

Abstract
The recent crystal structures of CC chemokine receptors 2 and 9 (CCR2 and CCR9) have provided structural evidence for an allosteric, intracellular binding site. The high conservation of residues involved in this site suggests its presence in most chemokine receptors, including the close homologue CCR1. By using [3H]CCR2-RA-[R], a high-affinity, CCR2 intracellular ligand, we report an intracellular binding site in CCR1, where this radioligand also binds with high affinity. In addition, we report the synthesis and biological characterization of a series of pyrrolone derivatives for CCR1 and CCR2, which allowed us to identify several high-affinity intracellular ligands, including selective and potential multitarget antagonists. Evaluation of selected compounds in a functional [35S]GTPγS assay revealed that they act as inverse agonists in CCR1, providing a new manner of pharmacological modulation. Thus, this intracellular binding site enables the design of selective and multitarget inhibitors as a novel therapeutic approach.
Introduction
Chemokines are chemotactic cytokines that control the migration and positioning of immune cells during physiological and pathological conditions by interacting with more than 20 different chemokine receptors.1 Chemokine receptors mainly belong to the class A of G protein-coupled receptors (GPCRs) and can be divided into four different subtypes, namely C, CC, CXC, and CX3C, according to the pattern of specific cysteine residues in their major endogenous chemokines.2 To exert their function, chemokines bind at the extracellular side of their receptors in a binding mechanism involving the N-terminal domain, extracellular loops, and the upper half of the transmembrane bundle.3,4 After activation, most chemokine receptors signal through heterotrimeric G proteins, mainly Gi/o class, and β-arrestins.2 CC chemokine receptors 1 (CCR1) and 2 (CCR2) are two of the 10 members of the CC subtype of chemokine receptors. CCR1 and CCR2 are expressed in a variety of immune cells, such as monocytes, dendritic cells, and T helper type-1 (TH1) cells, from where they regulate diverse inflammatory and homeostatic functions.5 Multiple chemokines activate these two receptors, including CCL3, CCL5, and CCL8 in the case of CCR1, and CCL2, CCL7, and CCL8 in the case of CCR2.2
Dysregulation of CCR1, CCR2, and their ligands has been linked to several inflammatory and immune diseases,6,7 which has resulted in many drug discovery efforts to develop small molecules that target these receptors.8,9 Several lines of evidence support a role for both CCR1 and CCR2 in the pathogenesis of diseases such as rheumatoid arthritis (RA) and multiple sclerosis (MS): increased expression of both receptors and their ligands in disease models and patients,10,11 protective effect of genetic knockout of CCR1 or CCR2 in disease models,12,13 and positive preclinical studies with chemokine-neutralizing monoclonal antibodies or small-molecule inhibitors of CCR1 or CCR2.14−16 Yet, only few clinical studies have shown promising results,17,18 while most of the drugs developed so far have failed in clinical trials due to lack of efficacy.8,9 In this regard, the development of multitarget drugs has been proposed as a strategy to overcome the lack of efficacy. Multitarget drugs are designed to specifically act on more than one drug target, which might be necessary in highly heterogeneous diseases, such as RA and MS, where more than one chemokine receptor is involved.19 The design of dual antagonists has been previously undertaken for CCR1/CCR3,20 CCR2/CCR5,21 CCR5/CXCR4,22 and CXCR1/CXCR2;23 however, no CCR1/CCR2 dual antagonists have so far been reported.
Recently, the crystal structures of CCR224 and CCR925 have revealed a novel allosteric binding site for small molecules in chemokine receptors. Both CCR2-RA-[R] in CCR2 and vercirnon in CCR9 bind in a pocket located in the intracellular surface of the receptors, partially overlapping with the binding site for G proteins and β-arrestins.24,25 These intracellular ligands can inhibit the receptors in a noncompetitive and insurmountable manner with regard to chemokine binding, as demonstrated previously in CCR2.26 This might result in higher efficacy even in the presence of a high local concentration of chemokines during a disease state. Together with the potential advantages of allosteric modulators of chemokine receptors, this intracellular binding site seems to be quite conserved among chemokine receptors, which suggests the presence of homologous pockets in other receptors such as CCR1.27 This conservation might provide an opportunity for the design of both selective and dual-targeting inhibitors of CCR1 and CCR2 as a novel approach to treat inflammatory and immune diseases.
For CCR2, several compounds belonging to different scaffolds have already been reported to bind to this intracellular binding site, including pyrrolone derivatives such as CCR2-RA-[R], sulfonamide derivatives, and 2-mercapto imidazoles.26,28 When tested for selectivity, some of these compounds also displayed a moderate activity on CCR1,29−31 suggesting that they might also bind to CCR1. Thus, we selected the pyrrolone scaffold to explore a potential intracellular binding site in CCR1. In our current study, we report the synthesis and the biological evaluation of novel and previously patented pyrrolone derivatives32,33 at both CCR1 and CCR2 in order to determine their selectivity and structure–affinity relationships (SAR) for both receptors. Finally, compounds were tested in a [35S]GTPγS binding assay in order to determine their functional effects in CCR1 and CCR2. Overall, our results provide evidence that CCR1 can also be targeted with intracellular allosteric modulators and that this binding site can be used for the design of multitarget compounds.
Results and Discussion
Synthesis of Pyrrolone Derivatives
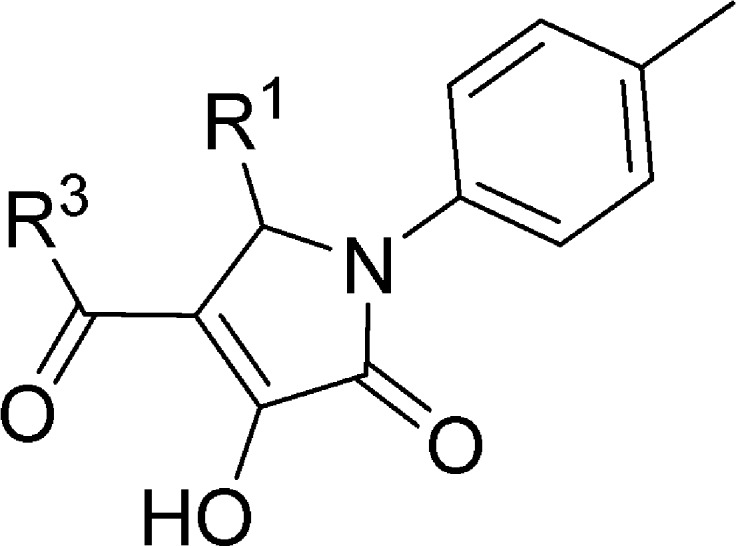
The racemic pyrrolones (6–24, 26–46) depicted in Scheme 1 were synthesized via a one-pot three-component condensation reaction, starting from the commercially available substituted aldehydes 1a–l, anilines 2a–q, and ethyl 2,4-dioxo-butanoates 3a–i in acetic acid33 (6–23, 26–46) or THF29 (24). The ethyl 2,4-dioxo-butanoates (3b–d,f,i), which were not commercially available, were prepared by a Claisen condensation starting from the methyl ketones (4b–d,f,i) and diethyl oxalate 5.34 Pyrrolone 25 was prepared via a transesterification of 24 by the use of p-toluenesulfonic acid in 2-propanol.
Scheme 1. Synthesis Route of Pyrrolones 6–48, with Different R1, R2, and R3 Substituents.

Reagents and conditions: (a) acetic acid, reflux for 2–4 h or THF, rt, overnight; (b) Na, EtOH, 0–20 °C, overnight; (c) p-toluenesulfonic acid, 2-propanol, reflux, 48 h.
Characterization of [3H]-CCR2-RA-[R] Binding on CCR1 and CCR2
[3H]-CCR2-RA-[R] is the (R)-isomer of [3H]-CCR2-RA, a high-affinity radioligand previously characterized in our group for CCR2.26 To avoid a possible effect of the lower-affinity isomer, we used the tritium-labeled (R)-isomer in the present study. As expected, [3H]-CCR2-RA-[R] binds with high affinity to osteosarcoma (U2OS) cells stably expressing CCR2b (U2OS-CCR2) as shown by saturation experiments (KD of 6.3 nM and Bmax of 2.6 pmol/mg, Supporting Information, Figure S1 and Table S1). Kinetic characterization showed that [3H]-CCR2-RA-[R] associates and dissociates in a biphasic manner (Supporting Information, Table S1), consistent with the previously reported [3H]-CCR2-RA kinetics.26 We had reported that [3H]-CCR2-RA binds with low affinity to CCR5 (KD of 100 nM),28 suggesting that CCR2-RA-[R] is a nonselective antagonist that can bind several chemokine receptors. In this regard, CCR1 is a close homologue of CCR2, with 61% amino acid similarity and 47% identity; furthermore, this amino acid similarity is >90% when only considering the amino acid residues involved in the intracellular binding site of CCR2-RA-[R] in CCR224 (Supporting Information, Figure S2). This prompted us to investigate the binding of [3H]-CCR2-RA-[R] in membrane preparations from U2OS cells stably expressing CCR1 (U2OS-CCR1). [3H]-CCR2-RA-[R] homologous displacement assays on U2OS-CCR1 yielded a KD of 13.5 nM and a Bmax of 6.1 pmol/mg (Figure 1a, Supporting Information, Table S1), suggesting the presence of an intracellular site in CCR1 and making it a suitable tool to study such binding pocket. Binding of [3H]-CCR2-RA-[R] to U2OS-CCR1 was also assessed in kinetic experiments at 25 °C. These experiments showed that [3H]-CCR2-RA-[R] associates and dissociates in a biphasic manner, similar to our findings in CCR2, but the association and dissociation rates were significantly higher in CCR1 than in CCR2 (Supporting Information, Figure S1 and Table S1).
Figure 1.

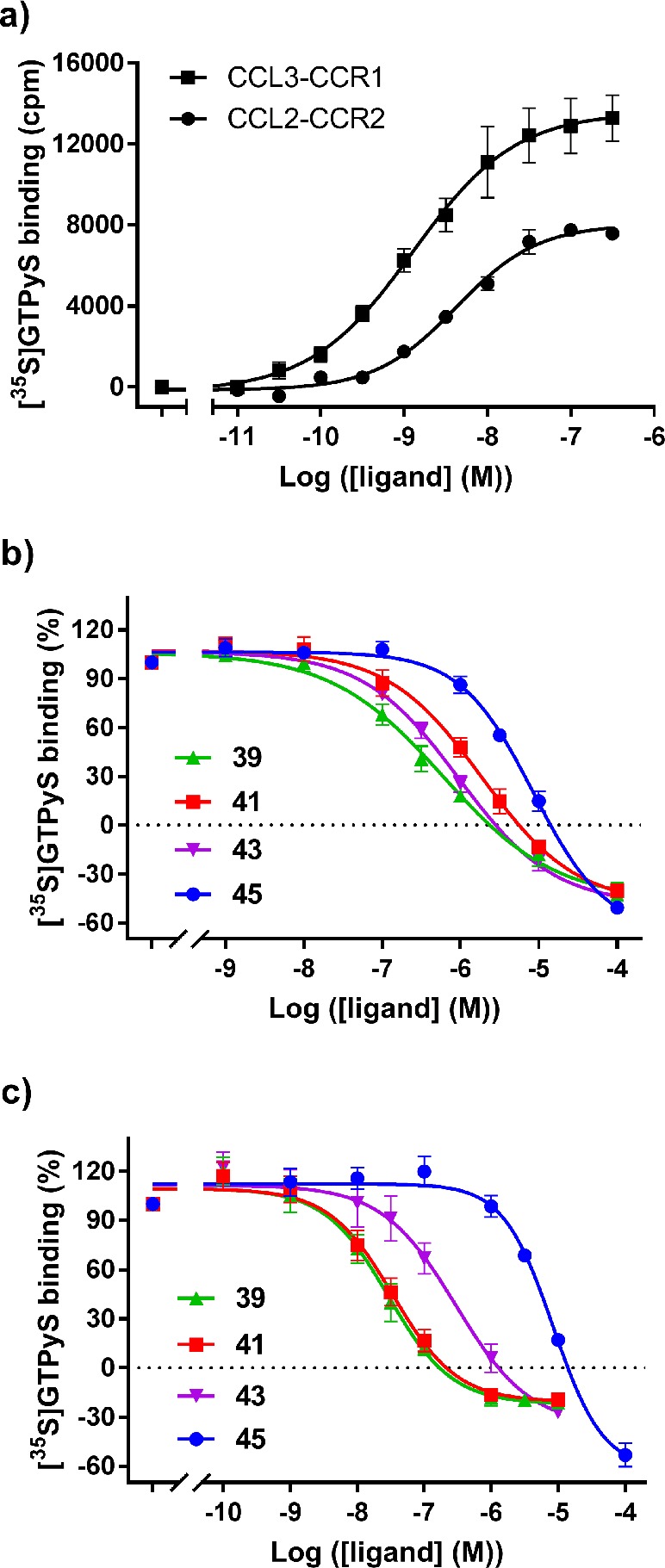
(a) Homologous displacement curves of 3, 6, and 12 nM [3H]-CCR2-RA-[R] specific binding by increasing concentrations of CCR2-RA-[R] in U2OS-CCR1 at 25 °C. (b) Displacement curves of 6 nM [3H]-CCR2-RA-[R] specific binding by increasing concentrations of SD-24, JNJ-27141491, and BX471 in U2OS-CCR1 at 25 °C. BX471 significantly enhanced the binding of [3H]-CCR2-RA-[R] up to 120%. Statistical significance between binding in absence (100%) and presence of 10 μM BX471 (116 ± 2%) was determined using an unpaired, two-tailed Student’s t-test with Welch’s correction. (c,d) Displacement curves of 6 nM [3H]-CCR2-RA-[R] specific binding by compounds 39, 41, 43, and 45 (b) in U2OS-CCR1 or (c) in U2OS-CCR2 at 25 °C. In the case of U2OS-CCR2, compound 45 did not displace more than 50% of [3H]-CCR2-RA-[R], thus only single-point data at 1 μM is shown. The dashed blue line corresponds to the nonlinear regression fit for compound 45 by GraphPad Prism 7.0. Data shown are mean ± SEM of at least three experiments performed in duplicate.
Overall, these findings allowed us to set up a [3H]-CCR2-RA-[R] competitive displacement assay on both U2OS-CCR1 and U2OS-CCR2 to determine the binding affinity (Ki) of unlabeled compounds. Using this assay, we first determined the ability of known ligands to displace this radioligand from CCR1, i.e., the CCR2 intracellular ligands SD-24 and JNJ-2714149126,28 and the CCR1 orthosteric antagonist BX47135 (Figure 1b). SD-24 and JNJ-27141491 fully displaced [3H]-CCR2-RA-[R] from CCR1 in a concentration-dependent manner, indicating that these compounds bind at the same binding site as CCR2-RA-[R]. SD-24 displaced the radioligand with a pKi of 7.45 ± 0.05 (Ki = 36 nM), while JNJ-27141491 displaced [3H]-CCR2-RA-[R] with a pKi of 6.9 ± 0.06 (Ki = 138 nM), consistent with previously reported activities in CCR1.30,31 To rule out that these compounds bind at the orthosteric binding site of CCR1, we also investigated the effect of BX471 in [3H]-CCR2-RA-[R] binding. As expected, BX471 was not able to displace the radioligand (Figure 1b); on the contrary, BX471 significantly enhanced the binding of [3H]-CCR2-RA-[R] by approximately 20% (116 ± 2% in the presence of 10 μM BX471), in a similar manner as previously reported with CCR2 orthosteric antagonists.24,26 This allosteric enhancement is consistent with two different binding sites in CCR1: the orthosteric binding site where BX471 binds and an intracellular pocket for CCR2-RA-[R], SD-24, and JNJ-27141491.
This [3H]-CCR2-RA-[R] assay was also used to determine the affinity of the synthesized pyrrolone derivatives. All pyrrolone derivatives 6–46 were first tested at a single concentration of 1 μM in both U2OS-CCR1 and U2OS-CCR2 (Tables 1–3). Compounds which displaced more than 50% of [3H]-CCR2-RA-[R] binding were further evaluated in this assay using at least six different concentrations of unlabeled compound in order to determine their binding affinity for the corresponding receptor subtypes (Figure 1c,d and Tables 1–3). Finally, we selected four compounds (39, 41, 43 and 45) to be tested in a functional [35S]GTPγS binding assay (Figure 3). The potency (pIC50) of these compounds was determined in the presence of an EC80 concentration of CCL3 (8 nM) or CCL2 (20 nM) in U2OS-CCR1 or U2OS-CCR2 membranes, respectively.
Table 1. Binding Affinities of Compounds 6–26 on Human CCR1 and Human CCR2.
| pKi ± SEM (Ki, nM)a or
displacement at 1 μM (%)b |
||||
|---|---|---|---|---|
| compd | R1 | R3 | CCR1 | CCR2 |
| 6 | c-hexyl | Me | 7.26 ± 0.04 (56) | 7.10 ± 0.03 (81) |
| 7 | c-heptyl | Me | 7.26 ± 0.03 (56) | 7.02 ± 0.06 (96) |
| 8 | c-octyl | Me | 7.24 ± 0.01 (57) | 6.79 ± 0.09 (170) |
| 9 | Ph | Me | 6.79 ± 0.04 (162) | 39% (38, 40) |
| 10 | 4-Me Ph | Me | 6.71 ± 0.06 (198) | 36% (42, 31) |
| 11 | 4-OMe Ph | Me | 6.27 ± 0.01 (541) | 5% (5, 5) |
| 12 | 4-Cl Ph | Me | 7.17 ± 0.01 (67) | 6.70 ± 0.08 (207) |
| 13 | 4-Br Ph | Me | 7.07 ± 0.07 (87) | 6.67 ± 0.03 (214) |
| 14 | 3-Me Ph | Me | 47% (51, 44) | 11% (14, 8) |
| 15 | 3-OMe Ph | Me | 28% (34, 22) | 0% (3, −3) |
| 16 | 3-Cl Ph | Me | 6.70 ± 0.01 (198) | 19% (25, 14) |
| 17 | 3-Br Ph | Me | 6.74 ± 0.02 (181) | 19% (20, 18) |
| 18 | c-hexyl | Et | 7.52 ± 0.01 (30) | 6.99 ± 0.06 (104) |
| 19 | c-hexyl | Pr | 7.54 ± 0.04 (29) | 6.86 ± 0.10 (144) |
| 20 | c-hexyl | Bu | 7.50 ± 0.004 (31) | 6.81 ± 0.05 (158) |
| 21 | c-hexyl | I-Pr | 7.39 ± 0.06 (42) | 6.50 ± 0.05 (316) |
| 22 | c-hexyl | c-Pr | 7.74 ± 0.08 (19) | 6.80 ± 0.05 (160) |
| 23 | c-hexyl | t-Bu | 7.66 ± 0.05 (22) | 6.81 ± 0.07 (158) |
| 24 | c-hexyl | OEt | 6.70 ± 0.01 (200) | 31% (36, 26) |
| 25 | c-hexyl | OiPr | 36% (45, 26) | 6% (10, 1) |
| 26 | c-hexyl | –Ph | 7.11 ± 0.01 (77) | 37% (45, 30) |
pKi and Ki (nM) values obtained from [3H]-CCR2-RA-[R] binding assays on U2OS membranes stably expressing human CCR1 or human CCR2. Values are means ± standard error of the mean (SEM) of at least three independent experiments performed in duplicate.
Percent of [3H]-CCR2-RA-[R] displacement by 1 μM compound. Values represent the mean of two independent experiments performed in duplicate.
Table 3. Binding Affinities of Compounds 43–46 on Human CCR1 and Human CCR2.

| pKi ± SEM
(Ki, nM)a or
displacement at 1 μM (%)b |
||||
|---|---|---|---|---|
| compd | R1 | R3 | CCR1 | CCR2 |
| 43 | c-hexyl | c-propyl | 8.27 ± 0.02 (5) | 7.82 ± 0.04 (15) |
| 44 | c-hexyl | Ph | 7.56 ± 0.04 (28) | 7.18 ± 0.03 (66) |
| 45 | 3-Br Ph | c-propyl | 7.30 ± 0.01 (50) | 45% (49, 42) |
| 46 | 3-Br Ph | Me | 7.19 ± 0.02 (65) | 6.67 ± 0.01 (216) |
pKi and Ki (nM) values obtained from [3H]-CCR2-RA-[R] binding assays on U2OS membranes stably expressing human CCR1 or human CCR2. Values are means ± standard error of the mean (SEM) of at least three independent experiments performed in duplicate.
Percent of [3H]-CCR2-RA-[R] displacement by 1 μM compound. Values represent the mean of two independent experiments performed in duplicate.
Figure 3.
(a) [35S]GTPγS binding upon stimulation of U2OS-CCR1 and U2OS-CCR2 by increasing concentrations of CCL3 and CCL2, respectively. In both cases, the response was corrected by subtracting the basal activity (approximately 8000 dpm for both CCR1 and CCR2). (b) Inhibition of CCL3-induced [35S]GTPγS binding by compounds 39, 41, 43, and 45 in U2OS-CCR1. (c) Inhibition of CCL2-induced [35S]GTPγS binding by compounds 39, 41, 43, and 45 in U2OS-CCR2. The level of basal activity in U2OS-CCR1 and U2OS-CCR2 is indicated by a dashed line. In all cases, data shown are mean ± SEM of at least three experiments performed in duplicate.
Docking of CCR2-RA-[R] in CCR1 and CCR2
To better understand the binding mode of CCR2-RA-[R] in both human CCR1 and CCR2b, we docked this compound into models of both receptors (Figure 2). In the case of CCR2, homology modeling was used to model the CCR2 residues between Ser2265x62 and Lys2406x32, which correspond to the M2 muscarinic acetylcholine receptor sequence in the CCR2b crystal structure (PDB 5T1A).24 These residues were modeled because this region is in close proximity to the CCR2-RA-[R] binding site. As expected from the sequence alignment (Supporting Information, Figure S2), CCR2-RA-[R] was predicted to bind to CCR1 in an overlapping binding site as the one reported in the crystal structure of CCR2,24 in a solvent-exposed intracellular pocket found between the intracellular ends of transmembrane segments 1–3, 6, 7, and helix 8 (Figure 2). The vinylogous carboxylic acid functionality makes similar interactions in CCR1 as in CCR2: the hydroxyl and the two carbonyl groups are involved in hydrogen-bond interactions with the side chain of Arg1313x50, and the backbone of Arg3078x49 and Phe3088x50 (Figure 2). A similar hydrophobic subpocket is also observed around the cyclohexyl moiety, which interacts with Ala6x33, Val/Leu6x36, Ile6x37, and Ile6x40. Interestingly, Val2446x36 in CCR2 is replaced by the bigger Leu2406x36 in CCR1, which pushes the ligand down against Arg1313x50, resulting in a slightly different binding orientation of CCR2-RA-[R] in this receptor (Figure 2). In addition, the exchange of Lys3118x49 in CCR2 by Arg3078x49 in CCR1 might also contribute to the stabilization of this slightly altered binding pose. This difference in orientation could result in CCR1 selectivity, as this orientation seems to open up the subpockets in the proximity of the cyclohexyl and the acetyl group of CCR2-RA-[R] in CCR1, allowing the introduction of bigger and more lipophilic substituents at these positions.
Figure 2.

Proposed binding mode of compound CCR2-RA-[R] in the homology models of CCR1 and CCR2, based on the crystal structure of CCR2 (PDB 5T1A).24 For CCR1, representative residues are shown as green “sticks” and for CCR2 as orange “sticks”. In all cases, oxygen and nitrogen atoms are represented in red and blue, respectively, and hydrogen bonds with dashed yellow lines. Residues are numbered based on the corresponding residue numbers and with structure-based Ballesteros–Weinstein numbers in superscript.37
Structure–Affinity Relationships (SAR)
Modifications Replacing the Cyclohexyl Group (R1, Table 1)
Several pyrrolone derivatives have been previously evaluated at CCR2,29,32,33,36 resulting in the identification of CCR2-RA-[R] as a hit compound for further development,29 but characterization of these compounds in CCR1 is mostly missing. Compound 6, previously reported and characterized in CCR2 by Zou et al. (2007),36 was selected as our starting point for the analysis of SAR in both CCR1 and CCR2. In our assay, compound 6 showed an affinity of 81 nM for CCR2 and a slightly higher affinity of 56 nM for CCR1 (Table 1). To note, the binding affinities reported previously for these pyrrolone derivatives were obtained with a 125I-CCL2 binding assay,29,36 resulting in lower affinities compared with our [3H]-CCR2-RA-[R] binding assay, as previously observed in our group.26 For our SAR study, we first examined different C5 substituents of the pyrrolone core (R1), as shown in Table 1. In line with previous studies,29 we found that increasing the size of the cycloalkyl group from cyclohexyl (6) to cycloheptyl (7) or cyclooctyl (8) resulted in a decrease in binding affinity for CCR2; however, the affinity for CCR1 was retained, indicating that bulkier groups are better tolerated in CCR1 than in CCR2 and providing an avenue for selectivity on CCR1 over CCR2. Previous studies showed that decreasing the size of the cycloalkyl group was also detrimental for CCR2,29 so we decided not to explore smaller ring sizes.
Substitution of the cycloalkyl group by a phenyl group (9) led to a great loss of CCR2 affinity (39% displacement at 1 μM), consistent with previously reported values showing a decreased affinity for an almost similar pair of compounds.36 Yet this substitution only led to a 3-fold decrease in CCR1 affinity (Ki of 162 nM), thus showing much higher selectivity for CCR1. Next, we explored the effect of N-aryl modifications in both affinity and selectivity (compounds 10–17), specifically the effect of para and meta substituents. In general, N-aryl groups on the R1 position resulted in increased selectivity toward CCR1, as most compounds did not displace more than 36% [3H]-CCR2-RA-[R] binding in CCR2 at a concentration of 1 μM. Only compounds 12 and 13, with halogen substitutions in para position (Cl and Br, respectively), regained CCR2 affinity (12, 207 nM; 13, 214 nM). Furthermore, para-substituted derivatives displayed significantly higher affinities compared with their meta-substituted analogues.
In the case of CCR1, introduction of a para-methyl moiety (10) resulted in a slight decrease in affinity compared with the unsubstituted 9; in contrast, the meta-substituted analogue (14) showed less than 50% displacement at 1 μM. Introduction of an electron-donating substituent (methoxy, 11 and 15) was not well tolerated in any position, as it led to an approximately 3-fold decrease in affinity when placed in para position (11, 541 nM) and a near complete loss of affinity when placed in meta position (15, 28% displacement at 1 μM). Halogen substituents in para position were also more favored in the case of CCR1, yielding higher affinities compared with the unsubstituted 9 and regardless of the halogen used (67 nM for R1 = 4-Cl phenyl (12), p < 0.0001 to 9; 87 nM for R1 = 4-Br phenyl (13); p = 0.0002 to 9). However, selectivity for CCR1 was notably reduced considering that these compounds displayed binding affinities of around 200 nM in CCR2. Although moving the halogens to the meta position (16 and 17) decreased the affinities more than 2-fold compared with their para analogues, selectivity for CCR1 was restored as these compounds showed less than 20% displacement of [3H]-CCR2-RA-[R] binding in CCR2. Together, the results for compounds 6–17 indicate that in CCR1 aliphatic groups yield higher affinities, while aromatic groups yield lower affinities but improved selectivity over CCR2.
Modifications to the Acetyl Group (R3, Table 1)
Previous modifications to the vinylogous carboxylic acid functionality in CCR2 showed detrimental effects in binding affinity.29,36 Indeed, mutagenesis and structural studies have shown crucial interactions of the hydroxyl and the two carbonyl groups with Glu3108x48, Lys3118x49, and Phe3128x50 (residues according to structure-based Ballesteros–Weinstein numbering37) in CCR2.24,28 Sequence alignment of CCR1 and CCR2 (Supporting Information, Figure S2) and our docking study (Figure 2) suggest similar interactions in CCR1, as only position 8.49 differs (arginine in CCR1 and lysine in CCR2). Therefore, we decided to keep the vinylogous carboxylic acid moiety and explore different modifications to the acetyl group at the R3 position (Table 1). A gradual increase in the length of the alkyl chain from a methyl group (6) to a butyl group (18–20) resulted in a ∼2-fold increase in CCR1 affinity (30 nM for R3 = ethyl (18), p = 0.0004 against 6; 29 nM for R3 = propyl (19), p = 0.0002 against 6; and 31 nM for R3 = butyl (20), p = 0.0010 against 6). In contrast, for CCR2, we observed a similar or a slight decrease in affinity. Introduction of a bulkier isopropyl group led to a decrease in affinity in both receptors, with a more drastic effect in CCR2 affinity. Replacing the isopropyl group with cyclopropyl (22) or tert-butyl (23) restored the affinity in CCR2 to values similar to compound 20 (22, 160 nM; 23, 158 nM); in CCR1, these modifications further improved the binding affinity to approximately 20 nM, yielding compounds with the highest affinity and selectivity observed in these series of R1 and R3 modifications (22, 19 nM; 23, 22 nM). These results suggest a larger hydrophobic subpocket in CCR1, able to accommodate larger and branched alkyl chains.
We also explored the effect of adding heteroatoms (oxygen in this case) between the carbonyl and an ethyl or isopropyl group (24 and 25, respectively). Overall, this led to a drastic drop in affinity for both receptors. This detrimental effect was most pronounced in compound 25, which displaced less than 40% of [3H]-CCR2-RA-[R] binding in CCR1 and less than 10% in CCR2. The transformation of the ketone into an ester might decrease the electron density on the carbonyl oxygen as well as the acidity of the adjacent protons, thus weakening or disrupting key hydrogen bonding interactions with Lys8x49 in CCR224,28 or Arg8x49 in CCR1. The need of an acidic function for intracellular antagonists has also been reported in a study with N-benzylindole-2-carboxylic acids, where the authors found a correlation between higher acidity and higher CCR2 affinity.38 Finally, replacing the methyl group in R3 with a phenyl group (26) had no effect on CCR1 affinity, while it only displaced 37% of [3H]-CCR2-RA-[R] binding in CCR2. Altogether, these findings indicate that bigger, more lipophilic groups in R3 are better tolerated in CCR1, while in CCR2 methyl is preferred.
Modifications to the Phenyl Ring (R2, Table 2)
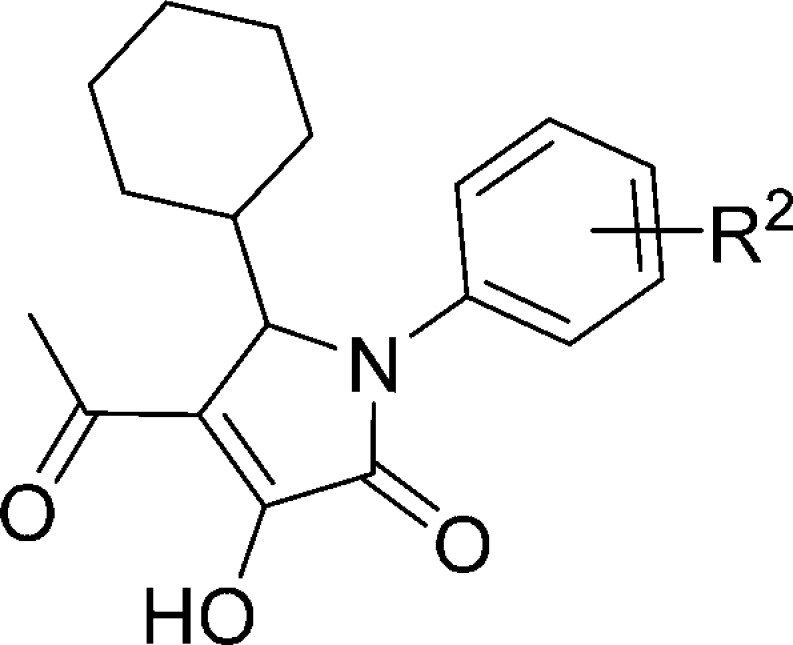
Table 2. Binding Affinities of Compounds 6, 27–42 on Human CCR1 and Human CCR2.
| pKi ± SEM (Ki, nM)a or
displacement at 1 μM (%)b |
|||
|---|---|---|---|
| compd | R2 | CCR1 | CCR2 |
| 27 | H | 42% (41, 42) | 45% (44, 45) |
| 6 | 4-Me | 7.26 ± 0.04 (56) | 7.10 ± 0.03 (81) |
| 28 | 4-Et | 6.78 ± 0.02 (168) | 7.19 ± 0.05 (66) |
| 29 | 4-OMe | 6.60 ± 0.07 (260) | 6.67 ± 0.05 (217) |
| 30 | 4-Cl | 7.41 ± 0.05 (40) | 7.73 ± 0.08 (19) |
| 31 | 4-Br | 7.62 ± 0.05 (24) | 7.80 ± 0.12 (17) |
| 32 | 4-CF3 | 6.86 ± 0.08 (144) | 7.04 ± 0.02 (92) |
| 33 | 3-Me | 6.31 ± 0.07 (500) | 6.58 ± 0.05 (265) |
| 34 | 3-F | 44% (45, 42) | 47% (48, 47) |
| 35 | 3-Cl | 6.28 ± 0.08 (541) | 6.62 ± 0.02 (239) |
| 36 | 3-CF3 | 25% (23, 27) | 6.54 ± 0.11 (305) |
| 37 | 2-F, 4-Me | 7.56 ± 0.10 (29) | 7.44 ± 0.05 (37) |
| 38 (CCR2-RA) | 2-F, 4-Cl | 7.82 ± 0.06 (15) | 8.00 ± 0.09 (11) |
| 39 | 2-F, 4-Br | 7.98 ± 0.04 (11) | 8.25 ± 0.02 (6) |
| 40 | 3,4-diMe | 7.37 ± 0.03 (43) | 7.75 ± 0.02 (18) |
| 41 | 3-Me, 4-Cl | 7.51 ± 0.01 (31) | 8.09 ± 0.08 (9) |
| 42 | 3-F, 4-Me | 7.32 ± 0.07 (49) | 7.24 ± 0.02 (57) |
pKi and Ki (nM) values obtained from [3H]-CCR2-RA-[R] binding assays on U2OS membranes stably expressing human CCR1 or human CCR2. Values are means ± standard error of the mean (SEM) of at least three independent experiments performed in duplicate.
Percent of [3H]-CCR2-RA-[R] displacement by 1 μM compound. Values represent the mean of two independent experiments performed in duplicate.
In addition, we explored different N-aryl modifications in the phenyl ring (R2, Table 2), starting with modifications in para position. Removing the methyl group in 6 yielded compound 27, with an unsubstituted phenyl group, which displaced less than 50% of the radioligand in both receptors. Increasing the size of the alkyl group from methyl (6) to ethyl (28) caused a 3-fold decrease in CCR1 affinity, while the affinity in CCR2 was maintained (28, 168 nM in CCR1 versus 66 nM in CCR2). Adding an electron-donating methoxy group was unfavorable for both receptors, as affinities dropped to 260 nM in CCR1 and 217 nM in CCR2. In contrast, an electron-withdrawing substituent (trifluoromethyl, 32) restored the affinity to 92 nM in CCR2, similar to our starting compound 6 and to 144 nM in CCR1. The substitution of the para-methyl group with halogens yielded derivatives with improved binding affinities in both receptors (30 and 31) but no gain in selectivity. Substitution with a chlorine (30) or bromine atom (31) led to a 4.5-fold increase in CCR2 affinity compared with 6, with Ki values around 20 nM regardless of the halogen. In the case of CCR1, the bromine atom (31) led to a 2-fold increase compared with 6 (31, 24 nM), while the smaller chlorine atom did not affect the affinity much (30, 40 nM). Although not synthesized in our study, Dasse et al. (2007)29 showed that the para-fluoro analogue performed worse in CCR2 than other para-halogen derivatives. In this regard, from fluoro to chloro there is an important increase in polarity (σ), lipophilicity (π), and size, whereas from chloro to bromo only lipophilicity and size increase.39,40 Taken together, these results suggest that lipophilicity and size of the halogen might be more important in CCR1 than in CCR2, while electronegativity or polarity could play a bigger role in CCR2.
Moving the substituents from the para to the meta position resulted in poor affinities for both receptors compared with their para-substituted analogues. In CCR1, the meta-methyl (33) and meta-chlorine (35) groups led to a 9-fold and 13-fold decrease in affinity, respectively; in CCR2, the affinities decreased 3-fold and 13-fold after the same substitutions. The addition of a trifluoromethyl group in meta position (36) also led to a 3-fold decrease in CCR2 affinity compared with its para-substituted analogue 32. In CCR1, 36 only displaced 25% of [3H]-CCR2-RA-[R] binding at a concentration of 1 μM, displaying the highest selectivity toward CCR2 in these series of modifications. Also detrimental was the addition of a fluorine group in meta position (34), which led to less than 50% displacement of [3H]-CCR2-RA-[R] binding in both receptors. Overall, substituents in the para position were more favored in both receptors, especially halogen substituents, yet none of the compounds displayed selectivity toward CCR1. Similarly as reported by Dasse et al. (2007),29 attempts to introduce different substituents in the ortho position were unsuccessful, thus we continued to explore different combinations of phenyl substituents.
As part of our SAR analysis, we synthesized compound 38 (also referred as CCR2-RA), which corresponds to the racemic mixture of the radioligand [3H]-CCR2-RA-[R] used in this study. This compound displayed an affinity of 15 nM in CCR1 and 11 nM in CCR2, similar to the KD values obtained in homologous displacement or saturation assays (Supporting Information, Table S1). Replacing the para-chloro group in 38 with a methyl moiety (37), while keeping the ortho-fluorine group, led to an expected decrease in affinity for both receptors, as compound 6 with a methyl group in para position performed worse than 30 with a chlorine atom in the same position. When the para substituent was replaced with a bromine atom (39), the affinity was restored to 11 nM in CCR1 and 6 nM in CCR2. Subsequent combinations of meta and para substituents (40–42) generated compounds with decreased CCR1 affinities compared with 38, as expected from the data on the monosubstituted meta analogues. Compound 41 displayed a slightly higher selectivity for CCR2 (9 nM in CCR2 versus 31 nM in CCR1). Overall, disubstituted derivatives performed better than the monosubstituted compounds in both receptors; however, no clear trend in selectivity was observed in these series.
In an attempt to improve both affinity and selectivity for CCR1, we decided to combine some of the best features observed at R1, R2, and R3 positions: a disubstituted phenyl ring with an ortho-fluoro and para-bromo moieties for R2 in order to retain the high affinity of 39, a cyclopropyl group or an unsubstituted phenyl ring at R3 (22 and 26) to gain selectivity, and a meta-bromo phenyl ring at R1 (17) to further improve selectivity for CCR1. These combinations resulted in four final compounds shown in Table 3 (43–46). To maintain a high affinity for CCR1, we kept the 2-fluoro-4-bromophenyl group at R2 constant, and we combined it with different R1 and R3 substituents. The combination with a cyclopropyl group at R3 position (43) led to the highest CCR1 affinity in our study (Ki of 5 nM), but selectivity over CCR2 was reduced compared with 22 (3-fold versus 8-fold). Replacing the cyclopropyl group at R3 by a phenyl group (44) decreased the affinity for CCR1 by more than 5-fold compared with 43. Compound 43, somewhat unexpectedly, bound to CCR2 with an affinity of 66 nM, more than 15-fold better than 26. Replacing the cyclohexyl group at R1 (43) by a 3-bromo-phenyl group (45) resulted in an improved selectivity over CCR2, as this compound did not displace more than 50% of [3H]-CCR2-RA-[R] binding at 1 μM, whereas it showed an affinity of 50 nM in CCR1. Finally, replacing the cyclopropyl with a methyl group at R3 (46) maintained the affinity for CCR1 and restored the affinity for CCR2 (65 nM in CCR1 and 216 nM in CCR2), with a concomitant loss of selectivity.
Functional Characterization of Selected Compounds
Following the SAR analysis, four compounds (39, 41, 43, and 45) were selected for further characterization in a G protein-dependent functional assay in order to assess their inhibitory potencies (pIC50) in both CCR1 and CCR2. The four compounds were selected based on their affinity and selectivity profile: compounds 43 and 39, with the highest affinity for either CCR1 or CCR2, respectively, compound 41, with higher selectivity toward CCR2, and compound 45, with higher selectivity toward CCR1. As a functional assay, we used a previously reported [35S]GTPγS binding assay on U2OS-CCR2 membranes, which had been applied in the functional characterization of several allosteric and orthosteric CCR2 ligands.26 Similarly as reported by Zweemer et al. (2013),26 CCL2 stimulated [35S]GTPγS binding in a concentration-dependent manner, displaying a potency of 5 nM in CCR2 (pEC50 = 8.3 ± 0.09, Figure 3a). Using the same assay conditions, we characterized the G protein activation of CCL3 in U2OS-CCR1 membranes. In this assay, CCL3 induced [35S]GTPγS binding in CCR1 with a higher potency than CCL2 in CCR2 (1.3 nM, pEC50 = 8.9 ± 0.06) and with a higher maximum effect (Emax) (Figure 3a). It should be noted that the potency of CCL3 in our study is lower than previously reported,41 which might be related to the differences in cell line and/or assay conditions.
For the antagonist assays, we used a submaximal EC80 concentration of CCL3 (8 nM) and CCL2 (20 nM) in CCR1 or CCR2, respectively, in order to evoke 80% stimulation of [35S]GTPγS binding. Although all compounds were able to inhibit CCL3- or CCL2-induced G protein activation, their potencies (IC50) ranged between 30 nM to 8 μM (Table 4 and Figure 3b,c). In CCR2, the potency of the compounds increased in the same order observed for affinity (Figure 3c, 45 < 43 < 41 < 39). In CCR1, 39 displayed the highest potency (590 nM), followed by 43 (950 nM), contrary to their binding affinity (Figure 3b, 43 > 39). In addition, the moderate selectivity observed in the binding assays was lost in this functional assay: except for 45, all compounds were more potent inhibitors of CCR2 than CCR1, as their potencies were 3-fold (43), 19-fold (39), or 48-fold (41) lower in CCR1. Upon comparison of potencies in the [35S]GTPγS assay and the affinities in the [3H]-CCR2-RA-[R] binding assay, we observed that all compounds displayed between 5 and 10-fold difference between assays in CCR2 (Tables 2–4), in agreement with previous characterization of CCR2-RA-[R] on this receptor.26 In contrast, all compounds displayed at least a 50-fold difference between assays when tested on CCR1. Such lack of correlation between apparent potencies and binding affinities in CCR1 might be dependent on the assay conditions used, G protein concentrations, or the chemokine used in this study; thus, further studies are warranted to fully characterize these ligands for their selectivity.
Table 4. Functional Characterization of Compounds 37, 39, 41, and 43 in U2OS-CCR1 and U2OS-CCR2 Using a [35S]GTPyS Binding Assay.
| inhibition
of [35S]GTPyS bindinga |
||||
|---|---|---|---|---|
| CCR1b |
CCR2c |
|||
| compd | pIC50 ± SEM (IC50, μM) | Hill slope | pIC50 ± SEM (IC50, μM) | Hill slope |
| 39 | 6.26 ± 0.10 (0.59)*** | –0.62 ± 0.05** | 7.57 ± 0.08 (0.03) | –0.94 ± 0.18 |
| 41 | 5.73 ± 0.09 (1.94)*** | –0.72 ± 0.08* | 7.47 ± 0.10 (0.04) | –0.88 ± 0.13 |
| 43 | 6.03 ± 0.04 (0.95) | –0.73 ± 0.02* | 6.54 ± 0.16 (0.33) | –0.80 ± 0.13 |
| 45 | 5.07 ± 0.05 (8.64) | –0.93 ± 0.01 | 5.06 ± 0.05 (8.77) | –1.20 ± 0.08 |
All values are means ± SEM of at least three independent experiments performed in duplicate. Unpaired t-test analysis with Welch’s correction was performed to analyze differences in pIC50 values between receptors, with differences noted as ***, p < 0.001. One-Way ANOVA with Dunnett’s posthoc test was performed to compare pseudo-Hill slopes against compound 45, which showed a pseudo-Hill slope of approximately unity in both receptors, with significant differences displayed as *, p < 0.05; **, p < 0.01.
Inhibition of CCL3-induced [35S]GTPyS binding in U2OS membranes stably expressing human CCR1. A concentration of 8 nM CCL3 was used in the assays to evoke an 80% response.
Inhibition of CCL2-induced [35S]GTPyS binding in U2OS membranes stably expressing human CCR2. A concentration of 20 nM CCL2 was used in the assays to evoke an 80% response.
In CCR1, all compounds behaved as inverse agonists, as they all significantly decreased the basal activity of CCR1 at the highest concentration tested (Supporting Information, Figure S3a). In this regard, it was previously demonstrated that CCR1 exhibits constitutive activity leading to ligand-independent G protein-activation, β-arrestin recruitment, and receptor internalization,42 which points to the development of inverse agonists as a potential therapeutic option for inflammatory diseases. Yet, only BX-47135 has been reported to act as inverse agonist in CCR1.42 This prompted us to further characterize these compounds as inverse agonists in CCR1 by measuring their inhibitory potency in absence of the agonist CCL3 (Supporting Information, Figure S3b and Table S2). Compounds 39 and 41 were more potent inverse agonists than antagonists, displaying a 3-fold and almost 10-fold higher potency, respectively, as inverse agonists. As such, their potencies as inverse agonists were more comparable to their binding affinities (Table 2 and Supporting Information, Table S2). In contrast, 43 and 45 showed similar potencies when measured in the absence or presence of CCL3 and thus displayed more than 130-fold difference between functional and binding assays (Table 2 and Supporting Information, Table S2). Interestingly, both compounds 43 and 45 have a cyclopropyl in the R3 position while 39 and 41 have a methyl group (Tables 2 and 3), which suggests that this larger group might be responsible for the difference in their efficacy and functional profile. Moreover, most compounds displayed pseudo-Hill slopes of less than unity in CCR1 when tested in the presence or absence of CCL3 (Table 4 and Supporting Information, Table S2), indicative of a more complex mechanism of inhibition, combining negative allosteric modulation and inverse agonism.43 Of note, the basal levels of constitutive activity in the [35S]GTPγS assay are very dependent on the assay conditions used, such as GDP concentrations. Yet, at a single concentration (100 μM) tested, all compounds consistently decreased the basal activity in CCR1 after varying GDP concentrations. For instance, compound 41 decreased basal activity by 22% (1 μM GDP), 26% (10 μM GDP), and 25% (20 μM GDP) (data not shown). To the best of our knowledge, these compounds represent the first intracellular ligands with demonstrated inverse agonism in CCR1. Both 45 and 43 decreased the basal activity of CCR2 to a similar or smaller level than in CCR1 (45, maximal decrease of 58%; 43, maximal decrease of 27%), indicative of inverse agonism (Supporting Information, Figure S3a). However, no constitutive activity has been reported for CCR2, with only one constitutively active mutant (CAM) described so far.44 In fact, Gilliland et al. (2013) showed that CCR2 was not able to induce ligand-independent cell migration or to constitutively associate with β-arrestin, pointing to a lack of constitutive activity.42 Moreover, several classes of orthosteric and allosteric CCR2 ligands did not show evidence of inverse agonism when previously tested in a similar [35S]GTPγS binding assay.26 Thus, the inverse agonism observed in this study might be the consequence of the expression level, ligand concentration, and/or assay conditions employed, so further research is warranted to investigate ligand-independent signaling in CCR2.
Conclusions
In this study, we have characterized [3H]-CCR2-RA-[R], a high-affinity intracellular antagonist previously described for CCR2,26 in both CCR1 and CCR2, which allowed us to conclude that this radioligand binds to CCR1 with a similar high affinity. By characterizing this radioligand in CCR1, we have provided evidence that CCR1 possesses an intracellular binding site that can be used for the design of noncompetitive compounds. In addition, this intracellular radioligand allowed us to explore the SAR of a series of pyrrolone derivatives in both CCR1 and CCR2. Although some of these derivatives had been previously described for CCR2, their characterization in CCR1 had not been reported. With the SAR analysis we learned that introduction of bulkier and more lipophilic groups at R1 and R3 positions was better tolerated in CCR1, allowing us to obtain better selectivity for this receptor. The high conservation between the intracellular pockets of CCR1 and CCR2 prevented us from finding high selectivity in these series of compounds but allowed us to find several potential dual-target antagonists. Finally, characterization of four selected compounds in a functional assay allowed us to determine their functional effects as antagonists in CCR2 and inverse agonists in the constitutively active CCR1, which opens up a novel avenue to modulate these receptors in inflammatory diseases. In addition, this highly conserved binding site might allow the design of both selective and multitarget inhibitors for chemokine receptors beyond CCR1 and CCR2.
Experimental Section
Chemistry: General Methods
All solvents and reagents were purchased from commercial sources and were of analytical grade. Demineralized water is simply referred to as H2O, as was used in all cases unless stated otherwise (i.e., brine). 1H NMR spectra were recorded on a Bruker AV 400 liquid spectrometer (1H NMR, 400 MHz) at ambient temperature. Chemical shifts are reported in parts per million (ppm), are designated by δ, and are downfield to the internal standard tetramethylsilane (TMS) in CDCl3. Coupling constants are reported in Hz and are designated as J. As a representative example of the obtained 1H NMR spectra, Supporting Information, Figure S4 shows the 1H NMR spectrum of compound 43. Analytical purity of the final compounds was determined by high pressure liquid chromatography (HPLC) with a Phenomenex Gemini 3 × C18 110A column (50 mm × 4.6 mm, 3 μm), measuring UV absorbance at 254 nm. Sample preparation and HPLC method was, unless stated otherwise, as follows: 0.3–0.8 mg of compound was dissolved in 1 mL of a 1:1:1 mixture of CH3CN/H2O/tBuOH and eluted from the column within 15 min, with a three component system of H2O/CH3CN/1% TFA in H2O, decreasing polarity of the solvent mixture in time from 80/10/10 to 0/90/10. All compounds showed a single peak at the designated retention time and are at least 95% pure. Liquid chromatography–mass spectrometry (LC–MS) analyses were performed using Thermo Finnigan Surveyor–LCQ Advantage Max LC–MS system and a Gemini C18 Phenomenex column (50 mm × 4.6 mm, 3 μm). The elution method was set up as follows: 1–4 min isocratic system of H2O/CH3CN/1% TFA in H2O, 80:10:10, from the fourth minute, a gradient was applied from 80:10:10 to 0:90:10 within 9 min, followed by 1 min of equilibration at 0:90:10 and 1 min at 80:10:10. Thin-layer chromatography (TLC) was routinely performed to monitor the progress of reactions, using aluminum coated Merck silica gel F254 plates. Purification by column chromatography was achieved by use of Grace Davison Davisil silica column material (LC60A 30–200 μm). Yields and reaction conditions were not optimized. Additionally, all compounds were screened using FAF-Drugs445,46 in order to detect potential pan-assay interference compounds (PAINS). None of the compounds was identified as PAINS after application of three different filters based on Baell et al.47
General Procedure for the Synthesis of Compounds 6–23, 26–46.33
The respective aldehyde 1a–l (1.0 equiv), aniline 2a–q (1.0 equiv), and ethyl 2,4-dioxo-butanoate analogue 3a–i (1.0 equiv) were dissolved in acetic acid (2.5 mL/mmol) and heated at 95 °C for 2–4 h under a nitrogen atmosphere. Upon completion of the reaction (TLC 1/7 EtOAct/petroleum ether), acetic acid was removed under reduced pressure, the residue was triturated with Et2O and stirred for 30 min, after which the pure product was collected by filtration.
4-Acetyl-5-cyclohexyl-3-hydroxy-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (6).33
Started from cyclohexane carboxaldehyde 1a (243 μL, 2.00 mmol, 1.00 equiv), 4-methylaniline (214 mg, 2.00 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (251 μL, 2.00 mmol, 1.00 equiv) in 5 mL of acetic acid. Yield: 287 mg, 46%, white solid. 1H NMR (400 MHz, DMSO): δ 7.38 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.4 Hz, 2H), 4.99 (d, J = 1.2 Hz, 1H), 2.43 (s, 3H), 2.32 (s, 3H), 1.83 (t, J = 11.2 Hz, 1H), 1.65–1.56 (m, 1H), 1.52–1.27 (m, 4H), 0.53 (qd, J = 12.4, 2.8 Hz, 1H) ppm. MS [ESI + H]+: 313.93.
4-Acetyl-5-cycloheptyl-3-hydroxy-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (7).32
Started from cycloheptylcarboxaldehyde 1b(48) (375 mg, 3.00 mmol, 1.00 equiv), 4-methylaniline 2b (321 mg, 3.00 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (377 μL, 3.00 mmol, 1.00 equiv) in 7.5 mL of acetic acid. Purified by recrystallization from a mixture of EtOAc and petroleum ether. Yield: 102 mg, 13%, off-white solid. 1H NMR (400 MHz, CDCl3): δ 7.26–7.22 (m, 4H), 4.95 (d, J = 1.6 Hz, 1H), 2.54 (s, 3H), 2.38 (s, 3H) ppm, 2.09–2.03 (m, 1H), 1.61–1.47 (m, 4H), 1.46–1.32 (m, 4H), 1.31–1.12 (m, 4H), 0.80 (qd, J = 10.8, 3.2 Hz, 1H) ppm. MS: [ESI + H]+: 328.13.
4-Acetyl-5-cyclooctyl-3-hydroxy-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (8).32
Started from cyclooctylcarboxaldehyde 1c (648 mL, 4.42 mmol, 1.00 equiv), 4-methylaniline 2b (474 mg, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of acetic acid. Purified by column chromatography using as eluent 1/6 EtOAc/petroleum ether. Yield: 118 mg, 8%, white solid. 1H NMR (400 MHz, CDCl3): δ 7.26–7.21 (m, 4H), 4.90 (d, J = 1.6 Hz, 1H), 2.53 (s, 3H), 2.37 (s, 3H), 2.22–2.14 (m, 1H), 1.62–1.52 (m, 1H), 1.50–1.15 (m, 13H) 0.89–0.78 (m, 1H) ppm. MS: [ESI + H]+: 342.20.
4-Acetyl-3-hydroxy-1-(4-methylphenyl)-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (9).32
Started from benzaldehyde 1d (449 mL, 4.42 mmol, 1.00 equiv), 4-methylaniline 2b (474 mg, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of acetic acid. Yield: 867 mg, 64%, off-white solid. 1H NMR (400 MHz, CDCl3): δ 7.28–7.24 (m, 5H), 7.22 (d, J = 6.0 Hz, 2H), 7.07 (d, J = 8.0 Hz, 2H), 5.75 (s, 1H), 2.49 (s, 3H), 2.16 (s, 3H) ppm. MS [ESI + H]+: 308.00.
4-Acetyl-3-hydroxy-5-(4-methylphenyl)-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (10)
Started from 4-methylbenzaldehyde 1e (521 mL, 4.42 mmol, 1.00 equiv), 4-methylaniline 2b (474 mg, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of acetic acid. Purified by recrystallization from acetone/hexanes. Yield: 257 mg, 18% yellowish solid. 1H NMR (400 MHz, DMSO-d6): δ 7.42 (d, J = 8.4 Hz, 2H), 7.12–7.04 (m, 4H), 6.98 (d, J = 8.0 Hz, 2H), 5.94 (s, 1H), 2.30 (s, 3H), 2.19 (s, 3H), 2.16 (s, 3H) ppm. MS [ESI + H]+: 322.00.
4-Acetyl-3-hydroxy-5-(4-methoxyphenyl)-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (11).49
Started from 4-methoxybenzaldehyde 1f (527 mL, 4.42 mmol, 1.00 equiv), 4-methylaniline 2b (474 mg, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of acetic acid. The desired product was obtained by column chromatography using a gradient of 1/6 EtOAc/petroleum ether to 1/3 EtOAc/petroleum ether, yielding 34 mg, 2% as an off-white solid. 1H NMR (400 MHz, DMSO-d6): δ 7.42 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 8.8 Hz, 2H), 6.73 (d, J = 8.8 Hz, 2H) 5.93 (s, 1H), 3.64 (s, 3H), 2.30 (s, 3H), 2.20 (s, 3H) ppm. MS [ESI + H]+: 337.80.
4-Acetyl-5-(4-chlorophenyl)-3-hydroxy-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (12).32
Started from 4-chlorobenzaldehyde 1g (621 mg, 4.42 mmol, 1.00 equiv), 4-methylaniline 2b (474 mg, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of acetic acid. The desired product was obtained by column chromatography using 1/6 EtOAc/petroleum ether as eluent, yielding 96 mg, 6% as a white solid. 1H NMR (400 MHz, DMSO- d6): δ 7.43 (d, J = 8.4 Hz, 2H), 7.30–7.18 (m, 4H), 7.08 (d, J = 8.4 Hz, 2H), 5.98 (s, 1H), 2.30 (s, 3H), 2.20 (s, 3H) ppm. MS [ESI + H]+: 342.00.
4-Acetyl-5-(4-bromophenyl)-3-hydroxy-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (13).32
Started from 4-bromobenzaldehyde 1h (818 mg, 4.42 mmol, 1.00 equiv), 4-methylaniline 2b (474 mg, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of acetic acid. Yield: 1.23 g, 72%, yellowish solid. 1H NMR (400 MHz, CDCl3): δ 7.39 (d, J = 8.8 Hz, 2H), 7.25 (d, J = 8.8 Hz, 2H), 7.11–7.08 (m, 4H), 5.73 (s, 1H), 2.27 (s, 3H), 2.23 (s, 3H) ppm. MS [ESI + H]+: 387.93.
4-Acetyl-3-hydroxy-5-(3-methylphenyl)-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (14)
Started from 3-methylbenzaldehyde 1i (600 mg, 5.00 mmol, 1.00 equiv), 4-methylaniline 2b (536 mg, 5.00 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (627 μL, 5.00 mmol, 1.00 equiv) in 12 mL of acetic acid. Yield: 560 mg, 35%, white solid. 1H NMR (400 MHz, DMSO- d6): δ 7.43 (d, J = 8.4 Hz, 2H), 7.11–7.05 (m, 3H), 7.02 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 7.2 Hz, 1H), 5.94 (s, 1H), 2.31 (s, 3H), 2.19 (s, 3H), 2.18 (s, 3H) ppm. MS [ESI + H]+: 321.93.
4-Acetyl-3-hydroxy-5-(3-methoxyphenyl)-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (15)
Started from 3-methoxylbenzaldehyde 1j (681 mg, 5.00 mmol, 1.00 equiv), 4-methylaniline 2b (536 mg, 5.00 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (627 μL, 5.00 mmol, 1.00 equiv) in 12 mL of acetic acid. Yield: 1.27 g, 75%, white solid. 1H NMR (400 MHz, DMSO- d6): δ 7.44 (d, J = 8.4 Hz, 2H), 7.12–7.07 (m, 3H), 6.79 (s, 1H), 6.75 (d, J = 0.8 Hz, 1H), 6.69 (dd, J = 8.0, 2.2 Hz, 1H), 5.97 (s, 1H), 3.65 (s, 3H), 2.32 (s, 3H), 2.20 (s, 3H) ppm. MS [ESI + H]+: 337.39.
4-Acetyl-5-(3-chlorophenyl)-3-hydroxy-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (16)
Started from 3-chlorobenzaldehyde 1k (703 mg, 5.00 mmol, 1.00 equiv), 4-methylaniline 2b (536 mg, 5.00 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (627 μL, 5.00 mmol, 1.00 equiv) in 12 mL of acetic acid. Yield: 619 mg, 36%, light-yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.28 (d, J = 8.4 Hz, 2H), 7.21–7.18 (m, 3H), 7.17–7.13 (m, 1H), 7.10 (d, J = 8.4 Hz, 2H), 5.75 (s, 1H), 2.29 (s, 3H), 2.27 (s, 3H) ppm. MS [ESI + H]+: 341.80.
4-Acetyl-5-(3-bromophenyl)-3-hydroxy-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (17)
Started from 3-bromobenzaldehyde 1l (925 mg, 5.00 mmol, 1.00 equiv), 4-methylaniline 2b (536 mg, 5.00 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (627 μL, 5.00 mmol, 1.00 equiv) in 12 mL of acetic acid. Yield: 993 mg, 51%, brown solid. 1H NMR (400 MHz, DMSO-d6): δ 7.48 (t, J = 1.6 Hz, 1H), 7.44 (d, J = 8.4 Hz, 2H), 7.34–7.30 (m, 1H), 7.20 (dt, J = 8.0, 1.6 Hz, 1H), 7.14 (t, J = 7.6 Hz, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.02 (s, 1H), 2.33 (s, 3H), 2.20 (s, 3H) ppm. MS [ESI + H]+: 386.67.
5-Cyclohexyl-3-hydroxy-4-propionyl-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (18)
Started from cyclohexane carboxaldehyde 1a (129 mg, 1.15 mmol, 1.00 equiv), 4-methylaniline 2b (123 mg, 1.15 mmol, 1.00 equiv), and ethyl 2,4-dioxohexanoate503b (198 mg, 1.15 mmol, 1.00 equiv) in 3 mL of acetic acid. Yield: 65 mg, 19%, white solid. 1H NMR (400 MHz, CDCl3): δ 7.31–7.23 (m, 4H), 4.96 (s,1H), 2.95–2.82 (m, 2H), 2.38 (s, 3H), 1.90 (t, J = 10.8 Hz, 1H), 1.66–1.54 (m, 4H), 1.43–1.41 (m, 1H), 1.17 (t, J = 7.2 Hz, 3H), 1.09–1.03 (m, 3H), 0.98–0.86 (m, 1H), 0.71–0.61 (m, 1H) ppm. MS [ESI + H]+: 328.13.
4-Butyryl-5-cyclohexyl-3-hydroxy-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (19).32
Started from cyclohexane carboxaldehyde 1a (605 μL, 5.00 mmol, 1.00 equiv), 4-methylaniline 2b (536 mg, 5.00 mmol, 1.00 equiv), and ethyl 2,4-dioxoheptanoate343c (198 mg, 1.15 mmol, 1.00 equiv) in 12 mL of acetic acid. Yield: 669 mg (39%) as a white solid. 1H NMR (400 MHz, DMSO): δ 7.39 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 5.02 (s, 1H), 2.89–2.70 (m, 2H), 2.32 (s, 3H), 1.84–1.78 (m, 1H), 1.61–1.32 (m, 7H), 0.97–0.80 (m, 6H), 0.80–0.73 (m, 1H), 0.57–0.48 (m, 1H) ppm. MS [ESI + H]+: 341.87.
4-Pentanoyl-5-cyclohexyl-3-hydroxy-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (20)
Started from cyclohexane carboxaldehyde 1a (266 mg, 2.37 mmol, 1.00 equiv), 4-methylaniline 2b (253 mg, 2.37 mmol, 1.00 equiv), and ethyl 2,4-dioxooctanoate343d (475 mg, 2.37 mmol, 1.00 equiv) in 5 mL of acetic acid. Yield: 237 mg, 28%, white solid. 1H NMR (400 MHz, DMSO): δ 12.02 (br s 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.24 (d, J = 7.6 Hz, 2H), 5.02 (s, 1H), 2.92–2.73 (m, 2H), 2.32 (s, 3H), 1.85–1.75 (m, 1H), 1.58–1.28 (m, 9H), 0.91–0.65 (m, 7H) 0.57–0.50 (m, 1H) ppm. MS [ESI + H]+: 356.00.
5-Cyclohexyl-3-hydroxy-4-isobutyryl-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (21)
Started from cyclohexane carboxaldehyde 1a (535 μL, 4.42 mmol, 1.00 equiv), 4-methylaniline 2b (474 mg, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxo-5-methylhexanoate 3e (823 mg, 4.42 mmol, 1.00 equiv) in 10 mL of acetic acid. Yield: 255 mg, 17%, white solid. 1H NMR (400 MHz, DMSO-d6): δ 12.07 (br s, 1H), 7.40 (d, J = 7.6 Hz, 2H), 7.24 (d, J = 7.6 Hz, 2H), 5.03 (d, J = 1.6 Hz, 1H), 3.44–3.41 (m, 1H), 2.32 (s, 3H), 1.80–1.70 (m, 1H), 1.62–1.59 (m, 1H), 1.46–1.37 (m, 4H), 1.09 (d, J = 6.8 Hz, 3H), 1.02 (d, J = 6.8 Hz, 3H), 0.97–0.77 (m, 4H), 0.59–0.53 (m, 1H) ppm. MS [ESI + H]+: 342.13.
5-Cyclohexyl-4-(cyclopropanecarbonyl)-3-hydroxy-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (22)
Started from cyclohexane carboxaldehyde 1a (605 μL, 5.00 mmol, 1.00 equiv), 4-methylaniline 2b (550 μL, 5.00 mmol, 1.00 equiv), and ethyl 4-cyclopropyl-2,4-dioxobutanoate513f (920 mg, 5.00 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 60 mg, 4%, white solid. 1H NMR (400 MHz, MeOD): δ 7.34 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 7.6 Hz, 2H), 5.01 (d, J = 2.0 Hz, 1H), 3.01–2.95 (m, 1H), 2.38 (s, 3H), 1.88 (t, J = 10.4 Hz, 1H), 1.72–1.64 (m, 1H), 1.60–1.48 (m, 3H), 1.41 (d, J = 11.2 Hz, 1H), 1.04–0.86 (m, 8H), 0.72–0.62 (m, 1H) ppm. MS [ESI + Na]+: 363.10.
5-Cyclohexyl-3-hydroxy-1-(4-methylphenyl)-4-pivaloyl-1,5-dihydro-2H-pyrrol-2-one (23).32
Started from cyclohexane carboxaldehyde 1a (121 μL, 1.00 mmol, 1.00 equiv), 4-methylaniline 2b (107 mg, 1.00 mmol, 1.00 equiv), and ethyl 5,5-dimethyl-2,4-dioxohexanoate 3g (175 μL, 1.00 mmol, 1.00 equiv) in 3 mL of acetic acid. Yield: 20 mg, 6%, white solid. 1H NMR (400 MHz, DMSO-d6): δ 7.41 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 8.4 Hz, 2H), 5.11 (d, J = 2.2 Hz, 1H), 2.32 (s, 3H), 1.63–1.58 (m, 2H), 1.52–1.46 (m, 3H), 1.31–1.28 (m, 1H) 1.25 (s, 9H) 1.01–0.69 (m, 4H), 0.69–0.59 (m, 1H) ppm. MS [ESI + H]+: 356.13.
Ethyl 2-Cyclohexyl-4-hydroxy-5-oxo-1-(4-methylphenyl)-2,5-dihydro-1H-pyrrole-3-carboxylate (24).29
Sodium 1,4-diethoxy-1,4-dioxobut-2-en-2-olate (1.25 g, 6.00 mmol) was dissolved in 25 mL of H2O, and 25 mL of Et2O was added. The mixture was acidified to pH 2 with 6 M HCl (aq) and was extracted with Et2O from the aqueous phase, dried over MgSO4, and concentrated in vacuo, yielding 1.05 g, 4.97 mmol, 83% diethyl 2-oxosuccinate as a yellow oil.52
Diethyl 2-oxosuccinate 3h (1.05 g, 4.97 mmol, 1.12 equiv) was added to a mixture of cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv) and 4-methylaniline 2b (474 mg, 4.42 mmol, 1.00 equiv) in 10 mL of dry THF and stirred at room temperature overnight. The reaction mixture was concentrated in vacuo, Et2O was added, and the white precipitate was collected by filtration. Yield: 400 mg, 26%, white solid. 1H NMR (400 MHz, CDCl3): δ 11.24 (s, 1H), 7.40 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.4 Hz, 2H), 5.00 (d, J = 1.8 Hz, 1H), 4.32–4.13 (m, 2H), 2.32 (s, 3H), 1.85–1.76 (m, 1H), 1.66–1.59 (m, 1H), 1.56–1.44 (m, 3H), 1.32 (d, J = 12.0 Hz, 1H), 1.26 (t, J = 7.2 Hz, 3H), 1.06–0.75 (m, 4H), 0.63–0.53 (m, 1H) ppm. MS [ESI + H]+: 344.07.
Isopropyl 2-Cyclohexyl-4-hydroxy-5-oxo-1-(4-methylphenyl)-2,5-dihydro-1H-pyrrole-3-carboxylate (25)
Ester 22 (343 mg, 1.00 mmol, 1.00 equiv) and p-toluenesulfonic acid (172 mg, 1.00 mmol, 1.00 equiv) were dissolved in 10 mL of 2-propanol, and the reaction mixture was refluxed for 48 h. The solvent was removed under reduced pressure, and the crude product was dissolved in 50 mL of EtOAc and washed 3× with H2O, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (4/1 EtOAc/petroleum ether) and yielded 150 mg, 9.5%, brownish solid. 1H NMR (400 MHz, CDCl3): δ 9.33 (s, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.4 Hz, 2H), 5.31–5.23 (m, 1H), 4.79 (d, J = 2.0 Hz, 1H), 2.37 (s, 3H), 1.88 (td, J = 9.6, 2.8 Hz, 1H), 1.73–1.66 (m, 1H), 1.63–1.50 (m, 3H), 1.38 (d, J = 6.8 Hz, 6H), 1.29–1.19 (m, 1H), 1.13–0.90 (m, 4H), 0.74–0.64 (m, 1H) ppm. MS [ESI + H]+: 357.93.
4-Benzoyl-5-cyclohexyl-3-hydroxy-1-(4-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (26)
Started from cyclohexane carboxaldehyde 1a (484 μL, 4.00 mmol, 1.00 equiv), 4-methylaniline 2b (428 mg, 4.00 mmol, 1.00 equiv), and ethyl 2,4-dioxo-4-phenylbutanoate533i (880 mg, 4.00 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 53 mg, 4%, white solid. 1H NMR (400 MHz, DMSO- d6): δ 7.85 (d, J = 7.6 Hz, 2H), 7.69–7.62 (m, 1H), 7.58–7.52 (m, 2H), 7.49 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 5.32 (s, 1H), 2.35 (s, 3H), 1.73–1.62 (m, 2H), 1.58–1.51 (m, 2H), 1.49–1.37 (m, 2H), 1.05–0.94 (m, 1H), 0.88–0.74 (m, 4H) ppm. MS [ESI + H]+: 375.93.
4-Acetyl-5-cyclohexyl-3-hydroxy-1-phenyl-1,5-dihydro-2H-pyrrol-2-one (27).32
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), aniline 2a (400 μL, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 1.00 g, 76%, yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.47–7.44 (m, 4H), 7.32–7.28 (m, 1H), 4.99 (d, J = 2.0 Hz, 1H), 2.54 (s, 3H), 1.97–1.90 (m, 1H), 1.69–1.66 (m, 1H), 1.59–1.48 (m, 3H), 1.45–1.41 (m, 1H), 1.10–0.99 (m, 3H), 0.92–0,86 (m, 1H), 0.63 (qd, J = 9.2, 3.6 Hz, 1H) ppm. MS [ESI + H]+: 300.07.
4-Acetyl-5-cyclohexyl-1-(4-ethylphenyl)-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (28).32
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 4-ethylaniline 2c (553 μL, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 134 mg, 9%, light-yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.32 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 6.4 Hz, 2H), 4.94 (s, 1H), 2.68 (q, J = 7.6 Hz, 2H) 2.54 (s, 3H), 1.95–1.90 (m, 1H), 1.68–1.66 (m, 1H), 1.67–1.51 (m, 3H), 1.43–1.41 (m, 1H), 1.26 (t, J = 7.6 Hz, 3H), 1.10–0.98 (m, 4H), 0.90–0.87 (m, 1H), 0.69–0.60 (m, 1H) ppm. MS [ESI + H]+: 328.13.
4-Acetyl-5-cyclohexyl-3-hydroxy-1-(4-methoxyphenyl)-1,5-dihydro-2H-pyrrol-2-one (29).32
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 4-methoxyaniline 2d (560 mg, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 805 mg, 56%, light-yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 7.41 (d, J = 8.8 Hz, 2H), 7.00 (d, J = 9.2 Hz, 2H), 4.96 (d, J = 1.2 Hz, 1H), 3.77 (s, 3H), 2.43 (s, 3H), 1.86–1.80 (m,1H), 1.60–1.58 (m, 1H), 1.40–1.37 (m, 2H), 1.35–1.32 (m, 2H), 0.99–0.91 (m, 3H), 0.87–0.75 (m, 1H), 0.58–0.55 (m, 1H) ppm. MS [ESI + H]+: 330.07.
4-Acetyl-1-(4-chlorophenyl)-5-cyclohexyl-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (30).32
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 4-chloroaniline 2e (544 mg, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 713 mg, 48%, light-yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.75 (s br, 1H), 7.42 (d, J = 8.8 Hz, 2H), 7.39 (d, J = 9.2 Hz, 2H), 4.95 (d, J = 2.0 Hz, 1H), 2.53 (s, 3H), 1.96–1.84 (m, 1H), 1.75–1.65 (m, 1H), 1.63–1.51 (m, 3H), 1.45–1.40 (m, 1H), 1.11–0.97 (m, 3H), 0.95–0.86 (m, 1H), 0.66 (qd, J = 12.4, 3.2 Hz, 1H) ppm. MS [ESI + H]+: 334.1.
4-Acetyl-1-(4-bromophenyl)-5-cyclohexyl-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (31).32
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 4-bromoaniline 2f (760 mg, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 910 mg, 53%, white solid. 1H NMR (400 MHz, CDCl3): δ 7.57 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.4 Hz, 2H), 4.96 (s, 1H), 2.53 (s, 3H), 1.97–1.87 (m, 1H), 1.73–1.65 (m, 1H), 1.62–1.49 (m, 3H), 1.45–1.37 (m, 1H), 1.15–0.97 (m, H), 0.95–0.86 (m, 1H), 0.66 (qd, J = 12.4, 3.2 Hz, 1H) ppm. MS [ESI + H]+: 378.1.
4-Acetyl-5-cyclohexyl-3-hydroxy-1-(4-(trifluoromethyl)phenyl)-1,5-dihydro-2H-pyrrol-2-one (32).32
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 4-trifluoromethylaniline 2g (556 μL, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 80 mg, 5%, white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.25 (s, 1H), 7.82 (s, 4H), 5.19 (s, 1H), 2.46 (s, 3H), 1.85 (t, J = 11.2 Hz, 1H), 1.60 (d, J = 10.8 Hz, 1H), 1.54–1.43 (m, 3H), 1.38 (d, J = 12.4 Hz, 1H), 1.05–0.74 (m, 4H), 0.52 (d, J = 12.4 Hz, 1H) ppm. MS [ESI + H]+: 369.07.
4-Acetyl-5-cyclohexyl-3-hydroxy-1-(3-methylphenyl)-1,5-dihydro-2H-pyrrol-2-one (33).29
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 3-methylaniline 2h (474 μL, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 511 mg, 37%, white solid. 1H NMR (400 MHz, CDCl3): δ 7.32 (t, J = 8.0 Hz, 1H), 7.26 (s, 1H), 7.19 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 7.6 Hz, 1H), 4.96 (d, J = 2.0 Hz, 1H), 2.53 (s, 3H), 2.40 (s, 3H), 1.95–1.88 (m, 1H), 1.69–1.65 (m, 1H), 1.59–1.50 (m, 3H), 1.45–1.39 (m,1H), 1.15–0.84 (m, 4H), 0.64 (qd, J = 12.4, 3.6 Hz, 1H) ppm. MS [ESI + H]+: 314.07.
4-Acetyl-5-cyclohexyl-1-(3-fluorophenyl)-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (34).32
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 3-fluoroaniline 2i (425 μL, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 226 mg, 16%, white solid. 1H NMR (400 MHz, CDCl3): δ 8.99 (s br, 1H), 7.45–7.34 (m, 1H), 7.29–7.25 (m, 1H), 7.21 (d, J = 8.0 Hz, 1H), 7.01 (td, J = 8.0, 2.0 Hz, 1H), 4.97 (d, J = 2.0 Hz, 1H), 2.55 (s, 3H), 1.94 (td, J = 12.0, 2.0 Hz, 1H), 1.70–1.67 (m, 1H), 1.62–1.53 (m, 3H), 1.47–1.40 (m, 1H), 1.15–0.97 (m, 3H), 0.96–0.84 (m, 1H), 0.66 (qd, J = 12.4, 3.6 Hz, 1H) ppm. MS [ESI + H]+: 318.27.
4-Acetyl-1-(3-chlorophenyl)-5-cyclohexyl-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (35).29
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 3-chloroaniline 2j (468 μL, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 805 mg, 55%, yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.92 (s br, 1H), 7.51 (t, J = 1.6 Hz, 1H), 7.38 (t, J = 8.0 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.29–7.25 (m, 1H), 4.96 (d, J = 2.0 Hz, 1H), 2.55 (s, 3H), 1.93 (td, J = 12.4, 2.0 Hz, 1H), 1.71–1.68 (m, 1H), 1.60–1.54 (m, 3H), 1.46–1.43 (m, 1H), 1.15–0.99 (m, 3H), 0.98–0.86 (m, 1H), 0.65 (qd, J = 12.4, 2.8 Hz, 1H) ppm. MS [ESI + H]+: 334.13.
4-Acetyl-5-cyclohexyl-3-hydroxy-1-(3-(trifluoromethyl)phenyl)-1,5-dihydro-2H-pyrrol-2-one (36)
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 3-trifluoromethylaniline 2k (552 μL, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 560 mg, 34%, brown solid. 1H NMR (400 MHz, DMSO-d6): δ 12.26 (br s, 1H), 7.98 (s, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.69 (t, J = 8.0 Hz, 1H), 7.64 (d, J = 7.6 Hz, 1H), 5.23 (d, J = 1.2 Hz, 1H), 2.45 (s, 3H), 1.86 (t, J = 11.2 Hz, 1H), 1.62–1.58 (m, 1H), 1.47–1.35 (m, 4H), 1.00–0.85 (m, 3H), 0.80–0.71 (m, 1H), 0.47 (qd, J = 12.8 Hz, 3.2 Hz, 1H) ppm. MS [ESI + H]+: 368.13.
4-Acetyl-5-cyclohexyl-1-(2-fluoro-4-methylphenyl)-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (37).29
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 2-fluoro-4-methylaniline 2l (499 μL, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 508 mg, 35%, white solid. 1H NMR (400 MHz, CDCl3): δ 9.35 (br s, 1H), 7.28–7.22 (m, 1H), 7.04–6.98 (m, 2H), 4.93 (d, J = 1.6 Hz, 1H), 2.52 (s, 3H), 2.39 (s, 3H), 1.98–1.91 (m, 1H), 1.69–1.63 (m, 1H), 1.57–1.41 (m, 4H), 1.13–1.02 (m, 3H), 0.92–0.82 (m, 1H), 0.62 (qd, J = 12.8 Hz, 3.2 Hz, 1H) ppm. MS [ESI + H]+: 332.1.
4-Acetyl-1-(4-chloro-2-fluorophenyl)-5-cyclohexyl-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (38).29
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 4-chloro-2-fluoroaniline 2m (490 μL, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 190 mg, 12%, light-yellow solid. 1H NMR (400 MHz, CDCl3): δ 9.07 (br s, 1H), 7.37 (t, J = 8.4 Hz, 1H), 7.28–7.22 (m, 2H), 4.96 (d, J = 2.0 Hz, 1H), 2.52 (s, 3H), 2.00–1.91 (m, 1H), 1.72–1.65 (m, 1H), 1.63–1.44 (m, 4H), 1.14–1.00 (m, 2H), 0.99–0.82 (m, 2H), 0.61 (qd, J = 12.8 Hz, 3.6 Hz, 1H) ppm. MS [ESI + H]+: 352.1.
4-Acetyl-1-(4-bromo-2-fluorophenyl)-5-cyclohexyl-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (39).32
Started from cyclohexane carboxaldehyde 1a (303 μL, 2.50 mmol, 1.00 equiv), 4-bromo-2-fluoroaniline 2n (475 μL, 2.50 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (395 μL, 2.50 mmol, 1.00 equiv) in 5 mL of AcOH. Purified by silica column chromatography using EtOAc/petroleum ether (1/6). The resulting impure product was stirred in diisopropylether, and the pure product was obtained by filtration. Yield: 36 mg, 4%, white solid. 1H NMR (400 MHz, CDCl3): δ 8.97 (s br, 1H, OH), 7.43–7.36 (m, 2H), 7.31 (t, J = 8.0 Hz, 1H), 4.96 (d, J = 1.2 Hz, 1H), 2.52 (s, 3H), 1.94 (t, J = 11.6 Hz, 1H), 1.68 (d, J = 13.2 Hz, 1H), 1.57–1.42 (m, 4H), 1.15–0.84 (m, 4H), 0.62 (qd, J = 12.4, 3.2 Hz, 1H) ppm. MS [ESI + H]+: 395.67.
4-Acetyl-5-cyclohexyl-1-(3,4-dimethylphenyl)-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (40).32
Started from cyclohexane carboxaldehyde 1a (303 μL, 2.50 mmol, 1.00 equiv), 3,4-dimethylaniline 2o (303 μL, 2.50 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (395 μL, 2.50 mmol, 1.00 equiv) in 5 mL of AcOH. Yield: 167 mg, 20%, white solid. 1H NMR (400 MHz, CDCl3): δ 9.06 (s br, 1H, OH), 7.21–7.16 (m, 2H), 7.10 (dd, J = 8.0, 1.6 Hz, 1H), 4.92 (d, J = 2.0 Hz, 1H), 2.53 (s, 3H), 2.29 (s, 3H), 2.28 (s, 3H),1.92 (t, J = 11.6 Hz, 1H), 1.73–1.65 (m, 1H), 1.60–1.49 (m, 3H), 1.42 (d, J = 12.4 Hz, 1H), 1.13–0.98 (m, 3H), 0.94–0.84 (m, 1H), 0.65 (qd, J = 12.0, 3.2 Hz, 1H) ppm. MS [ESI + H]+: 327.87.
4-Acetyl-1-(4-chloro-3-methylphenyl)-5-cyclohexyl-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (41).29
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 4-chloro-3-methylaniline 2p (626 mg, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 505 mg, 33%, white solid. 1H NMR (400 MHz, CDCl3): δ 9.39 (s br, 1H, OH), 7.40 (d, J = 8.4 Hz, 1H), 7.32 (d, J = 2.0 Hz, 1H), 7.17 (dd, J = 8.4, 2.4 Hz, 1H), 4.94 (d, J = 2.0 Hz, 1H), 2.55 (s, 3H), 2.42 (s, 3H), 1.93 (td, J = 12.0, 2.4 Hz, 1H), 1.72–1.64 (m, 1H), 1.60–1.49 (m, 3H), 1.46–1.34 (m,1H), 1.15–0.96 (m, 3H), 0.94–0.84 (m, 1H), 0.65 (qd, J = 12.8, 3.6 Hz, 1H) ppm. MS [ESI + H]+: 348.0.
4-Acetyl-5-cyclohexyl-1-(3-fluoro-4-methylphenyl)-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (42).32
Started from cyclohexane carboxaldehyde 1a (534 μL, 4.42 mmol, 1.00 equiv), 3-fluoro-4-methylaniline 2q (506 μL, 4.42 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (554 μL, 4.42 mmol, 1.00 equiv) in 10 mL of AcOH. Yield: 160 mg, 10%, white solid. 1H NMR (400 MHz, CDCl3) δ 7.24 (t, J = 8.0 Hz, 1H), 7.18 (dd, J = 6.8, 2.0 Hz, 1H), 7.09 (dd, J = 8.4, 2.0 Hz, 1H), 4.92 (d, J = 2.0 Hz, 1H), 2.54 (s, 3H), 2.30 (d, J = 1.6 Hz, 3H), 1.94 (td, J = 12.0, 2.0 Hz, 1H), 1.74–1.64 (m, 1H), 1.64–1.48 (m, 3H), 1.48–1.39 (m, 1H), 1.14–0.98 (m, 3H), 0.98–0.84 (m, 1H), 0.67 (qd, J = 12.8, 3.2 Hz, 1H). MS [ESI + H]+: 332.00.
1-(4-Bromo-2-fluorophenyl)-5-cyclohexyl-4-(cyclopropanecarbonyl)-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (43)
Started from cyclohexane carboxaldehyde 1a (242 μL, 2.00 mmol, 1.00 equiv), 4-bromo-2-fluoroaniline 2n (380 mg, 2.00 mmol, 1.00 equiv), and ethyl 4-cyclopropyl-2,4-dioxobutanoate513f (368 mg, 2.00 mmol, 1.00 equiv) in 5 mL of AcOH. Yield: 240 mg, 28%, white solid. 1H NMR (400 MHz, CDCl3): δ 7.43–7.34 (m, 3H), 5.06 (d, J = 1.6 Hz, 1H), 2.39–2.33 (m, 1H), 1.92 (t, J = 12.0 Hz, 1H), 1.72 (d, J = 12.4 Hz, 1H), 1.64–1.48 (m, 4H), 1.35–1.31 (m, 1H), 1.25–1.22 (m, 1H), 1.13–1.01 (m, 5H), 0.99–0.90 (m, 1H), 0.68 (qd, J = 12.4, 3.2 Hz, 1H) ppm. MS [ESI + H]+: 421.67.
4-Benzoyl-1-(4-bromo-2-fluorophenyl)-5-cyclohexyl-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (44)
Started from cyclohexane carboxaldehyde 1a (242 μL, 2.00 mmol, 1.00 equiv), 4-bromo-2-fluoroaniline 2n (380 mg, 2.00 mmol, 1.00 equiv), and ethyl 2,4-dioxo-4-phenylbutanoate533i (440 mg, 2.00 mmol, 1.00 equiv) in 5 mL of AcOH. Yield: 210 mg, 23%, white solid. 1H NMR (400 MHz, DMSO): δ 7.84 (d, J = 6.8 Hz, 2H), 7.78 (dd, J = 10.4, 2.0 Hz, 1H), 7.67–7.60 (m, 2H), 7.58–7.51 (m, 3H), 5.21 (d, J = 2.0 Hz, 1H), 1.64–1.44 (m, 6H), 1.00–0.70 (m, 5H) ppm. MS [ESI + H]+: 459.87.
1-(4-Bromo-2-fluorophenyl)-5-(3-bromophenyl)-4-(cyclopropanecarbonyl)-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (45)
Started from 3-bromobenzaldehyde 1l (233 μL, 2.00 mmol, 1.00 equiv), 4-bromo-2-fluoroaniline 2n (380 mg, 2.00 mmol, 1.00 equiv), and ethyl 4-cyclopropyl-2,4-dioxobutanoate513f (440 mg, 2.00 mmol, 1.00 equiv) in 5 mL of AcOH. Yield: 205 mg, 21%, off-white solid. 1H NMR (400 MHz, CDCl3): δ 7.40 (d, J = 6.8 Hz, 1H), 7.35–7.27 (m, 2H), 7.22 (d, J = 8.4 Hz, 1H), 7.19–7.11 (m, 2H), 7.07 (t, J = 8.0 Hz, 1H), 5.84 (s, 1H), 1.93–1.85 (m, 1H), 1.22–1.19 (m, 1H), 1.07–0.97 (m, 2H), 0.82–0.74 (m, 1H) ppm. MS [ESI + H]+: 495.67.
4-Acetyl-1-(4-bromo-2-fluorophenyl)-5-(3-bromophenyl)-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one (46)
Started from 3-bromobenzaldehyde 1l (291 μL, 2.50 mmol, 1.00 equiv), 4-bromo-2-fluoroaniline 2n (475 mg, 2.50 mmol, 1.00 equiv), and ethyl 2,4-dioxopentanoate 3a (395 mg, 2.50 mmol, 1.00 equiv) in 5 mL of AcOH. Purified by silica column chromatography using EtOAc/petroleum ether (1/19). Yield: 82 mg, 5%, yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.41 (dt, J = 7.6, 1.6 Hz, 1H), 7.33–7.24 (m, 2H), 7.22 (d, J = 8.4 Hz, 1H), 7.19–7.11 (m, 2H), 7.08 (t, J = 8.0 Hz, 1H), 5.74 (s, 1H), 2.17 (s, 3H) ppm. MS [ESI + H]+: 469.60.
In Vitro Characterization of Compound’s Activity
Chemicals and Reagents
[3H]-CCR2-RA-[R] (specific activity 59.6 Ci mmol–1), corresponding to the (R)-isomer of compound 38 ([3H]-(R)-4-acetyl-1-(4-chloro-2-fluorophenyl)-5-cyclohexyl-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one)), was custom-labeled by Vitrax (Placentia, CA). [35S]GTPγS (guanosine 5′-O-(3-[35S]thio)triphosphate), with a specific activity of 1250 Ci mmol–1, was purchased from PerkinElmer (Waltham, MA). CCR2-RA-[R], SD-24, and JNJ-27141491 were synthesized as described previously.30,36,54 BX471 was purchased from Cayman Chemical (Ann Arbor, MI, USA). Chemokine ligands CCL2 and CCL3 were purchased from PeproTech (Rocky Hill, NJ). Bovine serum albumin (BSA, fraction V) was purchased from Sigma (St. Louis, MO, USA). Bicinchoninic acid (BCA) and BCA protein assay reagent were purchased from Pierce Chemical Company (Rockford, IL, USA). Tango CCR1-bla and Tango CCR2-bla osteosarcoma (U2OS) cells stably expressing the human CCR1 or human CCR2b (U2OS-CCR1 or U2OS-CCR2, respectively) were obtained from Invitrogen (Carlsbad, CA). All other chemicals were obtained from standard commercial sources.
Cell Culture and Membrane Preparation
U2OS-CCR1 and U2OS-CCR2 were grown in a humidified atmosphere at 37 °C and 5% CO2 in McCoy’s 5A medium supplemented with 10% fetal calf serum, 2 mM glutamine, 0.1 mM nonessential amino acids (NEAAs), 25 mM HEPES, 1 mM sodium pyruvate, 100 IU/ml penicillin, 100 μg/mL streptomycin, 100 μg/mL G418, 50 μg/mL hygromycin, and 125 μg/mL zeocin (200 μg/mL zeocin for U2OS-CCR1). Cells were subcultured twice a week at a ratio of 1:3 to 1:8 on 10 cm Ø plates by trypsinization. For membrane preparation cells were subcultured on 15 cm Ø plates using dialyzed fetal calf serum. Membranes from U2OS-CCR1 and U2OS-CCR2 cells were prepared as described previously.26 Briefly, cells were detached from confluent 15 cm Ø plates by scraping them into 5 mL of phosphate-buffered saline (PBS), collected, and centrifuged for 5 min at 3000 rpm (700g). The pellets were resuspended in ice-cold 50 mM Tris-HCl buffer, pH 7.4, supplemented with 5 mM MgCl2, and homogenized with an Ultra Turrax homogenizer (IKA-Werke GmbH & Co. KG, Staufen, Germany). Membranes were separated from the cytosolic fraction by several centrifugation steps in an Optima LE-80 K ultracentrifuge (Beckman Coulter, Inc., Fullerton, CA) at 31000 rpm for 20 min at 4 °C. Finally, the membrane pellets were resuspended in 50 mM Tris-HCl buffer supplemented with 5 mM MgCl2, pH 7.4, divided into aliquots of 100 μL and 250 μL, and stored at −80 °C. Membrane protein concentrations were measured using a BCA protein determination with BSA as a standard.55
[3H]-CCR2-RA-[R] Binding Assays
[3H]-CCR2-RA-[R] (homologous) displacement assays in U2OS-CCR1 and U2OS-CCR2 were performed in a 100 μL reaction volume containing assay buffer (50 mM Tris-HCl, 5 mM MgCl2, and 0.1% CHAPS, pH 7.4), 6 nM [3H]-CCR2-RA-[R], 8–15 μg of membrane protein, and the competing ligand. Homologous displacement assays were carried out with three different concentrations of [3H]-CCR2-RA-[R], namely 3, 6, and 12 nM. In all cases, at least six concentrations of competing ligand were used and the reaction mixture was incubated for 120 min at 25 °C. Nonspecific binding was determined in the presence of 10 μM CCR2-RA-[R]. Total radioligand binding did not exceed 10% of the amount added to prevent ligand depletion. [3H]-CCR2-RA-[R] saturation binding assays in U2OS-CCR2 were also performed in a 100 μL reaction volume containing assay buffer, [3H]-CCR2-RA-[R] in 12 different concentrations ranging from 0.05 to 70 nM, and 15 μg of membrane protein. Nonspecific binding was determined in the presence of 10 μM JNJ-27141491 at four different concentrations of radioligand, namely 0.1, 0.4, 2.5, and 20 nM. In association assays, U2OS-CCR1 and U2OS-CCR2 membrane preparations were added to the reaction mix at different time points of incubation, ranging from 1 to 180 min incubation; in dissociation assays, membranes were first incubated with 6 nM [3H]-CCR2-RA-[R] for 90 min, and dissociation was initiated by the addition of 10 μM CCR2-RA-[R] at different time points, up to 150 min for CCR1 and 180 min for CCR2. For all experiments, incubations were terminated by dilution with ice-cold wash buffer (50 mM Tris-HCl buffer supplemented with 5 mM MgCl2 and 0.05% CHAPS, pH 7.4). Separation of bound from free radioligand was performed by rapid filtration through a 96-well GF/B filter plate using a PerkinElmer Filtermate harvester (PerkinElmer, Groningen, The Netherlands). Filters were washed 10 times with ice-cold wash buffer. Then 25 μL of Microscint scintillation cocktail (PerkinElmer, Groningen, The Netherlands) was added to each well and the filter-bound radioactivity was determined by scintillation spectrometry using the P-E 2450 Microbeta2 scintillation plate counter (PerkinElmer, Groningen, The Netherlands).
[35S]GTPγS Binding Assays
[35S]GTPγS binding assays were performed as described previously.26 Briefly, binding assays were performed in a 100 μL reaction volume containing assay buffer (50 mM Tris-HCl, 5 mM MgCl2, 100 mM NaCl, 1 mM EDTA, and 0.05% BSA, pH 7.4), 10 μM GDP, 10 μg of saponin, and 10 μg of membrane, either U2OS-CCR1 or U2OS-CCR2. To determine the EC50 value of CCL2 and CCL3, the membrane mixture was preincubated with increasing concentrations of chemokine for 30 min at 25 °C. To determine the IC50 values of the ligands, the membrane mixture was preincubated with increasing concentrations of the ligand of interest in the absence or presence of a fixed concentration of CCL2 (20 nM) or CCL3 (8 nM). Basal activity was determined in the absence of any ligand or chemokine. Finally, the mixture was incubated for another 90 min at 25 °C after the addition of 0.3 nM [35S]GTPγS in all cases. For all experiments, incubations were terminated by dilution with ice-cold 50 mM Tris-HCl, 5 mM MgCl2 buffer. Separation of bound from free [35S]GTPγS was performed as described under “[3H]-CCR2-RA-[R] binding assays”.
Data Analysis
All experiments were analyzed using GraphPad Prism 7.0 (GraphPad Software Inc., San Diego, CA, USA). The KD and Bmax values of [3H]-CCR2-RA-[R] in U2OS-CCR2 were calculated from saturation experiments by fitting the data to the equation Bound = (Bmax*[L])/([L] + KD), where Bmax is the maximum number of binding sites and KD is the concentration required to reach half-maximum binding at equilibrium conditions. In the case of U2OS-CCR1 membranes, the KD and Bmax values were calculated from homologous binding experiments by nonlinear regression analysis, using the “One Site–Homologous” model that assumes that unlabeled and labeled CCR2-RA-[R] have identical affinities. The (p)IC50 values of unlabeled ligands from [3H]-CCR2-RA-[R] binding assays were obtained by nonlinear regression analysis of the displacement curves and further converted into (p)Ki values using the Cheng–Prusoff equation.56 The (p)IC50 or (p)EC50 values from [35S]GTPγS curves were also obtained by nonlinear regression. The observed association rate constants (kobs,fast; kobs,slow) were calculated by fitting the data to a two-phase exponential association function; similarly, dissociation rate constants (koff,fast; koff,slow) were calculated using a two-phase exponential decay function. All values obtained are means ± standard error of the mean (SEM) of at least three independent experiments performed in duplicate, unless stated otherwise. Differences in kinetic rates and pIC50 values between receptors or between assay formats (in the absence or presence of chemokine) were analyzed using an unpaired, two-tailed Student’s t-test with Welch’s correction; differences in pKi values between compounds, in maximal [35S]GTPγS inhibition against basal activity or in pseudo-Hill slopes from [35S]GTPγS inhibition curves against compound 45, which showed a pseudo-Hill slope of approximately unity, were analyzed using a one-way ANOVA with Dunnett’s posthoc test. Significant differences are displayed as *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001.
Computational Receptor Modeling and Docking
All modeling was performed in the Schrodinger suite,57Figure 3b,c were made in a later version58 that includes the interaction and orientation of residues (e.g., backbone, side chain). As a starting point for the structure-based studies we used the recently published crystal structure of CCR2b in complex with both BMS-681 and CCR2-RA-[R] (PDB 5T1A).24 We replaced the sequence (CCR2b: sequence between L2265x62 and R2406x32) of the M2 muscarinic acetylcholine receptor, close to the intracellular binding site, by the CCR2b sequence using homology modeling59−61 and CCR5 as template (PDB 4MBS).62 A homology model of CCR1 was constructed on the basis of this CCR2b model. For both models, the knowledge-based scoring function was used. For the ligand docked, the pKa of the hydroxyl hydrogen was calculated to be 4.5 using Jaguar;63,64 therefore, the negatively charged protonation state was used. Compound CCR2-RA-[R] was docked in both models using Induced Fit docking.65,66 Visualizations were created using PyMOL;67 residues within 5 Å of the ligand and facing the binding site are shown.
Acknowledgments
We thank Gerard van Westen for providing the software licenses and the hardware required for the computational section of this study. Eelke B. Lenselink thanks the Agentschap Innoveren en Ondernemen (AIO) for financial support (AIO project 155028).
Glossary
Abbreviations Used
- CCR1
CC chemokine receptor 1
- CCR2
CC chemokine receptor 2
- GPCRs
G protein-coupled receptors
- SAR
structure–affinity relationships
- RA
rheumatoid arthritis
- MS
multiple sclerosis
- U2OS
human osteosarcoma cells
- [35S]GTPγS
guanosine 5′-O-(3-[35S]thio)triphosphate
- CAM
constitutive active mutant
- BCA
bicinchoninic acid
- NEAAs
nonessential amino acids
- PBS
phosphate-buffered saline
- SEM
standard error of the mean.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00605.
Equilibrium and kinetic characterization of [3H]-CCR2-RA-[R] in U2OS-CCR1 and U2OS-CCR2; sequence alignment of key residues in hCCR1 and hCCR2b; characterization of selected compounds as inverse agonists using a [35S]GTPγS binding assay; 1H NMR spectrum of compound 43 (PDF)
Homology models of CCR1 (PDB)
Homology models of CCR2b (PDB)
Molecular formula strings (CSV)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Griffith J. W.; Sokol C. L.; Luster A. D. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- Bachelerie F.; Ben-Baruch A.; Burkhardt A. M.; Combadiere C.; Farber J. M.; Graham G. J.; Horuk R.; Sparre-Ulrich A. H.; Locati M.; Luster A. D.; Mantovani A.; Matsushima K.; Murphy P. M.; Nibbs R.; Nomiyama H.; Power C. A.; Proudfoot A. E.; Rosenkilde M. M.; Rot A.; Sozzani S.; Thelen M.; Yoshie O.; Zlotnik A. International union of basic and clinical pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2014, 66, 1–79. 10.1124/pr.113.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleist A. B.; Getschman A. E.; Ziarek J. J.; Nevins A. M.; Gauthier P. A.; Chevigne A.; Szpakowska M.; Volkman B. F. New paradigms in chemokine receptor signal transduction: moving beyond the two-site model. Biochem. Pharmacol. 2016, 114, 53–68. 10.1016/j.bcp.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I. Chemokines and their receptors: insights from molecular modeling and crystallography. Curr. Opin. Pharmacol. 2016, 30, 27–37. 10.1016/j.coph.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Cotarelo P.; Gomez-Moreira C.; Criado-Garcia O.; Sanchez L.; Rodriguez-Fernandez J. L. Beyond chemoattraction: multifunctionality of chemokine receptors in leukocytes. Trends Immunol. 2017, 38, 927–941. 10.1016/j.it.2017.08.004. [DOI] [PubMed] [Google Scholar]
- Viola A.; Luster A. D. Chemokines and their receptors: drug targets in immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 171–197. 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- White G. E.; Iqbal A. J.; Greaves D. R. CC chemokine receptors and chronic inflammation—therapeutic opportunities and pharmacological challenges. Pharmacol. Rev. 2013, 65, 47–89. 10.1124/pr.111.005074. [DOI] [PubMed] [Google Scholar]
- Pease J.; Horuk R. Chemokine receptor antagonists. J. Med. Chem. 2012, 55, 9363–9392. 10.1021/jm300682j. [DOI] [PubMed] [Google Scholar]
- Szekanecz Z.; Koch A. E. Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 5–13. 10.1038/nrrheum.2015.157. [DOI] [PubMed] [Google Scholar]
- Haringman J. J.; Smeets T. J.; Reinders-Blankert P.; Tak P. P. Chemokine and chemokine receptor expression in paired peripheral blood mononuclear cells and synovial tissue of patients with rheumatoid arthritis, osteoarthritis, and reactive arthritis. Ann. Rheum. Dis. 2006, 65, 294–300. 10.1136/ard.2005.037176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltayeb S.; Berg A.-L.; Lassmann H.; Wallström E.; Nilsson M.; Olsson T.; Ericsson-Dahlstrand A.; Sunnemark D. Temporal expression and cellular origin of CC chemokine receptors CCR1, CCR2 and CCR5 in the central nervous system: insight into mechanisms of MOG-induced EAE. J. Neuroinflammation 2007, 4, 14. 10.1186/1742-2094-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fife B. T.; Huffnagle G. B.; Kuziel W. A.; Karpus W. J. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J. Exp. Med. 2000, 192, 899–906. 10.1084/jem.192.6.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottman J. B.; Slavin A. J.; Silva R.; Weiner H. L.; Gerard C. G.; Hancock W. W. Leukocyte recruitment during onset of experimental allergic encephalomyelitis is CCR1 dependent. Eur. J. Immunol. 2000, 30, 2372–2377. . [DOI] [PubMed] [Google Scholar]
- Brühl H.; Cihak J.; Schneider M. A.; Plachý J.; Rupp T.; Wenzel I.; Shakarami M.; Milz S.; Ellwart J. W.; Stangassinger M.; Schlöndorff D.; Mack M. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J. Immunol. 2004, 172, 890–898. 10.4049/jimmunol.172.2.890. [DOI] [PubMed] [Google Scholar]
- Amat M.; Benjamim C. F.; Williams L. M.; Prats N.; Terricabras E.; Beleta J.; Kunkel S. L.; Godessart N. Pharmacological blockade of CCR1 ameliorates murine arthritis and alters cytokine networks in vivo. Br. J. Pharmacol. 2006, 149, 666–675. 10.1038/sj.bjp.0706912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Cui L.; Gonsiorek W.; Min S.-H.; Anilkumar G.; Rosenblum S.; Kozlowski J.; Lundell D.; Fine J. S.; Grant E. P. CCR2 and CXCR4 regulate peripheral blood monocyte pharmacodynamics and link to efficacy in experimental autoimmune encephalomyelitis. J. Inflammation 2009, 6, 32. 10.1186/1476-9255-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dairaghi D. J.; Zhang P.; Wang Y.; Seitz L. C.; Johnson D. A.; Miao S.; Ertl L. S.; Zeng Y.; Powers J. P.; Pennell A. M.; Bekker P.; Schall T. J.; Jaen J. C. Pharmacokinetic and pharmacodynamic evaluation of the novel CCR1 antagonist CCX354 in healthy human subjects: implications for selection of clinical dose. Clin. Pharmacol. Ther. 2011, 89, 726–734. 10.1038/clpt.2011.33. [DOI] [PubMed] [Google Scholar]
- Tak P. P.; Balanescu A.; Tseluyko V.; Bojin S.; Drescher E.; Dairaghi D.; Miao S.; Marchesin V.; Jaen J.; Schall T. J.; Bekker P. Chemokine receptor CCR1 antagonist CCX354-C treatment for rheumatoid arthritis: CARAT-2, a randomised, placebo controlled clinical trial. Ann. Rheum. Dis. 2013, 72, 337–344. 10.1136/annrheumdis-2011-201605. [DOI] [PubMed] [Google Scholar]
- Horuk R. Promiscuous drugs as therapeutics for chemokine receptors. Expert Rev. Mol. Med. 2009, 11, e1. 10.1017/S1462399409000921. [DOI] [PubMed] [Google Scholar]
- Sabroe I.; Peck M. J.; Van Keulen B. J.; Jorritsma A.; Simmons G.; Clapham P. R.; Williams T. J.; Pease J. E. A small molecule antagonist of chemokine receptors CCR1 and CCR3. Potent inhibition of eosinophil function and CCR3-mediated HIV-1 entry. J. Biol. Chem. 2000, 275, 25985–25992. 10.1074/jbc.M908864199. [DOI] [PubMed] [Google Scholar]
- Junker A.; Kokornaczyk A. K.; Zweemer A. J.; Frehland B.; Schepmann D.; Yamaguchi J.; Itami K.; Faust A.; Hermann S.; Wagner S.; Schafers M.; Koch M.; Weiss C.; Heitman L. H.; Kopka K.; Wunsch B. Synthesis, binding affinity and structure-activity relationships of novel, selective and dual targeting CCR2 and CCR5 receptor antagonists. Org. Biomol. Chem. 2015, 13, 2407–2422. 10.1039/C4OB02397H. [DOI] [PubMed] [Google Scholar]
- Cox B. D.; Prosser A. R.; Sun Y.; Li Z.; Lee S.; Huang M. B.; Bond V. C.; Snyder J. P.; Krystal M.; Wilson L. J.; Liotta D. C. Pyrazolo-piperidines exhibit dual inhibition of CCR5/CXCR4 HIV entry and reverse transcriptase. ACS Med. Chem. Lett. 2015, 6, 753–757. 10.1021/acsmedchemlett.5b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriconi A.; Cesta M. C.; Cervellera M. N.; Aramini A.; Coniglio S.; Colagioia S.; Beccari A. R.; Bizzarri C.; Cavicchia M. R.; Locati M.; Galliera E.; Di Benedetto P.; Vigilante P.; Bertini R.; Allegretti M. Design of noncompetitive interleukin-8 inhibitors acting on CXCR1 and CXCR2. J. Med. Chem. 2007, 50, 3984–4002. 10.1021/jm061469t. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Qin L.; Ortiz Zacarías N. V.; de Vries H.; Han G. W.; Gustavsson M.; Dabros M.; Zhao C.; Cherney R. J.; Carter P.; Stamos D.; Abagyan R.; Cherezov V.; Stevens R. C.; IJzerman A. P.; Heitman L. H.; Tebben A.; Kufareva I.; Handel T. M. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 2016, 540, 458–461. 10.1038/nature20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald C.; Rappas M.; Kean J.; Dore A. S.; Errey J. C.; Bennett K.; Deflorian F.; Christopher J. A.; Jazayeri A.; Mason J. S.; Congreve M.; Cooke R. M.; Marshall F. H. Intracellular allosteric antagonism of the CCR9 receptor. Nature 2016, 540, 462–465. 10.1038/nature20606. [DOI] [PubMed] [Google Scholar]
- Zweemer A. J.; Nederpelt I.; Vrieling H.; Hafith S.; Doornbos M. L.; de Vries H.; Abt J.; Gross R.; Stamos D.; Saunders J.; Smit M. J.; IJzerman A. P.; Heitman L. H. Multiple binding sites for small-molecule antagonists at the CC chemokine receptor 2. Mol. Pharmacol. 2013, 84, 551–561. 10.1124/mol.113.086850. [DOI] [PubMed] [Google Scholar]
- Ortiz Zacarias N. V.; Lenselink E. B.; IJzerman A. P.; Handel T. M.; Heitman L. H. Intracellular receptor modulation: novel approach to target GPCRs. Trends Pharmacol. Sci. 2018, 39, 547–559. 10.1016/j.tips.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweemer A. J.; Bunnik J.; Veenhuizen M.; Miraglia F.; Lenselink E. B.; Vilums M.; de Vries H.; Gibert A.; Thiele S.; Rosenkilde M. M.; IJzerman A. P.; Heitman L. H. Discovery and mapping of an intracellular antagonist binding site at the chemokine receptor CCR2. Mol. Pharmacol. 2014, 86, 358–368. 10.1124/mol.114.093328. [DOI] [PubMed] [Google Scholar]
- Dasse O.; Evans J.; Zhai H.-X.; Zou D.; Kintigh J.; Chan F.; Hamilton K.; Hill E.; Eckman J.; Higgins P.; Volosov A.; Collart P.; Nicolas J.-M.; Kondru R. K.; Schwartz C. E. Novel, acidic CCR2 receptor antagonists: lead optimization. Lett. Drug. Des. Discovery 2007, 4, 263–271. 10.2174/157018007784619989. [DOI] [Google Scholar]
- Peace S.; Philp J.; Brooks C.; Piercy V.; Moores K.; Smethurst C.; Watson S.; Gaines S.; Zippoli M.; Mookherjee C.; Ife R. Identification of a sulfonamide series of CCR2 antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 3961–3964. 10.1016/j.bmcl.2010.04.142. [DOI] [PubMed] [Google Scholar]
- Buntinx M.; Hermans B.; Goossens J.; Moechars D.; Gilissen R. A.; Doyon J.; Boeckx S.; Coesemans E.; Van Lommen G.; Van Wauwe J. P. Pharmacological profile of JNJ-27141491 [(S)-3-[3, 4-difluorophenyl)-propyl]-5-isoxazol-5-yl-2-thioxo-2, 3-dihydro-1H-imidazole-4-carboxyl acid methyl ester], as a noncompetitive and orally active antagonist of the human chemokine receptor CCR2. J. Pharmacol. Exp. Ther. 2008, 327, 1–9. 10.1124/jpet.108.140723. [DOI] [PubMed] [Google Scholar]
- Zou D.; Dasse O.; Evans J.; Higgins P.; Kintigh J.; Kondru R.; Schwartz E.; Knerr L.; Zhai H. X.. Pyrrolidinone Derivatives. U.S. Patent US6,727,275B2, 2004.
- Zou D.; Dasse O.; Evans J.; Higgins P.; Kintigh J.; Kondru R.; Schwartz E.; Knerr L.; Zhai H. X.. Pyrrolidinone Derivatives. U.S. Patent Application US2003/0149081A1, 2003.
- Kopp M.; Lancelot J. C.; Dallemagne P.; Rault S. Synthesis of novel pyrazolopyrrolopyrazines, potential analogs of sildenafil. J. Heterocycl. Chem. 2001, 38, 1045–1050. 10.1002/jhet.5570380506. [DOI] [Google Scholar]
- Liang M.; Mallari C.; Rosser M.; Ng H. P.; May K.; Monahan S.; Bauman J. G.; Islam I.; Ghannam A.; Buckman B.; Shaw K.; Wei G. P.; Xu W.; Zhao Z.; Ho E.; Shen J.; Oanh H.; Subramanyam B.; Vergona R.; Taub D.; Dunning L.; Harvey S.; Snider R. M.; Hesselgesser J.; Morrissey M. M.; Perez H. D.; Horuk R. Identification and characterization of a potent, selective, and orally active antagonist of the CC chemokine receptor-1. J. Biol. Chem. 2000, 275, 19000–19008. 10.1074/jbc.M001222200. [DOI] [PubMed] [Google Scholar]
- Zou D.; Zhai H.-X.; Eckman J.; Higgins P.; Gillard M.; Knerr L.; Carre S.; Pasau P.; Collart P.; Grassi J. Novel, acidic CCR2 receptor antagonists: from hit to lead. Lett. Drug. Des. Discovery 2007, 4, 185–191. 10.2174/157018007780077381. [DOI] [Google Scholar]
- Isberg V.; de Graaf C.; Bortolato A.; Cherezov V.; Katritch V.; Marshall F. H.; Mordalski S.; Pin J.-P.; Stevens R. C.; Vriend G.; Gloriam D. E. Generic GPCR residue numbers – aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31. 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettle J. G.; Faull A. W.; Barker A. J.; Davies D. H.; Stone M. A. N-benzylindole-2-carboxylic acids: potent functional antagonists of the CCR2b chemokine receptor. Bioorg. Med. Chem. Lett. 2004, 14, 405–408. 10.1016/j.bmcl.2003.10.049. [DOI] [PubMed] [Google Scholar]
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch C.; Leo A.; Taft R. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Chou C. C.; Fine J. S.; Pugliese-Sivo C.; Gonsiorek W.; Davies L.; Deno G.; Petro M.; Schwarz M.; Zavodny P. J.; Hipkin R. W. Pharmacological characterization of the chemokine receptor, hCCR1 in a stable transfectant and differentiated HL-60 cells: antagonism of hCCR1 activation by MIP-1β. Br. J. Pharmacol. 2002, 137, 663–675. 10.1038/sj.bjp.0704907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland C. T.; Salanga C. L.; Kawamura T.; Trejo J.; Handel T. M. The chemokine receptor CCR1 is constitutively active, which leads to G protein-independent, β-arrestin-mediated internalization. J. Biol. Chem. 2013, 288, 32194–32210. 10.1074/jbc.M113.503797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz H. Hill coefficients, dose-response curves and allosteric mechanisms. J. Chem. Biol. 2010, 3, 37–44. 10.1007/s12154-009-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez Arias D.; Navenot J.-M.; Zhang W.-b.; Broach J.; Peiper S. C. Constitutive activation of CCR5 and CCR2 induced by conformational changes in the conserved TXP motif in transmembrane helix 2. J. Biol. Chem. 2003, 278, 36513–36521. 10.1074/jbc.M303739200. [DOI] [PubMed] [Google Scholar]
- Lagorce D.; Sperandio O.; Baell J. B.; Miteva M. A.; Villoutreix B. O. FAF-Drugs3: a web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015, 43, W200–W207. 10.1093/nar/gkv353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagorce D.; Douguet D.; Miteva M. A.; Villoutreix B. O. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep. 2017, 7, 46277. 10.1038/srep46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Cormier M.; Chardon A.; Blanchet J.; Rouden J.; Maddaluno J.; De Paolis M. An organocatalytic access to spiro[4.5]decanes and spiro[4.6]undecanes containing aminolactones and 3-aminopyrrolidines. Synthesis 2015, 47, 2549–2553. 10.1055/s-0034-1380146. [DOI] [Google Scholar]
- Koz’minykh V.; Igidov N.; Zykova S.; Kolla V.; Shuklina N.; Odegova T. Synthesis and pharmacological activity of 3-hydroxy-1,5-diaryl-4-pivaloyl-2,5-dihydro-2-pyrrolones. Pharm. Chem. J. 2002, 36, 188–191. 10.1023/A:1019832621371. [DOI] [Google Scholar]
- Kraïem J. B.; Amri H. Concise synthesis of α-(hydroxymethyl) alkyl and aryl vinyl ketones. Synth. Commun. 2013, 43, 110–117. 10.1080/00397911.2011.592626. [DOI] [Google Scholar]
- Frank R.; Bahrenberg G.; Christoph T.; Schiene K.; De V. J.; Damann N.; Frormann S.; Lesch B.; Lee J.; Kim Y. S.. Substituted Aromatic Carboxamide and Urea Derivatives as Vanilloid Receptor Ligands. Patent WO2010/127855A1, 2010.
- Carpino P. A.; Sanner M. A.. Cannabinoid Receptor Ligands and Uses Thereof. Patent WO2007/020502 A2, 2007.
- Chonan T.; Tanaka H.; Yamamoto D.; Yashiro M.; Oi T.; Wakasugi D.; Ohoka-Sugita A.; Io F.; Koretsune H.; Hiratate A. Design and synthesis of disubstituted (4-piperidinyl)-piperazine derivatives as potent acetyl-CoA carboxylase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3965–3968. 10.1016/j.bmcl.2010.04.134. [DOI] [PubMed] [Google Scholar]
- Doyon J.; Coesemans E.; Boeckx S.; Buntinx M.; Hermans B.; Van Wauwe J. P.; Gilissen R. A.; De Groot A. H.; Corens D.; Van Lommen G. Discovery of potent, orally bioavailable small-molecule inhibitors of the human CCR2 receptor. ChemMedChem 2008, 3, 660–669. 10.1002/cmdc.200700276. [DOI] [PubMed] [Google Scholar]
- Smith P. K.; Krohn R. I.; Hermanson G. T.; Mallia A. K.; Gartner F. H.; Provenzano M. D.; Fujimoto E. K.; Goeke N. M.; Olson B. J.; Klenk D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Cheng Y.-C.; Prusoff W. H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Maestro Release 2017-1; Schrödinger, LLC: New York, 2017.
- Maestro Release 2017-2; Schrödinger, LLC: New York, 2017.
- Prime Release 2017-1; Schrödinger, LLC: New York, 2017.
- Jacobson M. P.; Friesner R. A.; Xiang Z.; Honig B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. 10.1016/S0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- Jacobson M. P.; Pincus D. L.; Rapp C. S.; Day T. J.; Honig B.; Shaw D. E.; Friesner R. A. A hierarchical approach to all-atom protein loop prediction. Proteins: Struct., Funct., Genet. 2004, 55, 351–367. 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Tan Q.; Zhu Y.; Li J.; Chen Z.; Han G. W.; Kufareva I.; Li T.; Ma L.; Fenalti G.; Li J.; Zhang W.; Xie X.; Yang H.; Jiang H.; Cherezov V.; Liu H.; Stevens R. C.; Zhao Q.; Wu B. Structure of the CCR5 chemokine receptor–HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaguar Release 2017-1: Schrödinger, LLC: New York, 2017.
- Bochevarov A. D.; Harder E.; Hughes T. F.; Greenwood J. R.; Braden D. A.; Philipp D. M.; Rinaldo D.; Halls M. D.; Zhang J.; Friesner R. A. Jaguar: a high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. 10.1002/qua.24481. [DOI] [Google Scholar]
- Glide Release 2017-1; Schrödinger Suite Prime 2017-1 Induced Fit Docking Protocol; Schrödinger, LLC: New York, 2017.
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.8; Schrödinger, LLC: New York, 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.