

Abstract
Background:
B-cell maturation antigen (BCMA) is a tumour necrosis superfamily cell-surface receptor required for plasma cell survival. This study evaluated safety, tolerability and preliminary clinical activity of GSK2857916, a novel anti-BCMA antibody conjugated to microtubule-disrupting agent monomethyl auristatin-F, in patients with relapsed/refractory multiple myeloma (MM).
Methods:
This international, multicentre, open-label, first-in-human Phase 1 study comprised dose escalation (Part 1) and dose expansion (Part 2) phases. Adults with histologically or cytologically confirmed MM, Eastern Cooperative Oncology Group performance status 0/1, and progressive disease following stem cell transplant, alkylators, proteasome inhibitors and immunomodulators were recruited. In Part 1, patients received GSK2857916 (0 03–4 6 mg/kg) via 1-hour intravenous infusion. In Part 2, patients received the selected dose of GSK2857916 (3 4 mg/kg) every 3 weeks. Primary endpoints were maximum tolerated dose (MTD) and recommended Phase 2 dose (RP2D). All patients who received ≥1 dose were included in this prespecified administrative interim analysis (cut-off: 26 June 2017), which was performed for internal purposes. The study is ongoing (NCT02064387).
Findings:
Between July 2014 and February 2017, 73 patients were treated (Part 1 n=38; Part 2 n=35). No MTD was identified in Part 1. Based on safety/clinical activity, 3 4 mg/kg was selected as RP2D. Corneal events were common (42/73; 58%); most (37/42) were Grade 1/2 and did not result in treatment discontinuation in Part 2. The other most common Grade 3/4 events were thrombocytopenia (25/73; 34%) and anaemia (11/73; 15%). There were 12 treatmentrelated serious adverse events and no treatment-related deaths. Overall response rate at 3 4 mg/kg in Part 2 was 60% (21/35; 95% confidence interval: 42 1%–76 1%).
Interpretation:
At the identified RP2D, GSK2857916 is well tolerated and data suggest it has good clinical activity in heavily pretreated patients, thereby indicating that this may be a promising candidate for the treatment of relapsed/refractory MM.
Funding:
GlaxoSmithKline plc
INTRODUCTION
Recent progress in immunotherapy has significantly improved the prognosis for patients with multiple myeloma (MM).1 However, outcomes remain poor for those with relapsed and refractory disease. Patients who are refractory to both proteasome inhibitors (PI) and immunomodulatory drugs (IMiD) have an estimated survival of only 13 months.1 As such, the development of novel therapeutics to treat this disease is critical.
The normal function of B-cell maturation antigen (BCMA) is to promote the survival of B cells at later stages of differentiation, including plasma cells.2 Mice lacking expression of BCMA demonstrate a reduced number of long-lived bone marrow plasma cells but have an otherwise normal phenotype.3 BCMA membrane expression is present on a subset of normal late-stage B cells and is universally detected on normal and malignant plasma cells, including MM cells.2,4,5 Blocking of BCMA signalling has been shown to inhibit cell growth and survival in MM cells in preclinical studies.6 Soluble BCMA has been implicated in the reduction of polyclonal antibody levels and MM-associated immune deficiency.5 Higher BCMA levels are detected in patients with MM with progressive disease when compared with those with responsive disease, and correlate with reduced survival.7 Although the precise physiological implications of BCMA on myeloma cell growth and survival are unknown, the restricted expression profile of BCMA and its survival function in plasma cells make it an attractive target in this disease.
GSK2857916 is a humanised immunoglobulin (Ig)G1 monoclonal antibody-drug conjugate that binds specifically to BCMA.8 The parent antibody (GSK28579164) is conjugated to the tubulin polymerisation inhibitor monomethyl auristatin-F (MMAF) via a protease-resistant maleimidocaproyl linker. Upon binding to the cell surface, GSK2857916 is rapidly internalised and active cytotoxic drug (cys-mcMMAF) is released inside the cell.4 Additionally, the antibody is afucosylated, which increases binding to FcγRIIIa receptors, enhances recruitment and activation of immune effector cells, and enhances the killing of tumour cells by antibody-dependent cellular cytotoxicity.8 The potential immunogenic cell death mechanism has been shown to further induce macrophage-mediated phagocytosis.8 These various mechanisms of action result in significant in vitro and in vivo activity against myeloma cell lines and primary patient myeloma samples.4 Based on these encouraging non-clinical data, we undertook a Phase 1 first-in-human, open-label study to assess the safety and tolerability of GSK2857916 monotherapy in patients with relapsed/refractory MM.
METHODS
Study Design and Participants
This study was performed at 9 centres in the USA, Canada and the United Kingdom (for further details see Supplementary Appendix, p5). The study comprised two parts. The Part 1 dose-escalation phase assessed the safety and tolerability of GSK2857916 to identify the recommended Phase 2 dose (RP2D). Neuenschwander continual reassessment method (N-CRM)9 was utilised to inform the dose-escalation decisions. Part 2 evaluates the safety, tolerability, pharmacokinetics (PK) and preliminary clinical activity of the dose level identified in Part 1. Data for patients with MM are reported here; data for patients with BCMA-positive B-cell lymphoma (currently enrolling in Part 2) will be reported separately.
For Part 1, eligible adult (≥18 years of age) patients had histologically or cytologically confirmed MM, a European Cooperative Oncology Group performance status of 0 or 1, prior therapy with alkylators, PI and IMiD, had undergone stem cell transplant (if eligible) and refractory to the last line of treatment (defined as progressive disease on or within 60 days of completion of the last therapy) that included stem cell transplant. Those patients with a history of autologous stem cell transplant must have received the transplant >100 days prior to study enrolment and have no active infection. Female patients must be of non-childbearing potential or have a negative serum pregnancy test and use effective contraception throughout the study and for 60 days following last study treatment. Male patients with partners of childbearing potential must have had a vasectomy or use effective contraception throughout the study until 60 days following last study treatment. All patients must have adequate organ system function (see protocol, Supplementary Appendix, p19–285), and all prior treatment-related toxicities must be ≤Grade 1 at enrolment (except for alopecia and Grade 2 neuropathy).
For Part 2, patients met the eligibility criteria for Part 1, in addition to having measurable disease as defined by having at least one of the following: serum M-protein ≥0 5 g/dL; urine M-protein ≥200 mg/24h; serum free light chain (FLC) level ≥5 mg/dL and abnormal serum FLC ratio (<0 26 or >1 65); or biopsy proven plasmacytoma.
All patients had adequate hepatic (total bilirubin ≤1 5 times and aspartate transaminase and alanine transaminase ≤1 5 times the upper limit of the normal range), haematological (absolute neutrophil count ≥1 0 × 109/L, haemoglobin ≥8 0 g/dL, and platelets ≥50 × 109/L), and renal function (creatinine clearance ≥50 mL per minute for patients enrolled in Part 2) at screening.
There was no prospective selection of patients based on tumour, or soluble BCMA expression. Further details on inclusion and exclusion criteria are provided in the Supplementary Appendix, p3.
BMA117159 (ClinicalTrials.gov identifier: NCT02064387) was designed by the lead authors and sponsor; the study protocol summary is provided in the Supplementary Appendix, p19–285. It was conducted according to principles of Good Clinical Practice after approval by regulatory authorities and institutional review boards at each study site; no independent data monitoring committee was involved in this study. All patients provided written informed consent.
Information on GSK’s data sharing commitments and requesting access to anonymized individual participant data and associated documents can be found at www.clinicalstudydatarequest.com.
Procedures
GSK2857916 (GSK Manufacturing SpA, Parma, Italy; Baxter Oncology GmbH, Halle/Westphalia, Germany) at doses ranging between 0 03 mg/kg and 4 60 mg/kg was administered as a 1-hour intravenous infusion every 3 weeks for a maximum of 16 cycles. This schedule was chosen based on preclinical testing, patient convenience, and expected PK properties of an IgG1-based antibody-drug conjugate.11,12 The option to explore an additional dose-escalation cohort with a once-weekly dosing schedule was included in the protocol, but after evaluation of clinical and PK data from the every-3-week dosing cohort, it was not pursued. Prophylaxis for infusion-related reactions (IRRs) was not permitted for the first infusion to enable the assessment of the frequency and severity of IRRs with first dose, but could be administered for subsequent infusions at the discretion of the investigator. All patients received steroid eye drops with each infusion to mitigate corneal events, a known toxicity of MMAF (prednisolone phosphate 1% or dexamethasone 0 1% 4 times a day for 4 days, starting 1 day prior to each dose).13 Patients remained on treatment until progression, unacceptable toxicity, consent withdrawal, or completion of 16 doses of treatment. Dose modifications were allowed according to pre-defined criteria based on the nature of the event and toxicity grade.
Data on adverse events (AEs) were collected from the time of first dose administration until 30 days following discontinuation of study treatment. Ophthalmology examinations were conducted prior to the start of each cycle. Laboratory monitoring (haematology, clinical chemistry, urinalysis) was performed on screening, weekly during Cycle 1, then at the start of each subsequent cycle and end of study treatment. AEs were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 4 0.14
Clinical activity of GSK2857916 was assessed in accordance with the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.15 Disease assessment for evaluation of response was completed within 4 weeks prior to the first dose, and at the start of each cycle until the final study visit. In patients with extramedullary disease, assessment included imaging (e.g. computed tomography scan, magnetic resonance imaging, bone scan, plain radiography) prior to Cycles 5, 9 and 13, and physical examination (as indicated for palpable/superficial lesions) at the start of each cycle.
The PK of GSK2857916 (intact antibody-drug conjugate), unbound antibody, total antibody, and free cys-MMAF were assessed from blood samples taken pre-dose and at 1 hour (end of infusion) during Cycles 1–5. Additional samples were collected during Cycle 1 on Days 1, 8, 15 and 22 (Cycle 2 pre-dose).
Soluble BCMA (free and complex) was assessed using mesoscale discovery (MSD) immunoassays pre-dose, at end of infusion, and 24 hours post-end of infusion on Day 1, and on Days 8, 15 and 22; additional samples were collected every 21 days. MSD immuno-assays were validated and performed by GlaxoSmithKline (King of Prussia, PA, USA) (Supplementary Appendix, p3). During validation, the range of free soluble BCMA in healthy donors (n=25) was determined to be 3·23–10·68 ng/mL with a median value of 6·69 ng/mL (see Supplementary Appendix, p4).
Outcomes
The primary endpoints of the trial were to determine the safety, tolerability, maximum tolerated dose (MTD) and RP2D and schedule of GSK2857916. The MTD was defined as the dose which has the highest probability of having a dose-limiting toxicity (DLT) rate within the target toxicity interval and for which the probability that the DLT rate lies within the excessive toxicity or the unacceptable toxicity window is less than 25%. A DLT was considered if the event occurred with the first 21 days of treatment and its relationship to GSK2857916 cannot be excluded. Specific DLT criteria include albuminuria ≥2000 mg/24h (considered unrelated to disease progression); neutropenia (non-febrile, Grade 4 lasting ≥7 days; or febrile lasting ≥72 hours); Grade ≥3 thrombocytopenia; Grade ≥3 non-haematologic toxicity except for: nausea and vomiting, diarrhoea or skin reactions that can be controlled with medication, or clinically asymptomatic electrolyte abnormalities that can be controlled within 48 hours; and liver toxicity meeting pre-specified liver stopping criteria. Further details on DLTs can be found in the protocol, Supplementary Appendix, p19–285. RP2D was defined as MTD, or a dose lower then MTD that provides 95–100% receptor occupancy, with no evidence of target mediated disposition in the PK profile and demonstrating signs of clinical activity. Secondary endpoints were PK profile (single dose area under the curve, maximum serum concentration [Cmax], time to Cmax , clearance, steady-state volume of distribution [Vss], half-life [t½]; repeat dose Cmax and trough plasma concentration), the incidence of anti-drug antibodies, and clinical activity measured as overall response rate (ORR), defined as the percentage of subjects achieving confirmed partial response or better (≥PR) and clinical benefit rate, defined as the percentages of subjects with minimal response or better (≥MR). Data for some PK assessments and the incidence of anti-GSK2857916 will be reported separately. Exploratory endpoints included progression-free survival (PFS) and duration of response; time-to-progression and time-toresponse will be reported separately with the final study report. The dose for Part 2 expansion was chosen based on review of the totality of the safety, tolerability, activity and PK data during Part 1.
Statistical Analysis
The data cut (utilizing the flexible date as specified in the protocol, Section 13.6.2; Supplementary Appendix, p167), was performed when all patients with MM were enrolled, and had at least 4 months’ follow-up while on the study (data cut-off date: 26 June 2017).
Overall, the protocol had three prespecified futility analyses: after approximately 15, 22 and 30 patients are evaluable for response. However, due to a very fast accrual and frequent delays of dosing/assessments only the final one was performed, when 33 subjects were evaluable for response according to protocol criteria. At the time of this futility analysis, 19/33 responders were observed, which meant that the pre-specified futility threshold was passed (≥8 responders out of 33 evaluable subjects; as in Table 17 of the protocol [Supplementary appendix, p166]), and the study continued. Software used included SAS version 9.4, SAS version 9.2 (SAS Institute, Cary, CA, USA) for PK analyses, and Fixed and Adaptive Clinical Trial Simulator (FACTS) version 2.3 or higher for N-CRM model implementation.
The sample size for each dose level in Part 1 was determined by pre-defined criteria with a single subject cohort run-in (cohort size =1 for each dose level) until occurrence of a ≥Grade 2 toxicity, for which a relationship to GSK2857916 cannot be ruled out, and which occurs in one subject in Cycle 1 (21 days). At this point, the cohort size would be expanded to 3 or more subjects and the escalation will continue to follow the N-CRM procedure. Overall, it was anticipated that approximately 30 patients would be required to establish the MTD or RP2D in Part 1 of the study with testing up to the ~4·6 mg/kg dose where clinical activity was expected based on preclinical testing. An additional intermediate dose level of 2∙5 mg/kg was evaluated in Part 1 to investigate the emergent trend towards an increased frequency of higher grade corneal events in treated patients.
Demographics and safety data were descriptively summarised. ORR was reported with two-sided 95% exact confidence intervals (CI). Time-to-event endpoints, including PFS, duration of response, and time to response were analysed descriptively with the Kaplan–Meier method for Part 2 only, retrospectively. Subgroup analyses were performed retrospectively; the results should be interpreted with caution due to small sample sizes. All patients who received at least one dose of GSK2857916 were included in analyses. PK parameters were estimated using model-based population PK methods.
This study is registered with ClinicalTrials.gov, NCT02064387.
Role of the funding source
Study BMA117159 was funded by GlaxoSmithKline. The sponsor was involved in study design, collection and interpretation of data, and the writing of the report. All authors had full access to the data upon request, and were involved in data interpretation, manuscript preparation, revision, and final approval. The authors vouch for the accuracy of the data and adherence to the study protocol. The corresponding author had the final responsibility to submit for publication.
RESULTS
This is an ongoing study; enrolment of patients with MM took place from 29 July 2014 to 21 February 2017. As of the data cut-off date of 26 June 2017, 38 patients received at least one dose in Part 1 and 35 received at least one dose in Part 2 (Figure 1).
Figure 1. CONSORT diagram.
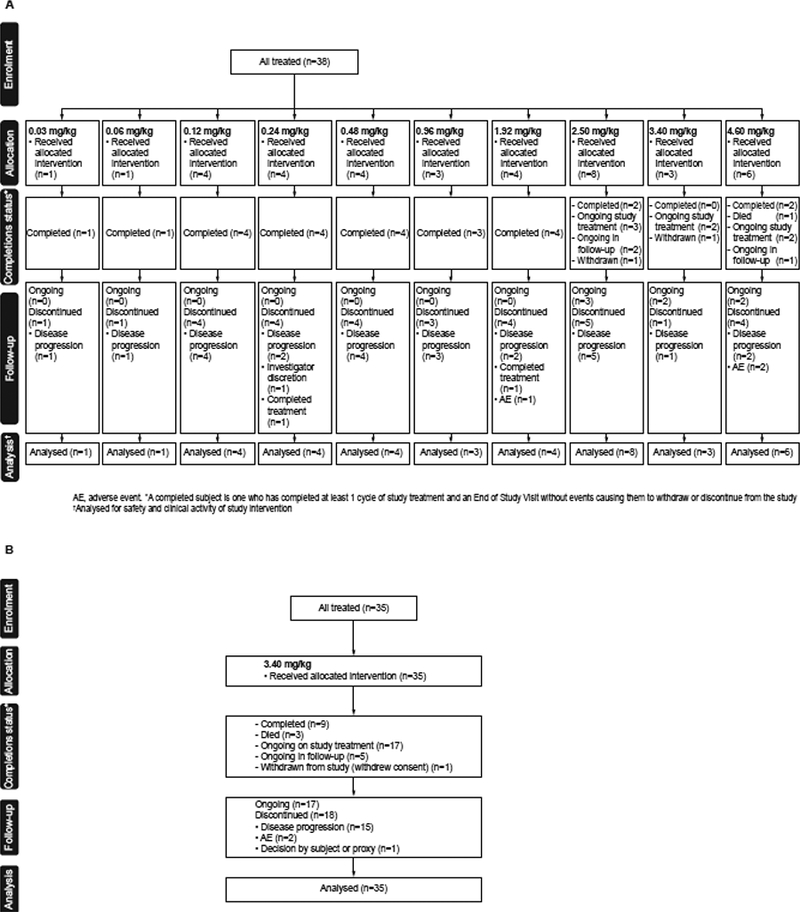
(A) Part 1 (Dose Escalation) and (B) Part 2 (Expansion Cohort)
The baseline characteristics and patient disposition for Part 1 and Part 2 are summarised in Table 1 and Supplementary Appendix, p6. Of the patients in Part 1, most (29/38 [76%] had received ≥5 lines of therapy, and 9/38 (24%) had received prior daratumumab. For patients in Part 2: the median time from diagnosis was 3·8 years (range: 1–10); the majority of patients were heavily pretreated, with 20 (57%) having received ≥5 prior lines of therapy (range: 1–>10); 31(89%) were double-refractory to a PI and IMiD and 13 (37%) were refractory to daratumumab; 12 (34%) had prior daratumumab and were also double refractory to PI and IMiD; and 31 (89%) had previously undergone stem cell transplant.
Table 1.
Patient baseline characteristics and disposition (Part 1 and Part 2)
| Part 1 | Part 2 | |
|---|---|---|
| Characteristic | n=38 | n=35 |
| Age, median (range), years | 60 (39–79) | 60 (46–75) |
| Sex, male / female, n (%) | 20 (53) / 18 (47) | 17 (49) / 18 (51) |
| Disease stage at diagnosis*I / II / III / unknown, % | 7 (18) /6 (16) / 8 (21) / 17 (45) |
9 (26) /5 (14) /5 (14) / 16 (46) |
| Myeloma light chain, n (%) | ||
| Kappa light chain | 34 (89) | 25 (71) |
| Lambda light chain | 4 (11) | 10 (29) |
| Myeloma immunoglobulin, n (%) | ||
| IgA | 8 (21) | 8 (23) |
| IgG | 29 (76) | 22 (63) |
| IgM | 0 | 1 (3) |
| Other | 1 (3) | 4 (11) |
| Genetics, n (%)† | ||
| del13 | Not Available | 5 (14) |
| del17p13 | Not Available | 6 (17) |
| t(11:14) | Not Available | 2 (6) |
| t(4:14) | Not Available | 3 (9) |
| t(14:16) | Not Available | 1 (3) |
| +1q21 | Not Available | 3 (9) |
| Other | Not Available | 15 (43) |
| Missing | Not Available | 11 (31) |
| Prior therapies, n (%) | ||
| Received ≥5 lines of therapy | 29 (76) | 20 (57) |
| Proteasome inhibitors, received / refractory | 38 (100) | 35 (100) / 34 (97) |
| Immunomodulatory drugs, received / refractory | 38 (100) | 35 (100) / 32 (91) |
| Pomalidomide, received / refractory | 31 (82) | 21 (60) / 20 (57) |
| Daratumumab, received / refractory‡ | 9 (24) | 14 (40) / 13 (37) |
| Carfilzomib, received / refractory | 23 (61) | 28 (80) / 26 (74) |
| Patient disposition, n (%) | ||
| Completed study | 25 (66) | 9 (26) |
| Died | 1 (3) | 3 (9) |
| Ongoing on study | 10 (26) | 22 (63) |
| On treatment | 7 (18) | 17 (49) |
| In follow-up | 3 (8) | 5 (14) |
| Withdrawn from study | 2 (5) | 1 (3) |
| Withdrew consent | 1 (3) | 1 (3) |
| Lost to follow-up | 1 (3) | 0 |
| Discontinued treatment | 31 (82) | 18 (51) |
| Disease progression | 25 (66) | 15 (43) |
| Completion of treatment | 2 (5) | 0 |
| Adverse event | 3 (8) | 2 (6) |
| Corneal Event | 2 (5) | 00 |
| Investigator discretion | 1 (3) | 1 (3) |
| Patient decision | 0 |
Assessed using the International Staging System classification15; sum of categories <100% due to rounding.
Multiple categories per patient possible; total may add to more than 100%; assessed using fluorescence in situ hybridisation.
Thirty-four percent of patients had prior daratumumab and were refractory to both immunomodulatory drugs and proteasome inhibitors.
Lactate dehydrogenase data at screening are not available.
For those patients in Part 2 in whom GSK2857916 was initiated at 3∙4 mg/kg, at the time of data cut-off, the overall median duration of follow-up was 6·6 months (interquartile range: 4∙7–8∙1) with a median administration of 5 infusions (range: 1–13); 22 (63%) patients were still on study (Table 1). Of those who discontinued treatment (n=18), 15 (43%) discontinued due to disease progression and 2 (6%) due to AEs.
In Part 1, doses ranging from 0·03 mg/kg to ~4·6 mg/kg, administered every 3 weeks, were explored. Detailed results of Part 1 are reported in the Supplementary Appendix, p7–10, p12–14. Overall, no dose-limiting toxic events were observed and thus no MTD was established (Supplementary Appendix, p7). The most common treatment-emergent AEs, SAEs, and corneal events for Part 1 are listed in Supplementary Appendix, p8–10. Dose reductions were necessary in 1/3 (33%) of patients receiving 0∙96 mg/kg, 2/4 (50%) of patients receiving 1∙92 kg/mg, 1/8 (13%) patients receiving 2∙50 mg/kg, 3/3 (100%) patients receiving 3∙40 mg/kg and 5/6 (83%) patients receiving 4∙60 mg/kg dose (Supplementary Appendix, p12). One patient out of 4 (25%) in the 1∙92 mg/kg group (limbal stem cell deficiency), and 2/6 patients in the 4∙60 mg/kg group discontinued treatment due to an AE(s) (foreign body sensation in eyes and thrombocytopenia [both in one patient] and hypercalcaemia). There was one death in Part 1, which was attributed to disease progression and not deemed treatment-related. Although patient numbers for individual dose groups were small, there was a trend towards increased frequency of Grade 3/4 corneal events with increasing dose (Supplementary Appendix, p10). Therefore, an additional dose level of 2·5 mg/kg, not included in the initial dose-escalation period, was later assessed to further characterise this apparent trend. Corneal events were still observed at 2·5 mg/kg, but no patients treated in this cohort had a response (n=8) as of the data cut-off date of 26 June 2017.
Based on estimated receptor saturation at doses ≥1·92 mg/kg (see PK findings, below), a 100% response rate observed in the 3 patients receiving 3·4 mg/kg, a lack of observed clinical activity in the 8 patients treated at 2∙5 mg/kg (see preliminary clinical activity findings, below), and low tolerability of the ~4·6 mg/kg dose (fevers, severe fatigue and headache), the recommended dose for Part 2 was set at 3·4 mg/kg.
For Part 2, the most common treatment-emergent AEs are listed in Table 2. All patients in Part 2 experienced at least one AE. The most common events occurring in ≥25% of patients were corneal events, thrombocytopenia (including the preferred term platelet count decreased), anaemia, aspartate aminotransferase increased, and cough. Corneal events included but were not limited to blurry vision, dry eyes, photophobia, and others (see Supplementary Appendix, p11). Grade 3/4 AEs were reported in 28 (80%) of 35 patients in Part 2 (Table 2). In Part 2, SAEs were reported in 14 (40%) patients, most commonly IRRs and lung infection (2 patients each). Five (14%) patients had SAEs that were considered related to the study drug by the investigator, including IRR (n=2), intracranial haemorrhage (n=1), lung infection and pyrexia (n=1), and pericardial effusion (n=1). The single patient with intracranial haemorrhage had a history of intracranial bleeding and pre-existing thrombocytopenia that worsened post treatment in the setting of disease progression.
Table 2.
All adverse events (Grade 3, 4 and 5) by percentage and Grade 1 or 2 events occurring in ≥10% of patients for Part 2
| Preferred Term, n (%) | Part 2 (n=35) | |||
|---|---|---|---|---|
| Grade | 1–2* | 3 | 4 | 5 |
| Vision blurred | 16 (46) | 0 | 0 | 0 |
| Dry eye | 11 (31) | 1 (3) | 0 | 0 |
| Cough | 9 (26) | 0 | 0 | 0 |
| Aspartate aminotransferase increased | 8 (23) | 2 (6) | 0 | 0 |
| Chills | 8 (23) | 0 | 0 | 0 |
| Nausea | 8 (23) | 0 | 0 | 0 |
| Photophobia | 8 (23) | 0 | 0 | 0 |
| Pyrexia | 8 (23) | 0 | 0 | 0 |
| Thrombocytopenia† | 8 (23) | 9 (26) | 3 (9) | 0 |
| Fatigue | 7 (20) | 0 | 0 | 0 |
| Alanine aminotransferase increased | 5 (14) | 0 | 0 | 0 |
| Anaemia | 5 (14) | 5 (14) | 0 | 0 |
| Back pain | 5 (14) | 1 (3) | 0 | 0 |
| Decreased appetite | 5 (14) | 0 | 0 | 0 |
| Diarrhoea | 5 (14) | 1 (3) | 0 | 0 |
| Headache | 5 (14) | 0 | 0 | 0 |
| Sinusitis | 5 (14) | 0 | 0 | 0 |
| Upper respiratory tract infection | 5 (14) | 0 | 0 | 0 |
| Arthralgia | 4 (11) | 0 | 0 | 0 |
| Constipation | 4 (11) | 0 | 0 | 0 |
| Lacrimation increased | 4 (11) | 0 | 0 | 0 |
| Dyspnoea | 3 (9) | 1 (3) | 0 | 0 |
| Epistaxis | 3 (9) | 0 | 0 | 0 |
| Hypercalcaemia | 3 (9) | 0 | 0 | 0 |
| Lymphocyte count decreased | 3 (9) | 0 | 0 | 0 |
| Musculoskeletal chest pain | 3 (9) | 0 | 0 | 0 |
| Pain in extremity | 3 (9) | 0 | 0 | 0 |
| Gamma-glutamyl transferase increased |
2 (6) | 2 (6) | 0 | 0 |
| Infusion-related reaction‡ | 2 (6) | 2 (6) | 0 | 0 |
| Lung infection | 2 (6) | 1 (3) | 0 | 0 |
| Urinary tract infection | 2 (6) | 1 (3) | 0 | 0 |
| Vomiting | 2 (6) | 0 | 0 | 0 |
| Neutrophil count decreased | 0 | 2 (6) | 0 | 0 |
| Visual acuity tests abnormal | 0 | 1 (3) | 0 | 0 |
| Keratitis | 1 (3) | 2 (6) | 0 | 0 |
| Eye pain | 1 (3) | 1 (3) | 0 | 0 |
| Eye disorder | 0 | 1 (3) | 0 | 0 |
| Retinal detachment | 0 | 1 (3) | 0 | 0 |
| Haematuria | 1 (3) | 1 (3) | 0 | 0 |
| Hyperkalaemia | 1 (3) | 0 | 0 | 0 |
| Hyperuricaemia | 1 (3) | 0 | 0 | 0 |
| Hypokalaemia | 1 (3) | 2 (6) | 0 | 0 |
| Hyponatraemia | 1 (3) | 0 | 0 | 0 |
| Hypotension | 1 (3) | 0 | 0 | 0 |
| Influenza | 1 (3) | 0 | 0 | 0 |
| Pain | 1 (3) | 0 | 0 | 0 |
| Pneumonia | 1 (3) | 1 (3) | 0 | 0 |
| Appendicitis | 0 | 1 (3) | 0 | 0 |
| Bacteraemia | 0 | 0 | 1 (3) | 0 |
| Cellulitis | 0 | 1 (3) | 0 | 0 |
| Cholecystitis infective | 0 | 0 | 1 (3) | 0 |
| Deep vein thrombosis | 0 | 1 (3) | 0 | 0 |
| Encephalopathy | 0 | 1 (3) | 0 | 0 |
| Fall | 0 | 1 (3) | 0 | 0 |
| Neutropenia | 0 | 1 (3) | 1 (3) | 0 |
| Pericardial effusion | 0 | 0 | 1 (3) | 0 |
Grade 1 or Grade 2 events occurring in 10% of patients.
Grouped term includes thrombocytopenia and platelet count decreased.
In Part 2, 8 (23%) patients had infusion-related reactions (as defined by a grouped term, including the preferred term infusion-related reaction); per protocol, premedication was not allowed for the first infusion.
In Part 2, 23 (66%) patients had AEs that led to dose reduction, and 25 (71%) patients had AEs that led to dose interruptions or delays (Supplementary Appendix, p12). Vision blurred was the most common AE that led to dose reduction, in 11/35 (31%) of patients, followed by thrombocytopenia (4/35; 11%) and keratitis (3/35; 9%). Two patients discontinued treatment due to AEs: thrombocytopenia (2 patients [6%]) and increased blood creatinine phosphokinase (1 patient [3%], who was one of the 2 patients who experienced thrombocytopenia). As of 26 June 2017, there were three deaths in Part 2, all of which were attributed to progressive myeloma and not deemed treatment-related.
AEs of clinical interest related to GSK2857916 included IRRs, thrombocytopenia, and corneal events. In order to fully evaluate the incidence and severity of IRRs, pre-medications were prohibited prior to the first infusion. In Part 2, 8 (23%) patients experienced IRRs; most (5/8) were Grade 1/2 and all occurred with the first dose. Following the first infusion, pre-medications were permitted and included paracetamol (n=8, 23%), antihistamines (n=7, 20%), steroids (n=2, 6%; dexamethasone, hydrocortisone sodium succinate), and sodium chloride (n=1, 3%). Any-grade and Grade 3/4 thrombocytopenia occurred in 20/35 (57%) and 12/35 (34%) of patients, respectively. The median time to first occurrence of thrombocytopenia was 7 days (range: 1–185) and the median duration for patients with a resolution date (n=9) was 8 days (range: 6–16). Two (6%) patients discontinued treatment and 7 (20%) required dose reduction or delays due to thrombocytopenia.
Corneal events were reported in 22/35 (63%) patients in Part 2 (Supplementary Appendix, p11). These were predominantly mild to moderate (Grade 1/2; 19/22 patients) (Supplementary Appendix, p11); 3 (9%) patients in Part 2 experienced Grade 3 corneal events (1 with keratitis, 1 with eye pain and keratitis, and 1 with dry eye). The median time to onset of corneal events was 23 days (range: 1–84) and the median duration for patients with a resolution date (n=13) was 30 days (range: 5–224). Eighty-nine percent of patients had corneal findings on ophthalmic examination characterised by a superficial punctate keratitis (n=27/35; 77%) often associated with epithelial (microcystic) oedema (n=22; 63%) and stromal oedema (n=5; 14%), or opacities (n=8; 23%). As of 26 June 2017, data on corneal examination were available for 13 patients with an end of treatment visit. Eleven of these 13 patients had corneal abnormalities on ophthalmic examination; most (9/11) were considered mild. While visual acuity assessed by Snellen method declined in most patients during treatment, by the end of treatment, possible or definite worsened vision (change from baseline in best-corrected visual acuity score ≥0·3) was evident in 3/13 and 5/12 patients with available data, in the right and left eye, respectively. Management of corneal events included dose reduction in 14 (40%) patients and/or dose interruptions or delays in 15 (43%) patients, and supportive measures, such as the use of artificial tears and increasing the duration or frequency of steroid eye drop treatment. No patient in Part 2 permanently discontinued study treatment due to a corneal event. This study is ongoing, hence full information on resolution of corneal events was not available at data cut-off.
The responses observed in Part 1 (including stratification by dose) and duration of study treatment are summarised in Supplementary Appendix p14. At doses between 0∙03 and 2∙5 mg/kg (n=29), there were two responses (one PR at 0∙96 mg/kg and one very good partial response [VGPR] at 1∙92 mg/kg), whereas 6/9 patients treated at 3∙4 and 4∙6 mg/kg responded (two PR, three VGPR, and one stringent complete response [sCR]), with four ongoing responses (duration of response range: 7∙98–13∙08 months; duration of study treatment range: 340+ to 400+ days) at time of data cut-off.
For Part 2, the confirmed ORR was 60% (95% CI: 42·1–76·1) with 1 (3%) patient achieving a sCR, 2 (6%) achieving a CR, 15 (43%) achieving a VGPR and 3 (9%) achieving a PR (Figure 2A and Figure 3A). Retrospective exploratory subgroup analyses revealed the ORR for those with high-risk cytogenetics (n=8) was 63% (95% CI: 24·5–91·5) (Figure 2B) and 42% (95% CI: 15·2–72·3) for those who received prior daratumumab and were double refractory to PI and IMiD (n=12). In addition, the ORR was 68·2% (95% CI: 45∙1–86∙1) in patients treated with ≤5 prior lines of treatment, and was 46·2% (95% CI: 19∙2–74∙9) in patients who had received >5 lines of therapy, (Figure 2B) (post hoc analysis).
Figure 2. Best responses to GSK2857916 (Part 2; 3∙4 mg/kg dose).
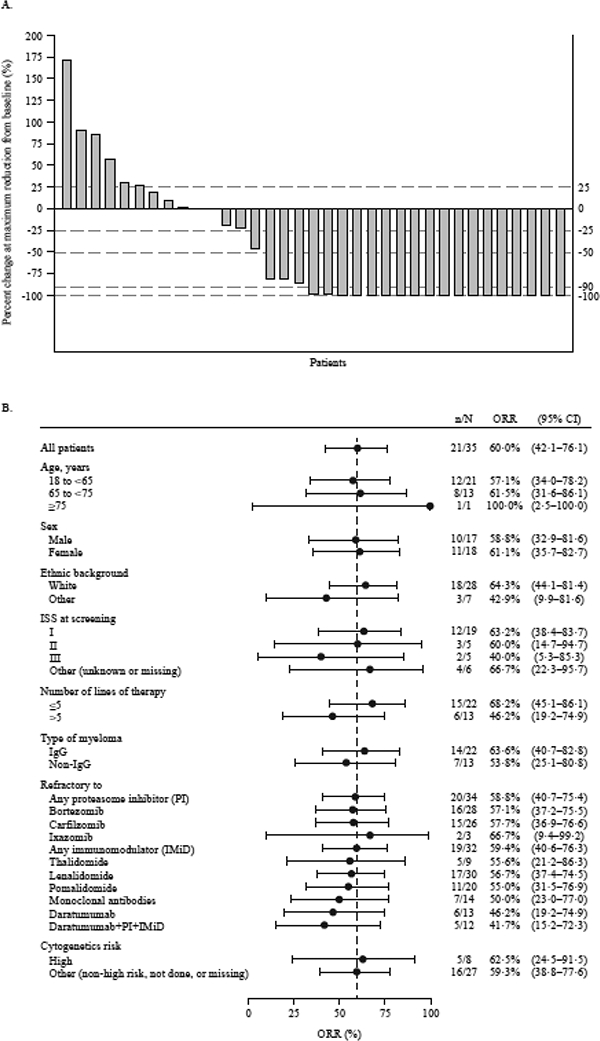
(A) Maximum percentage change of serum/urine M-protein or free light chain as compared with baseline values. For patients with measurable serum M-protein, serum values are depicted; for patients who are followed by urine M-protein, the urine values are depicted; and for patients who did not have measurable serum or urine M-protein and were followed by free light chains, the values for free light chain are depicted. (B) Forest plot of overall response rate by patient subgroup (Part 2). Patients with any of the following genetic abnormalities were considered high risk: t(4:14), del17p, t(14:16).
Figure 3. Response durations and effect of dose modifications.
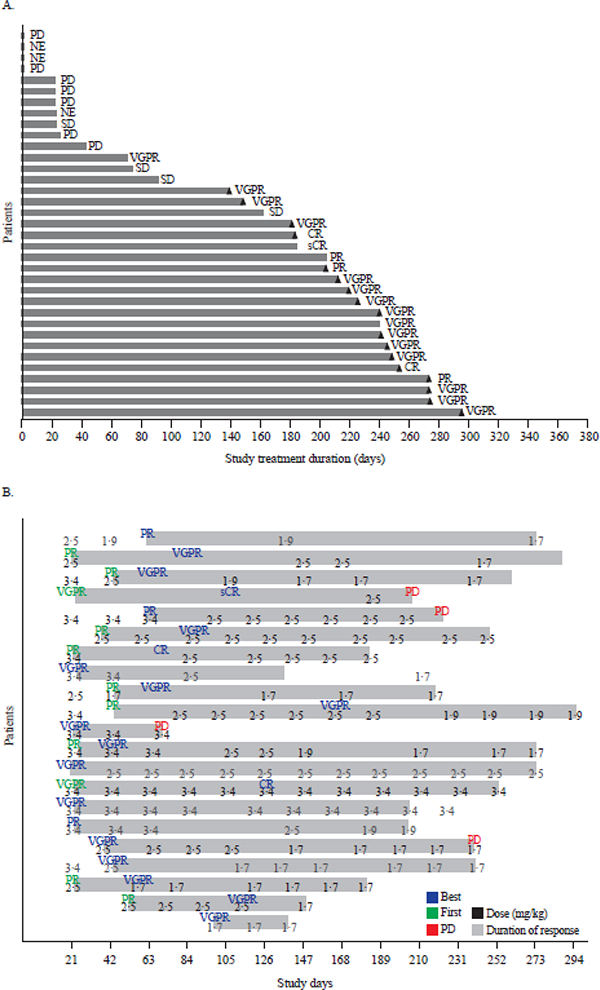
(A) Duration of study treatment by response in Part 2 (3∙4 mg/kg dose). Treatment duration counts the time difference between first dosing date and dosing end date without accounting for dosing interruptions. Triangles indicate ongoing patients. (B) Dose modifications in responding patients in Part 2. For each of the 21 responding patients, indicated in green font is initial response (PR or better); blue font, best response; red font, progressive disease; the numbers indicate dose for each infusion. CR: complete response, MR: minimal response, NE: not evaluable, PD: progressive disease, PR: partial response, sCR: stringent complete response, SD: stable disease, VGPR: very good partial response.
For Parts 1 and 2, maximum concentrations of GSK2857916, parent antibody, and total antibody were observed at end of infusion, whereas cys-mcMMAF concentrations peaked approximately 24 hours after dosing (Supplementary Appendix, p17). There was limited accumulation of GSK2857916 and cys-mcMMAF during subsequent cycles, and cys-mcMMAF molar ratio was <1% compared with GSK2857916. The PK of GSK2857916 were linear, dose proportional and well described using population PK methods with conventional allometry. GSK2857916 was cleared slowly with total plasma clearance of 0·37 L/d, and Vss was typical for a monoclonal antibody (4·2 L), implying confinement mainly in systemic circulation and interstitial space. The terminal phase half-life (t½; of GSK2857916 was 8–9 days (consistent with limited accumulation).
In Parts 1 and 2, at the end of the infusion, free soluble BCMA levels decreased by approximately 10-fold relative to baseline levels, with maximum reductions achieved at doses ≥1·92 mg/kg, and were restored to baseline after 7 days (Supplementary Appendix, p15). The levels of free soluble BCMA fell from pre-dose levels in a dose-dependent manner 24 hours after infusion. Soluble BCMA complex levels increased accordingly, with saturation reached at similar doses. Pre-dose (Cycle 1) free soluble BCMA levels were compared with response (as measured by reduction from baseline in serum and urine M-protein and serum FLC concentrations), but no clear relationship was evident (Supplementary Appendix, p16).
The median time to first response in Part 2 was 1·4 months (95% CI: 0·8–2·0). Despite dose interruptions and reductions for toxicities, responses were maintained and in some cases deepened (Figure 3B). With a median follow-up of 6∙6 months (interquartile range 4∙7–8∙1 months), the median PFS (post hoc analysis) was 7·9 months (95% CI: 3·1–not estimable) (Figure 4A), and the median duration of response was not estimable (Figure 4B); however, the 25th percentile for duration of response was 6∙7 months (95% CI: 1.6–not estimable). Survival data were immature at the data cut-off date.
Figure 4. Kaplan–Meier curves for progression-free survival and duration of response.
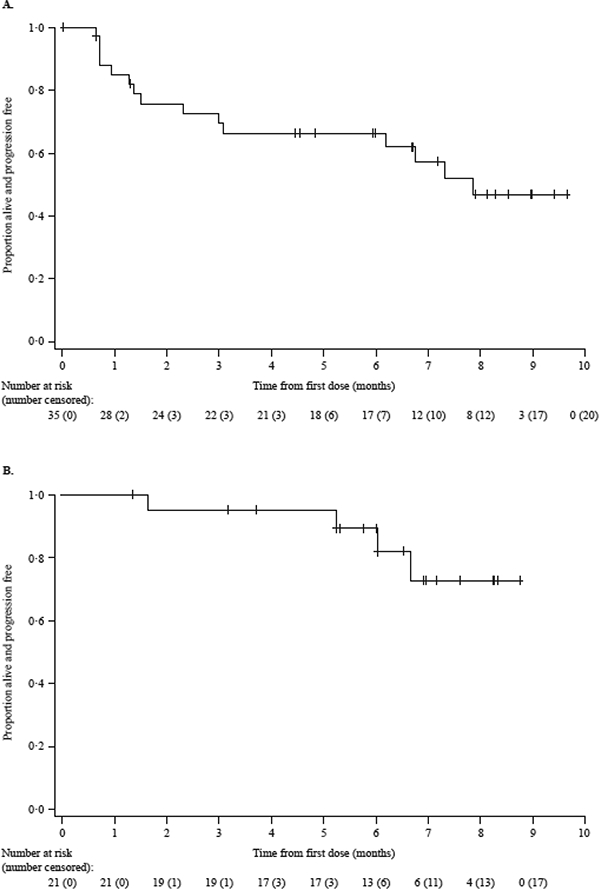
Progression-free survival (A) and duration of response (B) from Part 2 (dose expansion phase; 3∙4 mg/kg dose) are shown. The median progression-free survival was 7·9 months and the median duration of response was not estimable. Tick marks indicate censored data.
DISCUSSION
This ongoing, first-in-human study demonstrates that the novel anti-BCMA antibody-drug conjugate GSK2857916 has significant single-agent activity in patients with relapsed/refractory MM, with 21 (60%) of 35 patients achieving PR or better at the RP2D of 3·40 mg/kg. Patients were heavily pretreated, including 31/35 (89%) patients who were double-refractory to a PI and IMiD, and 13/35 (37%) patients who were refractory to daratumumab. In this context, the response depth, including 18/35 (51%) patients with VGPR or better, and median PFS of 7·9 months are notable (95% CI: 3.1 months–not estimable). Even among patients with prior daratumumab treatment and refractory to both PI and IMiD, the ORR was 42%.
The RP2D of 3∙4 mg/kg was selected based on the acceptable balance between clinical response and tolerability. The highest dose tested (4∙6 mg/kg) was considered to be not tolerated. Although only 3 patients received the 3∙4 mg/kg dose in Part 1, they all achieved a response (1 each of VGPR, sCR and PR). The dose selection is further supported by BCMA receptor saturation observed at doses ≥1∙92 mg/kg; although in a wider patient population, receptor saturation may potentially be affected by variability in soluble BCMA levels.
The depth and durability of responses seen with GSK2857916 in this population are promising and compare favourably with those described previously for any approved single agent, although the small sample size necessitates caution in data interpretation. Neither the antiSLAMF7 antibody elotuzumab nor the histone deacetylase inhibitor panobinostat had significant single-agent activity.16,17 In Phase 2 studies of carfilzomib monotherapy18 and pomalidomide alone or pomalidomide plus dexamethasone,19 responses were observed in only a minority of patients (24%, 18% and 33%, respectively) with median PFS rates of 3·7, 2·7 and 4·2 months, respectively. In the pivotal Phase 2 monotherapy study of the anti-CD38 antibody daratumumab, in which 64% of patients were refractory to lenalidomide and bortezomib, the ORR and median PFS at the approved 16 mg/kg dose were 36% and 5·6 months, respectively. Only 10% of patients achieved VGPR or better with daratumumab.20
The present results also demonstrate a manageable safety profile relative to currently available agents.21 Thrombocytopenia and corneal events were the most frequently reported AEs and are consistent with the known toxic effects of MMAF and other MMAF-linked antibody-drug conjugates.13 These are in contrast to the neurological and pulmonary toxicities attributed to brentuximab vedotin, which employs MMAE.22 The corneal events most often manifested as blurry vision, photophobia and dry eyes though a range of symptoms was possible, including increased lacrimation, pain and pruritus. Most events were Grade 1/2 and improved with dose interruptions and/or reduction. Dry eye and keratitis were the most common Grade 3 events; no Grade 4/5 events were reported and no patients discontinued because of corneal events in Part 2. Most patients did have corneal findings on examination, most commonly superficial punctate keratitis. For the 13 patients with end of treatment corneal examinations available as of 26 June 2017, the majority had findings classified as mild in severity. Furthermore, decline in visual acuity was found to have improved in the majority of those with available data at the end of the treatment visits. Importantly, while the lower starting doses of GSK2857916 tested in dose escalation were associated with lower response rates, reducing the dose to 2·5 or 1·7 mg/kg after achieving a response in Part 2 or delaying dosing to allow for recovery from toxicities did not appear to induce loss of response. Nevertheless, given the high rate of dose modifications, additional investigations into strategies to mitigate corneal events are warranted. Thrombocytopenia in most cases recovered between doses, and few bleeding events were reported. Infusions were administered over 1 hour and were associated with mostly Grade 1/2 IRRs.
An inherent limitation of Phase 1 study design is the necessarily small sample size; future later phase studies will provide more robust data on the clinical activity and safety profile of GSK2857916. As this is an ongoing study, additional data on potential long-term sequelae and also resolution of corneal events should be available on analysis of the final data. In addition, analyses of patients previously treated with daratumumab combinations will be performed in the future.
The target of GSK2857916 differentiates it from other monoclonal antibodies currently in the clinic for myeloma. BCMA is expressed universally on normal and malignant plasma cells, but not on non-haematopoietic cells or haematopoietic stem cells.23–25 Unlike SLAMF7 or CD38, the targets for elotuzumab and daratumumab, respectively, it is not expressed on natural killer cells, T cells, monocytes, red blood cells or lymphoid progenitor cells.8,24 In vitro testing of GSK2857916 against myeloma cells show that BCMA is recycled to the cell surface after binding and internalisation.4 Although previous studies have shown that gamma-secretase causes ongoing shedding of non-bound BCMA from plasma cells,26 whether overall BCMA expression on residual myeloma cells will be down-regulated after treatment, as has been described in occasional patients following BCMA chimeric antigen receptor (CAR) T cells,27,28 as well as with CD38 following daratumumab treatment,25 remains to be determined.
Published data indicate that high levels of soluble BCMA exist in patients with MM, particularly in those with advanced disease, which could potentially interfere with the binding of GSK2857916 to BCMA or act as a drug sink.5,7,26 This could theoretically affect both safety and activity, especially in patients with high soluble BCMA levels. Based on currently available data, there was no obvious relationship between reduction from baseline in M-protein (or FLC) measurement and pre-dose (Cycle 1) soluble BCMA levels.
The clinical activity of GSK2857916 expands on the initial promising reports targeting BCMA using CAR T cells,27,28 as well the preclinical activity of T cell-dependent bispecific antibodies against BCMA now entering the clinic,29 further validating BCMA as an attractive target for myeloma immunotherapy. However, unlike these approaches, GSK2857916 does not require a labour-intensive manufacturing process or treatment at a specialised centre, and is not associated with potentially severe toxicities such as cytokine release syndrome and encephalopathy.28,30
The activity of GSK2857916 may be a result of its ability to target myeloma cells via several different mechanisms of action. In addition to exerting direct anti-tumour activity against proliferating cells by delivering a potent anti-mitotic drug, it induces antibody-dependent cellular cytotoxicity via enhanced binding to multiple FcR-expressing immune effector cells, allowing it to target non-dividing myeloma cells. This utilisation of immune effector cells provides a rationale for combination with IMiDs; indeed, lenalidomide augmented the activity of GSK2857916 against MM cells in vitro.23 This mechanism also differentiates it from other antibody-drug conjugates under development for myeloma that have failed to demonstrate significant single-agent activity in myeloma, including lorvotuzumab mertansine (an anti-CD56maytansinoid)31 and indatuximab ravtansine (an anti-CD138-maytansinoid).32 Finally, data further suggest the potential of GSK2857916 to induce immunogenic cell death of myeloma cells,8 which may stimulate endogenous anti-myeloma immunity and thereby contribute to durability of responses observed in this heavily pretreated population.
In conclusion, this first trial of GSK2857916 provides evidence that targeting BCMA is effective in the treatment of advanced MM. GSK2857916 showed a favourable administration schedule and safety profile and potentially significant clinical activity in patients with MM with limited treatment options. Its target and mechanisms of action are distinctive, and additional monotherapy and combination studies are in development.
Supplementary Material
RESEARCH IN CONTEXT.
Evidence before this study:
New approaches to the treatment of relapsed refractory multiple myeloma (MM) are urgently needed. GSK2857916 provides such an advancement by targeting B-cell maturation antigen (BCMA), which is universally expressed in patients with MM. We searched PubMed with the terms “relapsed”, “myeloma”, “BCMA”, and “clinical trial” for studies published from 1 January 1990 to 1 January 2018. We identified multiple trials evaluating BCMA-targeted chimeric antigen receptor T cell and related adoptive cellular approaches but no published studies in humans of BCMA-targeted antibodies or antibody-drug conjugates.
Added value of this study:
This is the first Phase 1 trial of the novel anti-BCMA antibody-drug conjugate GSK2857916 in humans. The data demonstrate that GSK2857916 has significant activity in patients with heavily pretreated, relapsed/refractory MM, with 60% overall response rate and primary toxicities of corneal events and thrombocytopenia. The depth and durability of responses seen with GSK2857916 in this population are promising and compare favourably with those observed previously for any approved single agent. The results further validate BCMA as an attractive target for MM. GSK2857916 could potentially offer a new treatment option for patients with multiple myeloma that is steroid free and off the shelf with a convenient administration schedule.
Implications of all the available evidence:
These results provide strong evidence that targeting BCMA with an antibody-drug conjugate is effective in the treatment of advanced relapsed/refractory MM. GSK2857916 had a manageable safety profile, and potentially significant clinical activity in patients with MM with limited treatment options. Additional monotherapy and combination studies are in development.
ACKNOWLEDGEMENTS
Study BMA117159 was funded by GlaxoSmithKline. This research was additionally funded in part through a National Cancer Institute, National Institutes of Health Cancer Center Support grant (P30CA008748) to NL. RP is supported by the NIHR University College London Hospitals Biomedical Research Centre and this trial was supported in part by the NIHR UCLH Clinical Research Facility and the Cancer Research UK Experimental Cancer Medicine Centre. This material was presented in part at the 2016 and 2017 annual meetings of the American Society of Hematology, and at the 1st European Myeloma Network meeting 2018. We are grateful to the patients who participated in this study, the investigators and coordinators at the clinical sites, and the employees of GlaxoSmithKline who contributed to the design, implementation, and data analysis. Additional thanks to Timothy J. DiChiara, PhD, a consultant funded by GlaxoSmithKline, for editorial help with the manuscript (drafting and revising the manuscript based on author feedback); additional editorial support (revising the manuscript based on author feedback and adapting figures to journal guidelines) was provided by Leigh O’Connor, PhD, and Clare Slater PhD, CMPP, of Fishawack Indicia Ltd, UK, which was funded by GlaxoSmithKline. The authors would also like to thank Roxanne C. Jewell, E.J. Dettman (employees of GlaxoSmithKline), and Mary Pinder-Schenck (previous employee of GlaxoSmithKline) for their support in the development of the manuscript. Drug linker technology was licensed from Seattle Genetics, Inc (Bothell, WA, USA) and monoclonal antibody was produced using POTELLIGENT® Technology licensed from BioWa, Inc. (Princeton, NJ, USA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATIONS OF INTEREST
LDA has participated in speakers’ bureaux for Celgene, Takeda and Amgen. ADC is a consultant for and a member of an advisory board for GlaxoSmithKline and Celgene, is a member of an advisory board for Janssen and Bristol-Myers Squibb, has received research funding from Bristol-Myers Squibb, Celgene and Novartis. AH is an employee of and holds stocks/shares in GlaxoSmithKline and is a non-executive director and holds stocks in Imugene. NL has received research funding from GlaxoSmithKline; RP has received honoraria from Janssen, Takeda, Celgene, and Amgen, and travel support to attend meetings from Janssen, Takeda, and Celgene. PGR is a consultant for, and has received research funding from, Celgene, Takeda, and Jazz Pharmaceuticals, and is a member of the board of directors/advisory committee for Celgene, Jazz Pharmaceuticals, Janssen, and Millennium. HJS has received honoraria from Janssen, Celgene, and Amgen. ST is a consultant for and has received honoraria from Amgen and Celgene; has received honoraria from Takeda and AbbVie; is a consultant for Novartis; and has received research support from Janssen. PMV is a consultant for Amgen, Celgene, Janssen, Bristol-Myers Squibb, Novartis, Takeda, and Teneo-Bio, and has participated in speakers’ bureaux for Amgen, Celgene, and Janssen. KY is a consultant for Autolus, has received honoraria from Autolus, Amgen, Janssen, and Celgene, and has received research funding from Amgen, Janssen, Celgene, and Chugai. DJA, JBO, MMG, and ZH are employees of and hold stocks/shares in GlaxoSmithKline. SL was an employee of GlaxoSmithKline at the time of study conduct and holds stocks/shares in GlaxoSmithKline. BR and ENL declare no conflict of interest outside of the submitted work.
REFERENCES
- 1.Kumar SK, Dimopoulos MA, Kastritis E, et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: a multicenter IMWG study. Leukemia 2017; 31(11): 2443–8. [DOI] [PubMed] [Google Scholar]
- 2.Darce JR, Arendt BK, Wu X, Jelinek DF. Regulated expression of BAFF-binding receptors during human B cell differentiation. J Immunol 2007; 179(11): 7276–86. [DOI] [PubMed] [Google Scholar]
- 3.O’Connor BP, Raman VS, Erickson LD, et al. BCMA is essential for the survival of longlived bone marrow plasma cells. J Exp Med 2004; 199(1): 91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee L, Bounds D, Paterson J, et al. Evaluation of B cell maturation antigen as a target for antibody drug conjugate mediated cytotoxicity in multiple myeloma. Br J Haematol 2016; 174(6): 911–22. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez E, Gillespie A, Tang G, et al. Soluble B-Cell Maturation Antigen Mediates Tumor-Induced Immune Deficiency in Multiple Myeloma. Clin Cancer Res 2016; 22(13): 3383–97. [DOI] [PubMed] [Google Scholar]
- 6.Tai YT, Acharya C, An G, et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016; 127(25): 3225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez E, Li M, Kitto A, et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br J Haematol 2012; 158(6): 727–38. [DOI] [PubMed] [Google Scholar]
- 8.Tai YT, Mayes PA, Acharya C, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014; 123(20): 3128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neuenschwander B, Branson M, Gsponer T. Critical aspects of the Bayesian approach to phase I cancer trials. Stat Med 2008; 27(13): 2420–39. [DOI] [PubMed] [Google Scholar]
- 10.Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 2016; 17(8): e328–e46. [DOI] [PubMed] [Google Scholar]
- 11.Krop IE, Beeram M, Modi S, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol 2010; 28(16): 2698–704. [DOI] [PubMed] [Google Scholar]
- 12.Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 2010; 363(19): 1812–21. [DOI] [PubMed] [Google Scholar]
- 13.Eaton JS, Miller PE, Mannis MJ, Murphy CJ. Ocular Adverse Events Associated with Antibody-Drug Conjugates in Human Clinical Trials. J Ocul Pharmacol Ther 2015; 31(10): 589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.0. 2010. https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_40 (accessed April 2018).
- 15.Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol 2005; 23(15): 3412–20. [DOI] [PubMed] [Google Scholar]
- 16.Zonder JA, Mohrbacher AF, Singhal S, et al. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 2012; 120(3): 552–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolf JL, Siegel D, Goldschmidt H, et al. Phase II trial of the pan-deacetylase inhibitor panobinostat as a single agent in advanced relapsed/refractory multiple myeloma. Leuk Lymphoma 2012; 53(9): 1820–3. [DOI] [PubMed] [Google Scholar]
- 18.Siegel DS, Martin T, Wang M, et al. A phase 2 study of single-agent carfilzomib (PX171–003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012; 120(14): 2817–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with lowdose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood 2014; 123(12): 1826–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N Engl J Med 2015; 373(13): 1207–19. [DOI] [PubMed] [Google Scholar]
- 21.Gan HK, Reardon DA, Lassman AB, et al. Safety, Pharmacokinetics and Antitumor Response of Depatuxizumab Mafodotin as Monotherapy or in Combination with Temozolomide in Patients with Glioblastoma. Neuro Oncol 2017: doi: 10.1093/neuonc/nox202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gravanis I, Tzogani K, van Hennik P, et al. The European Medicines Agency Review of Brentuximab Vedotin (Adcetris) for the Treatment of Adult Patients With Relapsed or Refractory CD30+ Hodgkin Lymphoma or Systemic Anaplastic Large Cell Lymphoma: Summary of the Scientific Assessment of the Committee for Medicinal Products for Human Use. Oncologist 2016; 21(1): 102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaffer AL, Emre NC, Lamy L, et al. IRF4 addiction in multiple myeloma. Nature 2008; 454(7201): 226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res 2013; 19(8): 2048–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nijhof IS, Casneuf T, van Velzen J, et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016; 128(7): 959–70. [DOI] [PubMed] [Google Scholar]
- 26.Laurent SA, Hoffmann FS, Kuhn PH, et al. gamma-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun 2015; 6: 7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ali SA, Shi V, Maric I, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016; 128(13): 1688700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen AD, Garfall AL, Stadtmauer EA, et al. B-Cell Maturation Antigen (BCMA)-Specific Chimeric Antigen Receptor T Cells (CART-BCMA) for Multiple Myeloma (MM): Initial Safety and Efficacy from a Phase I Study. Blood 2016; 128(22): 1147-. [Google Scholar]
- 29.Hipp S, Tai YT, Blanset D, et al. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia 2017; 31(10): 2278. [DOI] [PubMed] [Google Scholar]
- 30.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014; 124(2): 188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berdeja JG. Lorvotuzumab mertansine: antibody-drug-conjugate for CD56+ multiple myeloma. Front Biosci (Landmark Ed) 2014; 19: 163–70. [DOI] [PubMed] [Google Scholar]
- 32.Ikeda H, Hideshima T, Fulciniti M, et al. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin Cancer Res 2009; 15(12): 4028–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.