

Abstract
Ribonucleotide reductase (RR) catalyses the rate-limiting step of dNTP synthesis, establishing it as an important cancer target. While RR is traditionally inhibited by nucleoside-based antimetabolites, we recently discovered a naphthyl salicyl acyl hydrazone-based inhibitor (NSAH) that binds reversibly to the catalytic site (C-site). Here we report the synthesis and in vitro evaluation of 13 distinct compounds (TP1-13) with improved binding to hRR over NSAH (TP8), with lower KD’s and more predicted residue interactions. Moreover, TP6 displayed the greatest growth inhibiting effect in the Panc1 pancreatic cancer cell line with an IC50 of 0.393 µM. This represents more than a 2-fold improvement over NSAH, making TP6 the most potent compound against pancreatic cancer emerging from the hydrazone inhibitors. NSAH was optimised by the addition of cyclic and polar groups replacing the naphthyl moiety, which occupies the phosphate-binding pocket in the C-site, establishing a new direction in inhibitor design.
Keywords: Ribonucleotide reductase, cancer, inhibitor, pancreatic, phosphate-binding
Introduction
Human ribonucleotide reductase (hRR) is a ubiquitous multi-subunit enzyme that is crucial for cell division and DNA repair1–4. RR catalyses the rate-determining step of dNTP synthesis by removing the 2′ hydroxyl of the ribose to generate a deoxyribose1. RR is critical for the maintenance of a balanced nucleotide pool in cells where imbalances lead to mutator phenotypes1,5,6. RR activity is maintained under tight regulation at the levels of transcription7, allostery1, cellular localisation8, and enzyme inhibition by Sml1 in Saccharomyces cerevisiae9. hRR is a multi-subunit enzyme consisting of a large subunit hRRM1, containing a catalytic site (C-site) and two allosteric sites known as the specificity site (S-site) and the activity site (A-site)1 (Figure 1). hRRM1 associates with the small subunit hRRM2 that houses a free radical essential for catalysis to form the holoenzyme10. ATP and dATP bind the A-site, inducing active and inactive hexamers, respectively11. Binding of dGTP, dTTP, dATP, or ATP to the S-site not only induces dimerisation of hRRM1 but also acts as a selection gate, regulating the relative Kcat/Km for the four NDP substrates: ADP, GDP, CDP, or UDP, respectively1.
Figure 1.

Structure of hRRM1 dimer.
Due to the crucial role hRR plays in replication, it is a major target for cancer chemotherapy12–14. One of the first described hRR inhibitors was hydroxyurea, which targets the di-iron cluster of the hRRM2 subunit to block catalysis15,16. The most common approach for developing inhibitors in the past few decades has involved modifying natural nucleoside substrates, resulting in the production of the clinically used drugs such as gemcitabine, fludarabine, clofarabine, and cladrabine17–23. Gemcitabine is a billion-dollar drug used as a front line treatment of pancreatic cancer24. Gemcitabine derives its main cytotoxicity through DNA chain termination25,26, irreversible inhibition of hRR27,28 and its interactions with several phosphate-binding proteins, such as deoxycytidine deaminase (dCMP deaminase)29, thymidylate synthase30, CTP-synthase31,32, and topoisomerase-133,34. The chain termination activity and lack of specificity exhibited by gemcitabine and other nucleoside-based drugs contribute to the toxic side effects that patients endure17–22. Thus, we have proposed that the development of a selective RR-targeted inhibitor may reduce toxic side effects and expand the therapeutic window35.
Our lab has set out to discover a new class of inhibitors that has higher selectivity for hRRM1, moving away from the nucleoside-based analogues and towards reversible, competitive, non-nucleoside inhibitors. An in silico drug screen of 350,000 compounds from the Cincinnati chemical library (formerly the Proctor and Gamble chemical library) was performed using a human RR structure complexed with the effector TTP and the substrate GDP (PDB code 3HND)35,36. From this screen came the discovery of our lead inhibitor, NSAH35. Crystallographic and kinetic studies indicated that NSAH binds reversibly to the C-site of hRR and acts as a competitive inhibitor. The x-ray crystal structure of NSAH bound to hRRM1 was determined to 2.7 Å resolution (PDB code 5TUS)35. Through chemical mimicry, NSAH occupies the space where the diphosphate ribose and base of the substrate bind in the C-site. NSAH adopts an S-shaped conformation, thermodynamically favouring the E-isomer. The crystal structure reveals that stabilisation of this conformation is due to a strong hydrogen bond between the carbonyl of NSAH and residues Ser217 and Cys218 and a hydrogen bond between the hydroxyl of the phenol of NSAH and residues Cys218 and Ser217.
This study also revealed that the salicyl acyl hydrazine moiety has a significant contributory role in the inhibitory activity against RR. NSAH inhibits cancer cell growth with IC50s within two-fold of gemcitabine and leads to the depletion of deoxypurine pools, a hallmark of cellular hRR inhibition. Unlike gemcitabine, NSAH demonstrated little measurable toxicity against normal mobilised peripheral blood progenitor cells, giving NSAH a higher therapeutic index than found for gemcitabine35. Nevertheless, medicinal chemistry and synthetic approaches can be used to improve the potency of NSAH and its target selectivity towards hRR. Indeed, a recent paper from our laboratory reported on a library of 25 NSAH analogues whose modifications were designed to target residues within the C-site of hRR that are important for interactions with natural substrates37. These results indicated that those analogues which demonstrated a 2–4 fold improved potency of cell-free hRR over NSAH all showed hydrogen bonding with Ser606, Thr607, and Ser217, residues that are known to hydrogen bond with natural substrates.
The present study investigated the structure activity relationship of a new library of compounds to explore the chemical diversity that can target a compound to the phosphate- and ribose-binding domains within the C-site. One structure-guided approach to this goal was to modify the naphthalene ring of NSAH by dissociating the fused ring, providing a biphenyl moiety with different types of substitutions (Group 1). Furthermore, the phenyl ring was replaced by thiophene and furan using a bioisosterism strategy (Figure 2(A)). To understand the effect of the linker on RR modulation, the hydrazide group was replaced by diacylhydrazine or thiazole (shown in purple, Figure 2(A), Group 2). Meanwhile, some polar rings such as pyridine, adenine, isatin and 2-pyridone were linked by hydrazide or diacylhydrazine to explore the structure-activity relationship. As a result, compounds TP1-13 (Figure 2(B)) were synthesised and relevant assays to characterise their interaction with hRRM1 were conducted in this study. Using docking studies to explore possible interactions with hRRM1, it was determined that this library of compounds displayed an increase in the interactions with the phosphate-binding region of the C-site, and the best binding compounds make use of strong interactions to either the phosphate-binding region or residues near loop 2. Cancer cell studies indicated that group 1 compounds showed the greatest potency in cells, where polar substituents incorporating electronegative elements distinguished the more cytotoxic compounds. In fact, the most potent inhibitors from this class demonstrated up to a two-fold improvement in potency against the growth inhibition of pancreatic cancer cells (Panc1) relative to NSAH. The results of this study will lead to the design of future generations of compounds that further improve on target hRR inhibition and cytotoxic efficacy.
Figure 2.

Schematic and structures of synthesised compounds.
Materials and methods
Synthesis and characterisation of TP1-13
Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were recorded with Bruker Fourier 500 NMR spectrometers, with chemical shifts in parts per million (δ) downfield from tetramethylsilane (TMS), the internal standard (Supplemental pages S10–35). High-resolution mass spectra (HRMS) were recorded with a JEOL (JMS-700) mass spectrometer (Supplemental pages S36–48). The purities of the final compounds were determined using an Agilent 1100 series HPLC system with a C-18 column (Agilent ZORBAX Eclipse XDB-C18 5 μm, 4.6 mm × 150 mm) and were found to be ≥95%. Flash column chromatography was conducted using silica gel (Merck Kieselgel 60, No. 9385, 230 − 400 mesh ASTM). All reactions were conducted under an atmosphere of dry N2.
(E)-2-Hydroxy-N'-(2-oxoindolin-3-ylidene)benzohydrazide (TP1)
A mixture of methyl salicylate (1.0 eq), N2H4 (1.1 eq), and EtOH was heated to reflux until the reaction complete. The reaction mixture was filtrated and washed with EtOH to afford a white solid. The resultant was re-suspended in EtOH, and then isatin (1.1 eq) was added. The resulting mixture was heated to reflux until the reaction was complete (12 h). The reaction was quenched by adding H2O and the resulting suspension was filtered and washed by EtOH to obtain TP1 (42%). mp = 320.4 °C (decomposed); 1H NMR (500 MHz, DMSO-d6) δ 6.92 (d, J = 8.0 Hz, 1H), 6.97 (t, J = 8.0 Hz, 1H), 7.01 (d, J = 8.5 Hz, 1H), 7.08 (t, J = 8.0 Hz, 1H), 7.35 (dt, J = 1.0, 8.0 Hz, 1H), 7.44 (dt, J = 1.5, 8.5 Hz, 1H), 7.57 (d, J = 7.5 Hz, 1H), 7.97 (dd, 1.0, 7.5 Hz, 1H), 11.13 (s, 1H), 11.64 (s, 1H), 14.35 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 111.34, 111.59, 115.75, 117.28, 117.51, 117.87, 120.14, 120.71, 120.95, 121.23, 122.35, 122.86, 124.17, 131.82, 131.85, 131.94, 133.26, 134.70, 134.93, 137.49, 139.03, 142.86, 144.35, 156.51, 157.14, 162.03, 163.36, 165.28. HRMS (ESI) for C15H12N3O3 [M + H]+ calculated 282.0879, found 282.0880.
(E)-2-Hydroxy-N'-((4-hydroxy-[1,1'-biphenyl]-3-yl)methylene)-benzohydrazide (TP2)
The title compound was obtained in 63% overall yield from compound 14 in a manner similar to that described for the preparation of TP1 (reaction time: 12 h): mp = 273.5 − 274.9 °C; 1H NMR (500 MHz, DMSO-d6) δ 6.96–7.01 (m, 2H), 7.05 (d, J = 8.5 Hz, 1H), 7.32 (t, J = 7.5 Hz, 1H), 7.43–7.48 (m, 3H), 7.62–7.65 (m, 3H), 7.88 (d, J = 2.0 Hz, 1H), 7.91 (dd, J = 1.0, 8.0 Hz, 1H), 8.76 (s, 1H), 11.34 (s, 1H), 11.80–11.86 (m, 1H), 12.11 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 116.15, 117.54, 117.78, 119.49, 126.62, 127.34, 127.77, 129.09, 129.37, 130.37, 132.00, 134.45, 139.90, 149.23, 157.61, 159.50, 165.07. HRMS (ESI) for C20H17N2O3 [M + H]+ calculated 333.1239, found 333.1241.
(E)-2-Hydroxy-N'-(2-hydroxy-5-(thiophen-3-yl)-benzylidene)-benzohydrazide (TP3)
The title compound was obtained in 70% overall yield from compound 14 in a manner similar to that described for the preparation of TP1 (reaction time: 12 h): mp = 282.9 − 284.6 °C; 1H NMR (500 MHz, DMSO-d6) δ 6.96–7.01 (m,3H), 7.46 (dt, J = 1.0, 8.0 Hz, 1H), 7.52 (d, J = 5.0 Hz, 1H), 7.61 (dd, J = 3.0, 4.5 Hz, 1H), 7.67 (dd, J = 2.0, 8.5 Hz, 1H), 7.75 (d, J = 1.5 Hz, 1H), 7.88–7.92 (m, 2H), 8.72 (s, 1H), 11.33 (s, 1H), 11.86–12.08 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ 116.17, 117.43, 117.78, 119.27, 119.49, 120.01, 126.47, 127.31, 127.45, 129.10, 129.97, 134.45, 141.22, 149.35, 157.24, 159.46, 165.05. HRMS (ESI) for C18H15N2O3S [M + H]+ calculated 339.0803, found 339.0804.
(E)-N'-(6-(furan-3-yl)-2-hydroxy-3-methoxybenzylidene)-2-hydroxybenzohydrazide (TP4)
The title compound was obtained in 61% overall yield from compound 14 in a manner similar to that described for the preparation of TP1 (reaction time: 12 h): mp = 228.3 °C (decomposed); 1H NMR (500 MHz, DMSO-d6) δ 3.83 (s, 3H), 6.70 (s, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.94–6.99 (m, 2H), 7.09 (d, J = 8.5 Hz, 1H), 7.44–7.48 (m, 1H), 7.81–7.85 (m, 3H), 8.79 (s, 1H), 11.71 (s, 1H), 12.18 (s, 1H),12.51 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 56.37, 112.98, 114.89, 115.62, 115.69, 117.84, 119.38, 120.65, 123.60, 126.44, 128.73, 134.61, 141.32, 143.96, 147.82, 149.33, 150.38, 159.79, 165.18. HRMS (ESI) for C19H17N2O5 [M + H]+ calculated 353.1137, found 353.1139.
(E)-2-Hydroxy-N'-((3-hydroxy-4-methoxy-[1,1'-biphenyl]-2-yl)methylene)-benzohydrazide (TP5)
The title compound was obtained in 58% overall yield from compound 14 in a manner similar to that described for the preparation of TP1 (reaction time: 12 h): mp = 257.9 − 259.4 °C; 1H NMR (500 MHz, DMSO-d6) δ 3.85 (s, 3H), 6.77 (d, J = 8.0 Hz, 1H), 6.91 (t, J = 7.5 Hz, 1H), 6.94 (d, J = 8.5 Hz, 1H), 7.12 (d, J = 8.5 Hz, 1H), 7.34–7.37 (m, 2H), 7.42–7.51 (m, 4H), 7.78 (dd, J = 1.0, 8.0 Hz, 1H), 8.54 (s, 1H), 11.64 (s, 1H), 12.16 (s, 1H), 12.56 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 56.39, 114.70, 115.34, 115.61, 117.82, 119.31, 120.82, 127.77, 128.67, 128.91, 130.36, 134.59, 135.97, 139.53, 147.90, 149.22, 150.16, 159.84, 165.30. HRMS (ESI) for C21H19N2O4 [M + H]+ calculated 363.1345, found 363.1347.
(E)-2-Hydroxy-N'-(2-hydroxy-3-methoxy-6-(thiophen-3-yl)benzylidene)-benzohydrazide (TP6)
The title compound was obtained in 75% overall yield from compound 14 in a manner similar to that described for the preparation of TP1 (reaction time: 12 h): mp = 229.5 °C; 1H NMR (500 MHz, DMSO-d6) δ 3.83 (s, 3H), 6.82 (d, J = 8.5 Hz, 1H), 6.91–6.97 (m, 2H), 7.08 (d, J = 8.5 Hz, 1H), 7.20 (d, J = 5.0 Hz, 1H), 7.44 (dt, J = 1.5, 8.5 Hz, 1H), 7.52(d, J = 1.5 Hz, 1H), 7.67 (dd, J = 3.0, 5.0 Hz, 1H), 7.81 (d, J = 8.0 Hz, 1H), 8.67 (s, 1H), 11.69 (s, 1H), 12.19 (s, 1H), 12.55 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 56.39, 114.72, 115.60, 115.64, 117.84, 119.35, 120.78, 124.66, 126.79, 128.71, 130.09, 130.63, 134.60, 139.82, 147.87, 149.24, 150.29, 159.83, 165.27. HRMS (ESI) for C19H16N2NaO4S [M + Na]+ calculated 391.0728, found 391.0731.
(E)-N'-(4–(6-amino-9H-purin-9-yl)benzylidene)-2-hydroxybenzohydrazide (TP7)
The title compound was obtained in 33% overall yield from compound 14 in a manner similar to that described for the preparation of TP1 (reaction time: 12 h): mp = 229.5 °C; 1H NMR (500 MHz, DMSO-d6) δ 6.95–7.01 (m, 2H), 7.41–7.47 (m, 3H), 7.90–7.97 (m, 3H), 8.08 (d, J = 8.5 Hz, 2H), 8.25 (s, 1H), 8.54 (s, 1H), 8.68 (s, 1H), 11.79 (s, 1H), 11.91 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 116.55, 117.74, 119.46, 119.88, 123.27, 128.74, 129.13, 133.39, 134.29, 137.00, 139.87, 148.14, 149.60, 153.73, 156.86, 159.38, 165.21. HRMS (ESI) for C19H16N7O2 [M + H]+ calculated 374.1365, found 374.1367.
(E)-2-Hydroxy-N'-((2-hydroxynaphthalen-1-yl)methylene)-benzohydrazide (TP8)
The title compound was obtained in 78% overall yield from compound 14 in a manner similar to that described for the preparation of TP1 (reaction time: 12 h): mp = 223.8 °C; 1H NMR (500 MHz, DMSO-d6) δ 6.97–7.04 (m, 2H), 7.23 (d, J = 9.0 Hz, 1H), 7.38–7.42 (m, 1H), 7.46 (t, J = 7.5 Hz, 1H), 7.59 (t, J = 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.91–7.94 (m, 2H), 8.30 (d, J = 8.5 Hz, 1H), 9.54 (s, 1H), 11.93 (s, 2H), 12.73 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 109.08, 116.17, 117.79, 119.37, 119.61, 121.43, 124.03, 128.21, 128.29, 129.24, 129.40, 132.18, 133.39, 134.49, 148.17, 158.62, 159.29, 164.48. HRMS (ESI) for C18H15N2O3 [M + H]+ calculated 307.1083, found 307.1085.
2–(5-(4-Methoxyphenyl)thiazol-2-yl)-phenol (TP9)
A mixture of 2-hydroxybenzonitrile (1.0 eq), O,O-diethyl dithiophosphate (2.0 eq), and H2O was stirred at 80 °C until reaction was complete (4 h). The reaction mixture was cooled, quenched with saturated NaHCO3, and extracted by ethyl acetate (3 times). The organic layer was collected and dried under reduced pressure. The resulting residue was dissolved in EtOH, and then 4-methoxyphenacyl bromide (1.0 eq) was added. The mixture was heated to reflux until the reaction was complete (4 h). The solvent was removed under reduced pressure. The resulting residue was purified by recrystallization with hexane and MeOH to afford a white solid TP9 (63%). mp = 95.2 − 96.8 °C; 1H NMR (500 MHz, DMSO-d6) δ 3.84 (s, 3H), 6.94 (dt, J = 1.0, 8.0 Hz, 1H), 7.00–7.03 (m, 3H), 7.33 (dt, J = 1.0, 7.5 Hz, 1H), 7.64 (s, 1H), 7.77 (dd, J = 1.0, 7.5 Hz, 1H), 7.84 (d, J = 9.0 Hz, 2H), 11.30 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 54.41, 109.78, 113.91, 116.96, 117.10, 119.32, 126.18, 126.94, 127.10, 131.36, 154.01, 156.33, 160.15, 168.19. HRMS (ESI) for C16H14NO2S [M + H]+ calculated 284.0745, found 284.0748.
(E)-N'-((2-Hydroxynaphthalen-1-yl)methylene)-4–(2-oxopyridin-1(2H)-yl)-benzohydrazide (TP10)
The title compound was obtained in 40% overall yield from compound 14 in a manner similar to that described for the preparation of TP1 (reaction time: 12 h): mp = 258.6 °C; 1H NMR (500 MHz, DMSO-d6) δ 6.36 (t, J = 6.5 Hz, 1H), 6.52 (d, J = 9.0 Hz, 1H), 7.25 (d, J = 9.0 Hz, 1H), 7.42 (t, J = 7.5 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 7.60–7.73 (m, 4H), 7.90 (d, J = 8.0 Hz, 1H), 7.94 (d, J = 9.0 Hz, 1H), 8.12 (d, J = 8.0 Hz, 2H), 8.26 (d, J = 8.0 Hz, 1H), 9.54 (s, 1H), 12.36 (s, 1H), 12.73 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 106.34, 109.02, 119.37, 121.11, 121.18, 124.04, 127.53, 128.28, 128.31, 128.89, 129.46, 132.11, 132.69, 133.31, 139.12, 141.29, 144.13, 147.63, 158.55, 161.51, 162.20. HRMS (ESI) for C23H18N3O3 [M + H]+ calculated 384.1348, found 384.1351.
N'-(2-Hydroxy-1-naphthoyl)-nicotinohydrazide (TP11)
The title compound was obtained in 55% overall yield from compound 14 in a manner similar to that described for the preparation of TP1 (reaction time: 3 h): mp = 227.1 − 228.6 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.04 (d, J = 7.5 Hz, 1H), 7.13 (d, J = 8.5 Hz, 1H), 7.35 (d, J = 9.0 Hz, 1H), 7.41 (t, J = 7.5 Hz, 1H), 7.49 (dt, J = 1.5, 9.0 Hz, 1H), 7.54 (dt, J = 1.0, 8.0 Hz, 1H), 7.88 (dd, J = 1.5, 7.5 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.5 Hz, 1H), 8.08 (d, J = 9.0 Hz, 1H), 10.41 (s, 1H), 10.80 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 103.07, 110.14, 117.64, 118.82, 120.29, 123.72, 124.17, 128.05, 128.58, 128.97, 129.26, 132.65, 133.94, 134.20, 156.83, 157.04, 161.88, 164.22. HRMS (ESI) for C17H13N3NaO3 [M + Na]+ calculated 330.0855, found 330.0850.
(E)-2-Hydroxy-N'-(pyridin-3-ylmethylene)-1-naphthohydrazide (TP12)
The title compound was obtained in 45% overall yield from compound 14 in a manner similar to that described for the preparation of TP1 (reaction time: 6 h): mp = 202.8 − 204.5 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.18 (d, J = 9.0 Hz, 1H), 7.31 (d, J = 7.5 Hz, 1H), 7.44–7.50 (m, 2H), 7.78 (d, J = 8.0 Hz, 1H), 7.82–7.86 (m, 2H), 8.22 (s, 1H), 8.35 (d, J = 8.0 Hz, 1H), 8.54 (d, J = 4.0 Hz, 1H), 8.85 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 114.48, 117.49, 123.11, 123.21, 124.14, 127.15, 127.92, 128.22, 130.92, 131.65, 132.00, 134.76, 145.03, 148.45, 149.99, 153.14, 166.39. HRMS (ESI) for C17H14N3O2 [M + H]+ calculated 292.1086, found 292.1088.
N'-(2-Hydroxybenzoyl)-nicotinohydrazide (TP13)
A mixture of methyl salicylate (1.0 eq), N2H4 (1.1 eq), and EtOH was heated to reflux until the reaction was complete (2 h). The reaction mixture was filtered and washed with EtOH to afford a white solid. To a mixture of the resulting product (1.0 eq), K2CO3 (1.2 eq) and p-dioxane nicotinoyl chloride (1.2 eq) was added and the resultant mixture was heated at 40 °C until the reaction was complete (4 h). The reaction mixture was quenched with H2O and extracted by ethyl acetate (3 times). The organic layer was collected, dried over MgSO4, and filtered. The resulting filtrate was dried under reduced pressure to obtain TP13 as a white solid (75%). mp = 216.6 − 217.7 °C; 1H NMR (500 MHz, DMSO-d6) δ 6.95–7.00 (m, 2H), 7.46 (t, J = 7.5 Hz, 1H), 7.57 (dd, J = 5.0, 8.0 Hz, 1H), 7.92 (d, J = 7.0 Hz, 1H), 8.26 (d, J = 8.0 Hz, 1H), 8.78 (d, J = 4.0 Hz, 1H), 9.08 (d, J = 1.0 Hz, 1H), 10.73 (s, 1H), 10.90 (s, 1H), 11.83 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 115.18, 117.84, 119.59, 124.19, 128.51, 128.94, 134.66, 135.75, 148.93, 153.06, 159.53, 164.62, 167.91. HRMS (ESI) for C13H12N3O3 [M + H]+ calcd 258.0879, found 258.0880.
Compound preparation and solubility determination
For each compound, stock solutions were made in 100% DMSO to assure full solubility. Spectra of standard solutions in 100% acetonitrile were taken using UV spectroscopy. An extinction coefficient was calculated from the plot of the absorbance vs drug concentration using Beer’s law (Supplemental Table 1).
KD values were determined in KD buffer [50 mM TRIS, pH 8.0, 5 mM MgCl2, 5% glycerol and 10 mM DTT]. Because the solubility of the compounds was limited in this buffer, the solubility was empirically determined as follows. Increasing amounts of a compound were dissolved in 1.5 ml of KD buffer. The solutions were mixed by vortex and centrifuged at 5000×g for 10 min, and the supernatant was promptly removed and absorbance measured by UV/Vis spectrophotometry. Using the extinction coefficients, the concentration of soluble drug was calculated for each total concentration (Supplemental Figure 1). Drug solubility measurements were carried out in duplicate. Plots of measured vs predicted concentrations were plotted in Origin graphing software. Samples having precipitates that did not pellet were excluded from the list of 13 compounds examined in this manuscript.
Protein expression and purification of hRRM1
The hRRM1 protein was expressed in Escherichia coli Bl21- codon plus (DE3)-RIL cells and purified using a peptide affinity column, as previously described in Fairman et al.38. The homogenous protein was pooled and concentrated to 0.2 mg/mL, quantified using UV spectroscopy.
KD determination by fluorescence quenching
The dissociation constant (KD) was measured for each of the compounds using a tryptophan florescence quenching assay, as described previously with hRRM1 at 0.2 mg/mL36. Tryptophan florescence spectra of hRRM1 were measured using a Horiba Fluoromax-4, 1155D-3113 FM spectrophotometer after exciting the sample at 295 nm. A background spectrum was recorded with protein in KD buffer followed by the incremental addition of compounds (0–150 µM) at room temperature. The data were fitted in Origin graphing software using a quadratic form of the equilibrium binding equation
where A is the amplitude of the reaction, c is the concentration of enzyme, x is the substrate concentration, y is the percent of fluorescence quenching and K is the KD. Measurements were made in duplicate to estimate error. KD’s were corrected for the measured concentration of soluble compound from the concentration of total added compound. Error of the corrected KD’s were calculated using the error propagation equation
Where σx is the error of the corrected KD, x is the corrected KD, σa is the error of the slope of the drug concentration plot, a is the slope of the drug concentration plot, σb is the error of the measured KD and b is the measured KD.
Cancer cell line growth inhibition assay
Growth inhibition assays were performed in the Translational Research Shared Resource of the Case Comprehensive Cancer Centre. Human pancreatic cancer cells (Panc1) were maintained in standard growth medium consisting of (RPMI1640 + 10% FBS + 2 mM glutamine + 100 U/mL penicillin, and 100 µg/mL streptomycin). Cells were monitored and shown to be negative for mycoplasma contamination using the Mycoplasma Detection kit (MycoAlert™, Lonza, Basel, Switzerland). For growth inhibition assays, cells were harvested by trypsinisation and seeded into 96-well tissue culture plates at 2500 cells/mL. The following day, triplicate wells were treated with an appropriate volume of 5x-inhibitor-containing medium. The cells were cultured for 3 additional days at 37˚C in a 5% CO2 humidified incubator. Cell growth was assessed by measuring total DNA content per well using an adaptation of the method of Labarca and Paigen39.
In silico docking studies
In silico docking of the compounds was performed using the Glide module of the Schrödinger 2017–3 modelling software suite as previously described36,40–42. The docking site for the C-site was defined as a 5 Å box centred on the lead compound NSAH (TP8) bound to the hRRM1 (PDB code 5TUS)37. Compounds were docked to the C-site using Glide SP. Hits were scored by a glide scoring function and examined for their interactions with the C-site residues using Maestro.
Results
Rationale for compound design and synthesis
Our previous study identified NSAH (TP8), containing a 2-hydroxybenzohydrazide moiety, as an RR modulator. Its structure contains three parts, namely: 2-hydroxybenzoyl group, hydrazide linkage, and naphthalene ring. In order to understand structure-activity relationships, specifically the influence of the hydrazide moiety, we designed diacylhydrazine-containing TP11 and TP13 and thiazole derivative TP9 (Figure 2(B)). Furthermore, hydrazide derivatives (TP1-8, TP10 and TP12) linked with biaryls or various heterocycles were also designed. Their syntheses are illustrated in Scheme 1A and 1B. Methyl salicylate (14) was reacted with hydrazine to obtain 2-hydroxybenzohydrazide (15) which was subjected to reaction with various aldehydes or isatin to give the corresponding products TP1-8 and TP10 (Figure 2(B)). Methyl 2-hydroxy-1-naphthoate (16) underwent a similar synthetic route to afford compound TP12 (Figure 2(B)). Meanwhile, the reaction of 15 and 17 with nicotinoyl chloride yielded, respectively, compounds TP13 and TP11, which possess a diacylhydrazine linkage (Figure 2(B)). The reaction of 2-hydroxybenzonitrile (18) with O,O-diethyl dithiophosphate, which generated the corresponding thioamide product, was followed by cyclisation with 4-methoxyphenacyl bromide to furnish compound TP9. These analogues were designed to enhance interactions with the C-site (Figure 1).
Scheme 1.

Synthetic approach to compounds TP1–13.
In silico compound docking interactions with hRRM1
Potential interactions of TP1-13 with hRRM1 were investigated using the Schrödinger docking software suite glide40–42. A hRRM1 dimer with lead compound NSAH bound to the C-site (PDB ID: 5TUS) was used as a model for the docking of the new compounds35. It was assumed that the primary site of binding for these compounds would be the C-site, because NSAH and all previously studied derivatives of NSAH have demonstrated a preference for the C-site35,37. Our studies showed that compounds TP2–7, 9–10 are longer and therefore extend beyond NSAH, allowing for the possibility of more interactions (Figure 3(A–D) and Supplemental Figure 1(B–F)). TP3 was not predicted to bind well to the phosphate-binding site in comparison to other new compounds (Figure 3(A), Supplemental Figure 2(C)). However, docking predicted it to have an increase in the number of strong interactions closer to the loop 2 region. TP7 was unique in that it was predicted to interact strongly in the phosphate-binding site with the residues: Thr607, Ser448, Ser606, and near the loop 2 region with residues: Pro294, Ala245, as well as to a residue to which NSAH exhibits strong hydrogen bonding (Ser217) (Figure 3(C), Supplemental Figure 2(G)). This is partly because TP7 extends to the outer extremities of the catalytic site (C-site). Thus, for all compounds except TP3, there was either an increase in the number of predicted C-site interactions and/or an increase in the number of strong binding interactions to the phosphate-binding site of the C-site, in comparison to the interactions that NSAH makes (Figures 3(A–D), Supplemental Figure 1(A–I), Supplemental Figure 2(A–M)).
Figure 3.

Predicted binding interactions of TP compounds at the C-site of hRRM1.
Investigation of compound binding to hRRM1 using intrinsic tryptophan fluorescence
TP1-13 were analysed for their ability to bind to hRRM1 and therefore quench its intrinsic tryptophan fluorescence. Using the criterion that ligands quenching at least 25% of the fluorescence have sufficient affinity for hRRM1, TP1-13 were characterised as binders of hRRM1. TP1, 4, 6, 7, 10, and 12 quenched at least 90% of the tryptophan fluorescence (Figure 4(A–B)). TP2, 5, 8, 9, 11, and 13 each quenched approximately 80% (Figure 4(C)), and TP3 quenched approximately 70% of tryptophan fluorescence. While percentage of quenching indicates the ability of a compound to bind to the enzyme, measuring the concentration of a compound needed to quench the fluorescence by half provides the KD, a measure of the affinity of a compound for the enzyme. Corrected KD’s (see Methods) ranged from 2.9 ± 0.39 – 22.0 ± 5.67 µM, where all of the new compounds have a lower KD than NSAH (22.0 ± 5.67 µM) (Table 1). The reduction in the KD indicates that the chemical modifications targeting the phosphate-binding site and loop 2 resulted in better binding to hRRM1 as compared to NSAH (Table 1).
Figure 4.

Quenching of tryptophan fluorescence of hRRM1 by TP ligands.
Table 1.
Summary of experimental data and molecular docking and pharmacokinetic data derived from the Schrödinger software.
| Chemical structure | Name | Molecular weight | Measured KD (µM) | Corrected KD (µM) | Cell based IC50 (µM) | Docking Score | LogP (o/w) | Membrane permeability: caco-2 (nm/sec) | Membrane permeability: MDCK (nm/sec) | Rotatable bonds | Hydrogen bond acceptors | Hydrogen bond donors |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group 1 | ||||||||||||
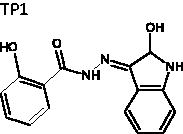 |
(E)-2-hydroxy-N'-(2-oxoindolin-3-ylidene)benzohydrazide | 281.08 | 14.97 ± 1.8 | 6.2 ± 0.77 | >10 | −6.055 | 2.556 | 191 | 82 | 4 | 3.25 | 1 |
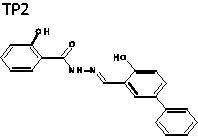 |
(E)-2-hydroxy-N'-((4-hydroxy-[1,1'-biphenyl]-3-yl)methylene)benzohydrazide | 332.12 | 36.02 ± 3.4 | 8.7 ± 2.4 | 2.400 | −6.294 | 4.163 | 358 | 163 | 7 | 3 | 2 |
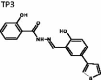 |
(E)-2-hydroxy-N'-(2-hydroxy-5-(thiophen-3-yl)benzylidene)benzohydrazide | 338.07 | 53.81 ± 8.0 | 5.3 ± 1.42 | 2.316 | −6.613 | 4.04 | 358 | 330 | 6 | 3 | 2 |
 |
(E)-N'-(6-(furan-3-yl)-2-hydroxy-3-methoxybenzylidene)-2-hydroxybenzohydrazide | 352.11 | 31.48 ± 1.7 | 9.3 ± 0.56 | 0.765 | −6.025 | 3.534 | 458 | 213 | 7 | 4.25 | 2 |
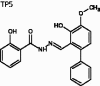 |
(E)-2-hydroxy-N'-((3-hydroxy-4-methoxy-[1,1'-biphenyl]-2-yl)methylene)benzohydrazide | 362.13 | 35.44 ± 1.9 | 4.8 ± 0.33 | 1.461 | −6.451 | 4.33 | 442 | 205 | 8 | 3.75 | 2 |
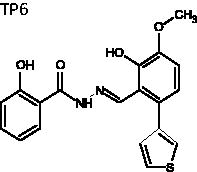 |
(E)-2-hydroxy-N'-(2-hydroxy-3-methoxy-6-(thiophen-3-yl)benzylidene)benzohydrazide | 368.08 | 30.0 ± 5.0 | 9.4 ± 1.6 | 0.393 | −6.461 | 4.29 | 437 | 406 | 7 | 3.75 | 2 |
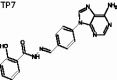 |
(E)-N'-(4-(6-amino-9H-purin-9-yl)benzylidene)-2-hydroxybenzohydrazide | 373.13 | 19.30 ± 1.96 | 4.0 ± 1.06 | >10 | −7.816 | 2.404 | 65 | 26 | 6 | 6.25 | 3 |
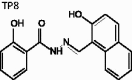 |
(E)-2-hydroxy-N'-((2-hydroxynaphthalen-1-yl)methylene)benzohydrazide | 306.32 | 31.29 ± 2.8 | 22.0 ± 5.67 | 1.075 | −6.293 (cis) | 3.505 | 471 | 219 | 6 | 3 | 2 |
| Group 2 | ||||||||||||
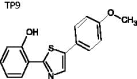 |
2-(5-(4-methoxyphenyl)thiazol-2-yl)phenol | 283.34 | 70.16 ± 8.8 | 9.9 ± 2.07 | >20 | −6.764 | 3.799 | 3064 | 2325 | 2 | 3 | 1 |
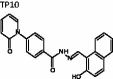 |
(E)-N'-((2-hydroxynaphthalen-1-yl)methylene)-4-(2-oxopyridin-1(2H)-yl)benzohydrazide | 383.13 | 36.76 ± 2.0 | 9.7 ± 1.36 | 2.920 | −5.907 | 3.707 | 351 | 160 | 5 | 6.25 | 2 |
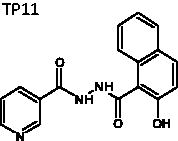 |
N'-(2-hydroxy-1-naphthoyl)nicotinohydrazide | 307.31 | 89.65 ± 4.9 | 4.1 ± 0.24 | >10 | −5.969 | 2.73 | 277 | 124 | 4 | 4.75 | 0.5 |
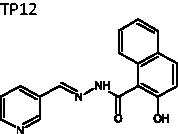 |
(E)-2-hydroxy-N'-(pyridin-3-ylmethylene)-1-naphthohydrazide | 291.30 | 27.44 ± 2.0 | 16.3 ± 1.3 | >10 | −5.230 | 3.362 | 620 | 295 | 5 | 3.75 | 1 |
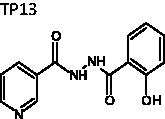 |
N'-(2-hydroxybenzoyl)nicotinohydrazide | 257.25 | 60.62 ± 3.1 | 2.9 ± 0.39 | >10 | −5.335 | 1.75 | 218 | 95 | 4 | 4.75 | 0.5 |
Inhibition of growth of Panc1 pancreatic cancer cells
Since gemcitabine is a core component of the current standard of care for pancreatic cancer, and we hoped to improve upon the therapeutic index for drugs treating this disease, we selected Panc1, a cell line derived from pancreatic cancer, for the study of the new compounds. Panc1 cells growing in 96-well plates were exposed to each compound continuously for three days, after which DNA content per well was measured as an indicator of cell growth. As shown in Figure 5 and Table 1, TP1, 7, 9, 11, 12, and 13 did not inhibit the growth of Panc1 cells, even at concentrations of 10 µM. Compounds TP2, 3, and 10 showed moderate growth inhibition with IC50’s between 2 and 3 µM (Figure 5 and Table 1). Compounds TP4–6, and 8 were the most potent against this cell line, with IC50’s below 1.5 µM (Figure 5 and Table 1). Overall, the results indicated that some compounds in group 1 have marked activity against the growth of Panc1 cells, whereas group 2 is mostly ineffective (Figure 5 and Table 1). TP6 demonstrated the greatest potency, with over a 2-fold decrease in IC50 over the lead compound NSAH (TP8) (Figure 5 and Table 1). A few of the compounds were also studied in other cell lines (A549 and HCT-116), showing similar potency as in Panc1 cells (Supplemental Figure 3). This finding suggests that these compounds do not target Panc1 cells specifically, but may be universally active against cancer cells, as would be expected since RR is found in all cells.
Figure 5.

Cancer growth-inhibitory activity of Panc1 pancreatic cancer cell line by TP compounds.
Discussion and conclusions
Decades of effort have been invested into elucidating the structure and physiological function of ribonucleotide reductase, in order to better target inhibitors for therapeutic intervention. While numerous diverse inhibitors have been developed to target hRR and several have been approved for clinical use, there have been few successful attempts to identify a non-nucleoside reversible inhibitor that targets the large subunit of hRR. In a recent paper by Ahmad et al., we reported on a rapid moderate-throughput screen that identified the first non-nucleoside inhibitor binding to the C-site, NSAH35,36. In a subsequent study by Huff et al., a library of compounds was produced, replacing the substituents on either side of the hydrazone moiety of NSAH with polar substituents to examine their effects on activity37. It was determined that replacing the phenol with polar substituents in the ortho position achieved the best activity against hRR. Replacement of the naphthalene with an indole provided similar activity. It was concluded that these derivatives favoured the C-site, just like NSAH, and all derivatives that showed improved activity against hRR in comparison to NSAH had increased interactions in the phosphate-binding site. However, these compounds had limited cellular activity.
Using the co-crystal structure of NSAH bound at the C-site of hRRM1 and its interactions (PDB code 5TUS) as a template in the present study, a small library of compounds was designed to increase the interactions with the C-site (Figure 2). It was anticipated that improved interactions would strengthen the specificity towards hRR and reduce off-target binding. The library consists of two distinct groups. For the Group 1 compounds, the naphthalene of NSAH was replaced in anticipation of creating more interactions with the residues that are important in binding the phosphates of the natural substrates (termed phosphate-binding site). For the Group 2 compounds, the hydrazine linker was modified and polar groups were added to these linkers. In order to determine how these modifications affected interaction with hRR, computational docking and experimental assays were conducted.
To determine the extent of binding of these compounds to the hRRM1 subunit, quenching of the fluorescence of the internal tryptophans of hRRM1 was measured. The KD’s ranged from 2.9 ± 0.39 – 22.0 ± 5.67 µM, indicating that all of the modified compounds bind more tightly than does NSAH (KD- 22.0 ± 5.67 µM) (Table 1). Examination of the docking of these compounds suggests that the overall decrease in KD is influenced by the predicted increase in interactions with the phosphate-binding site within the C-site of hRRM1 compared to the binding of NSAH. With the exception of TP3, all other analogues have stronger interactions with the phosphate-binding site than NSAH (Figure 3(B–D), Supplemental Figure 1(A–I), Supplemental Figure 2(A–M)). Apart from the increase in binding to the phosphate-binding region, no other single binding characteristic explains the trends in relative affinity seen with these 13 compounds; rather, a collection of interactions contribute to the observed binding pattern. All of the compounds with a KD less than 6.2 µM are predicted to make at least one strong hydrogen bond that could contribute to the tight binding to hRRM1. Within this group, TP7 and 11 are predicted to have the most hydrogen bonds, leading to stronger binding to hRRM1 (Figure 3(C) and Supplemental Figure 1(H)). TP7 has hydrogen bonds scattered throughout the length of the compound, interacting with the phosphate-binding site, at Cys218 and Ser217, and near loop 2 (Figure 3(C)). Our previous predictions for optimising interactions at the phosphate-binding site and with loop 2 appears to be validated by this study. TP11 and 13 are the only compounds in this library that have a linker composed of single bonds, allowing for free rotation (Figure 2(B)). With a few exceptions, compounds with a KD less than 9.4 µM form multiple predicted hydrogen bonds. TP3 is one of the exceptions, making fewer strong interactions than NSAH, and not interacting with the phosphate-binding site (Figure 3(A) and Supplemental Figure 2(C)). TP3’s lack of interactions with the phosphate-binding site is compensated by stronger interactions near loop 2. The trend that emerges from the study of this library is that the best hRRM1 binding compounds are either small and flexible (TP1, 11, 13) or long enough to reach loop 2 and/or the phosphate-binding site (TP3–7). Both of these groups of compounds’ options contain polar substituents, but there is a limit to the benefit of size and polarity, because some of the larger molecules had low solubility in the aqueous medium of the fluorescence assay.
A common challenge in drug development is that interaction with a target and inhibitory activity against a cell-free enzyme does not always predict how the drug will perform when tested in a cell or animal model. Thus, another approach we employed was to query the structures of the compounds in our library using QikProp, an algorithm of the Schrödinger suite to predict the cell permeability of these compounds in Caco-2 and MDCK cells (Table 1)43. With some exceptions, the algorithm was useful in predicting which compounds might be the most effective against Panc1 cells. With the exception of TP1 and 7, group 1 had the greatest potency in Panc1 cells with IC50’s below 2.5 µM (Figure 5 and Table 1). TP6, which had the greatest potency, had the greatest predictable permeability for both Caco-2 and MDCK cells within the top seven compounds (Table 1). TP4–6 and 8 have similar predicted permeabilities, with the exception of TP6, which has a greater permeability for MDCK cells (Table 1). The compounds with IC50’s between 2 and 3 µM are predicted to be less permeable than TP4–6 and 8 (Table 1). Out of the compounds that had cellular IC50’s greater than 10 µM (TP1, 7, 9, 11–13) only T9 and 12 had good predicted permeability, even greater than that of TP6 (Table 1). These results suggest that theoretical prediction of permeability does not always predict activity in cells. The lack of activity against cells could be due to an assortment of factors, including poor cell permeability, drug metabolism in the cell rendering them inactive, or being good substrates for efflux pumps.
TP4–6, which were the most potent against growth of Panc1 cells, have very similar structures (Figure 2(B)). These compounds maintain the phenol and the hydrazine linker of NSAH, but in place of the naphthalene is a guaiacol methoxyphenol linked to a furan, benzene, or thiophene. The docking of these compounds to the hRRM1 C-site provides evidence that they bind similarly to each other, revealing the importance of this modification. To further examine this modification and its contribution to enhanced binding and cellular potency, TP2 and 3 were compared. TP2 and 3 mimic TP5 and 6, respectively, but lack the methoxy group on the guaiacol methoxyphenol (Figure 2(B)). The removal of the methoxy appears to cause over a 2-fold decrease in potency in cells as well as the loss of a similar docking pattern (Table 1, Supplemental Figure 1(B) and Figure 3(A)). In the study by Huff et al., a few compounds contained a methoxy group, although on a different position, which caused a similar type of docking pattern37. However, while the methoxy seems to be an important entity and might be essential for better potency, it does not do so alone. TP9, which contains a methoxy group on a benzene ring, is not only ineffective in Panc1 cells but also has a completely different predicted pattern of binding to the C-site (Figure 5, Table 1 and Supplemental Figure 1(E) and 2(I)). This suggests that methoxylation of the benzene ring aids in improving cell and enzyme potency but acts in concert with other polar entities. The addition of the hydroxyls as well as the polar rings aids in the increase in potency. Considering the effects of TP4–6 in the cells, it might be suggested that a polar ring with an electronegative element might further increase the relative potency.
It is well known that phenolics have potentials for being chemotherapeutic agents; however, these compounds often have poor bioavailability in vivo44. Typically, phenolics are easily oxidised, leaving them vulnerable to chemical degradation or bacterial decomposition in the GI tract, most commonly by glucuronidation and sulphation. TP1–8, 13 contain phenolic acids, which proposes a risk of poor bioavailability when administered in vivo, eliminating an oral administration and restricting it to IV administration. As an oral compound is more desirable for patients for its ease of use, it might be beneficial to modify these compounds if they exhibit poor bioavailability in mouse studies. Modifying these compounds by converting the phenolic to esters or ethers might produce a pro-drug and increase the bioavailability of the phenolic metabolites.
A PAINS filter (http://cbligand.org/PAINS/), which is a now well-defined way to identify the presence of potentially problematic functional groups in analogue design, was used on our compound library. Despite our lead compound, NSAH, being a potential PAINS candidate, our studies have validated this compound as an RR inhibitor binding to the C-site of hRR1 and inhibiting RR in cells35,37. This validation adds confidence that TP1–13 would bind to the hRR1 C-site as docking to this site displayed favourable poses and possibly inhibits RR in cells. When developing compounds against RR, there is always a concern that these compounds contain chelating properties that may affect the Fe and free radical housed in the small subunit, a crucial element needed for the activity of RR. There have been many reports of agents that inhibit RR by this mechanism32,45–49. As this was a concern, NSAH was tested for its ability to chelate metals37. Results indicated that NSAH does not chelate Fe3+, the iron form present in RR, up to concentrations of 5 mM nor does it bind or sequester Mg2+ ions, which are also critical for RR activity. As these concerns are not an issue for the lead compound NSAH, we predict that these will not be an issue with TP1–13 either.
It is generally understood that targeting RR for cancer therapeutics is a challenging task as RR is present in all cells, normal and malignant. We have previously shown that the lead compound NSAH (TP8) was much less effective against blood progenitor cells than against cancer cell lines, demonstrating that NSAH has a higher therapeutic index than gemcitabine in the same cell-cell comparison35. This suggests that NSAH has selective activity in malignant cells, exploiting the greater proliferation in cancer cells50. Accordingly, the new class of inhibitors designed from NSAH might provide a superior, safer anticancer treatment. Results from this study indicate that replacing the naphthalene ring of NSAH with other cyclic ring structures containing methoxy and polar constituents results in analogues with superior activity against cancer cells. Future studies will elucidate cyclic and polar constituents which may provide further improvements in this class of RR inhibitors.
Supplementary Material
Funding Statement
This study was supported by NIH funding to P.I.: Dr. Chris G. Dealwis (R01GM100887), Case Comprehensive Cancer Centre CTSC pilot grant and Council to Advance Human Health. Drs. Lee and Dealwis were funded by the Taipei Medical School, Taiwan-CWRU grant. This research was supported in part by the Translational Research Shared resource of the Case Comprehensive Cancer Centre (P30CA043703) and Center for Scientific Review.
Acknowledgements
The authors thank Dr. William E. Harte (School of Medicine, CWRU) for insightful comments and suggestions during the development of this project. The authors thank Drs. J. Mieyal, R. Viswanathan, S. Huff, M. Kumar, and D. Wald, for useful discussions. This work made use of the High Performance Computing Resource in the Core Facility for Advanced Research Computing at Case Western Reserve University.
Disclosure statement
The authors report no declarations of interest and also declare no competing financial interest.
References
- 1.Brown NC, Reichard P. Role of effector binding in allosteric control of ribonucleoside diphosphate reductase. J Mol Biol 1969;46:39–55. [DOI] [PubMed] [Google Scholar]
- 2.Stubbe J, Ge J, Yee CS. The evolution of ribonucleotide reduction revisited. Trends Biochem Sci 2001;26:93–9. [DOI] [PubMed] [Google Scholar]
- 3.Jordan A, Reichard P. Ribonucleotide reductases. Annu Rev Biochem 1998;67:71–98. [DOI] [PubMed] [Google Scholar]
- 4.Kolberg M, Strand KR, Graff P, Andersson KK. Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta 2004;1699:1–34. [DOI] [PubMed] [Google Scholar]
- 5.Weinberg G, Ullman B, Martin DW. Jr,.: Mutator phenotypes in mammalian cell mutants with distinct biochemical defects and abnormal deoxyribonucleoside triphosphate pools. Proc Natl Acad Sci USA 1981;78:2447–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunz BA, Kohalmi SE, Kunkel TA, et al. International Commission for Protection Against Environmental Mutagens and Carcinogens. Deoxyribonucleoside triphosphate levels: a critical factor in the maintenance of genetic stability. Mutat Res 1994;318:1–64. [DOI] [PubMed] [Google Scholar]
- 7.Huang M, Zhou Z, Elledge SJ. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 1998;94:595–605. [DOI] [PubMed] [Google Scholar]
- 8.Yao R, Zhang Z, An X, et al. Subcellular localization of yeast ribonucleotide reductase regulated by the DNA replication and damage checkpoint pathways. Proc Natl Acad Sci USA 2003;100:6628–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 1998;2:329–40. [DOI] [PubMed] [Google Scholar]
- 10.Brown NC, Canellakis ZN, Lundin B, et al. Ribonucleoside diphosphate reductase. Purification of the two subunits, proteins B1 and B2. Eur J Biochem 1969;9:561–73. [DOI] [PubMed] [Google Scholar]
- 11.Holmgren A, Reichard P, Thelander L. Enzymatic synthesis of deoxyribonucleotides, 8. The effects of ATP and dATP in the CDP reductase system from E. coli. Proc Natl Acad Sci USA 1965;54:830–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jordheim LP, Guittet O, Lepoivre M, et al. Increased expression of the large subunit of ribonucleotide reductase is involved in resistance to gemcitabine in human mammary adenocarcinoma cells. Mol Cancer Ther 2005;4:1268–76. [DOI] [PubMed] [Google Scholar]
- 13.Jordheim LP, Seve P, Tredan O, Dumontet C. The ribonucleotide reductase large subunit (RRM1) as a predictive factor in patients with cancer. Lancet Oncol 2011;12:693–702. [DOI] [PubMed] [Google Scholar]
- 14.Shao J, Zhou B, Chu B, Yen Y. Ribonucleotide reductase inhibitors and future drug design. Curr Cancer Drug Targets 2006;6:409–31. [DOI] [PubMed] [Google Scholar]
- 15.Stearns B, Losee KA, Bernstein J. Hydroxyurea. A new type of potential antitumour agent. J Med Chem 1963;6:201. [DOI] [PubMed] [Google Scholar]
- 16.Donehower RC. An overview of the clinical experience with hydroxyurea. Semin Oncol 1992;19:11–9. [PubMed] [Google Scholar]
- 17.Huang P, Chubb S, Plunkett W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J Biol Chem 1990;265:16617–25. [PubMed] [Google Scholar]
- 18.Heinemann V, Xu YZ, Chubb S, et al. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2',2'-difluorodeoxycytidine. Mol Pharmacol 1990;38:567–72. [PubMed] [Google Scholar]
- 19.Parker WB, Shaddix SC, Chang CH, et al. Effects of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl)adenine on K562 cellular metabolism and the inhibition of human ribonucleotide reductase and DNA polymerases by its 5'-triphosphate. Cancer Res 1991;51:2386–94. [PubMed] [Google Scholar]
- 20.Parker WB, Shaddix SC, Rose LM, et al. Comparison of the mechanism of cytotoxicity of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl)adenine, 2-chloro-9-(2-deoxy-2-fluoro- beta-D-ribofuranosyl)adenine, and 2-chloro-9-(2-deoxy-2,2-difluoro- beta-D-ribofuranosyl)adenine in CEM cells. Mol Pharmacol 1999;55:515–20. [PubMed] [Google Scholar]
- 21.Griffig J, Koob R, Blakley RL. Mechanisms of inhibition of DNA Synthesis by 2-chlorodeoxyadenosine in human lymphoblastic cells. Cancer Res 1989;49:6923–8. [PubMed] [Google Scholar]
- 22.Avery TL, Rehg JE, Lumm WC, et al. Biochemical pharmacology of 2-chlorodeoxyadenosine in malignant human hematopoietic cell lines and therapeutic effects of 2-bromodeoxyadenosine in drug combinations in mice. Cancer Res 1989;49:4972–8. [PubMed] [Google Scholar]
- 23.Xie C, Plunkett W. Metabolism and actions of 2-chloro-9-(2-deoxy-2-fluoro-beta-D- arabinofuranosyl)-adenine in human lymphoblastoid cells. Cancer Res 1995;55:2847–52. [PubMed] [Google Scholar]
- 24.King RS. Gemcitabine. New first-line therapy for pancreatic cancer. Cancer Pract 1996;4:353–4. [PubMed] [Google Scholar]
- 25.Baker CH, Banzon J, Bollinger JM, et al. 2'-Deoxy-2'-methylenecytidine and 2'-deoxy-2',2'-difluorocytidine 5'-diphosphates: potent mechanism-based inhibitors of ribonucleotide reductase. J Med Chem 1991;34:1879–84. [DOI] [PubMed] [Google Scholar]
- 26.Gandhi V, Plunkett W. Modulatory activity of 2',2'-difluorodeoxycytidine on the phosphorylation and cytotoxicity of arabinosyl nucleosides. Cancer Res 1990;50:3675–80. [PubMed] [Google Scholar]
- 27.Wang J, Lohman GJ, Stubbe J. Enhanced subunit interactions with gemcitabine-5'-diphosphate inhibit ribonucleotide reductases. Proc Natl Acad Sci USA 2007;104:14324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heinemann V, Schulz L, Issels RD, Plunkett W. Gemcitabine: a modulator of intracellular nucleotide and deoxynucleotide metabolism. Semin Oncol 1995;22:11–8. [PubMed] [Google Scholar]
- 29.Heinemann V, Xu YZ, Chubb S, et al. Cellular elimination of 2',2'-difluorodeoxycytidine 5'-triphosphate: a mechanism of self-potentiation. Cancer Res 1992;52:533–9. [PubMed] [Google Scholar]
- 30.Honeywell RJ, Ruiz van Haperen VW, Veerman G, et al. Inhibition of thymidylate synthase by 2',2'-difluoro-2'-deoxycytidine (Gemcitabine) and its metabolite 2',2'-difluoro-2'-deoxyuridine. Int J Biochem Cell Biol 2015;60:73–81. [DOI] [PubMed] [Google Scholar]
- 31.Heinemann V. Ongoing selective internal radiation therapy-based studies in the treatment of liver-dominant metastatic colorectal cancer. Future Oncol (London, England) 2014;10:37–9. [DOI] [PubMed] [Google Scholar]
- 32.Heinemann V, Plunkett W. Modulation of deoxynucleotide metabolism by the deoxycytidylate deaminase inhibitor 3,4,5,6-tetrahydrodeoxyuridine. Biochem Pharmacol 1989;38:4115–21. [DOI] [PubMed] [Google Scholar]
- 33.Pourquier P, Gioffre C, Kohlhagen G, et al. Gemcitabine (2',2'-difluoro-2'-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clin Cancer Res 2002;8:2499–504. [PubMed] [Google Scholar]
- 34.Pourquier P, Pilon AA, Kohlhagen G, et al. Trapping of mammalian topoisomerase I and recombinations induced by damaged DNA containing nicks or gaps. Importance of DNA end phosphorylation and camptothecin effects. J Biol Chem 1997;272:26441–7. [DOI] [PubMed] [Google Scholar]
- 35.Ahmad MF, Alam I, Huff SE, et al. Potent competitive inhibition of human ribonucleotide reductase by a nonnucleoside small molecule. Proc Natl Acad Sci USA 2017;114:8241–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmad MF, Huff SE, Pink J, et al. Identification of non-nucleoside human ribonucleotide reductase modulators. J Med Chem 2015;58:9498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huff SE, Mohammed FA, Yang M, et al. Structure-guided synthesis and mechanistic studies reveal sweetspots on naphthyl salicyl hydrazone scaffold as non-nucleosidic competitive, reversible inhibitors of human ribonucleotide reductase. J Med Chem 2018;61:666–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fairman JW, Wijerathna SR, Ahmad MF, et al. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat Struct Mol Biol 2011;18:316–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Labarca C, Paigen K. A simple, rapid, and sensitive DNA assay procedure. Anal Biochem 1980;102:344–52. [DOI] [PubMed] [Google Scholar]
- 40.Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47:1739–49. [DOI] [PubMed] [Google Scholar]
- 41.Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 2004;47:1750–9. [DOI] [PubMed] [Google Scholar]
- 42.Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 2006;49:6177–96. [DOI] [PubMed] [Google Scholar]
- 43.Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Adv Drug Deliv Rev 2002;54:355–66. [DOI] [PubMed] [Google Scholar]
- 44.Gao S, Hu M. Bioavailability challenges associated with development of anti-cancer phenolics. Mini Rev Med Chem 2010;10:550–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lepoivre M, Flaman JM, Bobe P, et al. Quenching of the tyrosyl free radical of ribonucleotide reductase by nitric oxide. Relationship to cytostasis induced in tumor cells by cytotoxic macrophages. J Biol Chem 1994;269:21891–7. [PubMed] [Google Scholar]
- 46.Szalai VA, Brudvig GW. Reversible binding of nitric oxide to tyrosyl radicals in photosystem II. Nitric oxide quenches formation of the S3 EPR signal species in acetate-inhibited photosystem II. Biochemistry 1996;35:15080–7. [DOI] [PubMed] [Google Scholar]
- 47.Nyholm S, Mann GJ, Johansson AG, et al. Role of ribonucleotide reductase in inhibition of mammalian cell growth by potent iron chelators. J Biol Chem 1993;268:26200–5. [PubMed] [Google Scholar]
- 48.Hershko C. Control of disease by selective iron depletion: a novel therapeutic strategy utilizing iron chelators. Bailliere's Clin Haematol 1994;7:965–1000. [DOI] [PubMed] [Google Scholar]
- 49.Lundberg JH, Chitambar CR. Interaction of gallium nitrate with fludarabine and iron chelators: effects on the proliferation of human leukemic HL60 cells. Cancer Res 1990;50:6466–70. [PubMed] [Google Scholar]
- 50.Hakansson P, Hofer A, Thelander L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J Biol Chem 2006;281:7834–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.