed that mRNA and protein levels of FNDC5/irisin were reduced (Fig. 2a-c).
Figure 2. Brain FNDC5/irisin is reduced in AD models.
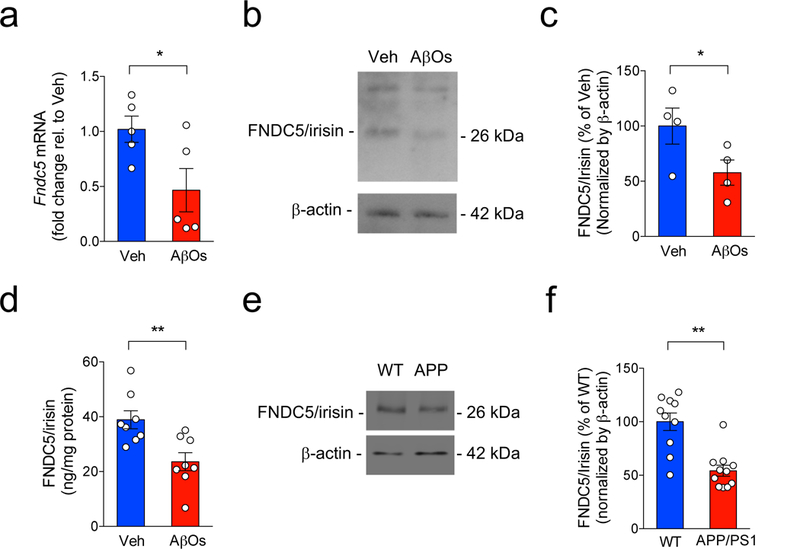
Fndc5 mRNA (a) and irisin levels (b,c) in control (Veh) and AβO-exposed human adult cortical slices after 12 h of treatment (N = 5 independent tissue donors for mRNA; 4 donors for protein levels; *p<0.05, paired Student’s t-test; two-sided). Irisin levels were normalized by β-actin. The experiments were repeated 3 times with similar results. See Source Data 3 for original data. (d) ELISA quantification of hippocampal irisin levels in WT C57BL/6 mice 24 h post-AβO infusion (N = 8 mice per group, **p<0.01, paired Student’s t-test; two-sided). The experiments were repeated 2 times with similar results. (e,f) Hippocampal levels of irisin in 13-16 month-old APP/PS1 ΔE9 mice (N = 10 for WT and 11 for APP/PS1 ΔE9, **p<0.01, paired Student’s t-test; two-sided). The experiments were repeated 2 times with similar results. See Source Data 4 for original data. Bars express mean ± SEM.
Consistent with previous reports18,24, we found that FNDC5/irisin is expressed in the mouse hippocampus and cortex of C57BL/6 mice, albeit at lower levels than in skeletal muscle (Extended Data Fig. 4a). Intracerebroventricular (i.c.v.) infusion of AβOs (10 pmol in a single dose)25,26 in C57BL/6 mice caused a significant reduction in hippocampal (Extended Data Fig. 4b,c), but not in skeletal muscle FNDC5/irisin mRNA (Extended Data Fig. 4d,e) after 24 h or 7 d. FNDC5/irisin protein levels detected by ELISA were reduced in the hippocampi of mice 24 h post-infusion of AβOs (Fig. 2d). FNDC5/irisin levels were also reduced in the hippocampi of 13-16 month-old APPswe/PS1ΔE9 mice (henceforth referred to as APP/PS1 ΔE9) (Fig. 2e,f), which develop amyloid pathology and memory deficits27. Taken together, our results indicate that FNDC5/irisin is expressed in the hippocampus, and is reduced in AD brains and CSF, as well as in experimental models of AD.
Because FNDC5/irisin levels are controlled by PGC-1α16, known to mediate synapse function and neuroprotection28, we investigated PGC-1α and PPARγ expression in AβO-infused mice. Expression of both PGC-1α (Extended Data Fig. 4f,g) and PPARγ (Extended Data Fig. 4h,i), but not of PPARα (Extended Data Fig. 4j,k), were reduced in mouse hippocampi 24 h and 7 days post-infusion of AβOs. PGC-1α protein levels were also reduced 7d post-infusion (Extended Data Fig. 4l,m).
Knockdown of brain FNDC5/irisin impairs synaptic plasticity and memory in mice
We infused C57BL/6 mice i.c.v. with lentiviruses harboring two different shRNA constructs that efficiently knockdown FNDC5 (Fig. 3a,b). This resulted in impaired maintenance of hippocampal long-term potentiation (LTP) (Fig. 3c,d) and memory in a novel object recognition (NOR) task (Fig. 3e). Mice infected with a control lentiviral vector (harboring an shRNA targeting luciferase) exhibited normal performance in the NOR task (Fig. 3e). Downregulation of brain FNDC5/irisin had no impact on performance in the radial arm water maze (RAWM) or contextual fear conditioning (CFC) tasks in WT mice (Fig. 3f,g). Control experiments showed that lentivirus-infected mice had no significant differences in motor activity or body weight (Extended Data Fig. 5a-c). Results thus indicate that while brain FNDC5/irisin does not contribute to CFC and RAWM memory, it influences hippocampal synaptic plasticity and NOR memory in C57BL/6 mice.
Figure 3. Downregulation of brain FNDC5/irisin impairs synaptic plasticity and object recognition memory in mice.
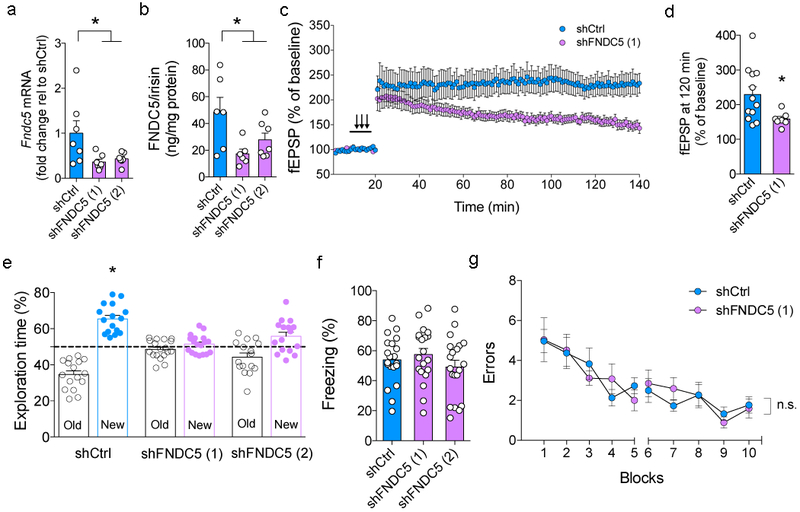
Two distinct shRNAs targeting Fndc5 (shFNDC5 1 or 2) or shCtrl were i.c.v. injected in C57BL/6 mice. Levels of Fndc5 mRNA (a) and FNDC5/irisin protein (b) in control (shCtrl) compared to shFNDC5 (1)- or shFNDC5 (2)-injected mice (N = 7 mice per group; *p<0.05, one-way ANOVA with Holm-Sidak correction) in the frontal cortex. Bars express mean ± SEM. (c) Average traces for field excitatory postsynaptic potentials (fEPSPs) in hippocampal slices from each experimental group (N =12 slices for shCtrl, 7 slices for shFNDC5 (1) obtained from 3-4 mice per group). Traces represent mean ± SEM per time. (d) fEPSP at 120 min. (N =12 slices for shCtrl, 7 slices for shFNDC5 obtained from 3-4 mice per group; *p<0.05, repeated measures two-way ANOVA with Holm-Sidak correction; two-sided). (e) Summary quantification of novel object discrimination in the NOR task in shCtrl, shFNDC5 (1)- or shFNDC5 -injected mice. *p<0.05, statistically different from 50% (chance level) (N = 16 mice for shCtrl; 18 for shFNDC5 (1), 16 for the shFNDC5 (2) group; one-sample t-test). (f) Contextual fear conditioning in shCtrl or shFNDC5-infused C57BL/6 mice (N = 20 mice per group; no significant difference was observed. One-way ANOVA followed by Holm-Sidak correction; two-sided). Bars express mean ± SEM. (g) shCtrl or shFNDC5(1)-infused C57BL/6 mice were assessed in a two-day radial arm water maze (RAWM) task and presented similar error profile across trials. Each block consisted of three consecutive trials. N = 9 shCtrl, 11 shFNDC5 (1); repeated measures two-way ANOVA. Values are presented as mean ± SEM.
Boosting brain FNDC5/irisin levels rescues synapse plasticity and memory defects in mouse models of AD
We next observed that recombinant irisin rescued impaired LTP in AβO-exposed hippocampal slices (Fig. 4a,b). Further, bilateral intrahippocampal infusion of recombinant irisin (75 pmol/site) prevented AβO-induced impairment in NOR and fear conditioning memory (Extended Data Fig. 6a,b).
Figure 4. Brain FNDC5/irisin rescues defective synaptic plasticity and memory in AD mice.
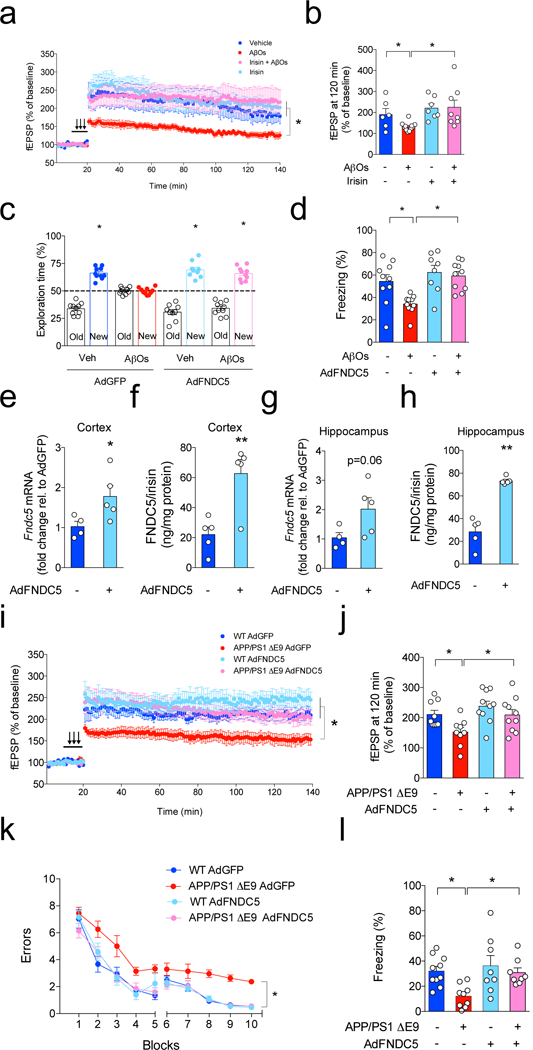
(a,b) LTP measurement in hippocampal slices (N = 6 slices for vehicle, 11 for AβOs, 7 for irisin, 8 for irisin + AβOs; slices were from 4 animals for each condition). (b) fEPSP at 120 min. (N = 6 slices for vehicle, 11 for AβOs, 7 for irisin, 8 for irisin + AβOs; *p<0.05, two-way ANOVA with Holm-Sidak correction; two-sided). Values are presented as mean ± SEM. (c,d) Effect of i.c.v injection of AdFNDC5 on memory impairment in AβO-infused C57BL/6 mice in the novel object recognition (c) and contextual fear conditioning (d) tasks (N = 10 mice for Veh GFP, 10 for AβOs GFP, 8 for Veh FNDC5, 10 for AβOs FNDC5; *p<0.05, statistically different from 50% (chance level) one-sample t-test). Bars are presented as mean ± SEM. (e,f) Cortical Fndc5 mRNA and protein expression in C57BL/6 mice 6 days after i.c.v. injection of AdGFP or AdFNDC5 vectors (N = 4 mice for AdGFP, 5 for AdFNDC5; Student’s t-test; *p<0.05). (g,h) Hippocampal Fndc5 mRNA and protein expression in C57BL/6 mice 6 days after i.c.v. injection of AdGFP or AdFNDC5 vectors (N = 4 mice for AdGFP, 5 for AdFNDC5; *p<0.05; Student’s t-test; two-sided. Bars are presented as mean ± SEM. (i-j) APP/PS1 ΔE9 mice (or WT littermates) were injected i.c.v. with an adenoviral vector expressing full-length FNDC5 (AdFNDC5) or green fluorescent protein (AdGFP, used as a control). (i) Average traces for field excitatory postsynaptic potentials (fEPSPs) in hippocampal slices from each experimental group (n = 10 slices for WT AdGFP, 9 APP/PS1 ΔE9 AdGFP, 10 WT AdGFP, 10 APP/PS1 ΔE9 AdFNDC5; from 4 animals per group). Traces represent mean ± SEM. (j) fEPSP at 120 min (n = 10 slices for WT AdGFP, 9 APP/PS1 ΔE9 AdGFP, 10 WT AdGFP, 10 APP/PS1 ΔE9 AdFNDC5; from 4 animals per group; *p<0.05, one-way ANOVA with Holm-Sidak correction; two-sided). (k,l) Effects of i.c.v. injection of AdFNDC5 on memory impairment in 3-4 month-old APP/PS1 ΔE9 mice in a two-day radial arm water maze (i) and in contextual fear conditioning (j) (N = 10 mice for WT AdGFP, 8 for APP/PS1 ΔE9 AdGFP, 11 for WT AdFNDC5, 10 for APP/PS1 ΔE9 AdFNDC5). (*p<0.05, Two-way ANOVA with Holm-Sidak correction; two-sided). Data are represented by mean ± SEM.
To further explore whether FNDC5/irisin could counteract the direct impact of AβOs on memory25,26, C57BL/6 mice were infected via i.c.v. with a control vector (AdGFP) or with AdFNDC5. Six days post-infection, mice received a single i.c.v. infusion of AβOs (or vehicle). Control measurements showed that neither viral infection nor AβO infusion caused any changes in speed or distance traveled, or in time spent at the center or periphery in an open field test (data not shown), indicating lack of effects on locomotor activity and anxiety. Brain expression of FNDC5/irisin blocked the AβO-induced alterations on both NOR (Fig. 4c) and CFC memory tests (Fig. 4d). Infection with AdFNDC5 led to increases in FNDC5/irisin mRNA expression and protein levels in the cortex (Fig. 4e,f) and hippocampus 6 days post-infection (Fig. 4g,h).
We further used a qPCR array to interrogate hippocampal gene expression related to synaptic plasticity in mice previously infected with either AdGFP or AdFNDC5, and subsequently infused with vehicle or AβOs. Amongst 84 genes analyzed, AdFNDC5 rescued AβO-induced changes in expression of 6 genes, whose expression levels were independently validated by qPCR. These genes included immediate early genes (Egr1, Egr4), and genes coding for the protein phosphatase calcineurin (Ppp3ca), neuronal pentraxin 2 (Nptx2), and subunits of AMPA and metabotropic glutamate receptors (Gria2, Grm2) (Extended Data Fig. 7), indicating possible protective actions of FNDC5/irisin against aberrant expression of synapse-related genes.
We next i.c.v-infused APP/PS1 ƊE9 mice27 or WT mice with the AdFNDC5 vector, or with AdGFP as a control, and evaluated synaptic plasticity and memory. Fourteen days post infection, after completion of behavioral experiments, acute hippocampal slices were prepared and CA3-CA1 synapses were stimulated for LTP induction. While hippocampal slices from APP/PS1 ƊE9 mice infected with AdGFP exhibited impaired LTP (Fig. 4i,j), slices from APP/PS1 ƊE9 mice i.c.v-infused with AdFNDC5 presented normal LTP (Fig. 4i,j). Brain expression of FNDC5/irisin rescued memory impairment in APP/PS1 ƊE9 mice in both radial arm water maze (RAWM) (Fig. 4k) and CFC (Fig. 4l). Similar results in LTP, RAWM and CFC were obtained with APPswe/PS1 M146L mice (henceforth referred to as APP/PS1 M146L), another mouse model of AD29 (Extended Data Fig. 8a-d). Control experiments showed that brain expression of FNDC5/irisin had no impact on locomotor activity, somatosensory behavior or body weight in APP/PS1 M146L mice (data not shown). Overall, results indicate that expression of FNDC5/irisin rescued hippocampal synaptic plasticity and memory in AD mice.
Neuroprotective actions of recombinant irisin in vitro
Because abnormal eIF2α phosphorylation and inhibition of protein synthesis have been recently described as key mechanisms driving synapse damage and memory failure in AD models10,26,30-32, we examined the effects of recombinant irisin on eIF2α-P and ATF4 levels in cultured primary hippocampal neurons. Irisin prevented AβO-induced elevation in eIF2α-P and ATF4 (Extended Data Fig 9a-c), as well as down-regulation of de novo protein synthesis in hippocampal neurons (Extended Data Fig. 9d,e). Control measurements revealed that total eIF2α immunoreactivity remained unchanged (not shown).
We further found that recombinant irisin prevented dendritic spine loss in cultured hippocampal neurons exposed to AβOs (Extended Data Fig. 9f,g). Additional experiments determined that recombinant irisin reduced AβO binding to neurons (Extended Data Fig. 9h,i). In vitro binding studies revealed no direct interaction between AβOs and recombinant irisin (Extended Data Fig. 9j), ruling out the possibility that blockade of binding to neurons might be caused by sequestration of AβOs by recombinant irisin added to the medium. In addition, surface FNDC5/irisin and AβOs did not co-localize in dendrites of hippocampal neurons (Extended Data Fig. 9k). Results further showed that FNDC5/irisin overexpression reduced hippocampal soluble Aβ42 levels in APP/PS1 M146L mice (Extended Data Fig. 9l), but not insoluble Aβ42 in the hippocampus or cortex (Extended Data Fig. 9m-9o),
We found that recombinant irisin stimulated the cAMP/PKA/CREB pathway in human cortical slices (Fig. 5a-c), a pathway that plays important roles in memory formation and has been found to be impaired in AD models33-35. Recombinant irisin further increased cAMP and pCREB in mouse hippocampal slices (Fig. 5d,e). Irisin-induced CREB phosphorylation was abolished by PKA inhibition with myristoylated PKI 14-22, a selective PKA inhibitor (Fig. 5e). We further found that PKA activity mediated protection against nuclear translocation of ATF4 induced by AβOs (Fig. 5f,g). Irisin further induced transient phosphorylation of extracellular signal-regulated kinase (pERK) in cultured neurons (data not shown). The effect of recombinant irisin was similar to forskolin, a direct activator of adenylyl cyclase (data not shown). Taken together, results provide initial clues into mechanisms by which recombinant irisin affords neuroprotection in experimental models of AD.
Figure 5. Irisin triggers brain cAMP/PKA/CREB signaling pathways.
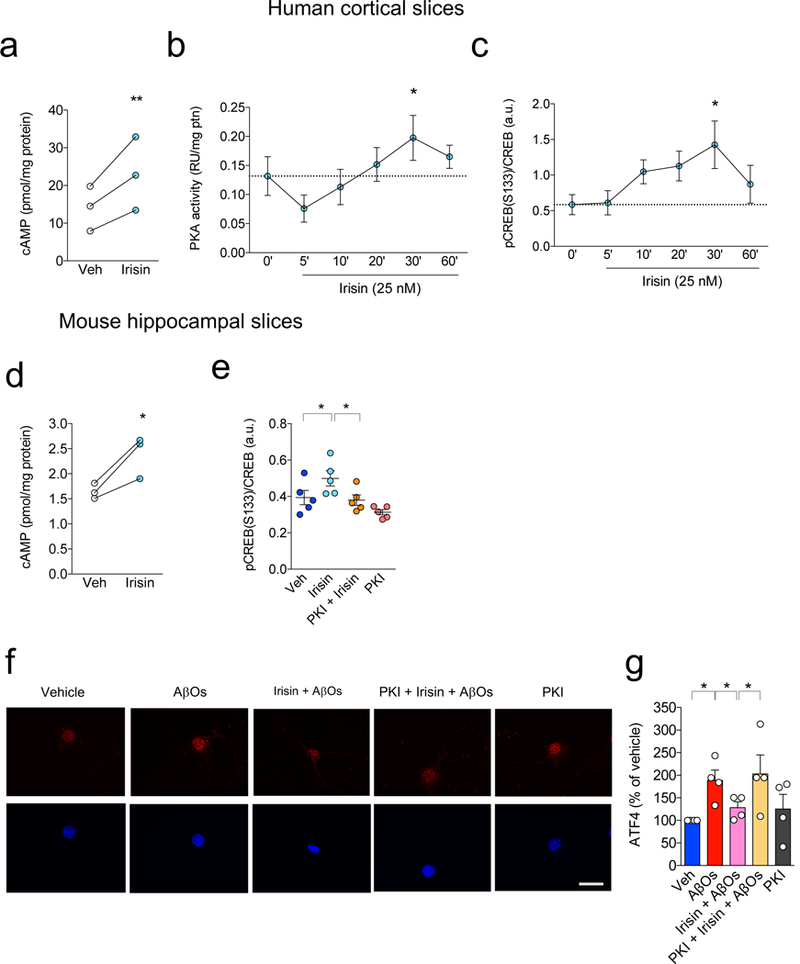
(a-d) Summary quantification of the effect of irisin (25 nM) on cyclic AMP (cAMP) accumulation (a), PKA activation (b) and CREB phosphorylation (c) in human cortical slices (N = 3 independent experiments with slices from different tissue donors for cAMP and pCREB, and 4 for PKA assay; *p<0.05; **p<0.01; repeated-measures ANOVA; two-sided). Data are represented by mean ± SEM. (d) Analysis of cyclic AMP (cAMP) accumulation induced by irisin (25 nM) in mouse hippocampal slices (N = 4 independent experiments with 6-8 slices from 4 independent animals; *p<0.05; paired Student’s t-test; two-sided), and (e) CREB activation (N = 2 independent experiments with 5 slices from 3 animals; *p<0.05; two-way ANOVA with Holm-Sidak correction; two-sided). Data are represented by mean ± SEM, n defined per slice. (f) Nuclear ATF4 levels (red) in primary cultures exposed to AβOs and/or irisin in the presence of PKI 14-22, a PKA selective inhibitor. Nuclei were counterstained in blue (DAPI). Scale bar = 10 μm. (g) Summary quantification of immunocytochemistry experiments (N = 4 experiments with independent neuronal cultures and AβO preparations; 30 images (from 2-3 coverslips) per experimental condition per experiment). *p<0.05; paired two-way ANOVA with Holm-Sidak correction, two-sided. Data are represented by mean ± SEM.
FNDC5/irisin mediates the protective actions of physical exercise on synaptic plasticity and memory in AD models
From a translational perspective, physical exercise could be a non-pharmacological strategy to increase hippocampal FNDC5/irisin in patients at risk of developing AD or in patients already exhibiting cognitive impairment. We thus initially tested whether a protocol of daily swimming (1 h/day, 5 d/week, 5 weeks) could protect mice from AβO-induced memory deficits and reduction in brain levels of FNDC5/irisin. Notably, exercised mice were protected from AβO-induced impairment in NOR memory both 24 h (Extended Data Fig. 10a) and 5 d (Extended Data Fig. 10b) following AβO infusion. Protection against AβO-induced memory deficits was also verified in the CFC paradigm (Extended Data Fig. 10c).
We further found that this exercise protocol prevented AβO-induced reductions in FNDC5/irisin mRNA (Extended Data Fig. 10d) and protein (Extended Data Fig. 10e) in mouse hippocampi. Moreover, consistent with a previous report18, hippocampal levels of FNDC5/irisin (Extended Data Fig. 10e) and BDNF (Extended Data Fig. 10f,g) were higher in exercised mice compared to sedentary animals.
We then asked whether brain FNDC5/irisin mediated the beneficial effect of physical exercise on synapse plasticity in APP/PS1 ΔE9 mice. We conducted an i.c.v. infusion of a lentiviral vector harboring FNDC5 shRNA and subsequently subjected mice to the exercise protocol. Results revealed that exercise improved LTP in APP/PS1 ΔE9 mice infused with an innocuous shRNA (targeting luciferase), but not in APP/PS1 ΔE9 mice exhibiting downregulated brain FNDC5/irisin expression (Fig. 6a,b). Additionally, we observed that downregulation of brain FNDC5/irisin did not exacerbate LTP impairment in sedentary APP/PS1 ΔE9 mice (Fig. 6a,b).
Figure 6. FNDC5/irisin mediates the beneficial effects of exercise on synaptic plasticity and memory.
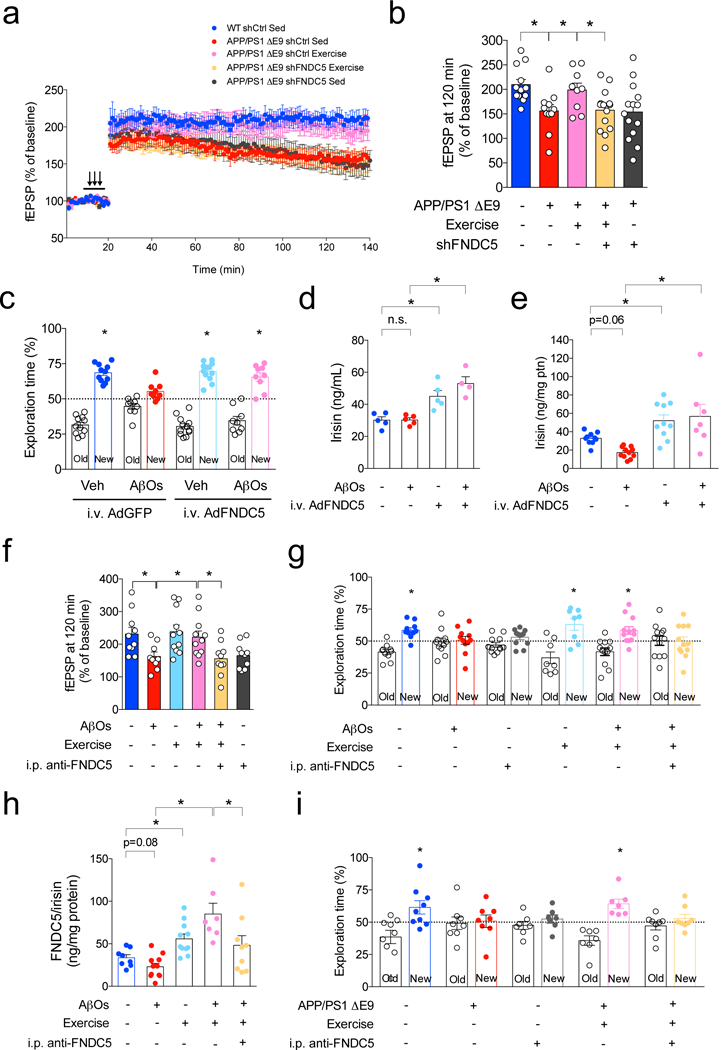
(a-b) APP/PS1 ΔE9 mice (or WT littermates) were injected i.c.v. with a lentiviral vector expressing an shRNA targeting FNDC5 (shFNDC5) or an shRNA targeting luciferase (shCtrl, used as a control), and were exercised. (a) Average traces for field excitatory postsynaptic potentials (fEPSPs) in hippocampal slices from each experimental group (N = 12 slices for WT shCtrl Sedentary (Sed), 11 for APP/PS1 ΔE9 shCtrl Sed, 9 for APP/PS1 ΔE9 shCtrl Exercised, 14 for APP/PS1 ΔE9 shFNDC5 Exercised, 12 for APP/PS1 ΔE9 shFNDC5 Sed; from 4 animals per group). Traces are represented by mean ± SEM. (b) fEPSP at 120 min. *p<0.05, Two-way ANOVA with Holm-Sidak correction. (N = 12 slices for WT shCtrl Sedentary (Sed), 11 for APP/PS1 ΔE9 shCtrl Sed, 9 for APP/PS1 ΔE9 shCtrl Exercised, 14 for APP/PS1 ΔE9 shFNDC5 Exercised, 12 for APP/PS1 ΔE9 shFNDC5 Sed; from 4 animals per group; *p<0.05, one-way ANOVA with Holm-Sidak correction; two-sided). Values are represented by mean ± SEM. (c) Effect of intravenously (i.v.) administered AdFNDC5 on NOR memory in AβO-infused mice 24 h after oligomer infusion (N = 11 Veh AdGFP; 9 AβOs AdGFP; 12 Veh AdFNDC5; 9 AβOs AdFNDC5; *p<0.05, statistically different from 50% (chance level) one-sample t-test). Values are represented by mean ± SEM. (d,e) Effect of i.v.AdFNDC5 administration on plasma (N = 5 mice for Veh AdGFP, 5 AβOs AdGFP, 5 Veh AdFNDC5, 4 AβOs AdFNDC5) (d) and hippocampal FNDC5/irisin levels (10 Veh AdGFP, 11 AβOs AdGFP, 10 Veh AdFNDC5, 7 AβOs AdFNDC5; *p<0.05, two-way ANOVA with Holm Sidak correction; two-sided). Values are represented by mean ± SEM. (e) in Veh- or AβO-injected C57BL/6 mice (10 Veh AdGFP, 11 AβOs AdGFP, 10 Veh AdFNDC5, 7 AβOs AdFNDC5; *p<0.05, two-way ANOVA with Holm Sidak correction; two-sided). Values are represented by mean ± SEM. (f,g) AD mice were intraperitoneally injected with an anti-FNDC5 antibody and exercised. They were tested on synaptic plasticity and NOR memory 72 h after AβO infusion. (f) Summary quantification of fEPSP at 120 min (N = 10 slices for Veh, 9 for AβOs, 11 for Exercise, 11 for Exercise + AβOs, 10 for anti-FNDC5, 10 for anti-FNDC5 + Exercise + AβOs; from 4 animals per group; *p<0.05, one-way ANOVA with Holm-Sidak correction; two-sided). Values are represented by mean ± SEM. (g) NOR performed 72 h after AβO infusion (N = 10 mice for Veh, 11 for AβOs, 12 for i.p. anti-FNDC5, 8 for Exercise, 12 for Exercise + AβOs, and 12 for i.p. anti-FNDC5 + Exercise + AβOs); *p<0.05; one-sample t-test. Values are represented by mean ± SEM. (h) Analysis of i.p. injections of anti-FNDC5 on exercise-induced hippocampal FNDC5/irisin in AβO-infused mice (N = 8 mice for Veh, 11 for AβOs, 11 for Exercise, 7 for Exercise + AβOs, and 9 for i.p. anti-FNDC5 + Exercise + AβOs). *p<0.05; one-way ANOVA with Holm-Sidak correction; two-sided. Values are represented by mean ± SEM. (i) APP/PS1 ΔE9 mice (or WT littermates) were injected i.p. with anti-FNDC5 (or an irrelevant IgG), and subjected to exercise. NOR memory was assessed 8 days after the first antibody injection and daily exercise session. Effect of anti-FNDC5 on the beneficial actions of exercise in APP/PS1 ΔE9 mice (N = 9 mice for WT, 8 APP/PS1 ΔE9, 7 WT i.p. anti-FNDC5, 8 Exercise APP/PS1 ΔE9, 7 APP/PS1 ΔE9 Exercise injected i.p. with anti-FNDC5). *p<0.05; one-sample t-test. Values are represented by mean ± SEM.
We next aimed to determine a potential role of peripheral FNDC5/irisin in the brain. We administered the AdFNDC5 vector into the caudal vein of mice to induce peripheral FNDC5/irisin expression, as previously described18. Peripheral administration of AdFNDC5 rescued NOR memory defects in AβO-infused mice (Fig. 6c). We next evaluated plasma levels of FNDC5/irisin in this group of mice and found unaltered levels in AβO-infused animals (Fig. 6d). As expected, plasma levels of FNDC5/irisin were increased in animals that received i.v. AdFNDC5 (Fig. 6d). Moreover, while hippocampal levels of FNDC5/irisin were decreased in AβO-infused mice, intravenous injection of AdFNDC5 resulted in increased hippocampal FNDC5/irisin levels and prevented the decrease triggered by AβOs (Fig. 6e). These results suggest that peripheral FNDC5/irisin can either reach the brain or trigger an increase in brain FNDC5/irisin. They further demonstrate that peripheral FNDC5/irisin affords protection against AβO-induced memory impairment.
Our second approach to investigate the role of peripheral FNDC5/irisin in synaptic plasticity and memory in AD models consisted in neutralizing peripheral FNDC5/irisin. To this end, we carried out intraperitoneal (i.p.) injections of anti-FNDC5 in either sedentary or exercised AβO-infused mice. This approach has been previously described to attenuate irisin-induced expression of pro-thermogenesis genes in mice16. Interestingly, i.p. administration of anti-FNDC5 blocked the protective actions of physical exercise against the impairments in synaptic plasticity and memory induced by AβOs (Fig. 6f,g). We further noted that i.p. administration of the anti-FNDC5 antibody per se impaired synaptic plasticity and performance in the NOR test (Fig. 6f,g). We next aimed to evaluate the effects of exercise and peripheral administration of anti-FNDC5 on hippocampal FNDC5/irisin levels. In accordance with results in Extended Data Fig. 10, we found that exercise prevented the decrease in hippocampal levels of FNDC5/irisin triggered by AβOs (Fig. 6h). Interestingly, we observed that administration of anti-FNDC5 led to a decrease in hippocampal FNDC5/irisin levels in exercised AβO-infused mice (Fig. 6h). Collectively these results indicate that peripheral irisin may reach the brain and mediate the neuroprotective actions of exercise in synaptic plasticity and memory in AD.
Lastly, we tested whether the approach to neutralize peripheral FNDC5/irisin using an anti-FNDC5 antibody would block the beneficial effects of exercise on memory in APP/PS1 ΔE9 mice. Our results showed that peripheral administration of the anti-FNDC5 antibody prevented the protective actions of exercise in APP/PS1 ΔE9 mice in the NOR test (Fig. 6i). Collectively, these results importantly demonstrate that FNDC5/irisin mediates the positive effects of exercise on synaptic plasticity and memory.
Discussion
AD is a neurological disorder primarily affecting memory, and with no cure to date. A variety of potential underlying mechanisms have been proposed to explain the pathogenesis of AD10,36. Considerable evidence indicates that memory impairment in AD is caused by synapse failure and loss2,37,38. Thus, therapies aimed at restoring or preserving synapse function and cognition are highly warranted.
Irisin was originally discovered as an exercise-induced myokine that shifts the adipose metabolism toward a thermogenic profile16,39. There has been some debate as to the nature and identity of FNDC5/irisin, as well as to its functional relevance in humans40. While concerns have been expressed regarding lack of specificity of anti-irisin antibodies41, sensitive approaches have been successfully employed to confirm the identity of irisin and to measure its circulating levels in humans17,20. Three complementary lines of evidence indicate the specificity of the anti-FNDC5 antibody and ELISA kit used in the current work: (1) FNDC5/irisin levels detected by ELISA showed the expected increase or decrease, respectively, when FNDC5/irisin was overexpressed by use of an adenoviral vector (in both brain and circulation) or when it was knocked down in the brain using a lentiviral vector; (2) surface labeling of FNDC5/irisin by anti-FNDC5 in cultured neurons was predictably reduced in neurons infected by a lentiviral vector harboring shRNA to knock down FNDC5/irisin; (3) mass spectrometry analysis identified peptides contained within FNDC5 in excised gel bands that were immunolabeled by the anti-FNDC5 antibody. By coupling immunodetection and mass spectrometry, we offer initial evidence that FNDC5/irisin is present in multiple forms with distinct apparent molecular weights in the brain, suggesting it may undergo post-translational modifications and/or exist in different aggregation states. Indeed, putative glycosylation sites are present in FNDC5/irisin21, and previous reports have suggested that shifts in electrophoretic mobility of FNDC5/irisin are due to glycosylation16,20.
An interesting study demonstrated that FNDC5/irisin is induced via PGC-1α in the mouse brain, and promotes BDNF expression18. Extending those previous findings and arguing for a physiological role of FNDC5/irisin in the human brain, we show that ex vivo human adult cortical slices express FNDC5/irisin and respond to exogenous recombinant irisin by activating the cAMP/PKA/CREB memory pathway42. Our in vitro findings further show that irisin blocks AβO-binding to neurons and prevents AβO-induced eIF2α-P and inhibition of protein synthesis. We and others have recently described these events as essential for synapse and memory failure in AD models, thereby pinpointing a potential downstream pathway by which irisin preserves memory10,26,30. We note that the cellular receptor(s) for irisin remain(s) to be identified, which limits current knowledge on downstream signaling mechanisms. The identification of the irisin receptor (s) and the detailed signaling mechanisms triggered in the periphery and in the brain are needed in the field.
The reduced brain and CSF levels of FNDC5/irisin in AD patients and in animal models reported here supports the notion that defective brain hormonal signaling in AD impact mechanisms related to memory formation and brain function3-5. It is noteworthy that we found a positive correlation between age and CSF irisin in control, non-demented subjects, but not in MCI and AD patients, suggesting that the increase in brain FNDC5/irisin with aging may be part of an endogenous mechanism to cope with the many challenges faced by the aging brain.
Irisin was reduced in the CSF of AD patients, but not in plasma, indicating a specific decrease in the CNS. Our finding that hippocampal FNDC5/irisin is reduced in moderate/late AD, but not in MCI, suggests that decreased FNDC5/irisin is not a likely cause of early cognitive impairment in AD, but may contribute to memory defects as disease progresses. Additional studies measuring irisin levels in the CSF during healthy aging and in patients with other neurological disorders are anticipated, and will help to determine when irisin levels decrease during the course of AD.
Blockade of either brain or peripheral FNDC5/irisin in mice impaired LTP and NOR memory, implicating FNDC5/irisin in physiological memory processes. However, future studies are warranted to address the precise physiological roles of brain and peripheral FNDC5/irisin in the formation and consolidation of different types of memories.
It is noteworthy that peripheral administration of the AdFNDC5 led to increases in hippocampal FNDC5/irisin and protected mice against memory impairment induced by AβOs. Most importantly, peripheral FNDC5/irisin is implicated in the preservation of hippocampal FNDC5/irisin levels, synaptic plasticity and memory in AD mice. Collectively, these results provide mechanistic information on the beneficial actions of FNDC5/irisin in the brain and suggest that a crosstalk between peripheral and central FNDC5/irisin influences synaptic plasticity and memory in mice.
Physical exercise has been previously shown to induce memory-related events in the brain43-45, and has been proposed as an approach to reduce the risk of AD, potentially bringing about significant benefits to subjects with MCI and early AD46-49. Many efforts to identify endogenous molecules responsible for the beneficial effects of exercise are underway. Brain PGC-1α and BDNF28,50, as well as peripheral cathepsin B and β-hydroxybutyrate51,52, have been described as important molecules acting as mediators of exercise-induced neuroprotection. Results presented here add FNDC5/irisin to this list.
Our findings suggest that FNDC5/irisin could comprise an attractive novel therapy aimed to prevent dementia in patients at risk, as well as to delay its progression in patients at later stages, including those who no longer can exercise. Many patients with dementia are disabled due to other age-related conditions or comorbidities (e.g., arthritis, heart disease, obesity, visual problems, depression) that preclude them from engaging in regular physical exercise. Therefore, the development of alternative approaches that build upon the beneficial effects of exercise in the brain may benefit those patients.
In conclusion, our results demonstrate that FNDC5/irisin levels are reduced in human AD brains and CSF as well as in AD mouse models, and that boosting either brain or peripheral FNDC5/irisin levels attenuates synaptic and memory impairments in AD mouse models. We further show that FNDC5/irisin is a novel mediator of the beneficial effects of exercise on synapse function and memory in AD models. Bolstering brain FNDC5/irisin levels, either pharmacologically or through exercise, may thus constitute a novel therapeutic strategy to protect/repair synapse function and prevent cognitive decline in AD.
Online Methods
Reagents
Aβ1-42 was from American Peptide (Sunnyvale, CA) or Anaspec (Fremont, CA). 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), DMSO, anti-ATF4 antibody (#WH0000468M1), myristoylated PKI 14-22, and poly-L-lysine were from Sigma-Aldrich (St. Louis, MO). Culture media, qPCR kits, Alexa-labeled Phalloidin, fluorescent secondary antibodies for immunocytochemistry, ProLong anti-fade reagent with DAPI were from Life Technologies (Carlsbad, CA). IRDye-conjugated antibodies for immunoblotting were from Licor (Lincoln, NE). Electrophoresis gels and reagents were from BioRad (Hercules, CA). SuperSignal chemiluminescence reagents and BCA protein assay kit were from Pierce (Deerfield, IL). Anti-eIF2α−P (phosphoS51) (#BML-SA405-100), anti-eIF2α (total) (#ADI-KAP-CP130), and PKA activity kits were from Enzo Life Sciences (Farmingdale, NY). The antibody against puromycin (12D10) (#MABE343) was obtained from EMD Millipore (Darmstadt, Germany). The antibody against β-tubulin III (Tuj1) (#MAB1195) was from R&D (Minneapolis, MN). ELISA kits for BDNF and phosphorylated CREB, and antibodies against FNDC5 (#ab131390), GAPDH (#ab9485) and β-actin (#ab8226) were from Abcam (Cambridge, MA). The antibody against BDNF (#sc546) was from Santa Cruz Biotechnology (Santa Cruz, CA). Aβ oligomer-sensitive antibody NU4 was a kind gift from Dr. William L. Klein (Northwestern University, IL). Recombinant irisin expressed in CHO cells was from Adipogen (Epalinges, Switzerland). Irisin ELISA kits were from Phoenix Pharmaceuticals (Rocky Hill, NJ). cAMP ELISA kits were from R&D Systems (Minneapolis, MN). Mouse synaptic plasticity PCR array kit (#PAM126Z) was from Qiagen (Hilden, Germany). Adenoviral vectors designed to express GFP or FNDC5 (AdGFP or AdFNDC5, respectively) were produced by Viraquest Inc. (North Liberty, IA) using the Gateway expression system (Life Technologies), as described16,18, and were kindly donated by Dr. Bruce Spiegelman (Harvard University, MA). High-titer viral particles (1.0 × 1011/mL) were diluted and used in reported experiments. MISSION lentiviral particles targeting Luciferase (control) or FNDC5 were obtained from Sigma-Aldrich (St Louis, MO).
NanoLC-LTQ Orbitrap mass spectrometry analysis of immunodetected FNDC5/irisin in mouse hippocampus
Mouse hippocampal homogenates (50 μg per lane) were resolved in 10% SDS-PAGE gels followed by electrotransfer of half of the gel (first 6 lanes) onto nitrocellulose membranes at 4 oC, constant 100 V for 90 minutes in Tris/Glycine transfer buffer (BioRad, cat #1610734) containing 20% (v/v) methanol. Blots were blocked in membrane blocking solution (Invitrogen, cat # 000105) for 1 hour at room temperature, incubated overnight at 4 oC with anti-FNDC5 polyclonal antibody (Abcam; catalog # ab131390; 1:1,000 dilution in blocking buffer), washed 3 times for 10 minutes with TBS-T, incubated for 1 hour at room temperature with peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Invitrogen; 1:50,000 dilution in blocking buffer), developed using Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific) and imaged on a Azure c600 (Azure Biosystems) imaging station. The positions of immunolabeled bands in the membranes were used to guide excision of the corresponding bands from unstained separate lanes from the other half of the gel (mirror) that had not been electrotransferred. Excised gel bands were subjected to in-gel digestion as described53. Commercial recombinant irisin expressed in CHO cells (Adipogen, cat #AG-40B-0136) digested in aqueous solution or digested/eluted from bands excised from an SDS-PAGE gel (run exactly as described above) were used as standards.
Mass spectrometry analyses were carried out at the Queen’s University (Kingston, ON, Canada) Mass Spectrometry facility. Samples containing tryptic fragments were analyzed on an Orbitrap Velos Pro (Thermo Scientific, Bremen, Germany) mass spectrometer coupled to a nano-LC system and nano-ESI source (Thermo Fischer Scientific). Eluent A was aqueous formic acid (0.1% v/v) and eluent B was HPLC grade acetonitrile containing 0.1% (v/v) formic acid. Samples (10 μL) were injected by the autosampler onto the trap column (C18, internal diameter 100 μm, length 20 mm, particle diameter 5 μm) (CMP Scientific) and were separated on an analysis column (C18, internal diameter 75 μm, length 100 mm, particle diameter 5 μm) (CMP Scientific) at a flow rate of 30 nL/min using two step gradients from 5 to 50% eluent B over 70 min, to 100% eluent B over 45 min, followed by 100% eluent B for 15 min. The transfer capillary temperature was set to 270 °C. An ion spray voltage of 2.0 kV was applied to a PicoTip™ on-line nano-ESI emitter (New Objective, Berlin, Germany). MS scans were acquired at an orbitrap resolution of 60,000 for an m/z range from 150 to 2,000. Following data acquisition, MS2 spectra were analyzed using Proteome Discoverer 1.4 (Thermo Fischer Scientific) with a precursor mass tolerance of 10 ppm and fragment mass tolerance of 0.8 Da. Alternatively, we manually searched for MS2 spectra corresponding to specific m/z values for peptides identified in recombinant irisin standards in the raw data for samples corresponding to each band excised from the SDS-PAGE gel.
Ex vivo human cortical slices
Adult human cortical slices were previously characterized, and were prepared as described23,54 with minor modifications. Healthy cortical tissue was obtained from patients with drug-resistant temporal lobe epilepsy subjected to surgical interventions for removal of epileptic foci. Donors were two males (aged 37 and 49 year-old) and three female (aged 18, 39 and 66 year-old). After dissection under sterile conditions, 400 μm-thick slices were prepared using a McIlwain tissue chopper and plated on Neurobasal A medium containing 2% B27, 500 μM glutamine, 5 ng/mL FGF2, 2 μM DHEAS, 1 ng/mL BDNF and 50 μg/ml gentamicin. After 7 days in culture at 37 °C and 5% CO2, slices were exposed to AβOs (500 nM) or the corresponding volume of vehicle for 12 h, and were processed for biochemical analyses thereafter. When present, irisin (25 nM) was added for the indicated time periods, and tissue was harvested for subsequent analyses.
Post-mortem human brain tissue
For Western blotting analyses, brain samples from non-cognitively impaired, early or late AD subjects (defined by combined pathological and cognitive assessment) were obtained from the University of Kentucky (UK) Alzheimer’s Disease Center biobank. Experimental procedures involving human tissue were in compliance with the UK Institutional Review board (IRB). Samples from Brodmann areas 21/22 (superior and mid-temporal gyri) were snap-frozen at autopsy as previously described. AD tissue was from symptomatic individuals with clinical and pathological features described in Supplementary Table 1. All subjects were categorized by MMSE as control (26-30), early AD (20-25), and late AD (13-17). Sample groups were separated so as to clearly define differences in content of proteins of interest and MMSE score.
Human cerebrospinal fluid and plasma
Cerebrospinal fluid (CSF) or plasma from control and AD patients were collected after extensive neuropsychological investigation supervised by a board-certified psychiatrist, as described55. The study cohort included males and females. Age (mean ± SD), in years, was distributed as follows: Controls (67.5 ± 4.9), MCI (71.5 ± 5.8), AD (72.5 ± 8.1), and LBD (71.6 ± 7.0). CSF was collected by lumbar puncture performed at 9AM in all cases. CSF was centrifuged, aliquoted and immediately frozen at −80 oC. Total blood was collected in heparin-coated tubes, and plasma was isolated by centrifugation. Irisin concentration was determined using a commercial ELISA kit (Phoenix Pharmaceuticals), according to manufacturer’s instructions.
Animals
Male C57BL/6 or Swiss mice were obtained from the animal facility at Federal University of Rio de Janeiro and were 2.5-3 month-old at the beginning of experiments. Male and female APPswe/PS1 ΔE9 mice on a C57BL/6 background27 were originally obtained from The Jackson Laboratories (http://research.jax.org/repository/alzheimers.html) and bred at our animal facility. Male and female double transgenic mice (APP/PS1 M146L) were obtained by crossing Tg2576 mice harboring mutant human APP (K670M:N671L) with mutant PS1 (M146L) mice29. Wild-type littermates were used as controls. All animals had their genotypes confirmed prior to use. Animals were housed in groups of five per cage with free access to food and water, under a 12 h light/dark cycle, with controlled room temperature and humidity. Experiments using C57BL/6 mice are presented in the main text, while biochemical and behavioral experiments in Swiss mice performed to validate and confirm our findings are presented as Extended Data Figures. All procedures followed the “Principles of Laboratory Animal Care” from the US National Institutes of Health.
Brain infusions in mice
For intracerebroventricular (i.c.v.) infusion of AβOs, mice were anesthetized with 2.5% isoflurane (Cristália; São Paulo, Brazil) using a vaporizer system (Norwell, MA) and were gently restrained only during the injection procedure, as described25,56. A 2.5 mm-long needle was unilaterally inserted 1 mm to the right of the midline point equidistant from each eye and 1 mm posterior to a line drawn through the anterior base of the eye25,26,56,57. 10 pmol AβOs (or vehicle) was injected in a final volume of 3 μL and the needle was kept in place for an additional 30 seconds to prevent backflow. Mice that showed signs of misplaced injections or any sign of hemorrhage were excluded from further analysis. Recombinant irisin (75 pmol per site) was bilaterally delivered into the hippocampal CA1 region (stereotaxical coordinates relative to Bregma: 2.0 mm AP, ± 1.5 mm ML, 1.5 mm DV), immediately before AβO injections. Adenoviral particles (1×108) harboring GFP (AdGFP) or FNDC5 (AdFNDC5) constructs were stereotaxically injected into the right lateral ventricle of WT C57BL/6 or APP/PS1 mice (coordinates relative to Bregma: 0.2 mm AP; 1.0 ML; 2.4 mm DV). Injections were performed in a volume of 2 μL infused over 30 s. Behavioral studies (see below) were carried out at least 6 days post injection. Lentiviral particles expressing shRNA against murine Fndc5 or Luciferase (control) were i.c.v.-injected as described above. Lentiviruses (titer of 1.0-2.0 × 109 particles/mL) were injected in a volume of 3 μL per animal. Behavioral and electrophysiological studies were carried out 4 weeks post-injections. AdFNDC5 or AdGFP (100 μL; 106 particles/μL) were i.v.-injected into the caudal vein six days prior to AβO infusions in mice anesthetized by isoflurane (2.5%) and gently restrained during the procedure.
Aerobic exercise in mice
Swiss mice were adapted to swimming for 10 min each day for 2 days to reduce water-induced stress. The duration of exercise was gradually increased until mice swam for 60 min, which was reached, on average, on the fifth day of training. Exercise sessions were performed during the light cycle and consisted of 60 min swimming sessions, 5 days per week for 5 weeks. Mice swam in groups of four in plastic barrels (60 cm depth × 45 cm diameter). Water temperature was maintained at ~ 24 °C. On the fifth week of training, animals were injected i.c.v. with AβOs (10 pmol) and the novel object recognition task (see below) was performed 24 h or 5 days post-injection of AβOs. Contextual fear conditioning (see below) was performed 6-7 days post-injection. Animals were euthanized by decapitation 1 h after the last test session. Since C57BL/6 are smaller than Swiss mice and less resistant to long exercise sessions, we adapted the protocol when using those mice. In a set of experiments, WT or APP/PS1 ΔE9 mice on a C57BL/6 background swam for 20 min for three weeks in groups of four before LTP recordings. In some experiments, WT C57BL/6 mice underwent exercise and were injected i.p. with anti-FNDC5 antibody or control IgG (5 mg) after the last two exercise sessions, and/or were infused i.c.v. with AβOs (10 pmol) in the last exercise session. Mice were assessed in the NOR test 24 h after oligomer injections. Alternatively, in experiments aimed at testing NOR memory, WT or APP/PS1 ΔE9 were subjected to daily swimming sessions for one week, and were injected i.p. with anti-FNDC5 antibody or non-specific IgG (5 μg) on the first, fourth and seventh days. NOR was performed 24 h after the last exercise session.
Behavioral Testing
Novel object recognition (NOR).
Object recognition tests were carried out in an open field arena measuring 0.3 (w) x 0.3 (d) x 0.45 (h) m, as described25,26. Total distance and velocity during the 5-minute open field session were recorded as measures of locomotor activity, and no differences were found among groups. Animals were trained in a 5 minute-long session during which they were placed at the center of the arena in the presence of two identical objects. Exploratory behavior (amount of time exploring each object) was recorded by trained researchers. One hour after training, animals were reinserted into the arena for the test session, in which one of the objects had been replaced by a different (novel) object. Again, the amounts of time spent exploring familiar and novel objects were measured. Animals that had a total exploration time below 10 (ten) seconds were excluded from NOR tests. Results are expressed as percentage of time exploring each object during the training or test session, and were analyzed using a one-sample Student’s t-test comparing the mean exploration time for each object with the fixed value of 50% (chance level).
Contextual fear conditioning.
Contextual fear conditioning was evaluated as described26,58. For experiments in APP/PS1 mice, animals were allowed to freely explore the conditioning chamber (0.4 × 0.25 × 0.3 m) for 2 min, after which a 2-sec shock stimulus (0.8 mA) was applied to the floor. Animals remained in the cage for 1 additional minute. After 24 h, mice were placed in the same cage and allowed to explore it for 5 min in the absence of electric shock. Freezing events were recorded during this period, and the percentage of time in freezing behavior was calculated for each animal by the Ethovision software (Noldus, Leesburg, VA). In experiments with irisin- or AdFNDC5-injected mice and corresponding controls, animals were allowed to freely explore the training chamber (0.25 × 0.25 × 0.25 m; Harvard Apparatus) for 3 minutes, after which they received two 2-sec long 0.35 mA foot-shocks with a 30 sec interval. Animals were removed from the cage after 30 s. Twenty-four hours thereafter, they were reinserted into the box for 5 min and total freezing time during test session was determined. Statistical significances of differences between groups were evaluated by two-way ANOVA followed by Holm-Sidak post-hoc test. Representative experiments are shown in the figures. After completion of fear conditioning experiments, animals were subjected to sensory threshold assessment to investigate potentially different susceptibilities to shock, and no significant differences in sensory response were found amongst experimental groups.
Two-day radial arm water maze (RAWM).
RAWM was performed in APP/PS1 mice as described59. Briefly, mice were tested for their ability to find a platform located in a defined arm of a six-arm maze along 30 trials in two days. At day 1, 5 training blocks were performed with alternation between visible and hidden platform. On day 2, all 5 blocks had the platform hidden. A training block comprises 3 trials of 60 s each. The number of entries into incorrect maze arms (errors) was recorded for each session, and the number of errors per block was averaged. Visual and motor abilities were assessed by recording swimming velocity and latency to reach a visible platform in an open swimming pool task.
Animal tracking was automatically performed by Ethovision software (Noldus).
Open field. Control experiment to assess locomotor and exploratory activity were performed in the same boxes as NOR, in which mice were allowed to freely move for 5 min. Total distance, mean velocity, percentage of time in the center or periphery were recorded and quantified using the ANY-maze software (Stoelting Co, Wood Dale, IL).
LTP measurements
Mice were euthanized by cervical dislocation followed by decapitation, and their hippocampi were immediately removed. Transverse hippocampal slices (400 μm) were cut, placed on infusion chambers on aCSF (124 mM NaCl, 4.4 mM KCl, 1 mM Na2HPO4, 25 mM NaHCO3, 2 mM MgCl2, 2mM CaCl2, 10 mM glucose) and allowed to recover for 90-120 min. For experiments with recombinant irisin and/or AβOs in slices from WT mice, AβOs (200 nM) and/or recombinant irisin (25 nM) were diluted in aCSF and perfused for 20 min before stimulus. For experiments with exercised mice that received anti-FNDC5 (or not), animals were exercised for three sessions and received an i.p. injection of anti-FNDC5 antibody (or non-specific IgG) after each session. An additional anti-FNDC5 injection was performed before slice preparation and perfusion with AβOs or vehicle. For experiments with adenovirus-mediated FNDC5 expression, WT or APP/PS1 ΔE9 mice were i.c.v.-injected with AdGFP (control) or AdFNDC5 14 days prior to euthanasia and slice preparation. Field EPSPs were recorded from hippocampal CA1 region according to established procedures34,60.
Aβ oligomers
Oligomers were prepared from synthetic Aβ1-42 and were routinely characterized by size-exclusion chromatography, as described23,25,61,62. For electrophysiology experiments in hippocampal slices, oligomers were prepared as previously reported60,63 and perfused at a final concentration of 200 nM in artificial cerebrospinal fluid (aCSF).
Mature hippocampal cultures
Primary rat hippocampal neuronal cultures were maintained in Neurobasal medium supplemented with B27 (Life Technologies, CA), glutamine, and antibiotics according to established procedures11,26,64, and were used after 18-21 days in vitro. Cultures were exposed to 500 nM Aβ oligomers or an equivalent volume of vehicle (2% DMSO in PBS) for the time intervals indicated in each experiment. When present, lentiviral vectors targeting FNDC5 (106/mL) were allowed to express for five days before cells were processed for immunocytochemistry. When present, recombinant irisin (25 nM) or forskolin (10 μM) was added to cultures 15 min before AβOs, and myristoylated PKI 14-22 (1 μM) was added 40 minutes before irisin.
Immunofluorescence
Cells were fixed in ice-cold 4% formaldehyde plus 4% sucrose for 10 min. AβO-sensitive (NU4 monoclonal antibody65) and FNDC5 immunolabeling were performed under non-permeabilizing conditions, while cells labeled for eIF2α-P (1:400), total eIF2α (1:400), GFAP (1:500), βIII-tubulin (1:500), or ATF4 (1:400) were permeabilized in 0.1% Triton X-100 for 5 min before primary antibody incubation, followed by Alexa-conjugated secondary antibodies (Life Technologies, CA). Nuclei were counterstained in DAPI-containing Prolong mounting solution. Coverslips were imaged on a Zeiss AxioObserver Z1 microscope. FNDC5, eIF2α−P, total eIF2α and ATF4 immunofluorescence intensities were each analyzed in at least 3 experiments (see Figure Legends) using independent neuronal cultures and AβO preparations. In each experiment, 20-30 images (from 2-3 coverslips) were acquired from each experimental condition. Histogram analysis of fluorescence at each pixel across the images was performed using NIH Image J66, as described26. When indicated, cell bodies were digitally removed from the images so that only immunostaining on dendritic processes was quantified. Statistical significances of differences between experimental groups were assessed by ANOVA followed by post-hoc Holm-Sidak test, and p-values are indicated in Figure Legends.
Spine density
After treatments, hippocampal neuronal cultures were fixed in ice-cold 4% formaldehyde plus 4% sucrose for 10 min and permeabilized in 0.1% Triton X-100 for 5 min. Cells were then blocked in 3% bovine serum albumin (BSA) for 1h and stained with Alexa-conjugated Phalloidin (2 U per coverslip), which labels F-actin, in 3% BSA for 2 h at room temperature. Coverslips were mounted in DAPI-containing Prolong Gold and stored in a dark and humid chamber overnight. Coverslips were imaged on a Zeiss AxioObserver Z1 microscope. Images were obtained from at least two coverslips per experimental condition for each experiment (three experiments with independent neuronal cultures). Two or three distal dendrite segments were isolated per neuron, and a researcher blind to experimental conditions manually determined the number of spines. Results are expressed as the mean number of spines per μm.
RNA extraction and quantitative RT-PCR
Total RNA was extracted from cultures or animal tissues using SV Total RNA Isolation System (Promega, CA), following manufacturer instructions. RNA from human brain tissue was extracted using RNeasy Lipid Tissue Mini Kit (Qiagen, Germany). Purity and integrity of RNA preparations were checked by the 260/280 nm absorbance ratio. Only preparations with 260/280 nm OD ratios higher than 1.8 and no signs of RNA degradation were used. RNA concentrations were determined by absorption at 260 nm. For qRT-PCR, one microgram of total RNA was used for cDNA synthesis using High Capacity cDNA Reverse Transcription kit (Applied Biosystems, CA). Quantitative expression analysis of target genes was performed on a 7500 Applied Biosystems (Foster City, CA) Real-Time PCR system with the Power SYBR Green kit. β-actin (actb) was used as an endogenous reference gene for data normalization. qRT-PCR was performed in 20 μL reaction volumes according to manufacturer’s protocols. Primer sequences are described in Supplementary Table 2. Cycle threshold (Ct) values were used to calculate fold changes in gene expression using the 2-ΔΔCt method67. Statistical significance of changes in expression was assessed using Student’s t-test or 2-way ANOVA, as indicated in Figure Legends.
PCR array
A PCR array kit (Qiagen, Germany; catalog # PAM126Z) was used to assess expression of 84 genes involved in mechanisms of synaptic plasticity, using cDNA obtained from hippocampi of AdFNDC5-infected (or AdGFP-infected) mice subsequently infused i.c.v. with AβOs (or vehicle). Hippocampi were dissected 7 days after AβO injection. The assay was conducted following manufacturer’s instructions, and results were initially analyzed using Volcano plots. Independent validation of differentially expressed genes was carried out by qPCR. Statistical significance of changes in expression was assessed using 2-way ANOVA.
Immunoblotting and in vitro plate binding assay
Hippocampi or mature hippocampal cultures were homogenized in RIPA buffer containing protease and phosphatase inhibitor cocktails, and were resolved on 4-20% polyacrylamide pre-cast gels (Invitrogen) with Tris/glycine/SDS buffer run at 200 V for 60 min at room temperature. The gel (30 μg total protein/lane) was electroblotted onto Hybond ECL nitrocellulose using 25 mM Tris, 192 mM glycine, 20% (v/v) methanol, pH 8.3, at 350 mA for 90 min at 4 °C. Membranes were blocked with commercial blocking solution (Licor) for 1 h at room temperature. For experiments with blocking peptide, anti-FNDC5 antibody was pre-incubated with molar ratio excess of recombinant irisin (5:1 or 10:1) for 3 h at room tempetature before incubation with membranes. Primary antibodies (anti-FNDC5, anti-synaptophysin, anti-PSD95 (1:1000), anti-β−actin monoclonal antibodies (1:20,000)) were diluted in blocking solution and incubated with the membranes overnight at 4 oC. After incubation with secondary anti-mouse or anti-rabbit IgGs (1:10,000 in TBS-T) for 60 min, membranes were washed, developed with SuperSignal West Femto Maximum Sensitivity substrate and imaged on photographic film. For Odyssey fluorescent detection, membranes were incubated with fluorescent IRDye-conjugated antibodies (1:10,000) for 60 min, washed and scanned in an Odyssey detector. Optical density determination for quantification was performed on ImageJ. In vitro plate binding assay was performed as described68.
Protein synthesis (SuNSET)
Non-radioactive metabolic labeling by puromycin incorporation was performed for detection of newly synthesized peptides, as described30,69. Briefly, hippocampal cultures were exposed to 1 μM puromycin for 30 min before experimental endpoints and harvested in RIPA buffer. Puromycin incorporation, used as a proxy of protein synthesis rate, was detected using 12D10 anti-puromycin antibody (1:20,000). Densitometric analysis was performed in whole lanes (from 10 to 180 kDa), and β-actin was used as a loading control.
Determination of cAMP levels
cAMP was measured radiometrically, as previously described70. Briefly, human cortical slices were exposed to irisin (25 nM; 45 min) in the presence of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX; 500μM) added 30 min prior to irisin.
ELISA
Tissue samples were homogenized in 100 mM Tris, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, supplemented with protease and phosphatase inhibitor cocktails. Blood was obtained by cardiac puncture, collected in heparin-coated low protein-adsorption plastic tubes, centrifuged, and plasma fractions were either freshly used for ELISA or immediately frozen in liquid nitrogen and stored at −80 oC until analysis. Soluble Aβ42 was measured in the supernatant, while insoluble Aβ42 was recovered by re-homogenizing the pellet in 6M guanidine hydrochloride for 1 h. Aβ42, BDNF, cAMP, CREB and irisin ELISA assays were performed according to kit manufacturer’s instructions, after sample dilution optimization.
Mouse genotyping
Animals were genotyped prior to studies using specific primers. APP/PS1 ΔE9 and APP/PS1 M146L genotyping were carried out according to standard protocols. Briefly, genomic DNA was extracted from sections of mice tails and subjected to amplification using specific forward and reverse primers for the respective transgenic construct. Thermal cycling consisted of 35 cycles at 94 °C for 1 min, 52 °C for 1 min, and 72 °C for 1 min. PCR products were resolved on a 1.5% agarose gel, evidencing specific bands for transgenic animals after ethidium bromide staining.
Statistical analysis
All analyses were performed with GraphPad Prism (La Jolla, CA) and datasets were assessed for normality and group variance prior to statistical testing. Values are expressed as mean ± SEM, unless otherwise stated. Two-tailed statistical tests are indicated in each Figure (* p < 0.05; ** p < 0.01), and post-test corrections were used every time multiple comparisons were performed (as indicated in each Figure).
Study design and approval
Sample size for each experiment was estimated by performing pilot studies and by previous experience with different experimental approaches. No algorithm or software was used to randomize animal subjects. Animal subjects were assigned to experimental groups by the researcher. Experiments involving irisin detection in human CSF, dendritic spine quantification in neuronal cultures, LTP experiments and behavioral experiments in mice were performed in a blind fashion. All procedures involving animal research were in compliance with international standards and were approved by the Institutional Animal Care and Use Committee of the Federal University of Rio de Janeiro (protocols # IBqM 022, # IBqM 041, # IBqM 055). Experiments performed at Columbia University Medical Center were approved by the Institutional Animal Care and Use Committee under protocol # AC-AAAE9652. Procedures involving human cortical tissue were approved by the National Committee for Research Ethics (CONEP) of the Brazilian Ministry of Health (protocol # 0069.0.197.000-05). Donors gave written informed consent for use of brain tissue that would otherwise have been discarded following surgeries. All experimental procedures involving post-mortem human brain tissue were in compliance with the UK Institutional Review Board (IRB). Experimental procedures involving human CSF were approved by the Committee for Research Ethics of Copa D’Or Hospital (Rio de Janeiro, Brazil; protocol # 47163715.0.0000.5249). Donors gave written informed consent for use of brain tissue or CSF. All animal and human studies have been performed according to international ethical regulations and standards. More details can be found at the Life Sciences Reporting Summary appended to this manuscript.
Extended Data
Extended Data Figure 1. Validation of anti-FNDC5 for detection of brain FNDC5/irisin.
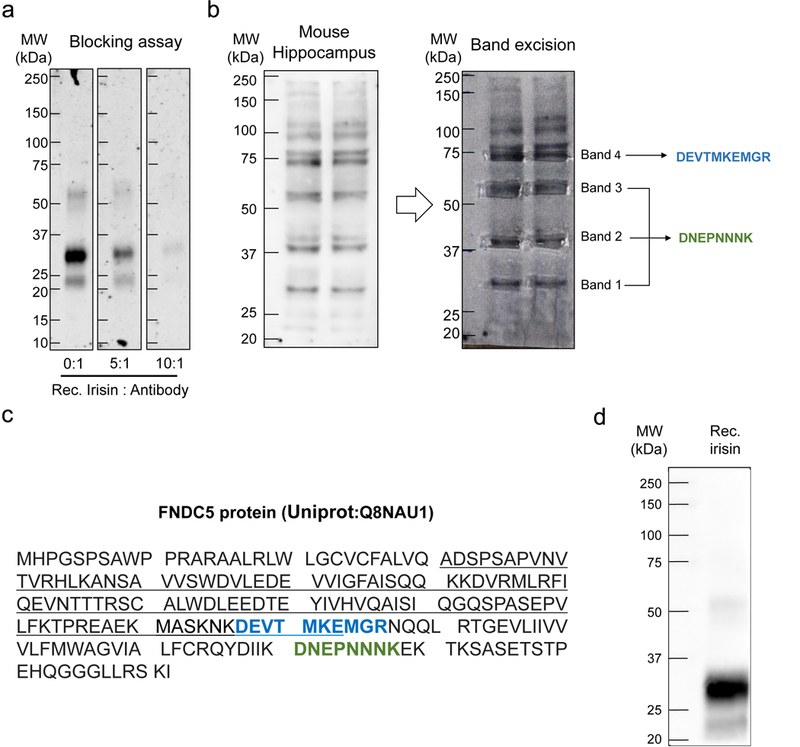
(a) Antibody blocking assay. Previous incubation of anti-FNDC5 (Abcam; ab131390) with increasing molar ratios of recombinant irisin (Adipogen; AG-40B-0136) reduces the signal of recombinant irisin detected by immunoblotting. The experiment was repeated 4 times with similar results. (b) Left panel: Immunoblot of mouse hippocampus homogenate probed with anti-FNDC5. Immunolabeled bands 1-4 (see “Results”) were used to guide excision of the corresponding bands from the other half of the SDS-PAGE gel that had not been electroblotted (right panel). Band excision was guided by overlaying the unstained gel onto an image of the developed immunoblot (right panel). Excised bands were subjected to in-gel tryptic digestion followed by mass spectrometry analysis, as described in Online Methods. Peptides identified by mass spectrometry in each band are indicated. (c) Full-length FNDC5 amino acid sequence. The sequence corresponding to irisin is underlined. Peptides identified by mass spectrometry in excised bands are highlighted in green (bands 1, 2 and 3) or blue (band 4). (d) Antibody used in the Phoenix ELISA kit (EK-067-29) recognizes recombinant irisin expressed in CHO cells (Adipogen; AG-40B-0136). The experiment was repeated 3 times with similar results. See Source Data 1 for original data.
Extended Data Figure 2. CSF and plasma irisin correlations.
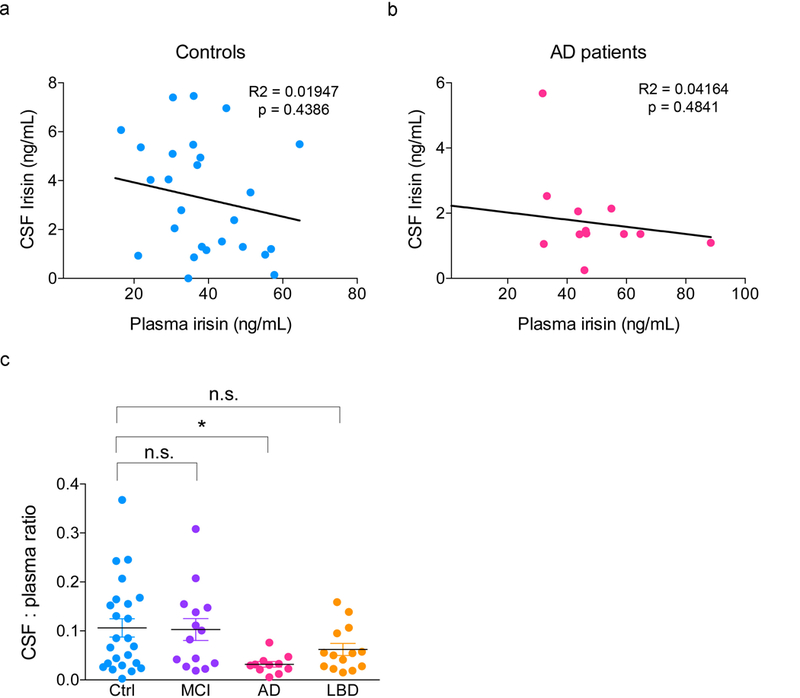
(a,b) CSF:plasma irisin levels correlation in controls (N = 26) (a) or AD patients (b) (N = 14; lines represent linear regression fits to the data; r2 and p values as indicated in the figure). (c) CSF to plasma irisin ratio is selectively reduced in AD patients, as compared to controls, MCI or LBD patients (N = 26 controls, 14 MCI, 11 AD, 13 LBD cases). Data are shown as mean ± SEM, *p<0.05; One-way ANOVA with Holm-Sidak post-test.
Extended Data Figure 3. AβOs reduce FNDC5/irisin levels in hippocampal neurons.
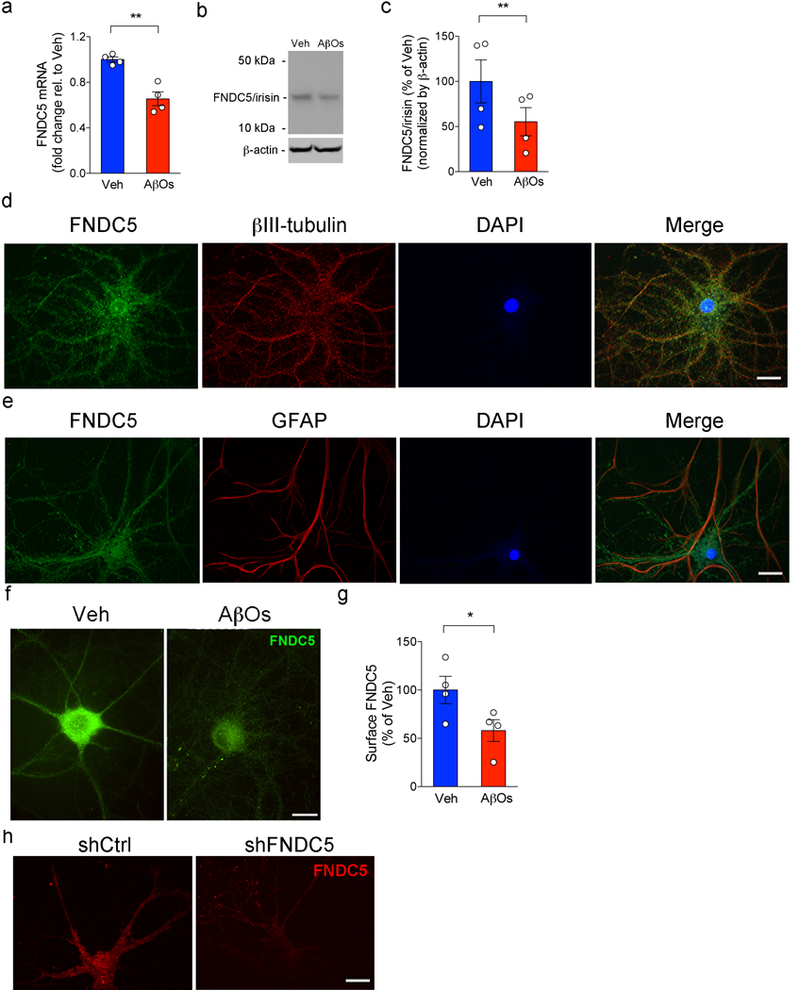
(a-c) Primary cultured hippocampal neurons were exposed to 500 nM AβOs for 24 h. Fndc5 mRNA (a) and FNDC5/irisin protein levels (b,c) in cultured hippocampal neurons exposed or not to AβOs (N = 4 experiments with independent neuronal cultures and AβO preparations; Date are shown as mean ± SEM,**p<0.01; paired Student’s t-test; two-sided). See Source Data 5 for original data. (d,e) Colocalization of surface FNDC5 immunoreactivity (green) with β-tubulin III (red) immunoreactivity in cultured hippocampal neurons (d). Colocalization of surface FNDC5 immunoreactivity (green) with glial fibrillary acidic protein (GFAP) (red) (e). The experiment was repeated 2 times with similar results in independent cultures. (f,g) Primary cultured hippocampal neurons were exposed to 500 nM AβOs for 24 h. (g) Summary quantification of surface FNDC5 immunoreactivity in cultured neurons (N = 4 experiments with independent neuronal cultures and AβO preparations; 30 images (from 2-3 coverlips) per condition per experiment. Data are shown as mean ± SEM *p<0.05, paired Student’s t-test; two-sided). (h) Surface FNDC5 immunoreactivity (red) in 18 DIV cultured hippocampal neurons after lentiviral knockdown of FNDC5 (shFNDC5) (N = 2 experiments with independent cultures; 30 images (from 2-3 coverslips) per condition per experiment). Scale bar = 10 mm.
Extended Data Figure 4. AβOs reduce hippocampal FNDC5, PGC-1α and PPARγ expression.
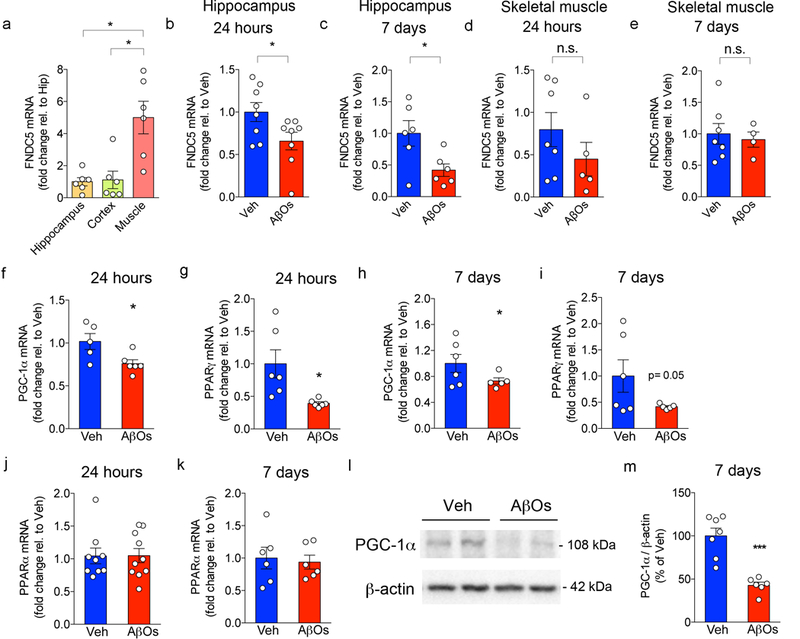
(a) Hippocampal, cortical and skeletal muscle (gastrocnemius) expression of FNDC5 in C57BL/6 mice (N = 6 per group). Data are shown as mean ± SEM; *p<0.05; paired one-way ANOVA with Holm-Sidak correction; two-sided. (b,c) Hippocampal FNDC5 mRNA in C57BL/6 i.c.v-infused with 10 pmol AβOs for 24 hours (N = 8 per group) (b) or 7 days (N = 5 per group) (c). Data are shown as mean ± SEM; *p<0.05; Student’s t-test; two-sided). (d,e) Skeletal muscle (gastrocnemius) FNDC5 mRNA in C57BL/6 i.c.v-infused with 10 pmol AβOs for 24 h (N = 7 Veh, 5 AβOs) (d) or 7 days (N = 7 Veh, 4 AβOs) (e). Data are shown as mean ± SEM; *p<0.05; Student’s t-test; two-sided). (f-k) Hippocampal mRNA levels of PGC-1α (f,h), PPARγ (g,i) and PPARα (j,k) were measured 24 h or 7 days after infusion, as indicated (N = 5 per group). mRNA levels were normalized by β-actin expression. (l,m) AβOs reduced hippocampal PGC-1α protein levels in C57BL/6 mice 7 days after infusion (N = 7 Veh, 6 AβOs). Data are shown as mean ± SEM; *p<0.05; Student’s t-test; two-sided). See Source Data 6 for original data.
Extended Data Figure 5. Lentiviral vectors expressing shFNDC5 did not cause motor impairment or affect body weight gain in mice.
(a) Distance traveled and (b) mean velocity of mice allowed to explore an open field arena for 5 min (N = 9 mice for shCtrl, 10 mice each for shFNDC5 (1) and (2) groups). Data are shown as mean ± SEM. (c) Body weight measured 0, 14 or 28 days after lentiviral injections (N = 10 mice per group; one-way ANOVA followed by Holm-Sidak post-test). Data are shown as mean ± SEM. No significantly statistical difference was found among groups.
Extended Data Figure 6. Intrahippocampal administration of recombinant irisin prevents AβO-induced memory impairment.
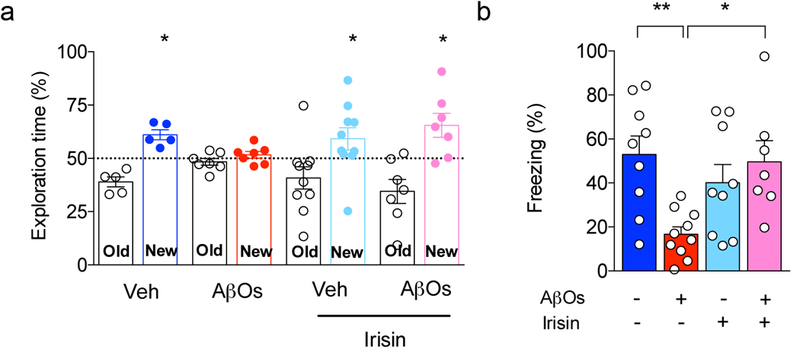
(a,b) Male Swiss mice (3 months old) were bilaterally injected with recombinant irisin (75 pmol per hippocampus) and received 10 pmol AβOs i.c.v. Novel object recognition (a) and contextual fear conditioning (b) tasks (tested 5 days post infusion of AβOs) (N = 9 mice for Veh, 10 for AβOs, 9 for irisin and 7 for AβOs + irisin). Data are shown as mean ± SEM. *p<0.05, Two-way ANOVA with Holm-Sidak post-test; two-sided.
Extended Data Figure 7. Validation of PCR array results by qPCR.
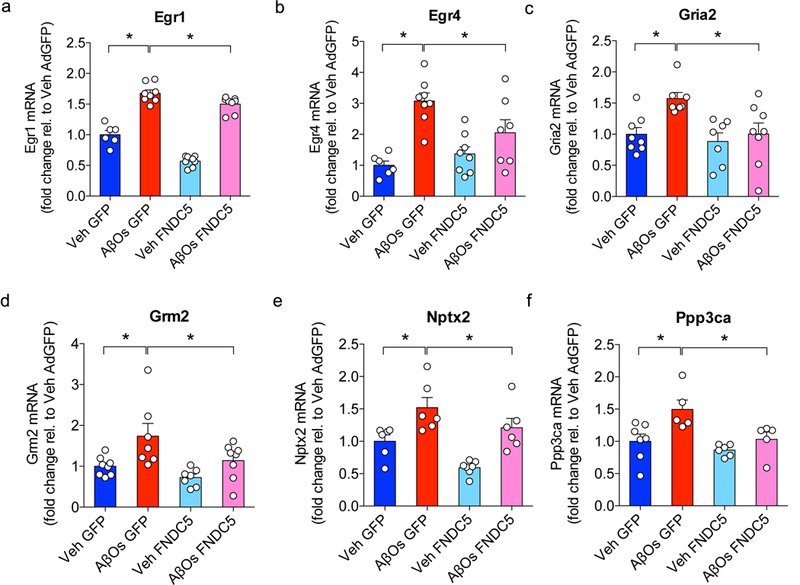
(a-f) AdFNDC5 expression of 6 synapse plasticity-related genes after AβO injection: (a) Egr1, (b) Egr4, (c) Gria2, (d) Grm2, (e) Nptx2, and (f) Ppp3ca. N = 6 mice per experimental group. Data are shown as mean ± SEM. *p<0.05; two-way ANOVA with Holm-Sidak post-test; two-sided.
Extended Data Figure 8. FNDC5/irisin rescues defective synaptic plasticity and memory in APP/PS1 M146L mice.
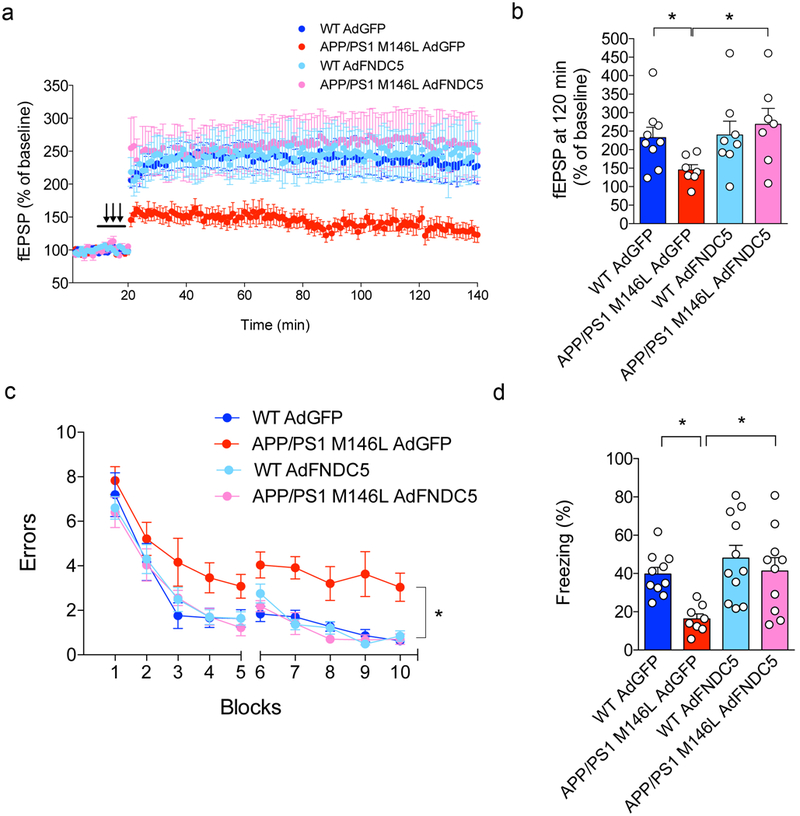
(a-d) APP/PS1 M146L mice (or WT littermates) were injected i.c.v. with an adenoviral vector expressing full-length FNDC5 (AdFNDC5) or green fluorescent protein (AdGFP, used as a control). Hippocampal slices were obtained and subjected to high-frequency stimulation for LTP recordings. (a) Field excitatory postsynaptic potentials (fEPSP) in hippocampal slices from each experimental group (N = 9 slices for Veh AdGFP, 7 for APP/PS1 M146L AdGFP, 8 for WT AdFNDC5, 7 for APP/PS1 M146L AdFNDC5; from 3-4 animals per group). (b) fEPSP at 120 min. (c,d) I.c.v.-injected AdFNDC5 rescued memory impairment in 3-4 months-old APP/PS1 M146L mice in 2-day radial arm water maze (c) and in contextual fear conditioning (d) (N = 10 mice for WT GFP, 8 for APP/PS1 GFP, 11 for WT FNDC5, 10 for APP/PS1 FNDC5). Data are shown as mean ± SEM. *p<0.05; two-way ANOVA with Holm-Sidak post-test; two-sided.
Extended Data Figure 9. Irisin counteracts AD-linked activation of cellular stress response and dendritic spine loss in hippocampal neurons.
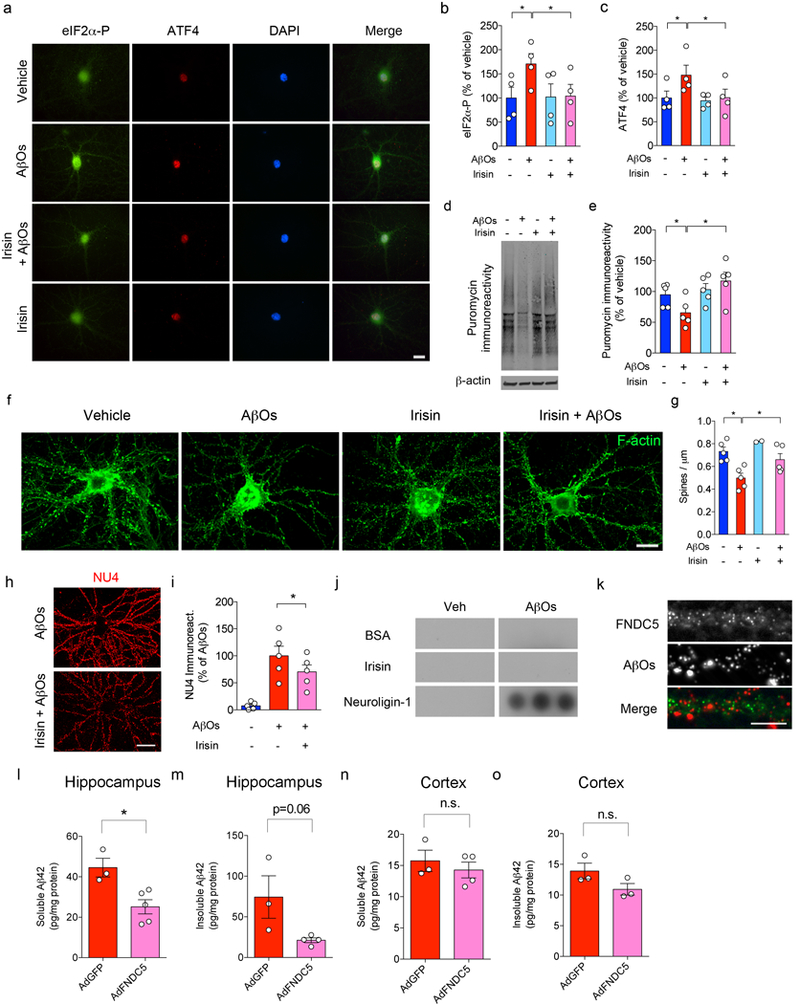
(a) Effect of irisin on AβO-induced increases in eIF2α-P (green) and upregulation of nuclear ATF4 (red). Nuclei were counterstained in blue (DAPI). Scale bar = 5 mm. (b,c) Summary quantification of immunocytochemistry experiments (N = 4 experiments with independent neuronal cultures and AβO preparations). *p<0.05; two-way ANOVA with Holm-Sidak correction; two-sided. Data are represented by mean ± SEM. (d,e) Summary quantification of of protein synthesis in hippocampal neurons, as measured by non-radioactive puromycin incorporation (SUnSET) normalized by β-actin levels (N = 4 experiments with independent hippocampal cultures and AβO preparations). *p<0.05; two-way ANOVA with Holm-Sidak correction; two-sided. Data are represented by mean ± SEM. (f,g) Representative images of dendritic spines in hippocampal neurons, as measured by F-actin labeling with Alexa-conjugated phalloidin (N = 5 experiments with independent neuronal cultures and AβO preparations). Scale bar = 20 mm. *p<0.05, two-way ANOVA. Data are shown as mean ± SEM. At least 30 neurons were analyzed per condition per experiment in immunocytochemistry experiments. (h,i) AβO binding to cultured hippocampal neurons, as detected by AβO-sensitive antibody NU4 (red), after treatment with recombinant irisin (25 nM). Scale bar = 10 mm. The experiments were repeated 5 times with similar results. (i) Summary quantification of 5 experiments with independent neuronal cultures and AβO preparations. Data are shown as mean ± SEM. *p<0.05; paired one-way ANOVA; two-sided. (j) AβO interaction with different proteins in a plate-binding assay. BSA was used as a negative control, while neuroligin-1 was used as a positive control (N = 3 experiments with independent AβO preparations)68. Representative dots were cropped from the same film. See Source Data 7 for original data. (k) Double immunocytochemistry co-localization between AβOs (red) and surface FNDC5 (green) in primary cultured hippocampal neurons (3 experiments with independent neuronal cultures and AβO preparations, with 20-25 images (from 2-3 coverslips) per experiment). Scale bar = 5 mm. The experiments were repeated 3 times with similar results. (l-o) Levels of soluble (l,n) and insoluble Aβ42 (m,o) in and hippocampus (N = 3 for AdGFP, 5 for AdFNDC5) and cortex (N = 3 for AdGFP, 4 for AdFNDC5) of APP/PS1 M146L mice. (m,o) Levels of insoluble Aβ42 in the hippocampus (N = 3 for AdGFP, 4 for AdFNDC5) (l) and cortex (N = 3 per group) (o) of APP/PS1 M146L mice. Data are shown as mean ± SEM. *p<0.05; Student’s t-test; two-sided.
Extended Data Figure 10. Exercise blocks AβO-induced memory impairment in mice.
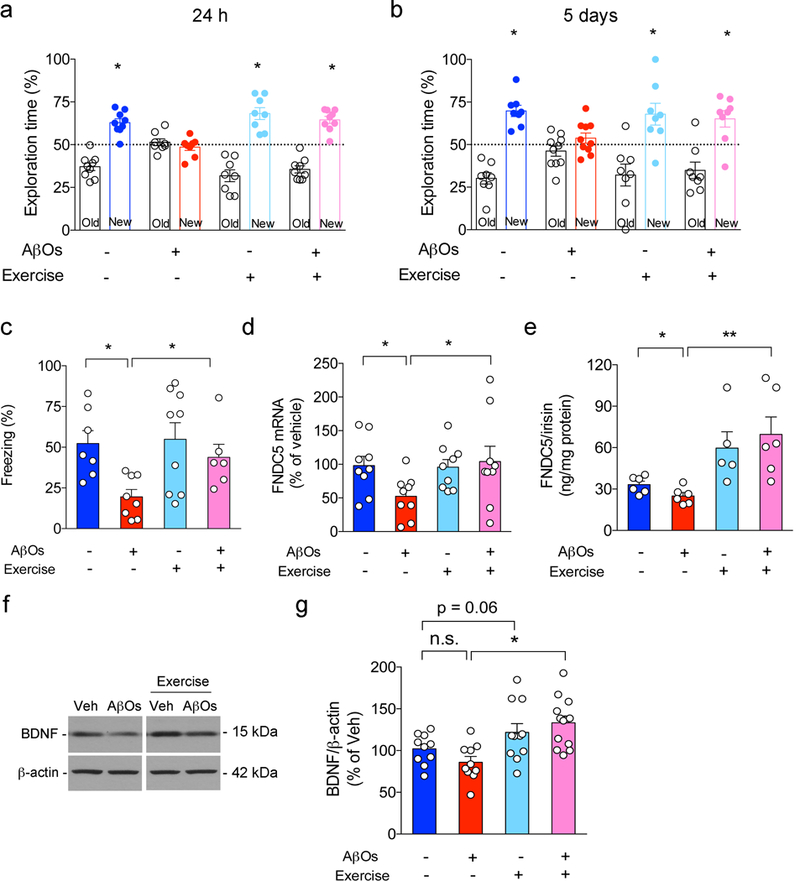
Effect of exercise (swimming; 5 weeks, 5 days/week, 1 h/day) on memory impairment induced by i.c.v. infusion of AβOs in Swiss mice. (a-c) Novel object recognition assessed 24 hours (N = 9 mice for vehicle, 8 for AβOs, 8 for exercise, 9 for exercise+AβOs) (a) or 5 days post infusion of AβOs (N = 8 mice for vehicle, 10 for AβOs, 8 for exercise and 8 for exercise+AβOs) (b). Data are shown as mean ± SEM. *p<0.05; one-sample Student’s t-test; (c) Contextual fear conditioning assessed 7 days post infusion of AβOs (N = 7 mice for vehicle, 8 for AβOs, 9 for exercised and 6 for exercised+AβOs). *p>0.05; two-way ANOVA followed by Holm-Sidak post-test; two-sided). (d,e) Hippocampal FNDC5 mRNA (d) (N = 9 mice per group) and FNDC5/irisin protein levels (e; measured by ELISA) after 5 weeks of exercise (N = 6 mice each for vehicle, AβOs and exercise+AβOs, and 5 for exercise) in male Swiss mice that received 10 pmol AβOs i.c.v. (f,g) Hippocampal BDNF levels in exercised mice (N = 10 mice for vehicle and AβOs, 11 for exercise, 13 for exercise+AβOs). Data are shown as mean ± SEM. *p<0.05; **p<0.01, two-way ANOVA followed by Holm-Sidak post-test; two-sided. Representative bands were cropped from the same membrane. See Source Data 8 for original data.
Supplementary Material
Acknowledgements
This work was supported by grants from Alzheimer’s Society Canada (to F.G.d.F.) and the Weston Brain Institute (to F.G.d.F.), National Institute for Translational Neuroscience (INNT/Brazil) (465346/2014-6 to STF and FGF), Human Frontiers Science Program (to F.G.d.F.), International Society for Neurochemistry (CAEN 1B to M.V.L.), National Institutes of Health (NIH-R01NS049442 to OA), Canadian Institutes of Health Research (CIHR MOP 136940 and MOP 89919 to V.F.P. and M.A.M.P.), and from the Brazilian funding agencies Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (451195/2017-5 to M.V.L., 406436/2016-9 to S.T.F., and 473324/2013-0 to F.G.d.F.) and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (202.817/2016 to M.V.L., 201.432/2014 to S.T.F. and 202.944/2015 to F.G.d.F.). R.L.F, G.B.d.F., G.C.K., F.C.R., J.R.C., D.B., and L.F.-G. were supported by fellowships granted by FAPERJ, CNPq or Comissão de Aperfeiçoamento de Pessoal de Nível Superior (CAPES/Brazil; financial code 001). S.M. was supported by an NIH T32 grant AG057461. We acknowledge the University of Kentucky Alzheimer’s Disease Center (UK-ADC) and its Neuropathology Core, which is supported by NIH/NIA P30 AG028383, for brain samples. We thank W.L. Klein (Northwestern University, IL) for the kind gift of oligomer-specific NU4 antibodies, B.M. Spiegelman (Harvard University, MA) for sharing AdGFP and AdFNDC5 adenoviral constructs, and J. Wang (Queen’s University, ON, Canada) for help with mass spectrometry analyses. We also thank A. Lepelley, M. Oliveira, M. Melo, A.C. Rangel and the CENABIO team for technical and/or administrative assistance.
Footnotes
Data availability
Mass spectrometry raw data (Figure 1; Extended Data Figure 1) and full blots (Figure 2; Extended Data Figures 3,4 and 10) are available as source data files accompanying this paper. Additional data that support the findings of this study are available from the corresponding author upon reasonable request. Requests of datasets obtained from human research will be subject to additional reviews steps by the Institutional Review Board who has granted permit for a particular research. Please contact the corresponding authors for additional information.
Competing Interests
The authors declare no competing interests.
References
- 1.Prince M, et al. The global prevalence of dementia: a systematic review and meta-analysis. Alzheimer’s & Dementia 9, 63–75 e62 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Ferreira ST, Lourenco MV, Oliveira MM & De Felice FG Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Frontiers in Cellular Neuroscience 9, 191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandez AM & Torres-Alemán I The many faces of insulin-like peptide signalling in the brain. Nature Reviews Neuroscience 13, 225–239 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Biessels GJ & Reagan LP Hippocampal insulin resistance and cognitive dysfunction. Nature Reviews Neuroscience 16, 660–671 (2015). [DOI] [PubMed] [Google Scholar]
- 5.McEwen BS Preserving neuroplasticity: Role of glucocorticoids and neurotrophins via phosphorylation. Proceedings of the National Academy of Sciences 112, 15544–15545 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.During MJ, et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nature Medicine 9, 1173–1179 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Chiu S-L, Chen C-M & Cline HT Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 58, 708–719 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grillo CA, et al. Hippocampal insulin resistance impairs spatial learning and synaptic plasticity. Diabetes 64, 3927–3936 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irving AJ & Harvey J Leptin regulation of hippocampal synaptic function in health and disease. Philos Trans R Soc Lond B Biol Sci 369, 20130155 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lourenco MV, Ferreira ST & De Felice FG Neuronal stress signaling and eIF2α phosphorylation as molecular links between Alzheimer’s disease and diabetes. Progress in Neurobiology 129, 37–57 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Bomfim TR, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Aβ oligomers. Journal of Clinical Investigation 122, 1339–1353 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Talbot K, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. Journal of Clinical Investigation 122, 1316–1338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Felice FG, Lourenco MV & Ferreira ST How does brain insulin resistance develop in Alzheimer’s disease? Alzheimer’s & Dementia 10, S26–S32 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Wadman M US government sets out Alzheimer’s plan. Nature 485, 426–427 (2012). [DOI] [PubMed] [Google Scholar]
- 15.De Felice FG Alzheimer’s disease and insulin resistance: translating basic science into clinical applications. Journal of Clinical Investigation 123, 531–539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boström P, et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481, 463–468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jedrychowski Mark P., et al. Detection and quantitation of circulating human irisin by tandem mass spectrometry. Cell Metabolism 22, 734–740 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wrann Christiane D., et al. Exercise Induces Hippocampal BDNF through a PGC-1α/FNDC5 Pathway. Cell Metabolism 18, 649–659 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen K, et al. Irisin protects mitochondria function during pulmonary ischemia/reperfusion injury. Science Translational Medicine 9, eaao6298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee P, et al. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metabolism 19, 302–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schumacher MA, Chinnam N, Ohashi T, Shah RS & Erickson HP The structure of irisin reveals a novel intersubunit beta-sheet fibronectin type III (FNIII) dimer: implications for receptor activation. Journal of Biological Chemistry 288, 33738–33744 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mucke L & Selkoe DJ Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harbor Perspectives in Medicine 2, 1–17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sebollela A, et al. Amyloid-β oligomers induce differential gene expression in adult human brain slices. Journal of Biological Chemistry 287, 7436–7445 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colaianni G, et al. The myokine irisin increases cortical bone mass. Proceedings of the National Academy of Sciences 112, 12157–12162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Figueiredo CP, et al. Memantine rescues transient cognitive impairment caused by high-molecular-weight Aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. Journal of Neuroscience 33, 9626–9634 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lourenco MV, et al. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metabolism 18, 831–843 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Jankowsky JL, et al. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomolecular Engineering 17, 157–165 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Cheng A, et al. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nature Communications 3, 1250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holcomb L, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nature Medicine 4, 97–100 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Ma T, et al. Suppression of eIF2α kinases alleviates Alzheimer’s disease–related plasticity and memory deficits. Nature Neuroscience 16, 1299–1305 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang W, et al. Repression of the eIF2α kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer’s disease. Neurobiology of Aging 41, 19–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trinh MA & Klann E Translational control by eIF2α kinases in long-lasting synaptic plasticity and long-term memory. Neurobiology of Learning and Memory 105, 93–99 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gong B, et al. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. Journal of Clinical Investigation 114, 1624–1634 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vitolo OV, et al. Amyloid β-peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proceedings of the National Academy of Sciences 99, 13217–13221 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaefer N, et al. The malleable brain: plasticity of neural circuits and behavior – a review from students to students. Journal of Neurochemistry 142, 790–811 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Katsnelson A, De Strooper B & Zoghbi HY Neurodegeneration: From cellular concepts to clinical applications. Science Translational Medicine 8, 364ps318 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Selkoe DJ Alzheimer’s disease is a synaptic failure. Science 298, 789–791 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Lepeta K, et al. Synaptopathies: synaptic dysfunction in neurological disorders. Journal of Neurochemistry 138, 785–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 63, 514–525 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Timmons JA, Baar K, Davidsen PK & Atherton PJ Is irisin a human exercise gene? Nature 488, E9–E10 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Albrecht E, et al. Irisin – a myth rather than an exercise-inducible myokine. Scientific Reports 5, 8889 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin KC & Kandel ER Cell adhesion molecules, CREB, and the formation of new synaptic connections. Neuron 17, 567–570 (1996). [DOI] [PubMed] [Google Scholar]
- 43.Suwabe K, et al. Rapid stimulation of human dentate gyrus function with acute mild exercise. Proceedings of the National Academy of Sciences 115, 10487–10492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Praag H, Fleshner M, Schwartz MW & Mattson MP Exercise, energy intake, glucose homeostasis, and the brain. Journal of Neuroscience 34, 15139–15149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neufer PD, et al. Understanding the cellular and molecular mechanisms of physical activity-induced health benefits. Cell Metabolism 22, 4–11 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Baker LD, et al. Effects of aerobic exercise on mild cognitive impairment: a controlled trial. Archives of Neurology 67, 71–79 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buchman AS, et al. Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology 78, 1323–1329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okonkwo OC, et al. Physical activity attenuates age-related biomarker alterations in preclinical AD. Neurology 83, 1753–1760 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muller S, et al. Relationship between physical activity, cognition, and Alzheimer pathology in autosomal dominant Alzheimer’s disease. Alzheimer’s & Dementia (2018), in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mattson Mark P. Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metabolism 16, 706–722 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moon HY, et al. Running-Induced Systemic Cathepsin B Secretion Is Associated with Memory Function. Cell Metabolism 24, 332–340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sleiman SF, et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. eLife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith RW, Wang J, Bucking CP, Mothersill CE & Seymour CB Evidence for a protective response by the gill proteome of rainbow trout exposed to X-ray induced bystander signals. Proteomics 7, 4171–4180 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Mendes ND, et al. Free-floating adult human brain-derived slice cultures as a model to study the neuronal impact of Alzheimer’s disease-associated Aβ oligomers. Journal of Neuroscience Methods 307, 203–209 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Drummond C, et al. Deficits in narrative discourse elicited by visual stimuli are already present in patients with mild cognitive impairment. Frontiers in Aging Neuroscience 7, 96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ledo JH, et al. Amyloid-β oligomers link depressive-like behavior and cognitive deficits in mice. Molecular Psychiatry 18, 1053–1054 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ledo JH, et al. Cross talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer’s amyloid-β oligomers in mice. Journal of Neuroscience 36, 12106–12116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trinchese F, et al. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. Journal of Clinical Investigation 118, 2796–2807 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alamed J, Wilcock DM, Diamond DM, Gordon MN & Morgan D Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nature Protocols 1, 1671–1679 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Puzzo D, et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer’s disease mouse model. Journal of Neuroscience 29, 8075–8086 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Madeira C, et al. d-serine levels in Alzheimer’s disease: implications for novel biomarker development. Translational Psychiatry 5, e561 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seixas Da Silva GS, et al. Amyloid-β oligomers transiently inhibits AMP-activated kinase and causes metabolic defects in hippocampal neurons. Journal of Biological Chemistry 292, 7395–7406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gong B, et al. Ubiquitin hydrolase Uch-L1 rescues β-amyloid-induced decreases in synaptic function and contextual memory. Cell 126, 775–788 (2006). [DOI] [PubMed] [Google Scholar]
- 64.De Felice FG, et al. Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. Journal of Biological Chemistry 282, 11590–11601 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Lambert MP, et al. Monoclonal antibodies that target pathological assemblies of Aβ. Journal of Neurochemistry 100, 23–35 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Abràmoff MD, Magalhães PJ & Ram SJ Image processing with Image J. Biophotonics International 2004, 1–7 (2004). [Google Scholar]
- 67.Livak KJ & Schmittgen TD Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔC(T)) method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 68.Brito-Moreira J, et al. Interaction of amyloid-β (Aβ) oligomers with neurexin 2α and neuroligin 1 mediates synapse damage and memory loss in mice. Journal of Biological Chemistry 292, 7327–7337 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schmidt EK, Clavarino G, Ceppi M & Pierre P SUnSET, a nonradioactive method to monitor protein synthesis. Nature Methods 6, 275–277 (2009). [DOI] [PubMed] [Google Scholar]
- 70.Beckman D, et al. Prion protein modulates monoaminergic systems and depressive-like behavior in mice. Journal of Biological Chemistry 290, 20488–20498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.