

Abstract
KCNE1 encodes a regulatory subunit of the KCNQ1 potassium channel-complex. Both KCNE1 and KCNQ1 are necessary for normal hearing and cardiac ventricular repolarization. Recessive variants in these genes are associated with Jervell and Lange-Nielson syndrome (JLNS1 and JLNS2), a cardio-auditory syndrome characterized by congenital profound sensorineural deafness and a prolonged QT interval that can cause ventricular arrhythmias and sudden cardiac death. Some normal-hearing carriers of heterozygous missense variants of KCNE1 and KCNQ1 have prolonged QT intervals, a dominantly inherited phenotype designated Romano-Ward syndrome (RWS), which is also associated with arrhythmias and elevated risk of sudden death. Co-assembly of certain mutant KCNE1 monomers with wild-type KCNQ1 subunits results in RWS by a dominant negative mechanism. This Mutation Update reviews variants of KCNE1 and their associated phenotypes, including biallelic truncating null variants of KCNE1 that have not been previously reported. We describe three homozygous nonsense mutations of KCNE1 segregating in families ascertained ostensibly for nonsyndromic deafness: c.50G>A (p.Trp17*), c.51G>A (p.Trp17*), and c.138C>A (p.Tyr46*). Some individuals carrying missense variants of KCNE1 have RWS. However, heterozygotes for loss of function variants of KCNE1 may have normal QT intervals while biallelic null alleles are associated with JLNS2, indicating a complex genotype-phenotype spectrum for KCNE1 variants.
Keywords: deafness, Jervell and Lange-Nielson syndrome, KCNE1, KCNQ1, prolonged-QT, Romano-Ward syndrome
1. BACKGROUND
Inherited predisposition to syncopal episodes and sudden death due to cardiac arrhythmias and arrest is a genetically heterogeneous disorder that can be transmitted as a recessive or dominant trait (Huikuri, Castellanos, & Myerburg, 2001; Katritsis, Gersh, & Camm, 2016; Roberts, 2006). Variants in at least 16 different genes are associated with an abnormal prolongation of ventricular repolarization known as long QT syndrome (LQTS1-LQTS15; MIM# 192500) (Alders & Christiaans, 1993; Grunert, Bodi, & Odening, 2017). LQTS has an incidence of approximately 1:2,000 births and occurs across various populations (Nakano & Shimizu, 2016; Priori et al., 2003; Priori et al., 2013; Splawski et al., 2000). Prolongation of the QT interval may also be one feature of a broader phenotype. A case in point is Jervell and Lange-Nielsen syndrome (JLNS, Long QT syndrome 1, LQTS type 1 MIM# 220400), which is inherited in an autosomal recessive manner and characterized by a prolonged QT interval and congenital, profound hearing loss (Jervell & Lange-Nielsen, 1957). JLNS type 1 is associated with biallelic pathogenic variants of KCNQ1, a gene discovered in a molecular genetic study of prolonged QT, which can be triggered by physical exertion (Neyroud et al., 1997). KCNQ1 encodes a slowly activating and slowly deactivating delayed-rectifier potassium channel (previous names are KvLQT1, Kv7.1, and KCNA8). KCNQ1 contributes to a potassium current that terminates the cardiac action potential (Priori et al., 2003; Wrobel, Tapken, & Seebohm, 2012). In the absence of a family history of a prolonged QT interval, the cardiac phenotype of a hearing or deaf individual may remain concealed until a syncopal episode occurs (Schwartz et al., 2001; Wang et al., 1996).
JLNS type 2 (MIM# 612347) is a less common cardio-auditory disorder that is also inherited as an autosomal recessive trait and associated with variants of KCNE1. The KCNE family of proteins has five members (KCNE1-KCNE5), all of which are small single-pass integral membrane proteins ranging in size from 103–177 residues. KCNE1 protein (previous names are LQT5, mink, and IsK) functions as a regulatory subunit of KCNQ1 (Schulze-Bahr et al., 1997; Splawski, Tristani-Firouzi, Lehmann, Sanguinetti, & Keating, 1997; Tranebjaerg, Samson, & Green, 1993; Tyson et al., 1997). KCNE1 co-assembles into a complex with KCNQ1, calmodulin and the lipid PIP2 (Sun & MacKinnon, 2017; Zaydman et al., 2013) and modulates the biophysical properties of the channel (Abbott, 2016; Jespersen, Grunnet, & Olesen, 2005; Sanguinetti et al., 1996; Wrobel et al., 2012). KCNE1 may also associate with other pore-forming alpha subunits, suggesting that the clinical manifestations of missense mutations of KCNE1 may not be limited to disruptions of currents ascribed to KCNQ1 (Abbott & Goldstein, 2002).
In the inner ear, the stria vascularis is an epithelial tissue that is involved in the formation and maintenance of the endocochlear potential of + 80 mV between the potassium-rich endolymph, a fluid that fills the scala media and bathes the apical surface of hair cells, and the perilymph which bathes the basolateral part of the hair cells (Figure 1A). Hair cells are the mechano-chemical transducers of sound. Normal strial function requires both KCNE1 and KCNQ1 and is required for hearing (Kang et al., 2008). To date, all reported variants of KCNE1 and KCNQ1 that are associated with deafness are recessive; carriers of heterozygous pathogenic variants of KCNE1 and KCNQ1 have normal hearing. Thus, the loss of function of KCNE1 in the inner ear is not compensated by any of the four KCNE1 paralogs (KCNE2-KCNE5) (McCrossan & Abbott, 2004). In contrast, individual carriers of some pathogenic variants of KCNE1 or KCNQ1, as well as variants of other LQTS-associated genes, may have prolongation of the QT interval, an autosomal dominant congenital disorder designated Romano-Ward syndrome (RWS, MIM# 192500) without any known inner ear phenotype (Romano, 1965; Ward, 1964) (Table 1). A dominant negative mechanism was demonstrated for RWS-associated variants of KCNQ1 and KCNE1. In a heterozygote, wild type and defective monomers can co-assemble to form an impaired KCNQ1 potassium channel complex, which can result in a prolonged QT interval (Schmitt et al., 2000; Splawski et al., 1997).
FIGURE 1.
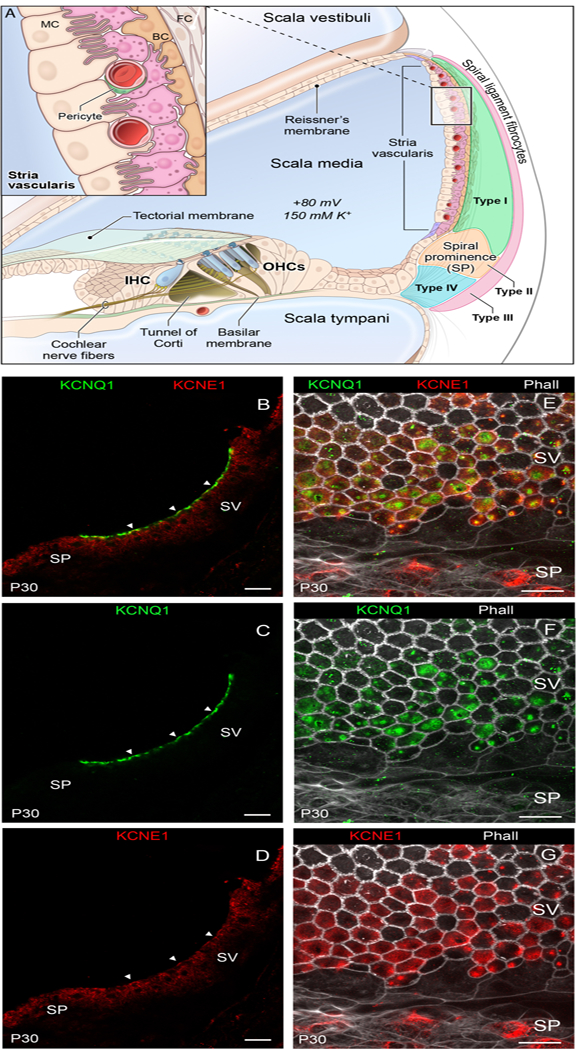
Localization of KCNQ1 and KCNE1 in the stria vascularis. A: Schematic cross section of the cochlear duct showing the position of the stria vascularis located in the lateral wall of the cochlea and the organ of Corti, showing outer hair cells (OHCs), which can amplify incoming sound waves, and inner hair cells (IHCs), which are responsible for the mechano-chemical transduction of sound. Inset depicts relationship between the major cell types of the stria vascularis that include the marginal cells (MC) facing the scala media, intermediate cells (IC) and basal cells (BC). B-D: Fluorescent immunohistochemistry of adult day 30 (P30) mouse mid-modiolar cross-sections depicting co-localization in the overlay (B) of KCNQ1 (green) (C) and KCNE1 (red) (D) to the luminal surface of the marginal cell facing the endolymph. Arrowheads point to examples of co-localization. E-G: Fluorescent immunohistochemistry of whole mounts of adult mouse stria vascularis depicting co-localization in the overlay (E) of KCNQ1 (green) (F) and KCNE1 (red) (G) on the luminal surface of the marginal cells. Spiral prominence (SP), stria vascularis (SV), phalloidin (Phall). Scale bars correspond to 20 micrometers in all panels.
Table 1.
Previously described and novel KCNE1 genetic variants
| No. | Protein variant | CADD score | Zygosity | Ascertainment phenotype | Country of origin | References |
|---|---|---|---|---|---|---|
| 1 | p.Leu3fs*4 | np | long QT | America | Kapplinger et al. (2009) | |
| 2 | p.Ser4fs* | np | long QT | America | Kapplinger et al. (2009) | |
| 3 | p.Asn5fs* | heterozygous | long QT | America | GeneDx | |
| 4 | p.Thr7Ile | 23.6 | compound heterozygous w/p.Asp76Asn | JLNS | Lebanon, North America, Europe | Schulze-Bahr et al. (1997); Splawski I et al. (2000) |
| 5 | p.Ala8Val | 0.807 | heterozygous | long QT | Japan | Ohno et al. (2007);Kapplinger et al. (2009) |
| 6 | p.Thr10Met | 0.015 | np | long QT | America | Kapplinger et al. (2009);Ng et al. (2013) |
| 7 | p.Leu16Pro | 13.12 | np | long QT | Japan | Yoshinaga et al. (2014) |
| 8 | p.Trp17* | homozygous | deafness, QT not evaluated | Pakistan | Current study; families lost to follow-up | |
| 9 | p.Trp17* | np | long QT | America | Kapplinger et al. (2009) | |
| 10 | p.Thr20Ile | 2.208 | heterozygous | long QT | France | Millat et al. (2009) |
| 11 | p.Gly25Val | 1.105 | heterozygous | long QT | Denmark | Olesen et al. (2012) |
| 12 | p.Ser28Leu | 24 | np | long QT | America | Kapplinger et al. (2009);Shim et al. (2005); Obeyesekere et al.(2012);Lieve et al. (2013) |
| 13 | p.Arg32His | 23 | np | long QT | America, North America, Europe | Kapplinger et al. (2009);Splawski I et al. (2000) |
| 14 | p.Arg36His | 6.212 | np | long QT | Italy | Napolitano et al. (2005) |
| 15 | p.Ser38Gly | 0.003 | heterozygous | sudden unexplained nocturnal death/noise induced HL | Netherlands, Germany, Canada China, Taiwan, Italy, Poland, America |
Paulussen et al. (2004);Van Laer et al. (2006);Han et al. (2014);Wugeti et al. (2015);Chang et al. (2014); |
| 16 | p.Tyr46* | homozygous | deafness, variable long QT |
Pakistan | Current study | |
| 17 | p.Val47Phe | 27.2 | compound heterozygous with p.Leu51His | long QT | North America, Europe, America | Splawski I et al. (2000); Bianchi et.al (1999) |
| 18 | p.Val47Ile | 10.08 | np | long QT (RWS) | African American, Caucasian | Ryan et al. (2012) |
| 19 | p.Leu51His | 35 | np | JLNS | North America, Europe, America | Splawski I et al. (2000); Bianchi et.al (1999) |
| 20 | p.Gly52Arg | 25.2 | heterozygous | long QT | China | Ma et al. (2003) |
| 21 | p.Phe53Ser | 24.9 | np | atrial fibrillation, early-onset | Italy | Napolitano et al. (2005) |
| 22 | p.Phe54Val | 24.3 | heterozygous | long QT | China | Liu et al. (2013) |
| 23 | p.Gly55Ser | 24.8 | np | long QT | America | Kapplinger et al. (2009) |
| 24 | p.Thr58Pro | 26.6 | np | long QT | America | Kapplinger et al. (2009) |
| 25 | p.Thr58Ile | heterozygous | long QT | GeneDx | ||
| 26 | p.TL58–59 PP# | np | JLNS | North America, Europe |
Splawski I et al. (2000) |
|
| 27 | p.Leu59Pro | 27.2 | np | JLNS | America | Kapplinger et al. (2009) |
| 28 | p.Gly60Asp | 29 | heterozygous | long QT | Denmark | Christiansen et al. (2014);Olesen et al. (2012) |
| 29 | p.Arg67Cys | 34 | np | long QT | America | Kapplinger et al. (2009) |
| 30 | p.Arg67His | 34 | np | long QT | America | Kapplinger et al. (2009);Lieve et al. (2013) |
| 31 | p.Arg67Leu | heterozygous | long QT | America | GeneDx | |
| 32 | p.Lys70Asn | 24.4 | heterozygous | long QT, >440ms | America | Lai et al. (2005) |
| 33 | p.Lys70Met | 27.3 | np | long QT (RWS) | America | Kapplinger et al. (2009) |
| 34 | p.His73Tyr | heterozygous | long QT | America | GeneDx | |
| 35 | p.Ser74Leu | 27.4 | np | long QT | North America, Europe | Splawski I et al. (2000) |
| 36 | p.Asn75fs*34 | np | long QT | America | Kapplinger et al. (2009) | |
| 37 | p.Asp76Asn | 24.2 | compound heterozygous with p.Thr7Ile | long QT | Lebanon, America, Denmark |
Schulze-Bahr et al. (1997); Splawski I et al. (2000);Lieve et al. (2013);Kapplinger et al. (2009);Christiansen et al. (2014) |
| 38 | p.Val80Ile | 11.48 | heterozygous | long QT | Spain, Finland | Riuró et al. (2015);Hietikko et al. (2012) |
| 39 | p.Tyr81Cys | 23.9 | heterozygous | long QT | Taiwan | Lai et al. (2005) |
| 40 | p.Ile82Phe | heterozygous | long QT | Korea | Chae (2017) Clin Chim Acta, 464:128–135 | |
| 41 | p.Glu83Lys | 23.8 | np | long QT | America, Denmark | Ghouse et al. (2015);Kapplinger et al. (2009) |
| 42 | p.Asp85Asn | 21.8 | np | long QT | Netherlands, Germany, Canada, Sweden | Paulussen et al. (2004);Van Laer et al. (2006);Yoshinaga et al. (2014) |
| 43 | p.Trp87Arg | 24 | heterozygous | long QT | North America, Europe, Finland, America | Splawski I et al. (2000);Hietikko et al. (2012); Bianchi et.al (1999) |
| 44 | p.Gln88* | heterozygous | long QT | America | GeneDx | |
| 45 | p.Asp91Glu | 22.3 | heterozygous | long QT | South Africa | Hedley et al. (2013) |
| 46 | p.Ala93Thr | 16.3 | heterozygous | long QT (RWS) | Denmark, Japan | Christiansen et al. (2014) |
| 47 | p.Arg98Trp | 28.1 | heterozygous | long QT (RWS) | North America, Europe, America, Japan | Splawski I et al. (2000);Ng et al. (2013);Ohno et al. (2007) |
| 48 | p.Val109Ile | 0.004 | heterozygous | long QT | Germany, Denmark | Schulze-Bahr et al. (2001);Ghouse et al. (2015) |
| 49 | p.Gln117* | np | long QT | America | Kapplinger et al. (2009) | |
| 50 | p.Thr125Met | 0.006 | np | long QT | America | Kapplinger et al. (2009); Cuenca et al. (2015) |
| 51 | p.Pro127Thr | 11.18 | np | long QT (RWS) | North America, Europe | Splawski I et al. (2000); Harmer et.al (2014) |
np = not provided
= nomenclature as reported in Splawski et al. (2000)
The exact nomenclature for TL58–59PP as reviewed by Mutalyzer is NM_000219.5: p.(Thr58_Leu59delinsProPro)
TL58–59PP reported by Tyson et al.,1997 was intended to indicate that prolines replaced threonine and leucine at residues 58 and 59.
This Mutation Update is focused on variants of KCNE1 and the associated cardiac and auditory phenotypes, and it complements more expansive reviews of inherited and acquired long QT syndromes (Roden & Viswanathan, 2005; Tranebjaerg, Samson, & Green, 2017; Tyson et al., 2000). Here, we spotlight the current knowledge of variants of KCNE1 associated with isolated long QT of RWS as well as the hearing loss and prolonged-QT of JLNS2. We also describe three previously unreported homozygous recessive, protein-truncating variants of KCNE1 segregating in consanguineous families ascertained for severe to profound deafness, indicating that KCNE1 should be included in screening panels for variants of genes reported to be associated with deafness. Lastly, the data reviewed here underscore the importance of a molecular genetic diagnosis, genetic counseling and a cardiologic assessment of the QT-interval for risk of sudden death for deaf individuals, as well as their heterozygous parents and normal-hearing relatives.
2. METHODS
The three families in this study (PKDF461, 4410 and 4502) were ascertained in Pakistan. Prior to inclusion, all persons in this study gave their written informed consent to participate in this study. For the protection of human subjects, IRB approvals for this study were obtained from the (1) National Centre of Excellence in Molecular Biology at the University of the Punjab and (2) Combined Neurosciences Blue Panel at the National Institutes of Health (OH-93-N-016 to TBF) for family PKDF461 members, and (3) Quaid-i-Azam University (IRB-QAU-153 to WA) and (4) Baylor College of Medicine and Affiliated Hospitals (H-17566 to SL) for members of families 4410 and 4502. At the initial time of ascertainment, deaf and hearing members of family PKDF461 did not report syncopal episodes and there were no sudden, unexplained deaths or other disorders, suggesting a nonsyndromic form of hearing loss. For family PKDF461 (Figure 2A), as an initial screen, at least three microsatellite markers were genotyped per locus to detect possible linkage to one of the reported deafness loci. Subsequently, an additional nineteen chromosome 21q markers were genotyped to refine an interval on chromosome 21q22.12 to 17.6 Mb, including 127 annotated genes and pseudogenes. The exons and flanking intronic DNA were Sanger sequenced for CLDN14 and TMPRSS3, two genes associated unambiguously with congenital nonsyndromic sensorineural hearing loss (Scott et al., 2001; Wilcox et al., 2001), as well as TSPEAR, a third gene also located in this 21q interval, but with a debatable association with deafness (Delmaghani et al., 2012; Peled et al., 2016) (Figure 2B). Because no likely pathogenic variants were uncovered in the coding sequence of these three chr21q-linked genes, a genomic DNA sample from affected individual V:4 of family PKDF461 was submitted to the New York Genome Center (NYGC) for whole genome sequencing at a mean genome-wide coverage of 30X depth. Sequence reads were mapped against the hg38 human reference sequence using Novoalign, with indel realignment, recalibration of base quality scores and variant calling was performed using a version of the Genome Analysis Toolkit (GATK) best practices pipeline. Variant annotation was performed with Annovar. Structural variant calls were generated using GenomeSTRiP (Genome STRucture in Populations). Alignments supporting individual variant calls were manually inspected using the Integrative Genomics Viewer (IGV). VCF files were imported into Ingenuity Variant Analysis (IVA, Qiagen) for further downstream analyses. For families 4410 and 4502, whole-exome sequencing was performed using the Roche NimbleGen SeqCap EZ Human Exome Library v.3.0, followed by 70 bp paired-end sequencing on a HiSeq2500/4000 instrument (Illumina Inc, San Diego, CA, USA). Reads were aligned to the hg19 human reference genome using the Burrows-Wheeler aligner (BWA). PCR duplicate removal and indel realignment was performed with Picard and GATK. Variants were called and recalibrated with GATK and were annotated with dbNSFP and ANNOVAR. Novel variants identified in this study were submitted to ClinVar (SCV000778831, SCV000778832 and SCV000778833).
FIGURE 2.
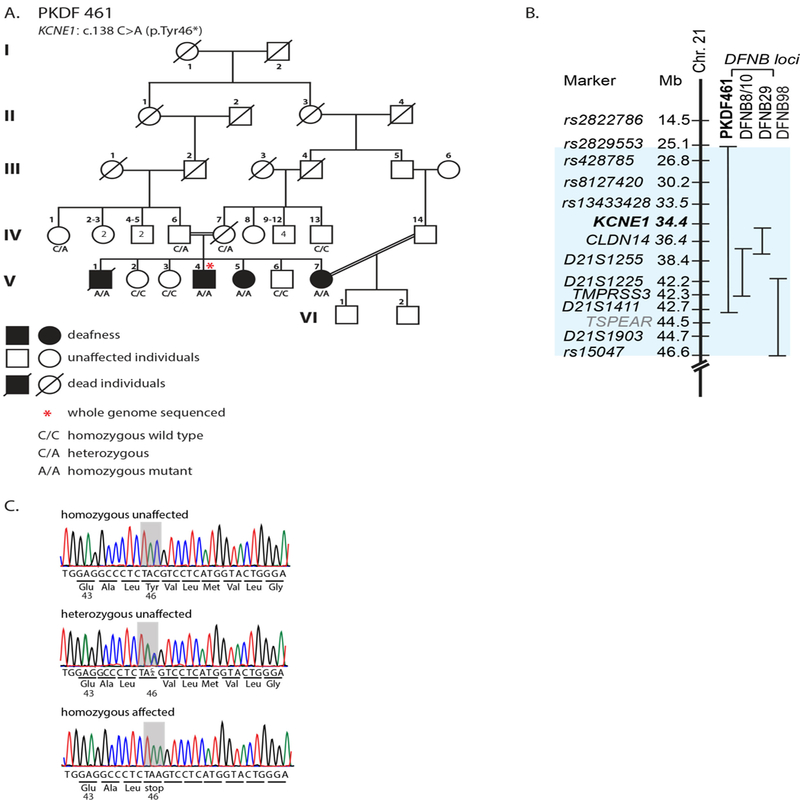
A: Pedigree of family PKDF461 segregating autosomal recessive hearing loss. A genomic DNA sample from individual V:4 was submitted for whole genome sequencing. Deaf male V:1 was said to have died from pulmonary tuberculosis at the age of twenty-two years. In retrospect, cardiac arrest may have been overlooked as clinicians at the time of his death were unaware of his KCNE1 genotype. There have been no reported syncopal episodes among the deaf individuals or p.Tyr46*carriers. A double horizontal line in the pedigree indicates a consanguineous union. B: Chromosome 21q linkage intervals indicate the locations of two previously reported recessive deafness loci, DFNB8/10 and DFNB29 and the location of the corresponding genes TMPRSS3 and CLDN14, respectively, relative to the location of KCNE1. The evidence supporting a recessive variant (c.1726_1728delGTCinsTT) of TSPEAR (light blue) associated with deafness (Delmaghani et al. 2012) has been called into question. Homozygosity for this variant was found in normal hearing individuals (Peled et al., 2016). C: Representative chromatograms of the genomic DNA sequences of three family members (V:2, IV:6 and V:4) who are homozygous for the reference allele, and heterozygous and homozygous for the KCNE1 c.138C>A variant. The affected codon is shaded in gray.
Nearly all of the variants of KCNE1 reported in peer-reviewed articles or databases were identified in subjects ascertained on the basis of a prolonged QT interval. Until the advent of massively parallel sequencing of the human exome and genome, clinicians relied on clinical clues for diagnoses. A small family segregating nonsyndromic hearing loss would most likely not have had further molecular genetic analyses. Since profound congenital deafness is an obvious phenotype, when the hearing status of a subject with long QT was not mentioned, we have assumed for this Mutation Update that hearing was within the normal range. For the majority of variants of KCNE1 reported to be pathogenic for long QT interval, the genotype or mode of inheritance was not specified. When clarification of the genotype was sought but not obtained from a communicating author, here we assumed that a subject was heterozygous for the variant and have classified the phenotype as RWS (Table 1).
The QT interval, measured by electrocardiogram (ECG) from the beginning of the QRS complex to the end of the T wave, represents ventricular depolarization and repolarization. The measurement of the QT interval may be either automated or manual, in the latter case performed by the physician using the individual lead with the longest interval. Several formulas have been proposed for heart rate correction of the QT interval. A widely used correction for heart rate is the Bazett’s formula that divides the measured QT by the square root of the RR interval (Bazett, 1920). The Fridericia’s formula divides the QT interval by the cube root of the RR interval instead, providing a more accurate correction for both elevated heart rate due to exertion and normal variation in resting heart rate (Friderica, 1920). Additionally, normal values for corrected QT (QTc) depend on gender and age. The following are the upper limits of a normal QT interval: women, 460 ms, men, 450 ms and children, 440 ms (Wagner et al., 2009). In our study, the QT interval was calculated manually from the start of the QRS complex to the end of the T wave which was determined by using the isoelectric baseline method. ECG tracings have a baseline that represents resting membrane potential and is referred to as the isoelectric line (Al-Khatib, LaPointe, Kramer, & Califf, 2003). Three consecutive QT intervals were measured and an average QT interval determined. The QTc interval was calculated using both Bazett and Fridericia formulas.
3. KCNE1 GENE STRUCTURE and FUNCTION
The human KCNE1 gene has seven annotated exons but only one is protein-coding (Figure 3A). Possible divergent biological properties and functions of the five transcription start sites of human KCNE1 remain unexplored (Lundquist et al., 2005). KCNE1 is a protein of only 129 residues (14.7 kDa) also known as MinK or IsK, which functions as a modulatory β-subunit of KCNQ1 (Chandrasekhar et al., 2011; Lvov, Gage, Berrios, & Kobertz, 2010). The extracellular N-terminus of KCNE1 has a conserved N-linked glycosylation site that mediates trafficking to the cell surface (Bas et al., 2011). Residues 44 to 66 of KCNE1 are predicted by TMHMM (Transmembrane Helix Prediction) to form a single-pass transmembrane domain, and the remainder of the protein extends into the cytosol (Figure 3B).
FIGURE 3.
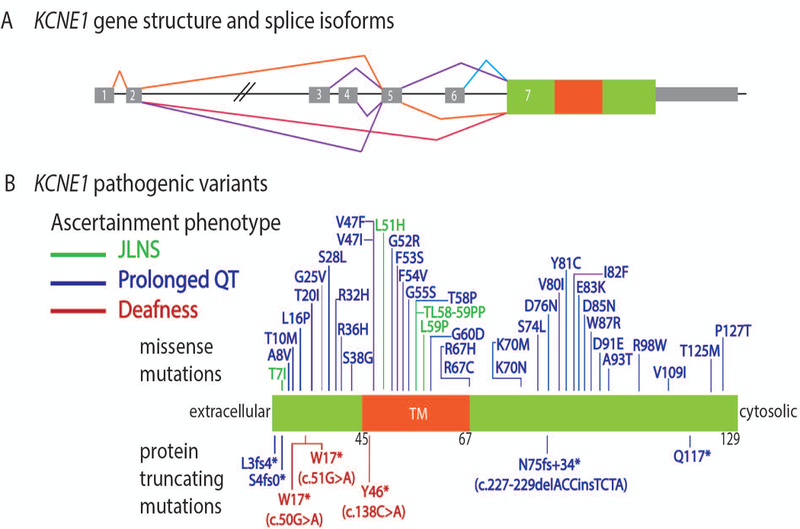
Schematic representation of the KCNE1 gene structure and the variants in patients that were ascertained initially with Jervell and Lange-Nielsen (JLNS), only prolonged QT or only nonsyndromic deafness because a cardiac abnormality was not suspected initially. A: KCNE1 gene structure showing alternative splice isoforms and the single protein coding exon 7. Gray rectangles are 5’ untranslated region (UTR) and 3’ UTR sequence. B: Variants of KCNE1 discovered in subjects ascertained for JLNS (green font), prolonged QT (blue font) or deafness (red font). The three novel homozygous truncating variants identified in this study are highlighted in red font. The location of residues 45 to 67 for the single-pass transmembrane domain (TM) is shown in orange.
KCNE1 subunits together with a KCNQ1 homo-tetramer form a slowly activating and slowly deactivating delayed-rectifier channel, which conducts potassium currents for myocardial repolarization (Jiang et al., 2009; Strutz-Seebohm et al., 2011). Each KCNQ1 subunit is 74.7 kDa (676 residues) and has six-transmembrane domains (Sun & MacKinnon, 2017). Transmembrane helices S1 to S4 are voltage sensors (Tombola, Pathak, & Isacoff, 2006) while S5 and S6 segments form the central pore domain (Sun & MacKinnon, 2017). Each of the four subunits of KCNQ1 has the potential to bind a KCNE1 subunit but the exact stoichiometry in vivo, that is whether two, three or four KCNE1 monomers are bound, remains unsettled (Murray et al., 2016; Nakajo, Ulbrich, Kubo, & Isacoff, 2010; Osteen et al., 2010). Coexpression of KCNE1 with KCNQ1 increases the outward current amplitude through KCNQ1 by one order of magnitude and also slows its activation by one order of magnitude (Sanguinetti et al., 1996).
The messenger RNAs for all five KCNE paralogs, including KCNE1, are detected in the heart (Lundquist et al., 2005). In vitro, all five of the KCNE proteins can individually complex with KCNQ1 tetramers and, depending on the cell type, modulate differently the gating kinetics, channel conductance and stability of this flexible potassium channel (Liin et al., 2015; Mazhari, Nuss, Armoundas, Winslow, & Marban, 2002; Murray et al., 2016; Nakajo & Kubo, 2015; Nakajo et al., 2010). In cardiomyocytes, channel gating of KCNQ1 is voltage-dependent. However, in the inner ear, KCNQ1 activation is voltage-independent, a switch in state that may depend on the interaction of the channel complex with membrane lipids (Liin et al., 2015; Zaydman et al., 2013). Whether one of the other four KCNE1 paralogs can partner with KCNQ1 in the heart and partially compensate for the loss of KCNE1 protein is unknown (Osteen et al., 2010).
4. VARIANTS OF KCNE1
The nomenclature for DNA and protein sequence variants of KCNE1 described here refers to accession numbers NM_000219.5 and NP_000210.2. Structural models and biochemical, physiological and genetic analyses have collectively revealed the critical importance of KCNE1 for cardiac and inner ear functions (Abbott, 2016). Although heterozygous pathogenic variants can be associated with RWS, usually there is a normal QT interval when the allele results in a loss of function (LOF).
The reported likely pathogenic variants of KCNE1 associated with JLNS2 and RWS all reside in its single protein-coding exon 7. Noncoding variants of KCNE1 have not yet been reported and would be experimentally challenging to associate with JLNS or RWS even if whole genome sequencing data were available. To date, only four variants of KCNE1 have been associated with Jervell and Lange-Nielson syndrome while 44 different, presumably heterozygous variants of KCNE1 are associated with RWS, nonsyndromic prolongation of the QT with normal hearing. A sudden unexplained nocturnal death was associated with one variant of KCNE1 (Figure 3B and Table 1). Forty-two of the reported 49 variants of KCNE1 associated with RWS are missense alleles, and seven are heterozygous truncating variants (Table 1). Not all protein-truncating variants are necessarily a total loss-of-function, as a premature translation termination in the single protein coding exon of KCNE1 probably does not result in nonsense mediated decay (NMD) of the mutant mRNA. Additionally, in reporting the variants and cardiac phenotype, many authors have not explicitly stated the diploid genotype (Table 1), nor have they comprehensively screened for and ruled out mutations in other long QT-associated genes. In this Update we assume that the truncating variants p.Trp17*and p.Gln117* of KCNE1 (Kapplinger et al., 2009), for example, were identified in heterozygous state and thus the phenotypic classification is RWS. This observation contrasts with our findings for family PKDF461, in which the two individuals heterozygous for a truncating variant of KCNE1 have QT intervals within the normal range (Table 2), suggesting that the cardiac phenotype of individuals carrying heterozygous truncating alleles cannot at present be reliably predicted from the genotype.
TABLE 2.
KCNE1 genotypes and QT measurements from electrocardiograms of PKDF461 family members
| Individual Number | Age (Yrs) |
Gender | KCNE1: p.Tyr46* | QT (msec) | HR (bpm) | QTcB (msec) |
QTcF (msec) |
|---|---|---|---|---|---|---|---|
| IV-6 | 49 | Male | +/− | 410 | 63 | 420 | 417 |
| IV-1 | 45 | Female | +/− | 400 | 78 | 456 | 437 |
| V-2 | 15 | Female | +/+ | 360 | 111 | 490 | 442 |
| V-5 | 13 | Female | −/− | 460 | 73 | 507 | 491 |
|
V-4 (Resting) |
18 | Male | −/− | 480 | 66 | 503 | 495 |
|
V-4 (under exercise) |
18 | Male | −/− | 360 | 119 | 507 | 452 |
| V-6 | 20 | Male | +/+ | 400 | 80 | 462 | 440 |
| V-7 | 28 | Female | −/− | 430 | 87 | 518 | 487 |
+ is the wild type allele, - is the p.Tyr46* nonsense mutation
B = Bazett’s formula, F = Fridericia’s formula
upper limit for normal range QT interval for women = 460 ms, man = 450 ms and for a child = 440 ms
Several heterozygous variants of KCNE1 or biallelic variants are of uncertain significance (VUS) and have been identified in individuals with RWS or JLNS (Supp. Table S1). As LQTS is genetically heterogeneous and not all LQTS-associated genes have been identified, a comprehensive cardiac examination alone cannot unequivocally establish a causal connection between a variant and phenotype. Aside from possible suppressing or enhancing modifiers in the genetic background of a subject, presently there is no uncomplicated biochemical test of KCNE1 function as the beta subunit of KCNQ1 that could suggest if a variant is sufficiently damaging to be disease-causing (Kapplinger et al., 2009; Obeyesekere et al., 2012; Sy et al., 2011). Presently it is unclear how much residual activity of KCNE1 in association with KCNQ1 is sufficient to not be disease-causing. Algorithms that consider evolutionary conservation of a residue and computational structural models can be used to estimate the functional consequences of KCNE1 variants (Kircher et al., 2014), but such predictions are not conclusive without experimental validation.
No pathogenic variants of KCNE1 associated with RWS or JLNS have been reported to be at high frequency in a particular population or ethnic group. However, a few pathogenic alleles or VUSs of KCNE1 are recurrent, having been reported in more than one unrelated individual. Heterozygosity for p.Thr10Met was reported in six RWS individuals with prolonged QT >480 ms (Kapplinger et al., 2009). The allele frequency of p.Thr10Met is 0.0002 in gnomAD, a database that, as of 2018, includes 125,748 and 15,708 genomes.
The KCNE1 p.Ser38Gly substitution is a common variant of doubtful pathogenicity. Serine 38 is designated as the wild type residue in UCSC Genome Browser http://genome.ucsc.edu/ and has overall frequency 0.36 in gnomAD, whereas the alternate glycine residue has frequency 0.64. Gly38 was associated with atrial fibrillation in a Chinese cohort of 108 patients (Lai et al., 2002), an observation that was replicated in the Chinese Han, indicating that “G38S” is a risk factor for atrial fibrillation (Yao, Ma, Xie, Liu, & Chen, 2012). In two other reports, p.Ser38Gly was labeled as “p.Gly38Ser”. Heterozygotes for p.Ser38Gly were reported in ten subjects with a long QT interval of ≥ 450 ms (Table 1), though with a minor allele frequency of 0.36, heterozygotes are the most common genotype in the population, assuming Hardy-Weinberg equilibrium. The p.Ser38Gly variant was also reported as a common polymorphism in a cohort from newborn hearing screening (Chang et al., 2014). Additionally, there is an aspartic acid at the homologous position in mouse, chimp, rhesus, rat, guinea pig and dog, which indicates a low level of evolutionary conservation of the serine at the 38th residue. Taken together, p.Ser38Gly appears to be a benign polymorphism.
In contrast, the KCNE1 recurrent variant p.Asp76Asn was reported in nine unrelated long-QT (440–480 ms) patients from the United States where LQTS diagnostic criteria is based on Schwartz-Moss score, where a score of more than four suggests high probability of LQTS (Kapplinger et al., 2009) and in twelve other subjects with long QT (Christiansen et al., 2014; Kapplinger et al., 2009; Lieve et al., 2013; Schulze-Bahr et al., 1997; Splawski et al., 1997). The p.Asp76Asn variant is likely to be pathogenic based upon electrophysiological data (Supp. Table S2). Compared to the wild type residue, the KCNE1 p.Asp76Asn variant reduces KCNQ1 current density as voltage dependence shifts to positive potentials, and there is a significant reduction of channel unitary conductance (Abbott & Goldstein, 2002). Genetic evidence also supports the conclusion that p.Asp76Asn is pathogenic. Its frequency in gnomAD is 6.856e-5, with 19 reported heterozygotes, mostly of European ancestry, and no homozygotes. Additionally, in compound heterozygosity with p.Thr7Ile, the p.Asp76Asn variant was reported in a JLNS patient that showed a prolongation of the QTc interval (470–520 msec) and congenital bilateral hearing loss (Schulze-Bahr et al., 1997).
Here we report three different homozygous nonsense mutations of KCNE1 that segregate with congenital hearing loss in three consanguineous Pakistani families. The PKDF461 family (Figure 2A), when ascertained, was considered to be segregating nonsyndromic hearing loss; other clinically relevant signs or symptoms were not evident from discussions with family members. Brief histories and physical examinations uncovered no clinically significant findings other than profound hearing loss. Three individuals (V:4, V:5 and V:7, Figure 4) all had profound, bilateral sensorineural hearing loss. In contrast, the audiograms for individuals with heterozygous mutations as described below show either normal hearing sensitivity (IV:1) or moderate sensorineural hearing loss with a pattern consistent with this individual’s history of noise exposure as a drummer (IV:6). Audiograms of wild type individuals (no mutations of KCNE1) show either normal hearing (V:6) or asymmetric hearing loss that is questionably sensorineural or mixed, and of unknown etiology (V:2). Together, these data indicate that profound congenital deafness is segregating as a recessive trait in family PKDF461.
FIGURE 4.
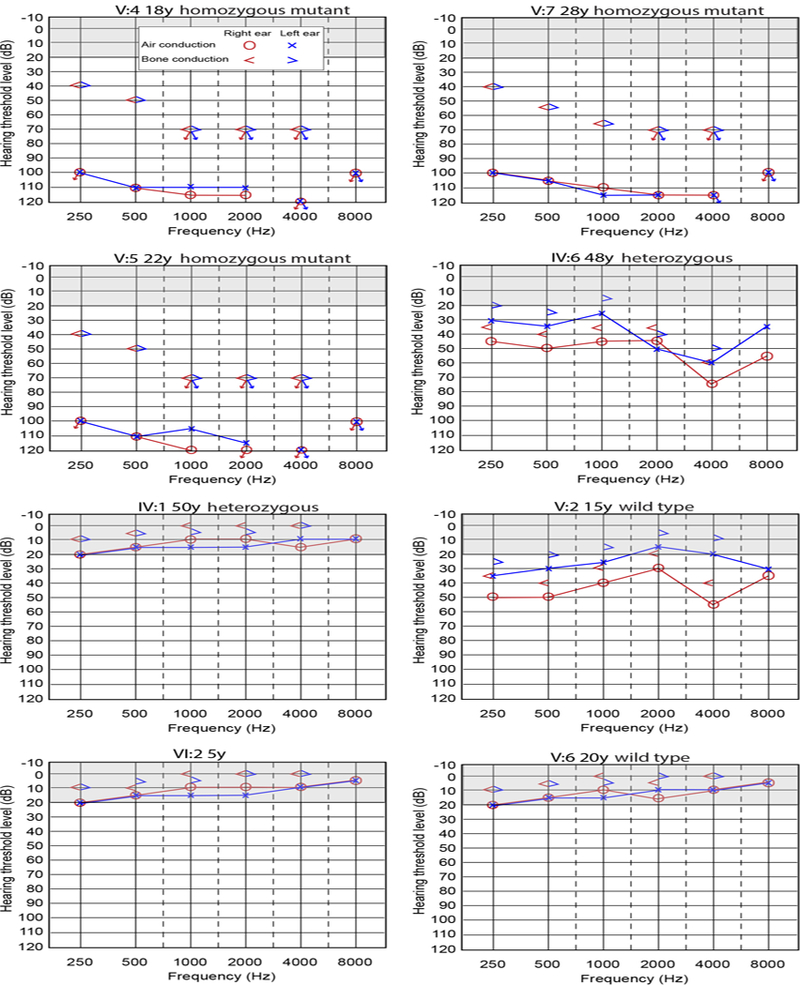
Audiometric data from members of family PKDF461 Audiograms from individual V:4, V:5 and V:7 indicate profound, bilateral sensorineural hearing loss. Audiograms from individuals V:6, VI:2, and IV:1 indicate normal hearing. Audiogram of individual IV:6, a forty-eight year-old drummer, shows increased hearing threshold in the frequency range of 3–6 kHz suggestive of noise-induced hearing loss (NIHL) (Halevi-Katz, Yaakobi, & Putter-Katz, 2015). Audiogram of individual V:2 indicates a mild low frequency hearing loss in the left ear and a mild to moderate hearing loss in the right ear, of unknown etiology. Arrows indicate no response at maximum audiometer output levels. Bone conduction thresholds for V:5 and V:7 at 250 and 500 Hz are considered responses to vibrotactile sensation and not an auditory sensation. Gray shaded region in the audiograms represent normal hearing range.
Using twenty-two microsatellite markers on chromosome 21q, we genetically fine mapped the deafness segregating in family PKDF461. Microsatellite D21S1411 is the distal boundary while SNP rs2829553 is the proximal linkage boundary of a 17.6 Mb homozygous interval shared among the four affected individuals of family PKDF461 (Figure 2B). This interval includes two previously reported nonsyndromic deafness loci, DFNB8/B10 (TMPRSS3) (Veske et al., 1996) and DFNB29 (CLDN14) (Ben-Yosef et al., 2001; Wilcox et al., 2001) and a disputed third deafness locus DFNB98 (TSPEAR) (Delmaghani et al., 2012; Peled et al., 2016). Sanger sequencing of all annotated exons of TMPRSS3, CLDN14 and TSPEAR did not reveal a coding or splice site variant that might explain the hearing loss segregating in family PKDF461. However, a homozygous noncoding regulatory variant disrupting transcription or splicing of one of these three genes could not be ruled out. Next, we performed whole genome sequencing for affected person V:4. Filtering for rare homozygous variants across the linkage interval, we identified a homozygous nonsense allele in KCNE1 (c.138C>A, p.Tyr46*). We confirmed by Sanger sequencing that this variant was co-segregating with deafness in all available family members (Figure 2C). Given the role of variants of this gene in JLNS-associated hearing loss, the assumption that the family was segregating nonsyndromic deafness was reconsidered.
The family members did not report a single syncopal episode among either deaf individuals or obligate heterozygous parents and their relatives. Twelve-lead electrocardiograms (ECGs) at standard settings of speed and amplitude were obtained in Pakistan. The ECGs were then evaluated by a cardiologist (A.B.) at the Clinical Center, National Institutes of Health in Bethesda, Maryland. In family PKDF461, homozygous p.Tyr46* deaf individuals at rest had QTcF intervals ranging from 487 to 495 ms and QTcB interval ranging from 503 to 518 ms with heart rate ranging from 66 to 87 beats per minute (bpm) (Table 2), putting them in the long QT range according to FDA consensus criteria (Nakano & Shimizu, 2016; Tranebjaerg et al., 2017). The two affected females, V:5 and V:7, had QTcF measurements of 491 and 487, and QTcB of 507 and 518 ms, respectively. However, deaf male V:4 had a long QTcF of 495 ms and QTcB of 503 ms at rest, but during exercise he had a shortening of the QT with a QTcF of 452 and QTcB of 507 ms when heart rate peaked at 119 bpm, which is the normal range. Two p.Tyr46* heterozygotes in family PKDF461 had QTcB and QTcF intervals in the normal range and also normal hearing. Apparently, these two p.Tyr46* KCNE1 heterozygotes do not have RWS. A possible explanation for an absence of a prolonged QT interval of an individual heterozygous for a loss-of-function (LOF) mutation of KCNE1 is that one copy of a wild type KCNE1 allele is sufficient for normal cardiac rhythm. In contrast, forty-two heterozygous substitutions of KCNE1 are reported to be associated with prolonged QT interval (RWS) but not hearing loss (Table 1). Apparently, KCNQ1 does not function adequately in the presence of certain heterozygous or biallelic amino acid substitutions of KCNE1, which presumably interact with the KCNQ1 tetramer and impairs its function by a dominant negative mechanism (Veitia, Caburet, & Birchler, 2018). Perhaps in heart, but not in the inner ear, one of the other four KCNE paralogs may partially substitute in the KCNQ1 complex when KCNE1 is absent (Jiang et al., 2009).
While ascertaining other families segregating nonsyndromic hearing loss, two more consanguineous families from Pakistan were identified and found to be segregating two additional biallelic truncating variants of KCNE1. Affected members were homozygous for one of two different nucleotide changes in the same codon, c.50G>A and c.51G>A. Both nonsense variants led to the same predicted protein truncation at residue 17 (p.Trp17*, Supp. Figure S1). As with PKDF461, family members did not mention cardiac arrhythmia, syncopal episodes, vestibular dysfunction, fainting spells or gross motor development delays. The ages of individuals V:4, V:5 and V:7 who are homozygous mutant for the KCNE1 variant in PKDF461 are 18, 13 and 28 years, respectively. Deaf members of family DEM 4502 did not report any history of sudden deaths, fainting or cardiologic assessments. However, after identifying the variant of KCNE1 segregating in these two families we attempted to re-contact them to inform the family of the molecular genetic data, gather cardiological data but because of a natural disaster that had occurred where they were living, we were unsuccessful.
The American College of Medical Genetics (ACMG) reported 59 medically actionable genes for which incidental findings of pathogenic or likely pathogenic variants should be reported to a patient (Kalia et al., 2017). Presently, KCNE1 is not listed as one of these 59 genes despite having the same cardiac phenotype as pathogenic variants of KCNQ1. We suggest that KCNE1 be added by the ACMG list because of the same life-threatening risk associated with mutant alleles of KCNE1 as has been reported for KCNQ1.
5. EXPRESSION OF KCNE1 IN THE STRIA VASCULARIS
In the stria vascularis of the cochlea, the KCNQ1 channel transports potassium ions into the endolymph, which bathes the apical surface of hair cells (Figure 1A), and contributes to the generation and maintenance of the +80 to +100 milli-volt endocochlear potential (EP) (Wangemann, 1995). The EP is required for signal transduction and depolarization of hair cells in response to deflection of the stereocilia following basilar membrane movement induced by sound waves. In the marginal cells of the stria vascularis, located in the lateral wall of the organ of Corti (Figure 1B–G), KCNE1 function is required for KCNQ1 channel function (Wangemann, 1995). From biochemical studies, KCNE2 to KCNE5 each can bind KCNQ1 and alter its channel properties (Abbott, 2016; Jiang et al., 2009). Whether wild type KCNE2, KCNE3, KCNE4 or KCNE5 have the potential to substitute for KCNE1 in the inner ear and restore hearing, either partially or entirely, could be experimentally answered by examining hearing in mice harboring inducible transgenes for each of the five KCNE paralogs expressed in a Kcne1-null background.
6. Kcne1 DEFICIENT MOUSE MODELS
Three different null alleles of mouse Kcne1 have been reported (Table 3). Two mouse lines have engineered deletions of the single protein-coding exon of Kcne1 (Kupershmidt et al., 1999; Vetter et al., 1996) and the third is a spontaneous p.Arg67* mutation, denoted as Kcne1pkr for “punk rocker” due to vestibular dysfunction manifested as a head-tossing phenotype (Letts et al., 2000). Homozygotes for each of the three Kcne1 alleles are deaf, and heterozygotes have normal hearing, recapitulating the auditory phenotype of humans with biallelic or heterozygous pathogenic variants of KCNE1 (Kupershmidt et al., 1999; Letts et al., 2000; Vetter et al., 1996). In the inner ear, homozygous Kcne1 deficient mice have a collapsed Reissner’s membrane and subsequently show degeneration of hair cells, confirming the importance of the role of KCNE1 in the mouse auditory system (Vetter et al., 1996).
TABLE 3.
Mouse models engineered to study KCNE1 function in the inner ear and heart
| Mutant allele of Kcne1 | Inner ear phenotype | Cardiac phenotype | Age | Reference |
|---|---|---|---|---|
| deletion of coding exon, replaced with neo-cassette | deaf, circling, head tossing | not evaluated | (P7, 10, 20, 42), 3,5 and 7mth | Vetter et al., 1996 |
| same allele as reported in Vetter et al., 1996 | not evaluated | longer QT interval at slow heart rates and a shorter QT interval at faster heart rates | 20 wk | Charpentier et al., 1998 |
| same allele as reported in Vetter et al., 1996 | not evaluated | longer QT at slow heart rates and shorter QT at faster heart rates | 3 wk and 12 wk | Drici et al., 1998 |
| same allele as reported in Vetter et al., 1996 | not evaluated | cardiac arrhythmogenicity | 3–6 mth | Balasubramaniam et al., 2003 |
| coding exon of Kcne1 deleted and substituted with a LacZ cassette | not evaluated | no prolonged QT interval at slow or fast heart rates | Neonates and adults | Kupershmidt et al., 1999 |
| Kcne1pkr spontaneous C>T (p.Arg67*) | deaf, circling, head tossing | not evaluated | 5wk | Letts et al., 2000 |
wk, week; mth, month; P, postnatal; LacZ, encoding E. coli β-galactosidase
Conclusions about a cardiac phenotype in the three Kcne1 mutant mouse models are less clear and differ among studies, perhaps for technical reasons. In one Kcne1 knockout mouse line, Drici and colleagues reported longer QT intervals at slow heart rates and a shorter QT interval at fast heart rates (Drici et al., 1998), similar to what we observed in one male subject of family PKDF461 who was tested both at rest and after exercise, as described above. In contrast, Charpentier and colleagues reported a QT interval for the same Kcne1 knockout mouse line that was indistinguishable from a wild type mouse, but they used microelectrodes to measure cardiac function in a restricted area of the myocardium (Charpentier, Merot, Riochet, Le Marec, & Escande, 1998). A third group reported that this same mouse model shows increased cardiac arrhythmogenicity after programmed electrical stimulation (Balasubramaniam, Grace, Saumarez, Vandenberg, & Huang, 2003). Kupershmidt and colleagues did not observe differences in ECGs in their independent mouse model compared to wild type controls (Kupershmidt et al., 1999). Cardiac rhythm in the Kcne1pkr mice was not tested (Letts et al., 2000). Regardless of any cardiac phenotypic differences among studies of homozygous mutant mice, the mouse has a normal heart rate of 500 to 700 beats per minute (BPM) (Ho et al., 2011). Such a high BPM increases the technical challenge of accurately measuring QT intervals, and further suggests that in some cases the mouse may not be a suitable model for human cardiac arrhythmias. In contrast, the inner ears of human and mouse are architecturally and functionally very similar to one another (Friedman, Dror, & Avraham, 2007). Mouse models of human hereditary deafness have been used extensively to study in vivo function of these genes.
7. MANAGEMENT AND CONCLUSIONS
The co-occurrence of profound hearing loss and a prolonged QT interval in Jervell and Lange-Nielsen syndrome justifies the initiation of a full evaluation of cardiac function in deaf individuals who lack a molecular genetic diagnosis. Individuals with possible hereditary deafness and their relatives who may carry heterozygous variants of KCNE1 and KCNQ1 are also at risk for prolonged QT and should similarly be investigated. Often, symptoms of arrhythmias remain occult until an affected individual has a serious cardiac event. For example, when families PKDF461, 4410 and 4502 were ascertained, information obtained from family members revealed no signs of vertigo, dizziness, syncope or sudden deaths. Therefore, we assumed prematurely that these families were segregating nonsyndromic deafness. Only after we identified a variant of KCNE1 in family PKDF461, did we obtain cardiology consultations and electrocardiograms from this family. In a study of 104 Pakistani children with moderate to severe sensorineural hearing loss, detailed history and screening electrocardiograms revealed LQTS in four children (3.8% of the deaf children), suggesting that mutations of KCNE1 and KCNQ1 may be significant but overlooked contributor to hearing loss. Moreover, evaluation of family members of these four individuals revealed LQTS in one normal hearing family member (Niaz, Rizvi, & Khurram, 2011). Accordingly, we suggest the following guidelines for management of individuals ascertained on the basis of congenital hearing loss in the initial absence of a molecular genetic diagnosis. Individuals who have bilateral, severe to profound congenital deafness of unknown etiology should have an ECG for assessment of the QT interval. Preventative treatment of life-threatening arrhythmias in these individuals may be indicated. Other medications prescribed to these patients should also be reviewed to eliminate, if possible, those that have the potential to prolong the QT interval (Arunachalam et al., 2018). Finally, KCNE1 and KCNQ1 should be added to the panel of deafness genes that are screened commercially and in academic institutions for patients assumed initially to have nonsyndromic deafness.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful for the participation of subjects in this study and thank Isabelle Roux PhD, Lisa Cunningham, PhD, Inna Belyantseva MD, PhD, Keri Richards MS and Penelope Friedman MD for advice and evaluation of clinical data. We dedicate this paper to the memory of Dr. Maria Bitner-Glindzicz who was the first to discover a variant of KCNE1 associated with JLNS. We thank Alan Hoofring, NIH Medical Arts, for panel A of Figure 1. This research was supported (in part) by the NIH Intramural Research Program funds to TBF (DC000048), NIDCD grant number Z1A-000046 to Dennis Drayna supporting SEB, and to MH (DC000088) and the Higher Education Commission of Pakistan to SR and WA, and the National Institute on Deafness and Other Communication Disorders grants R01 DC011651 and R01 DC003594 to SML. Written informed consents were obtained from all participants in this study. Institutional Review Board approval was obtained from the Combined Neurosciences Blue Panel at the National Institutes of Health (OH-93-N-016) and from National Centre of Excellence in Molecular Biology at the University of the Punjab. TBF takes responsibility for the integrity and accuracy of the data analyses. Research concept and study design: TBF, RF, SEB, SR, SML. Analyses and interpretation of the data: RF, SEB, RT, MH, RO, AUR, IS, MZA, AAB, AAK, CB, WA, AB, SML, SR, TBF. Drafting of the manuscript: RF, SEB, RT, TBF. All authors read and edited the manuscript. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Funding information
NIDCD/NIH Intramural Research Program funds to TBF (DC000048), Z1A-000046 to Dennis Drayna supporting SEB, and to MH (DC000088). The Higher Education Commission of Pakistan supports SR and WA. The National Institute on Deafness and Other Communication Disorders provides grants R01 DC011651 and R01 DC003594 to SML.
Footnotes
DISCLOSURE STAEMENT
The authors declare no conflict of interest.
REFERENCES
- Abbott GW (2016). KCNE1 and KCNE3: The yin and yang of voltage-gated K(+) channel regulation. Gene, 576(1 Pt 1), 1–13. 10.1016/j.gene.2015.09.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott GW, & Goldstein SA (2002). Disease-associated mutations in KCNE potassium channel subunits (MiRPs) reveal promiscuous disruption of multiple currents and conservation of mechanism. FASEB Journal, 16(3), 390–400. 10.1096/fj.01-0520hyp [DOI] [PubMed] [Google Scholar]
- Al-Khatib SM, LaPointe NM, Kramer JM, & Califf RM (2003). What clinicians should know about the QT interval. JAMA, 289(16), 2120–2127. 10.1001/jama.289.16.2120 [DOI] [PubMed] [Google Scholar]
- Alders M, & Christiaans I (1993). Long QT Syndrome. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews University of Washington, Seattle, WA. [Google Scholar]
- Arunachalam K, Lakshmanan S, Maan A, Kumar N, & Dominic P (2018). Impact of Drug Induced Long QT Syndrome: A Systematic Review. Journal of Clinical Medicine Research, 10(5), 384–390. 10.14740/jocmr3338w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam R, Grace AA, Saumarez RC, Vandenberg JI, & Huang CL (2003). Electrogram prolongation and nifedipine-suppressible ventricular arrhythmias in mice following targeted disruption of KCNE1. Journal of Physiology, 552(Pt 2), 535–546. 10.1113/jphysiol.2003.048249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bas T, Gao GY, Lvov A, Chandrasekhar KD, Gilmore R, & Kobertz WR (2011). Post-translational N-glycosylation of type I transmembrane KCNE1 peptides: implications for membrane protein biogenesis and disease. Journal of Biological Chemistry, 286(32), 28150–28159. 10.1074/jbc.M111.235168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazett HC (1920). An analysis of the time relationships or time-relations of electrocardiograms. Heart, 7, 353–380. [Google Scholar]
- Ben-Yosef T, Wattenhofer M, Riazuddin S, Ahmed ZM, Scott HS, Kudoh J, … Morell RJ (2001). Novel mutations of TMPRSS3 in four DFNB8/B10 families segregating congenital autosomal recessive deafness. Journal of Medical Genetics, 38(6), 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi L, Shen Z, Dennis AT, Priori SG, Napolitano C, Ronchetti E, … Brown AM (1999). Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Human Molecular Genetics, 8(8), 1499–1507. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar KD, Lvov A, Terrenoire C, Gao GY, Kass RS, & Kobertz WR (2011). O-glycosylation of the cardiac I(Ks) complex. Journal of Physiology, 589(Pt 15), 3721–3730. 10.1113/jphysiol.2011.211284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang RK, Lan YT, Silka MJ, Morrow H, Kwong A, Smith-Lang J, … Lin HJ (2014). Genetic variants for long QT syndrome among infants and children from a statewide newborn hearing screening program cohort. Journal of Pediatrics, 164(3), 590–595 10.1016/j.jpeds.2013.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier F, Merot J, Riochet D, Le Marec H, & Escande D (1998). Adult KCNE1-knockout mice exhibit a mild cardiac cellular phenotype. Biochemical and Biophysical Research Communications, 251(3), 806–810. 10.1006/bbrc.1998.9554 [DOI] [PubMed] [Google Scholar]
- Christiansen M, Hedley PL, Theilade J, Stoevring B, Leren TP, Eschen O, … Kanters JK (2014). Mutations in Danish patients with long QT syndrome and the identification of a large founder family with p.F29L in KCNH2. BMC Medical Genetics, 15, 31 10.1186/1471-2350-15-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenca S, Ruiz-Cano MJ, Gimeno-Blanes JR, Jurado A, Salas C, Gomez-Diaz I, … Inherited Cardiac Diseases Program of the Spanish Cardiovascular Research, N. (2016). Genetic basis of familial dilated cardiomyopathy patients undergoing heart transplantation. Journal of Heart and Lung Transplantation, 35(5), 625–635. 10.1016/j.healun.2015.12.014 [DOI] [PubMed] [Google Scholar]
- Delmaghani S, Aghaie A, Michalski N, Bonnet C, Weil D, & Petit C (2012). Defect in the gene encoding the EAR/EPTP domain-containing protein TSPEAR causes DFNB98 profound deafness. Human Molecular Genetics, 21(17), 3835–3844. 10.1093/hmg/dds212 [DOI] [PubMed] [Google Scholar]
- Drici MD, Arrighi I, Chouabe C, Mann JR, Lazdunski M, Romey G, & Barhanin J (1998). Involvement of IsK-associated K+ channel in heart rate control of repolarization in a murine engineered model of Jervell and Lange-Nielsen syndrome. Circulation Research, 83(1), 95–102. [DOI] [PubMed] [Google Scholar]
- Friderica L (1920). The duration of systole in the electrocardiogram of normal subjects and of patients with heart disease. Acta Medica Scandinavica, 53, 469–505. [Google Scholar]
- Friedman LM, Dror AA, & Avraham KB (2007). Mouse models to study inner ear development and hereditary hearing loss. International Journal of Developmental Biology, 51(6–7), 609–631. 10.1387/ijdb.072365lf [DOI] [PubMed] [Google Scholar]
- Ghouse J, Have CT, Weeke P, Bille Nielsen J, Ahlberg G, Balslev-Harder M, … Salling Olesen M (2015). Rare genetic variants previously associated with congenital forms of long QT syndrome have little or no effect on the QT interval. European Heart Journal, 36(37), 2523–2529. 10.1093/eurheartj/ehv297 [DOI] [PubMed] [Google Scholar]
- Grunert SC, Bodi I, & Odening KE (2017). Possible mechanisms for sensorineural hearing loss and deafness in patients with propionic acidemia. Orphanet Journal of Rare Diseases, 12(1), 30 10.1186/s13023-017-0585-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HG, Wang HS, Yin Z, Jiang H, Fang M, & Han J (2014). KCNE1 112G>a polymorphism and atrial fibrillation risk: a meta-analysis. Genetics and Molecular Research, 13(4), 8367–8377. 10.4238/2014.October.20.12 [DOI] [PubMed] [Google Scholar]
- Harmer SC, Mohal JS, Royal AA, McKenna WJ, Lambiase PD, & Tinker A (2014). Cellular mechanisms underlying the increased disease severity seen for patients with long QT syndrome caused by compound mutations in KCNQ1. Biochemical Journal, 462(1), 133–142. 10.1042/BJ20140425 [DOI] [PubMed] [Google Scholar]
- Hedley PL, Durrheim GA, Hendricks F, Goosen A, Jespersgaard C, Stovring B, … Corfield VA (2013). Long QT syndrome in South Africa: the results of comprehensive genetic screening. Cardiovascular Journal of Africa, 24(6), 231–237. 10.5830/CVJA-2013-032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietikko E, Kotimaki J, Okuloff A, Sorri M, & Mannikko M (2012). A replication study on proposed candidate genes in Meniere’s disease, and a review of the current status of genetic studies. International Journal of Audiology, 51(11), 841–845. 10.3109/14992027.2012.705900 [DOI] [PubMed] [Google Scholar]
- Ho D, Zhao X, Gao S, Hong C, Vatner DE, & Vatner SF (2011). Heart Rate and Electrocardiography Monitoring in Mice. Current Protocols in Mouse Biology, 1, 123–139. 10.1002/9780470942390.mo100159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huikuri HV, Castellanos A, & Myerburg RJ (2001). Sudden death due to cardiac arrhythmias. New England Journal of Medicine, 345(20), 1473–1482. 10.1056/NEJMra000650 [DOI] [PubMed] [Google Scholar]
- Jervell A, & Lange-Nielsen F (1957). Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. American Heart Journal, 54(1), 59–68. [DOI] [PubMed] [Google Scholar]
- Jespersen T, Grunnet M, & Olesen SP (2005). The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda, Md.), 20, 408–416. 10.1152/physiol.00031.2005 [DOI] [PubMed] [Google Scholar]
- Jiang M, Xu X, Wang Y, Toyoda F, Liu XS, Zhang M, … Tseng GN (2009). Dynamic partnership between KCNQ1 and KCNE1 and influence on cardiac IKs current amplitude by KCNE2. Journal of Biological Chemistry, 284(24), 16452–16462. 10.1074/jbc.M808262200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, … Miller DT (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genetics in Medicine, 19(2), 249–255. 10.1038/gim.2016.190 [DOI] [PubMed] [Google Scholar]
- Kang C, Tian C, Sonnichsen FD, Smith JA, Meiler J, George AL Jr., … Sanders CR (2008). Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry, 47(31), 7999–8006. 10.1021/bi800875q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, … Ackerman MJ (2009). Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm, 6(9), 1297–1303. 10.1016/j.hrthm.2009.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritsis DG, Gersh BJ, & Camm AJ (2016). A Clinical Perspective on Sudden Cardiac Death. Arrhythm Electrophysiol Rev, 5(3), 177–182. 10.15420/aer.2016:11:2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupershmidt S, Yang T, Anderson ME, Wessels A, Niswender KD, Magnuson MA, & Roden DM (1999). Replacement by homologous recombination of the minK gene with lacZ reveals restriction of minK expression to the mouse cardiac conduction system. Circulation Research, 84(2), 146–152. [DOI] [PubMed] [Google Scholar]
- Lai LP, Su MJ, Yeh HM, Lin JL, Chiang FT, Hwang JJ, … Huang SK (2002). Association of the human minK gene 38G allele with atrial fibrillation: evidence of possible genetic control on the pathogenesis of atrial fibrillation. American Heart Journal, 144(3), 485–490. [DOI] [PubMed] [Google Scholar]
- Lai LP, Su YN, Hsieh FJ, Chiang FT, Juang JM, Liu YB, … Lin JL (2005). Denaturing high-performance liquid chromatography screening of the long QT syndrome-related cardiac sodium and potassium channel genes and identification of novel mutations and single nucleotide polymorphisms. Journal of Human Genetics, 50(9), 490–496. 10.1007/s10038-005-0283-3 [DOI] [PubMed] [Google Scholar]
- Letts VA, Valenzuela A, Dunbar C, Zheng QY, Johnson KR, & Frankel WN (2000). A new spontaneous mouse mutation in the Kcne1 gene. Mammalian Genome, 11(10), 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieve KV, Williams L, Daly A, Richard G, Bale S, Macaya D, & Chung WK (2013). Results of genetic testing in 855 consecutive unrelated patients referred for long QT syndrome in a clinical laboratory. Genetic Testing and Molecular Biomarkers, 17(7), 553–561. 10.1089/gtmb.2012.0118 [DOI] [PubMed] [Google Scholar]
- Liin SI, Silvera Ejneby M, Barro-Soria R, Skarsfeldt MA, Larsson JE, Starck Harlin F, … Elinder F (2015). Polyunsaturated fatty acid analogs act antiarrhythmically on the cardiac IKs channel. Proceedings of the National Academy of Sciences of the United States of America, 112(18), 5714–5719. 10.1073/pnas.1503488112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Zhao Q, Su T, Tang S, Lv G, Liu H, … Cheng J (2013). Postmortem molecular analysis of KCNQ1, KCNH2, KCNE1 and KCNE2 genes in sudden unexplained nocturnal death syndrome in the Chinese Han population. Forensic Science International, 231(1–3), 82–87. 10.1016/j.forsciint.2013.04.020 [DOI] [PubMed] [Google Scholar]
- Lundquist AL, Manderfield LJ, Vanoye CG, Rogers CS, Donahue BS, Chang PA, … George AL Jr. (2005). Expression of multiple KCNE genes in human heart may enable variable modulation of I(Ks). Journal of Molecular and Cellular Cardiology, 38(2), 277–287. 10.1016/j.yjmcc.2004.11.012 [DOI] [PubMed] [Google Scholar]
- Lvov A, Gage SD, Berrios VM, & Kobertz WR (2010). Identification of a protein-protein interaction between KCNE1 and the activation gate machinery of KCNQ1. Journal of General Physiology, 135(6), 607–618. 10.1085/jgp.200910386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Lin C, Teng S, Chai Y, Bahring R, Vardanyan V, … Hui R (2003). Characterization of a novel Long QT syndrome mutation G52R-KCNE1 in a Chinese family. Cardiovascular Research, 59(3), 612–619. [DOI] [PubMed] [Google Scholar]
- Mazhari R, Nuss HB, Armoundas AA, Winslow RL, & Marban E (2002). Ectopic expression of KCNE3 accelerates cardiac repolarization and abbreviates the QT interval. Journal of Clinical Investigation, 109(8), 1083–1090. 10.1172/JCI15062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrossan ZA, & Abbott GW (2004). The MinK-related peptides. Neuropharmacology, 47(6), 787–821. 10.1016/j.neuropharm.2004.06.018 [DOI] [PubMed] [Google Scholar]
- Millat G, Kugener B, Chevalier P, Chahine M, Huang H, Malicier D, … Rousson R (2009). Contribution of long-QT syndrome genetic variants in sudden infant death syndrome. Pediatric Cardiology, 30(4), 502–509. 10.1007/s00246-009-9417-2 [DOI] [PubMed] [Google Scholar]
- Murray CI, Westhoff M, Eldstrom J, Thompson E, Emes R, & Fedida D (2016). Unnatural amino acid photo-crosslinking of the IKs channel complex demonstrates a KCNE1:KCNQ1 stoichiometry of up to 4:4. Elife, 5 10.7554/eLife.11815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K, & Kubo Y (2015). KCNQ1 channel modulation by KCNE proteins via the voltage-sensing domain. Journal of Physiology, 593(12), 2617–2625. 10.1113/jphysiol.2014.287672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K, Ulbrich MH, Kubo Y, & Isacoff EY (2010). Stoichiometry of the KCNQ1 - KCNE1 ion channel complex. Proceedings of the National Academy of Sciences of the United States of America, 107(44), 18862–18867. 10.1073/pnas.1010354107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y, & Shimizu W (2016). Genetics of long-QT syndrome. Journal of Human Genetics, 61(1), 51–55. 10.1038/jhg.2015.74 [DOI] [PubMed] [Google Scholar]
- Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, … Leonardi S (2005). Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA, 294(23), 2975–2980. 10.1001/jama.294.23.2975 [DOI] [PubMed] [Google Scholar]
- Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, … Guicheney P (1997). A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nature Genetics, 15(2), 186–189. 10.1038/ng0297-186 [DOI] [PubMed] [Google Scholar]
- Ng D, Johnston JJ, Teer JK, Singh LN, Peller LC, Wynter JS, … Program, N. I. H. I. S. C. C. S. (2013). Interpreting secondary cardiac disease variants in an exome cohort. Circulation: Cardiovascular Genetics, 6(4), 337–346. 10.1161/CIRCGENETICS.113.000039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niaz A, Rizvi SF, & Khurram D (2011). Prevalence of long QT syndrome and other cardiac defects in deaf-mute children. Journal of Ayub Medical College, Abbottabad, 23(1), 5–8. [PubMed] [Google Scholar]
- Obeyesekere MN, Sy RW, Klein GJ, Gula LJ, Modi S, Conacher S, … Krahn AD (2012). End-recovery QTc: a useful metric for assessing genetic variants of unknown significance in long-QT syndrome. Journal of Cardiovascular Electrophysiology, 23(6), 637–642. 10.1111/j.1540-8167.2011.02265.x [DOI] [PubMed] [Google Scholar]
- Ohno S, Zankov DP, Yoshida H, Tsuji K, Makiyama T, Itoh H, … Horie M (2007). N- and C-terminal KCNE1 mutations cause distinct phenotypes of long QT syndrome. Heart Rhythm, 4(3), 332–340. 10.1016/j.hrthm.2006.11.004 [DOI] [PubMed] [Google Scholar]
- Olesen MS, Bentzen BH, Nielsen JB, Steffensen AB, David JP, Jabbari J, … Schmitt N (2012). Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Medical Genetics, 13, 24 10.1186/1471-2350-13-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen JD, Gonzalez C, Sampson KJ, Iyer V, Rebolledo S, Larsson HP, & Kass RS (2010). KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proceedings of the National Academy of Sciences of the United States of America, 107(52), 22710–22715. 10.1073/pnas.1016300108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, … Aerssens J (2004). Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long QT syndrome patients. Journal of Molecular Medicine (Berlin, Germany), 82(3), 182–188. 10.1007/s00109-003-0522-z [DOI] [PubMed] [Google Scholar]
- Peled A, Sarig O, Samuelov L, Bertolini M, Ziv L, Weissglas-Volkov D, … Sprecher E (2016). Mutations in TSPEAR, Encoding a Regulator of Notch Signaling, Affect Tooth and Hair Follicle Morphogenesis. PLoS Genet, 12(10), e1006369 10.1371/journal.pgen.1006369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, … Cappelletti D (2003). Risk stratification in the long-QT syndrome. New England Journal of Medicine, 348(19), 1866–1874. 10.1056/NEJMoa022147 [DOI] [PubMed] [Google Scholar]
- Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, … Asia Pacific Heart Rhythm, S. (2013). Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace: European Pacing, Arrhythmias, and Cardiac Electrophysiology, 15(10), 1389–1406. 10.1093/europace/eut272 [DOI] [PubMed] [Google Scholar]
- Riuro H, Campuzano O, Berne P, Arbelo E, Iglesias A, Perez-Serra A, … Brugada R (2015). Genetic analysis, in silico prediction, and family segregation in long QT syndrome. European Journal of Human Genetics, 23(1), 79–85. 10.1038/ejhg.2014.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R (2006). Genomics and Cardiac Arrhythmias. Journal of the American College of Cardiology, 47(1), 9–21. 10.1016/j.jacc.2005.08.059 [DOI] [PubMed] [Google Scholar]
- Roden DM, & Viswanathan PC (2005). Genetics of acquired long QT syndrome. Journal of Clinical Investigation, 115(8), 2025–2032. 10.1172/JCI25539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano C (1965). Congenital Cardiac Arrhythmia. Lancet, 1(7386), 658–659. [DOI] [PubMed] [Google Scholar]
- Ryan JJ, Kalscheur M, Dellefave L, McNally E, & Archer SL (2012). A KCNE1 missense variant (V47I) causing exercise-induced long QT syndrome (Romano Ward). International Journal of Cardiology, 156(2), e33–35. 10.1016/j.ijcard.2011.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, & Keating MT (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature, 384(6604), 80–83. 10.1038/384080a0 [DOI] [PubMed] [Google Scholar]
- Schmitt N, Schwarz M, Peretz A, Abitbol I, Attali B, & Pongs O (2000). A recessive C-terminal Jervell and Lange-Nielsen mutation of the KCNQ1 channel impairs subunit assembly. EMBO Journal, 19(3), 332–340. 10.1093/emboj/19.3.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Bahr E, Schwarz M, Hauenschild S, Wedekind H, Funke H, Haverkamp W, … Isbrandt D (2001). A novel long-QT 5 gene mutation in the C-terminus (V109I) is associated with a mild phenotype. Journal of Molecular Medicine (Berlin, Germany), 79(9), 504–509. 10.1007/s001090100249 [DOI] [PubMed] [Google Scholar]
- Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, … Funke H (1997). KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nature Genetics, 17(3), 267–268. 10.1038/ng1197-267 [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, … Bloise R (2001). Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation, 103(1), 89–95. [DOI] [PubMed] [Google Scholar]
- Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry A, Chrast R, … Antonarakis SE (2001). Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nature Genetics, 27(1), 59–63. 10.1038/83768 [DOI] [PubMed] [Google Scholar]
- Shim SH, Ito M, Maher T, & Milunsky A (2005). Gene sequencing in neonates and infants with the long QT syndrome. Genet Test, 9(4), 281–284. 10.1089/gte.2005.9.281 [DOI] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, … Keating MT (2000). Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation, 102(10), 1178–1185. [DOI] [PubMed] [Google Scholar]
- Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, & Keating MT (1997). Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nature Genetics, 17(3), 338–340. 10.1038/ng1197-338 [DOI] [PubMed] [Google Scholar]
- Strutz-Seebohm N, Pusch M, Wolf S, Stoll R, Tapken D, Gerwert K, … Seebohm G (2011). Structural basis of slow activation gating in the cardiac I Ks channel complex. Cellular Physiology and Biochemistry, 27(5), 443–452. 10.1159/000329965 [DOI] [PubMed] [Google Scholar]
- Sun J, & MacKinnon R (2017). Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell, 169(6), 1042–1050 10.1016/j.cell.2017.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sy RW, van der Werf C, Chattha IS, Chockalingam P, Adler A, Healey JS, … Krahn AD (2011). Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation, 124(20), 2187–2194. 10.1161/CIRCULATIONAHA.111.028258 [DOI] [PubMed] [Google Scholar]
- Tombola F, Pathak MM, & Isacoff EY (2006). How does voltage open an ion channel? Annual Review of Cell and Developmental Biology, 22, 23–52. 10.1146/annurev.cellbio.21.020404.145837 [DOI] [PubMed] [Google Scholar]
- Tranebjaerg L, Samson RA, & Green GE (1993). Jervell and Lange-Nielsen Syndrome. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, & Ledbetter N (Eds.), GeneReviews(R) University of Washington, Seattle, WA. [Google Scholar]
- Tranebjaerg L, Samson RA, & Green GE (2017). Jervell and Lange-Nielsen Syndrome. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews((R)) University of Washington, Seattle, WA. [Google Scholar]
- Tyson J, Tranebjaerg L, Bellman S, Wren C, Taylor JF, Bathen J, … Bitner-Glindzicz M (1997). IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Human Molecular Genetics, 6(12), 2179–2185. [DOI] [PubMed] [Google Scholar]
- Tyson J, Tranebjaerg L, McEntagart M, Larsen LA, Christiansen M, Whiteford ML, … Bitner-Glindzicz M (2000). Mutational spectrum in the cardioauditory syndrome of Jervell and Lange-Nielsen. Human Genetics, 107(5), 499–503. [DOI] [PubMed] [Google Scholar]
- Van Laer L, Carlsson PI, Ottschytsch N, Bondeson ML, Konings A, Vandevelde A, … Van Camp G (2006). The contribution of genes involved in potassium-recycling in the inner ear to noise-induced hearing loss. Human Mutation, 27(8), 786–795. 10.1002/humu.20360 [DOI] [PubMed] [Google Scholar]
- Veitia RA, Caburet S, & Birchler JA (2018). Mechanisms of Mendelian dominance. Clinical Genetics, 93(3), 419–428. 10.1111/cge.13107 [DOI] [PubMed] [Google Scholar]
- Veske A, Oehlmann R, Younus F, Mohyuddin A, Muller-Myhsok B, Mehdi SQ, & Gal A (1996). Autosomal recessive non-syndromic deafness locus (DFNB8) maps on chromosome 21q22 in a large consanguineous kindred from Pakistan. Human Molecular Genetics, 5(1), 165–168. [DOI] [PubMed] [Google Scholar]
- Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, … Barhanin J (1996). Inner ear defects induced by null mutation of the isk gene. Neuron, 17(6), 1251–1264. [DOI] [PubMed] [Google Scholar]
- Wagner GS, Macfarlane P, Wellens H, Josephson M, Gorgels A, Mirvis DM, … Heart Rhythm S (2009). AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part VI: acute ischemia/infarction: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society: endorsed by the International Society for Computerized Electrocardiology. Circulation, 119(10), e262–270. 10.1161/CIRCULATIONAHA.108.191098 [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, … Keating MT (1996). Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nature Genetics, 12(1), 17–23. 10.1038/ng0196-17 [DOI] [PubMed] [Google Scholar]
- Wangemann P (1995). Comparison of ion transport mechanisms between vestibular dark cells and strial marginal cells. Hearing Research, 90(1–2), 149–157. [DOI] [PubMed] [Google Scholar]
- Ward OC (1964). A New Familial Cardiac Syndrome in Children. Journal of the Irish Medical Association, 54, 103–106. [PubMed] [Google Scholar]
- Wilcox ER, Burton QL, Naz S, Riazuddin S, Smith TN, Ploplis B, … Friedman TB (2001). Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell, 104(1), 165–172. [DOI] [PubMed] [Google Scholar]
- Wrobel E, Tapken D, & Seebohm G (2012). The KCNE Tango - How KCNE1 Interacts with Kv7.1. Frontiers in Pharmacology, 3, 142 10.3389/fphar.2012.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wugeti N, Yu-Jun G, Juan S, & Mahemuti A (2015). Correlation analysis between the delayed rectifier potassium channel KCNE1 (G38S) polymorphism and atrial fibrillation among the senior Uygur population in Xinjiang. Genetics and Molecular Research, 14(4), 15906–15912. 10.4238/2015.December.7.1 [DOI] [PubMed] [Google Scholar]
- Yao J, Ma YT, Xie X, Liu F, & Chen BD (2012). Association of KCNE1 genetic polymorphisms with atrial fibrillation in a Chinese Han population. Genetic Testing and Molecular Biomarkers, 16(11), 1343–1346. 10.1089/gtmb.2012.0149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinaga M, Kucho Y, Sarantuya J, Ninomiya Y, Horigome H, Ushinohama H, … Horie M (2014). Genetic characteristics of children and adolescents with long-QT syndrome diagnosed by school-based electrocardiographic screening programs. Circ Arrhythm Electrophysiol, 7(1), 107–112. 10.1161/CIRCEP.113.000426 [DOI] [PubMed] [Google Scholar]
- Zaydman MA, Silva JR, Delaloye K, Li Y, Liang H, Larsson HP, … Cui J (2013). Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proceedings of the National Academy of Sciences of the United States of America, 110(32), 13180–13185. 10.1073/pnas.1305167110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.