

SUMMARY
Protein degradation is known to be a key component of expression regulation for individual genes, but its global impact on gene expression has been difficult to determine. We analyzed a parallel gene expression dataset of yeast meiotic differentiation, identifying instances of coordinated protein-level decreases to identify new cases of regulated meiotic protein degradation, including of ribosomes and targets of the meiosis-specific anaphase-promoting complex adaptor Ama1. Comparison of protein and translation measurements over time also revealed that, although meiotic cells are capable of synthesizing protein complex members at precisely matched levels, they typically do not. Instead, the members of most protein complexes are synthesized imprecisely, but their protein levels are matched, indicating that wild-type eukaryotic cells routinely use post-translational adjustment of protein complex partner levels to achieve proper stoichiometry. Outlier cases, in which specific complex components show divergent protein-level trends, suggest timed regulation of these complexes.
In Brief
Eisenberg et al. leverage global translation and protein data to identify cases of regulated protein degradation in meiosis. Analyses of temporal trends reveal that members of protein complexes can be synthesized at ideal stoichiometry but that they are usually made imprecisely and their levels adjusted by degradation.
Graphical Abstract
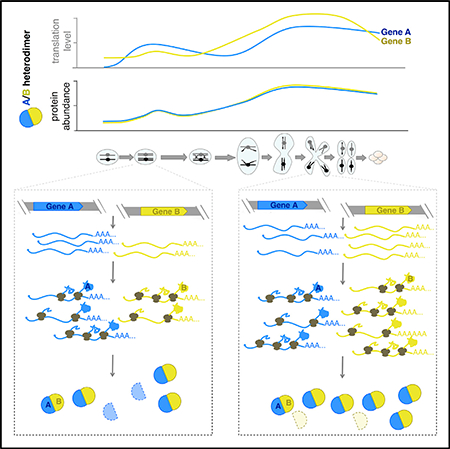
INTRODUCTION
The protein complement of a cell defines its structure and function and is determined by the relative rates of synthesis and degradation for each protein present. The mechanisms and specificity determinants of synthetic processes in gene expression, especially transcription, have been well studied. In addition, the basic classes of mechanisms by which proteins are degraded within cells, including through regulated ubiquitin-based protein turnover, have been defined. However, it remains difficult to systematically determine the impact of regulated protein degradation in an unperturbed system, even at steady state. Comprehensively assessing the timing and specificity of protein degradation mechanisms in the context of cellular differentiation is an even greater challenge, but also particularly critical, as the transitions between sequential cellular stages require waves of both synthesis of new proteins and removal of pre-existing proteins.
Early examples of regulated degradation were identified by single-gene analyses, such as the case of Cyclin during the cell cycle and meiosis (Evans et al., 1983). Cyclin protein synthesis was observed to be constitutive, but the protein level fluctuated, leaving regulated protein degradation as the remaining explanation for the protein expression pattern observed. These observations ultimately led to the discovery of the conserved anaphase-promoting complex/cyclosome (APC/C)-based specificity mechanism, which is responsible for a key event in cell division (Irniger et al., 1995; King et al., 1995; Sudakin et al., 1995). Here we apply a similar approach, based on examining genome-wide protein patterns during a natural process to identify cases of protein degradation more globally. We recently generated a complex dataset that enabled these analyses, and that includes deep and matched mRNA-, translation-, and protein-level measurements during the natural process of meiotic differentiation in budding yeast (Cheng et al., 2018). These analyses allowed the identification of genes that we propose to be targets of the Ama1-APC/C and revealed degradation and re-synthesis of ribosomal proteins following meiosis in maturing gametes.
We also found strong and widespread evidence for post-translational adjustment of the relative levels of protein complex components over the natural process of meiotic differentiation. While such regulation has been shown to occur in mutant conditions that create an imbalance among macromolecular complex components (Abovich et al., 1985; Dephoure et al., 2014; Gorenstein and Warner, 1977; Ishikawa et al., 2017; Lam et al., 2007; Papp et al., 2003; Sung et al., 2016a; Torres et al., 2010; Warner, 1977; Warner et al., 1985), the effect that we observe is widespread in wild-type cells subjected to no external perturbations. Our analyses show that although eukaryotic cells are capable of synthesizing binding partners with effectively perfectly matched levels, they typically synthesize them at only roughly similar levels and fine-tune their stoichiometry through protein degradation. Finally, we find that divergence of protein-level trends of specific protein complex members from their partners is suggestive of functional differences for these members, including regulatory roles.
RESULTS
Protein Degradation Inferred from Comparison of Protein and Translation Data
Although our comprehensive dataset of gene expression through meiotic differentiation (Figure 1A) was previously analyzed only for transcriptional and translational regulation (Cheng et al., 2018), we found that comparison of translation and protein patterns could also be used to infer post-translational regulation of gene expression that accurately captures expected regulation. For example, the protein for cyclin-dependent kinase (CDK) Cdc28 is required through much of meiosis and was present for a period of at least 3 hr after translation ceased, suggesting protein stability during this time frame (Figure 1B). In contrast, protein levels of the synaptonemal complex (SC) component Zip1 declined in concert with a decrease in translation, which is consistent with the known active degradation of this protein in late prophase (Figure 1C; Sourirajan and Lichten, 2008). In an extreme case of such regulation, the CDK inhibitor Sic1 showed decreased protein levels, despite high ongoing translation during meiotic S phase and prophase, which is consistent with the critical known regulation of this protein (Figure 1D; Dirick et al.,1998).
Figure 1. Regulated Protein Degradation Can Be Detected by Analysis of Protein Levels during Meiosis.

(A) Schematic of meiotic gene expression experiment. Illustrations representing vegetative growth or meiotic stage are used to depict sample identity throughout figures. Left-hand vegetative cells are exponentially growing, and far-right cells are in nutrient-poor sporulation medium. Meiotic stages are noted above central portion of illustration and time in sporulation medium is noted directly below.
(B–D) Comparison of translation, assayed by ribosome footprint density (blue) and protein, assayed by quantitative mass spectrometry (black) are shown over time points for (B) Cdc28 (green box highlights a period of inferred protein stability; RPKM, reads per kilobase million); (C) Zip1 (pink box highlights a period of inferred protein instability that matches known regulation); and (D) Sic1 (pink box highlights a period of inferred protein instability that matches known regulation).
(E) Protein fold changes between sequential time points were calculated for genes (n = 4,464) quantified by mass spectrometry (i.e., first column is 1.5 hr/0 hr protein abundance ratio, and so on). Values were subjected to hierarchical clustering. A cluster containing known Ama1 targets are noted at middle right.
See also Figure 2.
Protein-level Co-clustering Identifies Additional Candidate Targets of Ama1
We reasoned that we should be able to identify cases of coordinated degradation by simply looking for groups of proteins that show similar timing and degree of protein-level decrease between sequential meiotic time points. To this end, we calculated the ratio of the protein level at each time point to that of the previous time point for every protein quantified and performed hierarchical clustering (Figure 1E). This analysis revealed a variety of discrete patterns of protein-level change over meiosis. Most prominent in the degree of change observed was a group of genes for which protein abundance increased prior to and early in the meiotic divisions and then rapidly decreased. This cluster included the Polo kinase Cdc5, the mid-meiotic master transcription factor Ndt80, and the prospore membrane leading edge component Ssp1, which are known targets of the meiotic APC/C activator Ama1 (Figure 1E; Diamond et al., 2009; Okaz et al., 2012). The cluster contained 42 genes overall, including several with well-defined roles during the first meiotic division, but no characterized mechanism of protein degradation.
Ama1 activates APC/C-mediated degradation during multiple periods in meiosis, including late in the first meiotic division (Diamond et al., 2009; Okaz et al., 2012; Tan et al., 2011). Thus far, Ama1 targets have been identified on a gene-by-gene basis. We sought to determine whether temporal protein expression patterns similar to Cdc5, Ndt80, and Ssp1 could indicate Ama1-dependent degradation. We focused on two genes in the Cdc5/Ndt80/Ssp1 cluster (Figure 1E), RNA-binding proteins Pes4 and Mip6, based on their known meiotic function and our ability to construct C-terminally tagged versions of their encoded proteins that could be visualized by western blot. Because clustering was based on similar overall protein-level changes over time, gene groupings were likely to share both synthesis and degradation mechanisms. The former was already clear, as all genes in this group are predicted targets of Ndt80, which activates its own transcription along with a large set of other targets in late prophase to enable the meiotic divisions (Chu and Herskowitz, 1998). Matched mRNA sequencing (mRNA-seq) data were consistent with this regulation, showing overlapping patterns of mRNA accumulation for all five genes (Figure 2A). Translation patterns were also generally similar for these genes, which is consistent with no reported strong translational regulation for their mRNAs (Figure 2B). Protein accumulation patterns were similarly comparable for these genes, as expected, but the later decrease in protein levels was even more similar, with nearly identical downward slopes in late meiosis I (MI) (Figure 2C). The degree of similarity of these protein down-slopes mirrored that of their highly coordinated mRNA up-slopes and suggested that the degradation of these proteins may be mediated by the same temporally restricted mechanism (Figures 2A and 2C).
Figure 2. Protein Co-clustering Is Predictive of Shared Degradation Regulation by Ama1.

(A) mRNA levels over time for known and predicted Ama1 targets. The green box highlights a period of matched mRNA induction timing, consistent with known transcriptional co-regulation.
(B) Translation levels over time for known and predicted Ama1 targets.
(C) Protein levels over time for known and predicted Ama1 targets. The pink box highlights a period of decrease in protein levels that is matched in timing and degree, suggesting co-regulation.
(D–H) Western blot analysis and quantification for protein levels of meiotic proteins, with and without AMA1. (D) Known Ama1-dependent degradation target Ssp1, (E) known Ama1-dependent degradation target Ndt80, (F) predicted Ama1 target Pes4, (G) predicted Ama1 target Mip6, and (H) Stu2.
We observed the expected wild-type pattern of protein accumulation for Ndt80 and Ssp1, and the relative persistence of epitope-tagged protein in cells deleted for AMA1 (Figures 2D, 2E, S1A, S1B, S1F, S1G, S1K, and S1L). We found that Pes4 and Mip6 also showed the persistence of high protein levels past the normal stage of degradation in ama1Δ cells, suggesting that their degradation is at least partially dependent on Ama1-APC/C in mid-meiosis, as predicted by cluster analysis (Figures 1E, 2F, 2G, S1H, and S1I). We confirmed that this effect could not be explained by generally delayed meiotic progression in ama1Δ cells (Figures S1C, S1D, S1M, S1N) and further compared to a protein, Stu2, which has a similar overall protein pattern to the other candidates but was not in the same discrete subcluster as Cdc5, Ssp1, Ndt80, Pes4, and Mip6 (Figure 1E). We found no change in Stu2 protein persistence in ama1Δ cells compared to wild-type, suggesting that the effect that we observe is specific (Figures 2H, S1E, S1J, and S1O). While Pes4 and Mip6 were not previously identified as targets of regulated degradation, these proteins have a temporally restricted role, mediating the translational repression of several Ndt80 targets prior to late meiosis II (MII; Jin et al., 2017). We conclude that the Ama1-APC/C has targets beyond those previously characterized and that its activity during the meiotic divisions may coordinate the destruction of diverse proteins that are important for MI but unneeded during MII.
Substructure in Protein Clustering of Genes Co-regulated for Protein Synthesis Suggests Post-translational Adjustment Based on Physical Interactions
As the comparison of protein-level changes among genes that were co-regulated for synthesis was informative for identifying Ama1-dependent proteins, we wondered whether such analyses could more generally elucidate cases of regulated protein degradation. We turned to a group of genes that we previously identified as showing highly similar protein synthesis patterns (Brar et al., 2012) and that are enriched for roles in recombination and SC formation, two processes involved in the physical linkage of “mom” and “dad” chromosomes before MI segregation (Figure S2, left). For the complex components that we were able to quantify by mass spectrometry, protein abundance patterns showed greater diversity than translation level patterns. In addition, functional subgroups emerged from the protein measurements that had more similar patterns than the group as a whole (Figure S2, right), and protein abundance-level clusters correlate with known physical interactions. For example, recombination and SC factors, which interact directly or indirectly with chromatin, clustered separately from the genes that are not thought to be involved in these processes (Figure S2, bold versus plain text). Furthermore, proteins that are known structural components of the SC, which associate with chromatin (and each other) during meiosis, formed a subcluster in the protein abundance data that is distinct from more direct regulators of recombination (Figure S2, orange versus purple; reviewed in Cahoon and Hawley, 2016; Zickler and Kleckner, 2015). Finally, this grouping included three characterized heterodimers, and each pair clustered together in the protein but not the translation data (Figure S2, green; Humphryes et al., 2013; Pochart et al., 1997; Shinohara et al., 2008). Since protein abundance measurements integrate translation and protein degradation, we propose that the more precise matching of protein levels for interaction partners results from the degradation of uncomplexed subunits.
This conclusion is consistent with a predominant model for protein complex assembly and homeostasis that is based on mutant analyses. It has been widely reported that members of stable protein complexes are present with equivalent steady-state protein levels and that this balanced stoichiometry is important for cellular fitness (Burke et al., 1989; reviewed in Harper and Bennett, 2016; Veitia and Potier, 2015). Experiments in which one protein complex component was decreased in expression, for example, revealed resultant decreases in other complex components (Abovich et al., 1985; Stevens and Davis, 1998). Overexpression of a single protein complex component can result in decreased protein stability for this component, resulting from its proteasome-mediated degradation (Abovich et al., 1985; Ishikawa et al., 2017; Stevens and Davis, 1998; Sung et al., 2016a; Warner et al., 1985). Aneuploid cells carrying an extra chromosome showed expression from this chromosome that was proportionally increased at the protein level for most genes, with the exception of protein complex members, which were dampened at the protein level relative to expectations based on mRNA and translation measurements (Dephoure et al., 2014). In addition, a pulsed mass spectrometry study in mammalian cell lines revealed an initial rapid proteasome-dependent phase of degradation of a subset of proteins, which were enriched for protein complex membership, suggesting that the oversynthesis of some complex members leaves a subset of newly synthesized proteins uncomplexed and thus unstable (McShane et al., 2016).
While it is established that protein-level adjustment through degradation can occur in diverse mutant and perturbed conditions, its prevalence in wild-type cells under natural conditions has been difficult to assess. It is known that members of most protein complexes do not show highly correlated mRNA levels (Gandhi et al., 2011; Liu et al., 2009), and in isolated natural cases, it has been shown that protein complex components are synthesized out of stoichiometry and adjusted to similar levels by protein degradation (Blikstad et al., 1983; Lehnert and Lodish, 1988; Mueller et al., 2015). It has also been reported, however, that wild-type cells tend to synthesize protein complex components in proportion to the stoichiometry seen in final complexes, perhaps precluding the need for regulation at the level of degradation (Li et al., 2014; Taggart and Li, 2018). Coherently reconciling results from individual gene studies and large-scale studies, as well as between regulation in wild-type and perturbed cellular conditions, has been challenging. Quantitatively comparing the levels of mRNA, translation, and protein in parallel for complex partners in wild-type cells should address this problem, but this requires comparing sequencing- and mass spectrometry-based measurements. These two fundamentally different types of measurements show different dynamic ranges of detection, and by traditional mass spectrometry, precise direct comparison of measurements between proteins with different physical properties is difficult.
Precisely Matched Complex Component Synthesis Is Possible but Not Pervasive during Meiotic Differentiation
We reasoned that our dataset provided a unique opportunity to assess the degree of post-translational adjustment of levels of stable protein complex members in wild-type cells because of several advantageous properties of the data. First, we measured mRNA, translation, and protein from matched extracts, allowing their direct comparison. Second, we measured nearly 80% of the annotated yeast proteome. Third, we made measurements for several sequential time points, which allowed analysis of trends and correction for differences in dynamic ranges of detection for mass spectrometry and sequencing data. As a simple first test, we examined the patterns of mRNA, translation, and protein for several heterodimer pairs. We found that in the case of TUB1/TUB2—the primary tubulin heterodimer genes—levels of mRNA, translation, and protein over time were remarkably well matched for both components (Figure 3A). The degree of similarity was striking given that these genes are encoded on separate chromosomes and show disparate promoter, 5′ UTR, and open reading frame (ORF) sequences. Furthermore, TUB1 mRNA is subject to splicing, while TUB2 is not. This suggests that eukaryotic cells are capable of synthesizing complex components in surprisingly precise stoichiometry, even when the genes encoding them have different cis-regulatory regions and sequences. However, in the cases of three other heterodimer pairs initially investigated in detail—Rbg1/Tma46, Pob3/Spt16, and Gtr1/Gtr2—we observed some similarity between mRNA and translation for the two genes, but much higher similarity at the protein level, suggesting that meiotic cells synthesize these protein partners somewhat imprecisely and that their levels are adjusted post-translationally to achieve 1:1 stoichiometry (Figures 3B–3D).
Figure 3. Meiotic Cells Are Capable of Perfect Synthesis Matching of Heterodimer Partners, but It Is Uncommon.

(A–F) Z-score plots show gene expression level trends of mRNA levels (left), translation levels (middle), and protein levels (right) over all time points for pairs of genes, including (A) heterodimer Tub1 and Tub2, (B) heterodimer Rbg1 and Tma46, (C) heterodimer Pob3 and Spt16, (D) heterodimer Gtr1 and Gtr2, (E) sequential enzymes involved in purine nucleotide biosynthesis Ade1 and Ade2, and (F) sequential enzymes involved in histidine biosynthesis His2 and His5.
(G) Correlation coefficients for translation and protein between annotated heterodimer partners are shown at left. Yellow represents higher, blue represents lower. The same scaling is used at upper right to compare to a subset of sequential enzymes in biosynthetic pathways. Below right, a summary of trends for heterodimers and adjacent enzymes in biosynthetic pathways. Heterodimer partners show a greater protein than translation correlation, while sequential biosynthetic enzymes show the opposite trend.
(H) Cumulative distribution plots for the translation and footprint correlations in (G). Translation correlations are indistinguishable for the heterodimers and biosynthetic pathway genes, but protein correlations are significantly higher for the heterodimers as assessed by the Kolomogorov-Smirnov (K-S) test.
See also Figures S2, S3, and S4.
To determine the degree to which these examples reflect general trends, we isolated data for all of the genes noted as members of heterodimeric complexes in the Saccharomyces genome database (https://www.yeastgenome.org). We restricted our analysis to cases in which we were able to quantify both heterodimer members at all levels (mRNA, translation, and protein) for every time point and for which the heterodimer did not clearly involve prevalent partner substitution by either entirely different proteins or their paralogous partner. This last filter was a necessary simplification for some heterodimers, but the determination of “prevalence” was sometimes nuanced. For example, TUB3 was not included in our heterodimer analysis of TUB1/TUB2 because—although it is known that Tub3 can substitute for Tub1 in interacting with Tub2—Tub3 was synthesized at very low levels during meiosis, ranging from only 7% to 17% of those of Tub2. Our approach resulted in a list of 37 high-confidence heterodimer pairs (Figure 3G, left). The two heterodimers that make up the annotated tetramer for the Ndc80 kinetochore complex were also included (Spc24/Spc25 and Ndc80/Nuf2), because the temporal meiotic regulation of this complex (discussed below) has been studied in detail (Chen et al., 2017; Chia et al., 2017; Miller et al., 2012). Among these 39 cases, we found that 9 showed a pattern similar to that seen for Tub1/Tub2, with correlation coefficients for both translation and protein for the 2 genes that were higher than 0.95. Of the remaining 30 cases, 26 (87%) showed patterns suggestive of post-translational adjustment of protein levels, with protein-level correlations for the two genes exceeding translation-level correlations, by a large margin in some cases (Figures 3G, left, 3H, and S3C). This result suggests a model by which heterodimer partners are usually synthesized imprecisely and adjusted by degradation of unbound partners in unperturbed wild-type cells.
We considered the possibility that the higher protein compared to translation or mRNA correlations could simply reflect the differences in sequencing and mass spectrometry measurements, or the instantaneous nature of translation versus steady-state nature of protein abundance measurements. To determine whether protein measurements simply tended to correlate better than translation measurements for technical reasons, we examined gene pairs that should have similar expression patterns, but not because of stable physical association. We reasoned that sequential enzymes in linear biosynthetic pathways represent such a class of gene pairs and would be expected to show correlated expression, but not necessarily physical protein association. We again excluded cases in which paralogs serve major redundant roles and were again restricted to cases in which both sequential pathway components were quantified in our dataset at all levels. We included 21 protein pairs that fit these criteria without repetition of either protein in the pair in the analysis (Figure 3G, right). This set of pathway partners showed a starkly different pattern of translation- and protein-level correlation than was seen for heterodimers (examples in Figures 3E and 3F, right side of 3G and 3H, and S3A–S3C). In two cases, Trp3/Trp5 and Lys4/Lys12, expression correlation at both translation and protein levels was high and roughly equivalent. Only 7 of the remaining 19 (37%) cases showed a higher protein correlation than translation correlation for the pairs. Subsequent analysis of these cases revealed that at least two of these sets (Erg25/Erg26 and Erg6/Erg2), although not annotated as heterodimers in the Saccharomyces genome database, are in fact reported to associate physically in a complex (Baudry et al., 2001; Mo and Bard, 2005). The remaining 12 gene pairs (57%) showed a poorer correlation between protein levels compared to translation levels (Figures 3G, right, 3H, S3A, and S3B). Only the difference between protein correlations for the two types of gene pairs is significant (p < 0.001), suggesting that the high protein-level correlation observed for complex components is not simply an artifact of our measurements (Figure 3H). We further confirmed that differences in fold changes, absolute changes, or average gene expression levels for gene pairs measured did not skew protein and translation correlation coefficients in our data (Figures S3D–S3G and S4A–S4F).
Poorly Matched Protein Complex Component Levels Suggest Regulatory Roles
Of the 39 heterodimers that we examined in the full dataset, only 4 (13.3%) showed protein levels that were more poorly correlated over meiosis than translation levels (Figure 3G). This set of genes are candidates for future study of potentially interesting biological regulation in meiosis, as this is the pattern seen for the Ndc80/Nuf2 heterodimer (Figure 3G), which reflects the recent finding that Ndc80 complex activity is silenced early in meiosis by specific downregulation of Ndc80 protein levels (Chen et al., 2017; Chia et al., 2017; Miller et al., 2012). The downregulation of Ndc80 results in an inactive outer kinetochore during meiotic prophase because Ndc80 acts as a linchpin component. When it is present, the complex is active; when it is not present, the complex is not (Chen et al., 2017; Chia et al., 2017; Miller et al., 2012). When we analyzed their protein levels side by side, as determined by mass spectrometry, all four Ndc80 complex components showed patterns that are consistent with this reported regulation (Figure S5A). Spc24, Spc25, and Nuf2 showed similar protein-level trends to one another over meiosis, but Ndc80 alone showed low levels early in meiosis that rose before the first meiotic division. At the translation level, there was poorer correlation among Spc24, Spc25, and Nuf2 than was seen at the protein level, while Ndc80 was similarly poorly correlated with all three at both the protein and translation levels (Figure S5A).
Trends in Regulation of Heterodimers Also Apply to Multiprotein Complexes
Analysis of stable multisubunit protein complexes for which we were able to quantify most or all of the components also revealed evidence of degradation of free monomers (Figures 4A–4E, 4I, 4J, S5B, and S5C). Comparison of translation and protein patterns over meiosis for the oligosaccharyltransferase (OST), chaperonin containing TCP-1 (CCT), F1F0 ATPase, histone deacetylase (HDA), endoplasmic reticulum membrane protein complex (EMC), Ccr4-Not, exosome, translocon, and prefoldin complexes revealed a trend similar to that observed for heterodimers (Figures 4A–4E, 4I, 4J, S5B, and S5C). Although in some cases expression was already well correlated at the translation level, in all of the cases analyzed, the correlation was even higher at the protein level. A control comparison of linear biosynthetic pathway members did not show a similar trend, as exemplified by the heme, pyrimidine, ergosterol, histidine, and nucleotide biosynthesis pathways (Figure 4F–4H, S5D, and S5E). Interestingly, although most complex members showed better matching of protein than translation level patterns, outlier components were also observed in two cases analyzed (the exosome and the Ccr4-Not complex), as seen for the Ndc80 complex (Figures 4I, 4J, and S5A). This suggests a mechanism for meiotic regulation of complex activity through regulation of the levels of one or more key members, as discussed above for the Ndc80 complex.
Figure 4. Members of Multiprotein Complexes Show Higher Agreement of Protein Levels during Meiosis than Translation, While Members of Biosynthetic Pathways Do Not.

(A–J) Z-score plots to show gene expression level trends of translation levels (middle) and protein levels (right) are shown over all time points for groups of genes, including all quantified members of representative protein complexes and biosynthetic pathways: (A) the OST complex, (B) the CCT complex, (C) the F1F0 ATPase complex, (D) the HDA complex, (E) the EMC complex, (F) the heme biosynthesis pathway, (G) the pyrimidine biosynthesis pathway, (H) the ergosterol biosynthesis pathway, (I) the Ccr4-Not complex, and (J) the exosome complex.
See also Figure S5.
A more detailed investigation of the exosome components revealed that the three protein components (Mpp6, Rrp6, and Lrp1) that did not show high protein level correlation with the others are known to be non-constitutive complex members (Figures 4J and 5A). Mpp6 is an accessory component of the nuclear exosome, and Rrp6/Lrp1 associate tightly as a subcomplex that also associates specifically with the nuclear exosome (Feigenbutz et al., 2013; Synowsky et al., 2009). These three genes clustered tightly with the core exosome components at the mRNA and translation levels, but they are clearly distinct from core exosome components in protein-level clustering, with the closely physically associated Rrp6/Lrp1 showing much tighter protein-level correlation with each other than at either other level of expression measurement (Figure 5A). These patterns support specific post-translational adjustment of protein levels based on physical association of protein complex components.
Figure 5. Among Highly Correlated Protein-level Trends for Complex Members, Outliers Suggest Non-constitutive Association.

(A) Hierarchical clustering of levels of exosome complex components (from Figure 4J) for mRNA (left), translation (middle), and protein (right). Mpp6 (red) is a non-constitutive component and clusters far from others at the protein level. Rrp6/Lrp1 (green) is a non-constitutive heterodimer; these genes cluster closely to each other but separate from the core exosome complex at the protein level.
(B) Hierarchical clustering of protein data for proteasome components and accessory factors. Matched mRNA (far left) and translation levels (middle). Note two discrete protein-level clusters—one with all 20S components except Pre9 and the other cluster with all 19S components. Far right, a proteasome illustration is color coded to match gene names to its left.
(C) All 19S regulatory (orange) and 20S core (blue) proteasome members were analyzed together, scaled to the maximum value measured for each. The means and SDs (bars) are shown for mRNA (left), translation (middle), and protein (right). Note protein divergence between the two groups of genes at late time points (represented by gray box), suggesting synthesis co-regulation for all but independent post-translational adjustment for two complexes at late stages.
See also Figure 4.
Analysis of proteasome components similarly revealed a specific protein-level discordance between core and regulatory subunits. We were able to quantify all 33 26S proteasome subunits at every level in our gene expression dataset, as well as 4 associated factors. All 26S proteasome subunits showed highly similar patterns of mRNA accumulation and translation, which is consistent with their transcriptional co-regulation (Figures 5B and 5C) (Mannhaupt et al., 1999; Sato et al., 2009). It was not possible, based on clustering of mRNA or translation measurements, to distinguish between subcomplexes (Figures 5B and 5C). In contrast, the protein-level patterns for this set of 33 26S genes showed 2 major groupings, which corresponded almost perfectly with the 19S regulatory particle and the 20S proteasome core (Figures 5B and 5C). The 19S components showed a decrease in protein levels that precedes the decrease seen for 20S subunits by ~3 hr during the meiotic program (Figures 5B and 5C). The core proteasome can be activated independent of the 19S regulatory particle (Schmidt et al., 2005), and the regulation observed late in the meiotic program suggests a natural context for this role. The sole exception to the distinct patterns of protein expression for the core and regulatory subunits was Pre9, which clustered with the regulatory particle subunits rather than the proteasome core, of which it is a reported member (Figure 5B). Pre9 has been reported to be unlike other 20S components in multiple studies. It is known that Pre9 directly interacts with the tails of Rpt2 and Rpt6 (the 2 proteins that it clusters between in Figure 5B) to mediate 19S and 20S association, and the interaction with Rpt6 is the basis for specificity of the 19S/20S register (Park et al., 2013). Pre9 is also the only non-essential 20S member (Kusmierczyk et al., 2008; Velichutina et al., 2004), suggesting a role that differs from the other core members. It is unclear what this role is, but it is worth noting that cells deleted for PRE9 have been shown to have specific meiotic defects, attributed to the proteasome’s association with meiotic chromosomes during recombination (Ahuja et al., 2017).
All of the proteasome-associated proteins analyzed that did not fit into one of the two major clusters are known to have additional roles (Figure 5B). The 19S regulatory particle component Sem1 is known to be part of other, non-proteasome complexes (Kragelund et al., 2016), and neither Cic1 or Blm10/PA200 are thought to constitutively or exclusively associate with the proteasome. Cic1 associates with the proteasome but also with preribosomal particles (Harnpicharnchai et al., 2001; Jäger et al., 2001), and Blm10/PA200 is involved in both core particle assembly and mature particle activation (Fehlker et al., 2003). The protein-level distinctions among proteasomal genes are interesting in light of their high degree of known transcriptional co-regulation, and this suggests robust post-translational adjustment of protein levels; but it also demonstrates that protein measurements taken over time can be surprisingly sensitive in identifying functional distinctions among groups of proteins with identical control of synthesis.
Ribosomal Proteins Are Highly Co-regulated at All Levels, Degraded Late in the Meiotic Program
The ribosome is another large multiprotein complex with known co-regulation of component synthesis. In contrast to the proteasome, clustering of ribosomal protein (RP) levels did not reveal subclustering based on any reported physical feature of the ribosome or large versus small subunit identity (Figure 6A). Rather, 72 of the 98 RP-encoding genes that we quantified at the protein level showed extremely similar patterns over the meiotic program at every level of gene expression measured. The other 26 showed slightly different protein-level patterns, the basis of which we do not yet understand (Figure 6A). We also noted that protein-level patterns for all RP genes examined indicated protein degradation late in MII and re-synthesis in spores (Figure 6A). Comparison of the mRNA, translation, and protein abundance measurements for this group of genes revealed a signature of protein degradation similar to what we observed for the known degradation target Sic1 (Figure 1D), with translation actually increased in late meiosis while protein levels decrease (Figure 6B). In spores, subsequently, protein levels increased to a level similar to early meiotic cells, while translation (and mRNA levels) remained high (Figure 6B). While the transcriptional uptick in RP genes had previously been seen during sporulation (Chu et al., 1998), its association with protein degradation was not evident.
Figure 6. RPs Are Actively Degraded Late in the Meiotic Program.

(A) Hierarchical clustering of protein levels was performed for all RP genes quantified (right), and is compared to matched translation (middle) and mRNA (left). Values shown are z-score normalized.
(B) Quartile analysis of all RPs at all levels of expression. Pink shading represents period late in meiosis when transcription and translation increase but protein decreases, a hallmark of active degradation.
(C) A strategy to identify active protein degradation and re-synthesis after spore wall formation. This approach uses heterozygous GFP tagging of the protein of interest, in this case Rpl26b, in diploid cells. Before spore formation, protein from both alleles is in the cytosol. After spore formation, if a protein is degraded and re-synthesized, then the fluorescent signal should decrease in spores that inherited the untagged allele and should increase in spores that inherited the tagged allele. This is observed for Rpl26b, but not histone protein Htb1. Inset numbers represent frame numbers for 20-min intervals; scale bar represents 2 μM.
(D) Quantification of the fluorescence over time for the two cells in (C), starting when spore individualization begins. Note the decrease in GFP signal in two spores and the increase in the other two.
(E) Quantification of additional cells (n = 10 tetrads) from the experiment in (C) and (D) and a similar experiment using heterozygous RPL29-GFP. Error bars represent SD. p values determined by paired t test: *p = 0.033, ***p < 0.001.
See also Figure S6.
To confirm this regulation independently, we GFP-tagged the RP gene RPL26B in a strain carrying mCherry-tagged histone H2B (encoded by HTB1). Both tags were heterozygous in diploid cells. Thus, during meiotic stages in which the cytosol was continuous, before spore packaging, we expect to see homogeneous green cytosolic and red nuclear signals. Following spore individualization, we would expect to see red signal remaining in all four spore nuclei if histones are stable, suggesting that they were synthesized before spore packaging (Figure 6C). In contrast, if RPs were degraded and re-synthesized in spores, then only the two spores carrying the RPL26B-GFP allele should be green and the other two spores should lose GFP signal relative to earlier stages (Figure 6C). Indeed, this was what we observed. We saw evidence that Htb1 continues to be synthesized in spores, resulting in an increase in signal in the two spores that presumably carry the HTB-mCherry allele, but in the case of RPL26B, we observed an increase in signal in two spores and a decrease in the other two (Figures 6C and 6D). This was observed and quantified for individual cases and was a general trend among cells of this genotype (Figures 6D and 6E). The loss of GFP signal in two spores was not due to photobleaching resulting from time-lapse imaging, as cells on the same microfluidic plate that were not previously imaged showed similar relative levels of GFP in a 2:2 bright:dim ratio as those that were imaged over a period of time (Figure S6). A similar effect could be seen in cells carrying a heterozygous RPL29-GFP allele (Figures 6E and S6). We concluded that ribosomes are actively degraded and re-synthesized at the end of the yeast meiotic program.
DISCUSSION
Important individual examples of regulated protein degradation during meiotic differentiation are well characterized, but it has been challenging to determine the pervasiveness of this mode of regulation in meiosis. We report signatures in matched global quantitative mass spectrometry and ribosome profiling data that allowed us to identify specific, natural, and previously unidentified cases of regulated protein degradation during the yeast meiotic program. These signatures include periods of stable or even increased translation of a given gene, while protein abundance decreases, as well as periods of particularly rapid decline in protein levels for groups of genes in concert. These trends are sensitively detected in our dataset because cells undergoing meiotic differentiation do not display the type of dilution due to cell growth and division that is a major contributor to protein-level decline during mitotic growth (Christiano et al., 2014).
Ama1, a meiosis-specific APC/C subunit, is important for meiotic progression (Cooper et al., 2000). We observed that all three previously identified Ama1 targets that we were able to quantify in our mass spectrometry dataset showed patterns of protein-level change during meiosis that were strikingly similar to one another as well as to a small group of other proteins (Figure 1E). We hypothesized that this group included new Ama1 targets and confirmed that Pes4 and Mip6, two members of this cluster with meiotic roles during a precise window, are degraded in an Ama1-dependent manner (Figure 2). This is an unorthodox approach for the identification of potential E3 targets, but at least in this case, it seems to allow specific and sensitive detection based on the ability to follow natural protein patterns over time in an unperturbed system.
Analyses of protein data also revealed previously unrecognized, coordinated degradation and re-synthesis of RP subunits following gamete (spore) formation (Figure 6). Why would cells expend energy to degrade RPs and concomitantly re-synthesize them? We propose two explanations. First, this could be a mechanism of cellular quality control. It has been shown that the abnormal nucleolar morphology observed in aged yeast cells is reset in all four gametes by the process of meiotic differentiation (Unal et al., 2011). It is possible that this nucleolar morphology reflects defective rRNA synthesis or processing, and thus resultant ribosomes may be of poor quality. Because gamete quality is important for an organism’s genome stability on an evolutionary scale and the proteins synthesized in a gamete provide critical functions, including mediating gene expression, the destruction and re-synthesis of a gamete’s ribosomes may be a mechanism of ensuring gamete integrity. Second, it is possible that ribosome composition or modification is altered in meiosis relative to mitotic growth and that these alterations must be reset after meiosis. The translation of upstream ORFs (uORFs) within 5′ leaders of thousands of mRNAs is dramatically upregulated during meiosis, even in cases in which an apparently identical transcript is present under mitotic and meiotic conditions (Brar et al., 2012). If meiotic modification to the core translation machinery contributes to this effect, then destruction of this machinery could enable cells to return to mitotic translation patterns.
The most surprising finding from our analyses was that members of stable protein complexes are typically synthesized with imprecise stoichiometry that is adjusted post-translationally (Figures 3, 4, and 5). Effectively perfect translation matching is occasionally seen in our dataset, including for the Tub1/Tub2 heterodimer, demonstrating that cells are capable of very precisely matched synthesis levels, even for proteins encoded from genomically distant genes with dissimilar regulatory regions (Figures 3A and 3G), yet this is not the norm (Figures 3, 4, and 5). This conclusion differs from that of recent studies that reported matched synthesis as the rule among protein complex components (Li et al., 2014; Taggart and Li, 2018). There are significant biological and analytical differences between our study and theirs. The conclusion that synthesis rates were matched for protein partners was determined using estimation of absolute translation levels from ribosome profiling data of cells in rich, steady-state growth conditions (Li et al., 2014; Taggart and Li, 2018). Our study, in contrast, was of cells undergoing meiotic differentiation. Natural developmental processes, including meiosis, involve proteome remodeling over time, and this may influence cellular strategies for protein complex regulation. Furthermore, our study used analyses of trends in matched series of protein and translation measurements to enable the quantitative comparison of these two different types of data. Despite these key differences, we argue that the data in all of these studies are actually consistent with our conclusions; we find that some heterodimers do show “perfect” synthesis matching and most show a positive correlation between translation patterns over time. It is not that we observe decoupled synthesis rates for complex components, but simply that protein-level patterns match better than translation patterns for most cases analyzed here, suggesting an important role for both levels of regulation in eukaryotes.
Post-translational adjustment of levels of protein complex partners has been previously observed in a variety of experimental systems by perturbation of normal cellular homeostasis through gene (or chromosome) overexpression (Abovich et al., 1985; Dephoure et al., 2014; Gorenstein and Warner, 1977; Ishikawa et al., 2017; Lam et al., 2007; Papp et al., 2003; Sung et al., 2016a; Torres et al., 2010; Warner, 1977; Warner et al., 1985). Unincorporated complex components have been shown to be unstable in several contexts as a result of proteasome-dependent degradation. For example, if a single RP is overexpressed relative to others, then excess subunits are degraded by a mechanism that depends on the E3 ligase Tom1 (_Sung et al., 2016b). It has previously been challenging to determine, however, whether the post-translational adjustment of levels of protein complex components occurs naturally in wild-type, unperturbed cells, or simply occurs as a fail-safe for mutant conditions. Our study argues that the former is the case.
The extremely high degree of protein-level correlation for most protein complex components also enables the sensitive detection of outlier components, which may be involved in regulatory roles. For example, the Ndc80 kinetochore complex components are well matched at the protein level, with the exception of the namesake component Ndc80, which is low early in meiosis and increases before the first meiotic division (Figure S5A). The mechanism and importance of this regulation during meiosis have been shown (Chen et al., 2017; Chia et al., 2017; Miller et al., 2012). In such cases, a single outlier component can act as a linchpin for complex activity, an efficient mechanism for rapid complex activation or inactivation. Analysis of similar outlier cases for other complexes seems promising for uncovering other similar types of regulation (Figures 4I, 4J, 5, and S5B) (de Lichtenberg et al., 2005).
The findings reported here regarding protein complex component regulation are informative in considering the cellular balance between perfection and efficiency. Yeast cells undergo meiosis in the absence of glucose or amino acids, and this is therefore a context in which cellular economy of resources is extremely important. The prevalence of imprecise synthesis of protein complex partners and subsequent degradation of unpartnered subunits even in these conditions implies a general advantage to this strategy, relative to perfect synthesis matching. Cases such as the tubulin heterodimer reveal that eukaryotic cells are capable of nearly perfect co-synthesis of partner proteins, and the rarity of this regulation suggests something interesting about the cases in which it does occur. In the case of Tub1/Tub2, for which it is known that stoichiometric imbalance leads to toxicity (Burke et al., 1989), the disadvantage of post-translational buffering seems clear. Investigation of the few other cases that we identified as showing extremely well-matched synthesis may yield new insights into the features and cellular roles of these components, as well as broader principles of cellular resource management in gene expression regulation.
STAR+METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gloria Brar (gabrar@berkeley.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast growth and sporulation
All yeast strains used were diploid Saccharomyces cerevisiae of the SK1 background. Strains used in this study are listed in the key resources table. For meiotic time courses, yeast were inoculated into YEPD overnight, then diluted to OD600 0.2 into buffered YTA and grown for 12 hours. Cells were washed in water and resuspended in sporulation media supplemented with 0.02% raffinose. Time points were taken at indicated times.
METHOD DETAILS
Western blotting
Western blotting was performed using a standard TCA-based protocol. Briefly, 2.5 OD units of culture were treated with 5% TCA at 4C for at least 10 min. Samples were then washed with 1 mL acetone. Acetone was aspirated and pellets were dried overnight at RT. Lysates were made by adding 100 mL protein lysis buffer [50 mM TE, 3 mM DTT, 1.1 mM PMSF (Sigma), 1 μM pepstatin A, 1X protease inhibitor cocktail (Roche)] and 1 volume acid-washed glass beads (Sigma), and bead-beating for 5 min at RT. 3X SDS loading buffer was added and samples were boiled for 5 min. Beads were pelleted by centrifugation and 5 mL supernatant was loaded onto 4–12% Bis-Tris polyacrylamide gels. Following electrophoresis, proteins were transferred using a semi-dry transfer apparatus (Trans-Blot Turbo, BioRad). The following antibodies were used: mouse anti-V5 (Invitrogen, 1:2,000), rabbit anti-hexokinase (Stratech, 1:10,000), anti-mouse and anti-rabbit secondaries (Li-Cor, 1:15,000). Primary antibody incubation was overnight, secondary for 1–2 hr. Blots were visualized and quantified using a Li-Cor system.
Meiotic cell staging
The meiotic stage of a cell was determined based on its DAPI morphology by fluorescent microscopy. 200 cells were counted per strain per time point.
Heterodimer analyses
Heterodimers were defined as annotated in the Saccharomyces Genome Database (SGD; https://www.yeastgenome.org). Only cases in which both partners were quantified for mRNA, translation, and protein were analyzed and cases were excluded in which there was a reported major alternative interactor (typically a paralog). Three additional cases were excluded for which one partner appeared to be especially lowly expressed (a mean translation RPKM < 20). Z-scores were calculated and the correlation over all 10 samples for the pair of genes was determined.
Linear pathway analyses
Linear biosynthetic pathways were identified based on literature searching and SGD confirmation. Only cases in which both partners were quantified for mRNA, translation, and protein were analyzed and cases were excluded in which there was a reported major alternative interactor (typically a paralog). Also, each gene was only analyzed in one pairing (either with its upstream or downstream partner, decisions about which to use were based on maximization of possible partners to analyze). Unlike the heterodimer analyses, no cases needed to be excluded based on especially low expression of either gene (a mean translation RPKM < 20). Z-scores were calculated and the correlation over all 10 samples for the pair of genes was determined.
Heterozygous RP-GFP imaging
After two hours in SPO media, 100 uL of cells were placed in a CellASIC ONIX Microfluidic Plate (Y04D) and maintained with fresh SPO media at 2psi using the CellASIC ONIX Microfluidic Perfusion System (CellASIC Corp., Hayward, CA, USA). The cells were held at 30 C using a thermostatic system for the microscope stage.
Cells were imaged using a DeltaVision microscope with a 60x/1.42 oil-immersion objective (DeltaVision, GE Healthcare, Sunnyvale, CA) and filters: FITC (EX475/28, EM525/48) and mCherry (EX575/25, EM625/45). Images were acquired using the softWoRx software (softWoRx, GE Healthcare) with z stacks of 8 slices with 0.5 mm spacing. For time lapse imaging, images were taken every 20 minutes for 12 hours. After 24 hours, images were taken from the same points that had been imaged during the time-lapse portion, as well as points from the same wells that had not been imaged previously.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
A Kolomogorov-Smirnov (K-S) test was used to determine significance for the differences between cumulative distribution plots shown in Figure 3H. A two-tailed t test was used to determine significance between fluorescence intensities for the data presented in Figure 6E.
Heterozygous RP-GFP quantification
All images were deconvolved using softWoRx software accompanying the DeltaVision microscope, and maximum-intensity projections were generated using ImageJ/FIJI image processing software (RRID:SCR_002285; Schneider et al., 2012). Mean intensity of signal from the cells was measured using the “measure” tool in FIJI, and was divided by the background signal from the same image.
DATA AND SOFTWARE AVAILABILITY
Genome-wide data analyzed here were generated previously (Cheng et al., 2018). In short, mRNA levels were assayed by mRNA-seq, translation measurements were assayed by ribosome profiling, and protein levels were assayed by quantitative mass spectrometry (TMT10) for 8 time points during the meiotic program and two vegetative time points (one in rich media and one in sporulation media matched to meiotic samples). All measurements showed high reproducibility, with R values ranging from 0.935 to 0.992. All original data can be found at NCBI GEO: GSE108778 and MassIVE: MSV000081874. Processed data used for analyses here are in Table S1.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-V5 antibody | Invitrogen | Cat#46–0705 |
| Rabbit anti-hexokinase antibody | Stratech | Cat#H2035 |
| Anti-rabbit secondary | Li-Cor | Cat#925–68071 |
| Anti-mouse secondary | Li-Cor | Cat#925–32210 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PMSF | Sigma | Cat#78830 |
| Pepstatin A | Sigma | Cat #P4265 |
| Protease Inhibitor Cocktail | Roche | Cat#29384100 |
| Experimental Models: Organisms/Strains | ||
| BrÜn1061 (MATa/α ndt80::NDT80–3V5::KANMX) | This paper | N/A |
| BrÜ n11983 (MATa/α stu2::STU2–3V5:KanMX ama1::HISMX) | This paper | N/A |
| BrÜn11985 (MATa/α ndt80::NDT80–3V5::KANMX ama1::HISMX) | This paper | N/A |
| BrÜn12076 (MATa/α stu2::STU2–3V5:KanMX) | This paper | N/A |
| BrÜn13016 (MATa/α pes4::PES4–3V5:KanMX/ ama1::HISMX) | This paper | N/A |
| BrÜn13018 (MATa/α pes4::PES4–3V5:KanMX) | This paper | N/A |
| BrÜn13024 (MATa/α mip6::MIP6–3V5:KanMX ama1::HISMX) | This paper | N/A |
| BrÜn13026 (MATa/α mip6::MIP6–3V5:KanMX) | This paper | N/A |
| BrÜn13712 (MATa/α ssp1::SSP1–3V5:KanMX ama1::HISMX) | This paper | N/A |
| BrÜn13714 (MATa/α ssp1::SSP1–3V5:KanMX) | This paper | N/A |
| BrÜn7085 (MATa/α rpl29::RPL29-HTA-GFP:KanMX/RPL29 htb1::HTB1-mCherry-HISMX6/HTB1) | This paper | N/A |
| BrÜn7087 (MATa/α rpl26b::RPL26B-HTA-GFP:KanMX/RPL29 htb1:: HTB1-mCherry-HISMX6/HTB1) | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.net/Downloads |
Highlights.
The synthesis of most protein complex components during meiosis is imprecisely matched
The levels of most protein interaction partners are post-translationally adjusted
Ribosomal proteins are degraded and re-synthesized late in the meiotic program
Analysis of meiotic protein levels over time points to additional Ama1-APC/C targets
ACKNOWLEDGMENTS
Thanks to Nick Ingolia and Elçin Ünal for helpful comments on this manuscript. This work was funded by investigator awards from the NIH (DP2-GM-119138), the Alfred P. Sloan Foundation (FG-2016-6229), and the Pew Charitable Trusts (00029624), and by Bowes Foundation funding to G.A.B. A.H. was funded by National Science Foundation pre-doctoral fellowship DGE 1752814. A.R.E. and A.H. were funded in part by NIH training grants to the Molecular and Cell Biology department (T32 GM 0007232 and T32 HG 00047). M.J., S.H., and A.K. were funded by the NIH (R35-GM-128802) and Columbia University start-up funding.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.008.
REFERENCES
- Abovich N, Gritz L, Tung L, and Rosbash M (1985). Effect of RP51 gene dosage alterations on ribosome synthesis in Saccharomyces cerevisiae. Mol. Cell. Biol 5, 3429–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja JS, Sandhu R, Mainpal R, Lawson C, Henley H, Hunt PA, Yanowitz JL, and Börner GV (2017). Control of meiotic pairing and recombination by chromosomally tethered 26S proteasome. Science 355, 408–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry K, Swain E, Rahier A, Germann M, Batta A, Rondet S, Mandala S, Henry K, Tint GS, Edlind T, et al. (2001). The effect of the erg26–1 mutation on the regulation of lipid metabolism in Saccharomyces cerevisiae. J. Biol. Chem 276, 12702–12711. [DOI] [PubMed] [Google Scholar]
- Blikstad I, Nelson WJ, Moon RT, and Lazarides E (1983). Synthesis and assembly of spectrin during avian erythropoiesis: stoichiometric assembly but unequal synthesis of alpha and beta spectrin. Cell 32, 1081–1091. [DOI] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, and Weissman JS (2012). High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke D, Gasdaska P, and Hartwell L (1989). Dominant effects of tubulin overexpression in Saccharomyces cerevisiae. Mol. Cell. Biol 9, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon CK, and Hawley RS (2016). Regulating the construction and demolition of the synaptonemal complex. Nat. Struct. Mol. Biol 23, 369–377. [DOI] [PubMed] [Google Scholar]
- Chen J, Tresenrider A, Chia M, McSwiggen DT, Spedale G, Jorgensen V, Liao H, van Werven FJ, and Ünal E (2017). Kinetochore inactivation by expression of a repressive mRNA. eLife 6, e27417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Otto GM, Powers EN, Keskin A, Mertins P, Carr SA, Jovanovic M, and Brar GA (2018). Pervasive, coordinated protein level changes driven by transcript isoform switching during meiosis. Cell 172, 910–923.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia M, Tresenrider A, Chen J, Spedale G, Jorgensen V, Ünal E, and van Werven FJ (2017). Transcription of a 5′ extended mRNA isoform directs dynamic chromatin changes and interference of a downstream promoter. eLife 6, e27420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiano R, Nagaraj N, Fröhlich F, and Walther TC (2014). Global proteome turnover analyses of the yeasts S. cerevisiae and S. pombe. Cell Rep. 9, 1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, and Herskowitz I (1998). Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol. Cell 1, 685–696. [DOI] [PubMed] [Google Scholar]
- Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, and Herskowitz I (1998). The transcriptional program of sporulation in budding yeast. Science 282, 699–705. [DOI] [PubMed] [Google Scholar]
- Cooper KF, Mallory MJ, Egeland DB, Jarnik M, and Strich R (2000). Ama1p is a meiosis-specific regulator of the anaphase promoting complex/cyclosome in yeast. Proc. Natl. Acad. Sci. USA 97, 14548–14553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lichtenberg U, Jensen LJ, Brunak S, and Bork P (2005). Dynamic complex formation during the yeast cell cycle. Science 307, 724–727. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Hwang S, O’Sullivan C, Dodgson SE, Gygi SP, Amon A, and Torres EM (2014). Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. eLife 3, e03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond AE, Park J-S, Inoue I, Tachikawa H, and Neiman AM (2009). The anaphase promoting complex targeting subunit Ama1 links meiotic exit to cytokinesis during sporulation in Saccharomyces cerevisiae. Mol. Biol. Cell 20, 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirick L, Goetsch L, Ammerer G, and Byers B (1998). Regulation of meiotic S phase by Ime2 and a Clb5,6-associated kinase in Saccharomyces cerevisiae. Science 281, 1854–1857. [DOI] [PubMed] [Google Scholar]
- Evans T, Rosenthal ET, Youngblom J, Distel D, and Hunt T (1983). Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 33, 389–396. [DOI] [PubMed] [Google Scholar]
- Fehlker M, Wendler P, Lehmann A, and Enenkel C (2003). Blm3 is part of nascent proteasomes and is involved in a late stage of nuclear proteasome assembly. EMBO Rep. 4, 959–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenbutz M, Jones R, Besong TMD, Harding SE, and Mitchell P (2013). Assembly of the yeast exoribonuclease Rrp6 with its associated cofactor Rrp47 occurs in the nucleus and is critical for the controlled expression of Rrp47. J. Biol. Chem 288, 15959–15970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi SJ, Zenklusen D, Lionnet T, and Singer RH (2011). Transcription of functionally related constitutive genes is not coordinated. Nat. Struct. Mol. Biol 18, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorenstein C, and Warner JR (1977). Synthesis and turnover of ribosomal proteins in the absence of 60S subunit assembly in Saccharomyces cerevisiae. Mol. Gen. Genet 157, 327–332. [DOI] [PubMed] [Google Scholar]
- Harnpicharnchai P, Jakovljevic J, Horsey E, Miles T, Roman J, Rout M, Meagher D, Imai B, Guo Y, Brame CJ, et al. (2001). Composition and functional characterization of yeast 66S ribosome assembly intermediates. Mol. Cell 8, 505–515. [DOI] [PubMed] [Google Scholar]
- Harper JW, and Bennett EJ (2016). Proteome complexity and the forces that drive proteome imbalance. Nature 537, 328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphryes N, Leung W-K, Argunhan B, Terentyev Y, Dvorackova M, and Tsubouchi H (2013). The Ecm11-Gmc2 complex promotes synaptonemal complex formation through assembly of transverse filaments in budding yeast. PLoS Genet. 9, e1003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irniger S, Piatti S, Michaelis C, and Nasmyth K (1995). Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell 81, 269–278. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Makanae K, Iwasaki S, Ingolia NT, and Moriya H (2017). Post-Translational Dosage Compensation Buffers Genetic Perturbations to Stoichiometry of Protein Complexes. PLoS Genet. 13, e1006554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S, Strayle J, Heinemeyer W, and Wolf DH (2001). Cic1, an adaptor protein specifically linking the 26S proteasome to its substrate, the SCF component Cdc4. EMBO J. 20, 4423–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Zhang K, Sternglanz R, and Neiman AM (2017). Predicted RNA Binding Proteins Pes4 and Mip6 Regulate mRNA Levels, Translation, and Localization during Sporulation in Budding Yeast. Mol. Cell. Biol 37, e00408–e00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, and Kirschner MW (1995). A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 81, 279–288. [DOI] [PubMed] [Google Scholar]
- Kragelund BB, Schenstrøm SM, Rebula CA, Panse VG, and Hartmann-Petersen R (2016). DSS1/Sem1, a Multifunctional and Intrinsically Disordered Protein. Trends Biochem. Sci 41, 446–459. [DOI] [PubMed] [Google Scholar]
- Kusmierczyk AR, Kunjappu MJ, Funakoshi M, and Hochstrasser M (2008). A multimeric assembly factor controls the formation of alternative 20S proteasomes. Nat. Struct. Mol. Biol 15, 237–244. [DOI] [PubMed] [Google Scholar]
- Lam YW, Lamond AI, Mann M, and Andersen JS (2007). Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr. Biol 17, 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnert ME, and Lodish HF (1988). Unequal synthesis and differential degradation of alpha and beta spectrin during murine erythroid differentiation. J. Cell Biol 107, 413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G-W, Burkhardt D, Gross C, and Weissman JS (2014). Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C-T, Yuan S, and Li K-C (2009). Patterns of co-expression for protein complexes by size in Saccharomyces cerevisiae. Nucleic Acids Res 37, 526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannhaupt G, Schnall R, Karpov V, Vetter I, and Feldmann H (1999). Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett. 450, 27–34. [DOI] [PubMed] [Google Scholar]
- McShane E, Sin C, Zauber H, Wells JN, Donnelly N, Wang X, Hou J, Chen W, Storchova Z, Marsh JA, et al. (2016). Kinetic Analysis of Protein Stability Reveals Age-Dependent Degradation. Cell 167, 803–815.e21. [DOI] [PubMed] [Google Scholar]
- Miller MP, Unal E, Brar GA, and Amon A (2012). Meiosis I chromosome segregation is established through regulation of microtubule-kinetochore interactions. eLife 1, e00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo C, and Bard M (2005). A systematic study of yeast sterol biosynthetic protein-protein interactions using the split-ubiquitin system. Biochim. Biophys. Acta 1737, 152–160. [DOI] [PubMed] [Google Scholar]
- Mueller S, Wahlander A, Selevsek N, Otto C, Ngwa EM, Poljak K, Frey AD, Aebi M, and Gauss R (2015). Protein degradation corrects for imbalanced subunit stoichiometry in OST complex assembly. Mol. Biol. Cell 26, 2596–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okaz E, Arguüello-Miranda O, Bogdanova A, Vinod PK, Lipp JJ, Markova Z, Zagoriy I, Novak B, and Zachariae W (2012). Meiotic prophase requires proteolysis of M phase regulators mediated by the meiosis-specific APC/CAma1. Cell 151, 603–618. [DOI] [PubMed] [Google Scholar]
- Papp B, Päl C, and Hurst LD (2003). Dosage sensitivity and the evolution of gene families in yeast. Nature 424, 194–197. [DOI] [PubMed] [Google Scholar]
- Park S, Li X, Kim HM, Singh CR, Tian G, Hoyt MA, Lovell S, Battaile KP, Zolkiewski M, Coffino P, et al. (2013). Reconfiguration of the proteasome during chaperone-mediated assembly. Nature 497, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochart P, Woltering D, and Hollingsworth NM (1997). Conserved properties between functionally distinct MutS homologs in yeast. J. Biol. Chem 272, 30345–30349. [DOI] [PubMed] [Google Scholar]
- Sato Y, Sakamoto K, Sei M, Ewis AA, and Nakahori Y (2009). Proteasome subunits are regulated and expressed in comparable concentrations as a functional cluster. Biochem. Biophys. Res. Commun 378, 795–798. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Haas W, Crosas B, Santamaria PG, Gygi SP, Walz T, and Finley D (2005). The HEAT repeat protein Blm10 regulates the yeast proteasome by capping the core particle. Nat. Struct. Mol. Biol. 12, 294–303. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Oh SD, Hunter N, and Shinohara A (2008). Crossover assurance and crossover interference are distinctly regulated by the ZMM proteins during yeast meiosis. Nat. Genet. 40, 299–309. [DOI] [PubMed] [Google Scholar]
- Sourirajan A, and Lichten M (2008). Polo-like kinase Cdc5 drives exit from pachytene during budding yeast meiosis. Genes Dev. 22, 2627–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens RC, and Davis TN (1998). Mlc1p is a light chain for the unconventional myosin Myo2p in Saccharomyces cerevisiae. J. Cell Biol. 142, 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, and Hershko A (1995). The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell 6, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung M-K, Reitsma JM, Sweredoski MJ, Hess S, and Deshaies RJ (2016a). Ribosomal proteins produced in excess are degraded by the ubiquitin-proteasome system. Mol. Biol. Cell 27, 2642–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung M-K, Porras-Yakushi TR, Reitsma JM, Huber FM, Sweredoski MJ, Hoelz A, Hess S, and Deshaies RJ (2016b). A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. eLife 5, e19105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synowsky SA, van Wijk M, Raijmakers R, and Heck AJR (2009). Comparative multiplexed mass spectrometric analyses of endogenously expressed yeast nuclear and cytoplasmic exosomes. J. Mol. Biol. 385, 1300–1313. [DOI] [PubMed] [Google Scholar]
- Taggart JC, and Li GW (2018). Precise stoichiometric production of protein complexes in eukaryotes without widespread feedback regulation. Cell Syst. Published online December 2018. 10.1016/j.cels.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan GS, Magurno J, and Cooper KF (2011). Ama1p-activated anaphase-promoting complex regulates the destruction of Cdc20p during meiosis II. Mol. Biol. Cell 22, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, Dunham MJ, and Amon A (2010). Identification of aneuploidy-tolerating mutations. Cell 143, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal E, Kinde B, and Amon A (2011). Gametogenesis eliminates age-induced cellular damage and resets life span in yeast. Science 332, 1554–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitia RA, and Potier MC (2015). Gene dosage imbalances: action, reaction, and models. Trends Biochem. Sci 40, 309–317. [DOI] [PubMed] [Google Scholar]
- Velichutina I, Connerly PL, Arendt CS, Li X, and Hochstrasser M (2004). Plasticity in eucaryotic 20S proteasome ring assembly revealed by a subunit deletion in yeast. EMBO J. 23, 500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JR (1977). In the absence of ribosomal RNA synthesis, the ribosomal proteins of HeLa cells are synthesized normally and degraded rapidly. J. Mol. Biol 115, 315–333. [DOI] [PubMed] [Google Scholar]
- Warner JR, Mitra G, Schwindinger WF, Studeny M, and Fried HM (1985). Saccharomyces cerevisiae coordinates accumulation of yeast ribosomal proteins by modulating mRNA splicing, translational initiation, and protein turnover. Mol. Cell. Biol 5, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zickler D, and Kleckner N (2015). Recombination, Pairing, and Synapsis of Homologs during Meiosis. Cold Spring Harb. Perspect. Biol 7, a016626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.