

Abstract
Neurofibromatosis type 1 (NF1) is a cancer predisposition disorder that results from inactivation of the tumor-suppressor Neurofibromin, a negative regulator of RAS signaling. NF1 patients present with a wide range of clinical manifestations and the tumor with highest prevalence is cutaneous neurofibroma (cNF). Most patients harboring cNF suffer greatly from the burden of those tumors, which have no effective medical treatment. Ironically, none of the numerous NF1 mouse models developed so far recapitulate cNF. Here, we discovered that Hoxb7 serves as a lineage marker to trace the developmental origin of cNF neoplastic cells. Ablating Nf1 in the Hoxb7 lineage faithfully recapitulates both human cutaneous and plexiform neurofibroma. In addition, we discovered that modulation of the Hippo pathway acts as a “modifier” for neurofibroma tumorigenesis. This mouse model opens the doors for deciphering the evolution of cNF to identify effective therapies, where none exist today.
Keywords: Neurofibromatosis Type 1, NF1, Dermal Neurofibroma, Cutaneous Neurofibroma, Peripheral Nerve Sheath Tumor, Plexiform Neurofibroma, Schwann Cells, Tumor Cells of Origin, Tumor Microenvironment, Neurofibromin, Hippo pathway, Hoxb7
Introduction
Neurofibromatosis type 1 (NF1) is one of the most common genetic disorders of the nervous system and occurs in about 1 in 3000 births. More than 25 years ago, independent laboratories discovered that mutations in the NF1 gene were responsible for causing NF1 in patients (1, 2). Subsequent genetic and biochemical analysis reveal that the protein product, neurofibromin, acts as a tumor suppressor and negative regulator of the RAS signaling pathway. The clinical spectrum of NF1 is very broad and affects many organ systems. Interestingly, the origin of most of the tissues affected in NF1 patient can be traced back to neural crest cells, a stem cell population originating from the separation of the neural tube and ectoderm at the end of the neurulation phase in embryologic development. Examples of cells with a neural crest origin include Schwann cells, melanocytes, and glial cells. Indeed, loss of NF1 function in the Schwann cell lineage leads to neurofibroma formation, NF1-/- melanocytes lead to café-au-lait macules (CALM) (3) and Lisch nodules (iris hamartoma), NF1-/- in glial cells leads to astrocytomas (4).
There are several subtypes of neurofibromas. Most clinicians subdivide them according to their distinctive location in relation to the skin dermis and propensity for malignancy. Dermal or cutaneous neurofibromas (cNF) are strictly confined to the dermis and do not progress to frank malignant sarcoma lesions. They can be as numerous as thousands of tumors that cover most of the body surface. Consequently, daily activities such as showering, being confronted to public and mobility can be a significant burden. The only effective treatment options are surgical approaches, which are not practical in patients with numerous cNFs (5). Neurofibromas involving the internal nerve plexus are called plexiform neurofibromas (pNF). They are located below the dermis and have an increased risk of malignancy. In addition to their specific locations, the onset is drastically different. pNF is congenital but cNF rarely appears before puberty. When a cNF is observed on a relatively large area of skin but still confined in the dermis and it is perfectly covered by skin pigmentation, it is called a diffuse cutaneous neurofibroma (diffuse cNF) (6, 7). In this case, the skin is much thicker, the predominant body location is the head and neck, upper arms and the pigment are localized in the dermis. Diffuse cNF is different from diffuse plexiform neurofibroma which shares most of the diffuse cNF features except that it also involves the nerve plexus under the skin. Other sub-types of pNF include para-spinal and nodular. These are well circumscribed sphere-like tumors that grows in the dorsal root ganglion (DRG) and peripheral nerve, respectively. Importantly, it is currently not known what causes different types of neurofibromas to develop. Thus, multiple subtypes of both cNF and pNF are described in the literature and vary according to their gross morphology, pigmentation, body location and propensity to malignancy. The development of a cNF mouse model that faithfully recapitulates the human clinical scenario would allow researchers to study the progression of cNF and test potential anti-neurofibroma drugs, which is not feasible at the moment.
The report of the NF1 gene sequence started the race to build the first NF1 mouse model. Functional inactivation of Nf1 in all cell types is embryonically lethal in mice but Nf1+/− littermates (mimicking the genetic context of NF1 in patients) live long enough for spontaneous tumor formation studies (8). Disappointingly, the cardinal features of NF1 such as Lisch nodules, skin pigmentation and neurofibromas did not develop in Nf1+/− mice (8). These results suggest that loss of function of the second Nf1 allele may be a rate limiting step in the cell of origin of neurofibroma. Coupled with the advent of the Cre-lox technology, dramatically increasing the number of Nf1-/- cells in a tissue-specific manner in Nf1+/− mice yields a robust mouse model of neurofibroma (9). However, all neurofibromas were of the plexiform type in this model. This suggests that cNFs have a different developmental origin than pNF.
As a first step toward the identification of the cell of origin of cNF, Le et al. 2009 (10) ablated Nf1 at adult stage in a localized manner in the skin by taking advantage of the topical activation of a non-specific inducible system (CMV-Cre-ERT2;Nf1f/-). It resulted in cNF resembling tumors found in NF1 patients and demonstrated that the cell of origin of cNF persists at adult stage and are present in the skin vicinity. One well established source of neural crest-derived stem cells residing in the adult dermis are skin-derived precursors (SKPs) (11). To further investigate the cell of origin of cNF, Le et al. 2009 (10) isolated SKPs from CMV-Cre-ERT2;Nf1f/- mice and successfully generated neurofibromas in an orthotopic mouse model. At this point, the next logical step was to build a germline mouse model using a Cre driver specific to SKPs to definitively identify the sub-population of stem cells responsible for cNF. However, SKPs comprise heterogeneous populations and as such SKP-specific Cre don’t exist. As SKPs originate from the neural crest, several neural crest-specific Cre driver (Wnt-1, Pax-3 and Mpz) were bred to Nf1f/- mice to investigate their ability to drive neurofibroma development. Unfortunately, deletion of Nf1 in those model resulted in premature death, precluding any neurofibroma formation analysis (12).
We reasoned that the spatial expression (identity and number of cells involved) was not adequate as it may have hit a too broad and vital population of cells (13) although the timing (before neural crest cells bifurcate into cNF and pNF cell of origin) of the Cre tested was accurate (14). Therefore, our strategy was to identify and take advantage of mouse neural crest Cre lines that are expressed in the sub-population of SKPs that give rise to cNF while sparing most of the vital neural-crest derived cells, as a balance between the right time and the right location. Here, we report the use of Hoxb7-Cre mice to lineage trace the specific cell of origin identity for cNF. We discovered that ablating Nf1 in the Hoxb7 lineage-derived cells faithfully recapitulates both human cutaneous and plexiform neurofibroma. In addition, we discovered that the modulation of the Hippo pathway acts as a “modifier” to the development of neurofibroma.
Results
Hoxb7 lineage-derived cells populate nerve ending in the dermis
To identify a putative cNF cell of origin lineage marker, we extensively searched the literature for a mouse Cre line with the highest chance of being expressed in a restricted population of neural crest cells giving rise to cNF. Classic studies on the morphogenesis of IX-X ganglionic complexes suggest that the cells expressing the HOXB7 protein derive from migrating neural crest cells (15). To lineage trace the Hoxb7 cell progeny, we crossed Hoxb7 mice with R26-LacZ and submitted E9.5 – E12.5 whole-mount embryos to X-Gal staining (Figure 1A). At E9.5, the neuropore has recently closed, which coincides with the initial delamination of neural crest cells. We observed HOXB7 expression mainly in the trunk neural crest as well as some punctate in the cranial region (Figure 1A). At E10.5–11.5, some migratory neural crest cells stop at the future site of what will be the DRG while other cells further migrate along the peripheral nerve. Note the apparent lower signal intensity due to the thickening of the skin at this developmental stage. At E12.5, some Hoxb7 lineage derived cells start to “re-appear”, especially in the cranial region (Figure 1A). This is due to the migratory Hoxb7 lineage derived cells have now reach the skin surface; hence the signal is visible once again. To confirm the results obtain with X-Gal staining, we performed immunohistochemistry using anti-HOXB7 antibodies on histological sections from the chest and cranial level at E12.5. HOXB7 is expressed specifically in a subset of cells in the DRG (Figure 1A, middle and lower panels).
Figure 1. Hoxb7 lineage-derived cell populate nerve ending in the dermis.
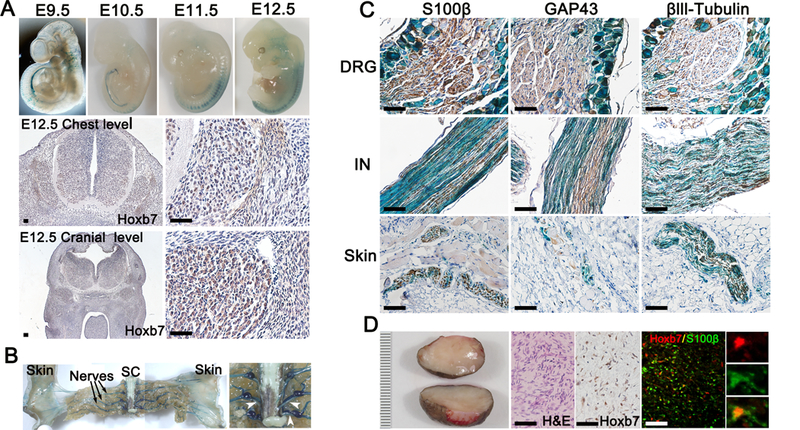
(A-C) X-Gal staining was performed on E9.5 – E12.5 embryos (A) and adult mice (B-C) with genotype Hoxb7-Cre;LacZ. (A) Immunohistochemistry using anti-HOXB7 antibodies on transectional cut of E12.5 embryo at the chest (middle panels) and cranial (lower panels) level. (B) Gross dissection of adult mice peripheral nervous system. Black arrow indicates dorsal sensory nerves. White arrow heads indicates ventral roots. SC=Spinal Cord. (C) Immunohistochemistry using Schwann cell (S100β, GAP43) and neuronal (βIII tubulin) markers on histological section of dorsal root ganglion (DRG), intercostal nerve (IN) and skin X-Gal counterstained (blue). (D) Typical representation of human cNF (left panel). Scale bar in millimeters. Hematoxylin and eosin (H&E) and immunohistochemistry using anti-HOXB7 antibodies on human cNF (middle panels). Immunofluorescence on human cNF using anti-HOXB7 and anti-S100β antibodies. Scale bar = 50μm.
Next, we determined the fate of HOXB7 cells postnatally. As expected, cells originating from Hoxb7-derived lineage populated the DRG, the peripheral nerve as well as the nerve ending in the skin (Figure 1B, left panel). In agreement with our observation in Figure 1A, we obtained very little signal from the ventral motor nerve as compared to the dorsal sensory nerve when we studied the whole-mounted Hoxb7-Cre;LacZ X-Gal stained mice (Figure 1B, right panel). Histological sections of DRG, intercostal nerve (IN) and skin confirm these findings. The LacZ reporter overlapped with the Schwann cell (S100β, GAP43) and the neuronal (βIII-tubulin) markers in the peripheral nervous system from DRG to dermis twigs (Figure 1C). Finally, we confirmed the expression of HOXB7 in Schwann cell from human cNF (Figure 1D). As the signal from the Schwann cell marker S100β is cytoplasmic and HOXB7 is nuclear, higher magnification images confirm the co-localization of Schwann cell marker S100β and HOXB7 (Figure 1D, insert). Altogether, these data suggest that cells from the Hoxb7 lineage may be the cell of origin of cNF.
Hoxb7 lineage derived Nf1-/- SKPs give rise to neurofibroma
To demonstrate that the Hoxb7 lineage in the skin can give rise to neurofibromas, we crossed the Hoxb7-Cre;LacZ mice to Nf1f/f to generate Hoxb7-Cre;Nf1f/f;LacZ. Next, we isolated, cultured and implanted SKPs from Hoxb7-Cre;Nf1f/f;LacZ newborn and control littermate into the sciatic nerves of nude mice (Figure 2A) (10, 16). After 3–4 months, mice implanted with SKPs from Hoxb7-Cre;Nf1f/f;LacZ mice consistently generate neurofibroma that are LacZ positive (Figure 2A) whereas none develop using cells derived from control littermates. The histological hallmarks of neurofibroma such as disorganized nerve structure, hypercellular Schwann cell and abundant collagen matrix were faithfully recapitulated (Figure 2A). To further prove that the cells of origin of cNF are derived from the Hoxb7 lineage, we decided to purify the Hoxb7 lineage positive SKPs by FACS. To do so, we crossed Hoxb7-Cre;Nf1f/f to the fluorescent reporter mice R26-YFP, isolated, cultured and submitted these SKPs to FACS analysis. However, the fraction of Hoxb7 lineage positive SKPs recovered was about 10%, precluding the direct testing of this sub-population of cells in our in vivo neurofibromagenesis assay as it required a larger amount of cells. This was consistent with the PCR analysis of DNA from bulk Hoxb7-Cre;Nf1f/f SKPs showing a faint band for Nf1 ablation (Figure 2A). As an alternative approach, we decided to use the bulk population of SKPs isolated from the Hoxb7-Cre;Nf1f/f;YFP mice and quantified the number of YFP positive cells before and after implantation (in the final neurofibroma tumor). We predicted that if the cNF cells of origin are truly derived from the Hoxb7 lineage, then there should be an accumulation (enrichment) of YFP+ cells in neurofibroma. As expected, up to 80% of neurofibroma cells were YFP+ (Figure 2B), providing additional support to our hypothesis that Hoxb7 lineage-derived cells can give rise to neurofibroma when Nf1 is deleted and that the cell of origin of cNF is within the Hoxb7 lineage population of SKPs.
Figure 2. Hoxb7 lineage derived Nf1−/− SKPs give rise to neurofibroma.
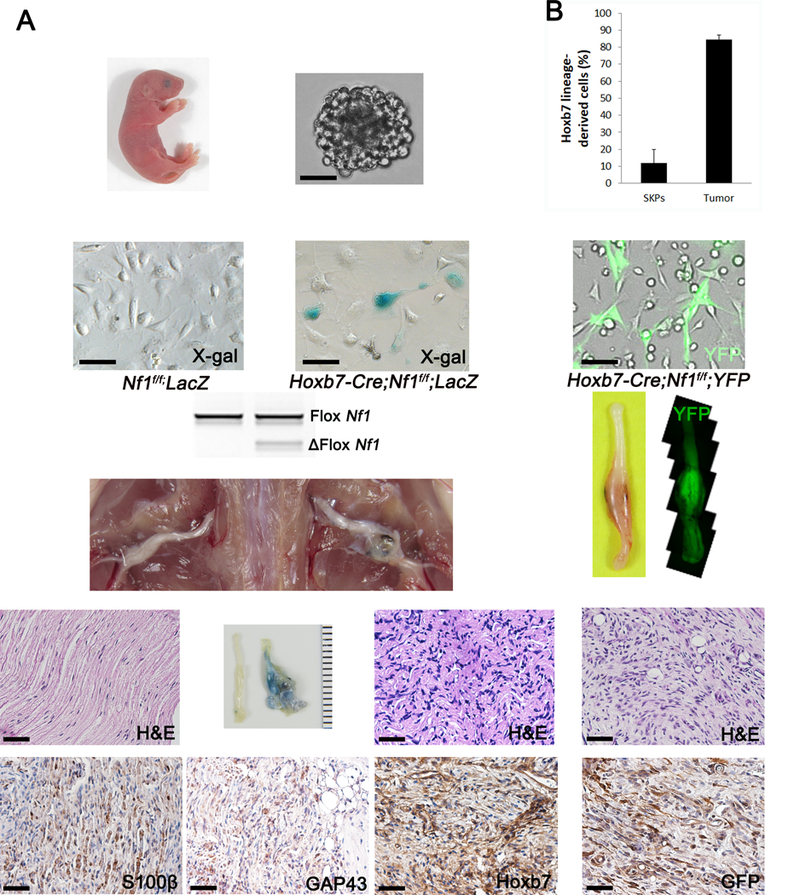
(A) Diagram of experimental design: Isolation of SKPs from dorsal skin of newborns pups with Nf1f/f;LacZ (left panel), Hoxb7-Cre;Nf1f/f;LacZ (middle panel), Hoxb7-Cre;Nf1f/f;YFP (right panel). Nf1f/f;LacZ- SKPs and Nf1−/−;LacZ+ or YFP+ SKPs were implanted on the left and right sciatic nerve of nude mice, respectively. H&E and immunohistochemistry to measure expression of Schwann cell marker (S100β, GAP43) and HOXB7 (HOXB7, GFP) were performed on histological sections of the right sciatic nerve. Scale bar = 50μm. (B) Estimation of enrichment of Hoxb7 lineage derived cells in neurofibroma. YFP+ and YFP- cells from SKPs harvested from Hoxb7-Cre;Nf1f/f;YFP were analyzed by FACS to determine the basal level of YFP+ cells. YFP+ cells were evaluated in SKP-induced neurofibroma by immunohistochemistry.
Ablation of Nf1 in Hoxb7 lineage cells gives rise to classic diffuse cutaneous neurofibroma
To definitively demonstrate that cell derived from the Hoxb7 lineage are the cell of origin of cNF, we analyzed the skin phenotype of a cohort of Hoxb7-Cre;Nf1f/f (n=16) and Hoxb7-Cre;Nf1f/- (n=9) mice hereafter called H7;Nf1mut mice. Strikingly, a large fraction (16 out of 25, 64%) of H7;Nf1mut mice developed skin lesions by 1 year of age (Supplemental Table 1). The skin lesions had a relatively large surface of very thick skin, were fully pigmented and developed mainly in the head and neck and upper limb areas (Figure 3A), all characteristics reminiscent of the diffuse type of neurofibroma. To determine whether or not these lesions were restricted to the dermis (diffuse cutaneous neurofibroma) or connected to a nerve plexus below the skin (diffuse plexiform neurofibroma), we carefully dissected the dorsal skin and lifted it up. All tumors examined (16 out of 16, 100%) were exclusively confined to the dermis, as expected for diffuse cNF (Figure 3B). Thus, the H7;Nf1mut mouse model completely recapitulates the characteristic body location, skin thickness, pigmentation and cutaneous restriction of human diffuse cNF shown in Figure 3C, D.
Figure 3. Ablation of Nf1 in Hoxb7 lineage cells give rise to diffuse cutaneous Neurofibroma.
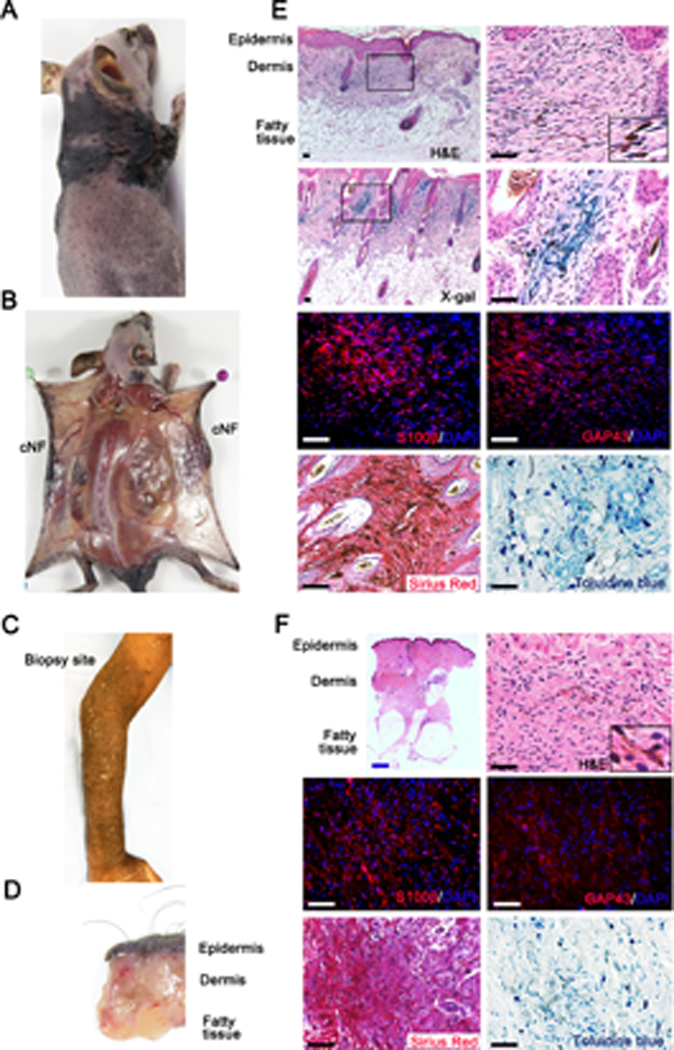
(A) H7;Nf1mut mouse model bearing diffuse cNF. (B) Dissection of H7;Nf1mut mice showing that tumors are contained within the dermis. (C) NF1 patient with diffuse cNF of the forearm. (D) Biopsy of human diffuse cNF showing that tumor is contain within the dermis. (E-F) Histological characterization of murine (E) and human (F) diffuse cNF. Histochemistry was performed with X-Gal staining (Hoxb7 lineage tracing in mice), H&E, Sirius red (collagen), toluidine blue (mast cells), and immunofluorescence with anti-S100β and anti-GAP43 (Schwann cell markers). Black and white scale bar = 50μm. Blue scale bar = 1mm.
Next, we performed histological evaluation of tumor specimens to confirm the skin lesion from the H7;Nf1mut to be of the diffuse cNF type. Hematoxylin and eosin (H&E) staining revealed hypercellularity in the dermis with cells harboring wavy nuclei typical of Schwann cells mixed with collagen fibers (Figure 3E). X-Gal staining indicated that these LacZ+ cells derived from Hoxb7 lineage (Figure 3E, 2nd row panels). Immunofluorescence with Schwann cell-specific marker (S100β, GAP43) and immunohistochemistry with Sirius Red further confirmed these results along with mast cell staining (toluidine blue) (Figure 3E). In addition, accumulation of dermal pigmentation (Figure 3E, upper right panel insert) was observed. Importantly, all these features completely recapitulate human clinical samples of diffuse cNF (Figure 3F). Furthermore, as it is known that the nerve microenvironment plays an important role in neurofibroma development (17, 18), immunostaining for the neuronal marker beta-III tubulin reveals consistent presence of nerve fibers in both mouse and human cutaneous neurofibroma tumors (Supplemental Figure 1). Whereas topical application of tamoxifen in adult CMV-Cre-ERT2;Nf1f/- gave rise to discrete cNF (10), biallelic Nf1 inactivation in a larger population of the cell of origin (Hoxb7) embryonically gave rise to diffuse cNF.
Ablation of Nf1 in Hoxb7 lineage cells also gives rise to plexiform neurofibroma
Hoxb7 lineage-derived cells do not solely populate nerve endings in the dermis, and hence may not be restricted to generate diffuse cNF upon Nf1 deletion. Our Hoxb7 lineage tracing experiment (Figure 1) indicated an overlapping DRG and peripheral nerve staining pattern compared to PLP-Cre-ERT2;LacZ, a mice model used to trace the cell of origin of pNF (16). This suggests that ablation of Nf1 in Hoxb7 lineage-derived cells could trigger the development of pNF in addition to cNF, a phenomenon very frequent in NF1 patients. Some H7;Nf1mut mice began to show clinical signs characteristic of pNF development (scruffy fur, limping, limb paralysis) (16) as early as 5 months (Supplemental Table 1). To quantify pNF tumor burden, we performed a whole spinal cord dissection by carefully preserving peripheral nerves and DRGs in the cohort previously mentioned in Figure 3. About half of the H7;Nf1mut mice showed characteristic enlargement of DRGs and peripheral nerves (Figure 4A, Supplemental Table 1). Histological characterization of both enlarged DRGs and peripheral nerves confirmed the distinguishing characteristics of pNF such as the presence of neoplastic Schwann cell in an abundant collagen matrix (Figure 4B). Interestingly, a very similar number of mice developed either only diffuse cNF, only pNF or both diffuse cNF and pNF (Supplemental Table 1). However, the similar mean age at death for the group of mice developing exclusively cNF or exclusively pNF (Supplemental Table 1) suggests that additional factors may play a role in the development of cNF and pNF. It is also possible that given longer time, these mice will develop both diffuse cNF and pNF. To our knowledge, the H7;Nf1mut mouse is the first model to recapitulate both human cNF and pNF.
Figure 4. Ablation of Nf1 in Hoxb7 lineage cells give rise to plexiform Neurofibroma.
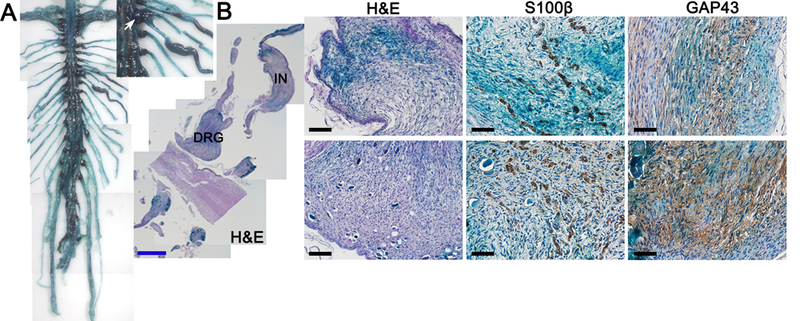
(A) Dissected spinal cord and peripheral nerves from H7;Nf1mut. White arrow = DRG. Black arrow head = intercostal nerve. (B) X-Gal staining and immunohistochemistry with anti-S100β and anti-GAP43 (Schwann cell markers) on intercostal nerve (IN) (upper panels) and dorsal root ganglion (DRG) (lower panels) and from H7;Nf1mut. Black scale bar = 50μm. Blue scale bar = 1mm.
Hippo pathway acts as a modifier of cutaneous neurofibroma
Although H7;Nf1mut mice develop classic diffuse cNF, they do not develop multiple discrete cNF that cover the entire body surface. Therefore, we investigated potential modifiers that could favor the formation of classic cNF. The Hippo pathway regulates cell growth and has been implicated in a number of cancer types (19). Recent whole exome sequencing and pathway analysis of human cutaneous neurofibromas revealed somatic mutations in 3 genes (RASSF1A, SFN (encode the protein 14–3-3) and DLG4) that belong to the Hippo pathway, warranting further investigation of the Hippo pathway in the context of NF1 patients (20). In fact, Neurofibromatosis Type 2 (NF2) patients, which harbor a mutation in a key upstream regulator of the Hippo pathway, can develop some clinical features of NF1 such as cutaneous neurofibromas (21, 22). Therefore, we hypothesized that the Hippo pathway may be a modifier of neurofibromagenesis. To extend the work of Faden et al. (20), we performed mutation analysis on a whole genome sequencing dataset of 33 cutaneous neurofibromas collected from 9 individual NF1 patients (23) and found multiple germline mutation in genes involved in the Hippo pathway (Figure 5A). Very interestingly, one particular tumor had a very high number of variant across the whole exome including 30 sporadic DNA mutations in the Hippo pathway (Figure 5B). These results encouraged us to evaluate the activity of the key effectors of the Hippo pathway (YAP and TAZ). We detected a high expression level of YAP and TAZ in the nucleus of both human discrete and diffuse cNF (Figure 5C). To make sure that the downstream targets of YAP and TAZ are transcribed in cNF, we confirmed that the gene expression level of AXL and CTGF, two established genes modulated by the Hippo pathway (24), were upregulated compared to normal skin margin of NF1 patients (Figure 5D). Thus, the Hippo pathway is dysregulated in at least some human cNF tumors.
Figure 5. Hippo pathway act as a modifier of cutaneous neurofibroma.
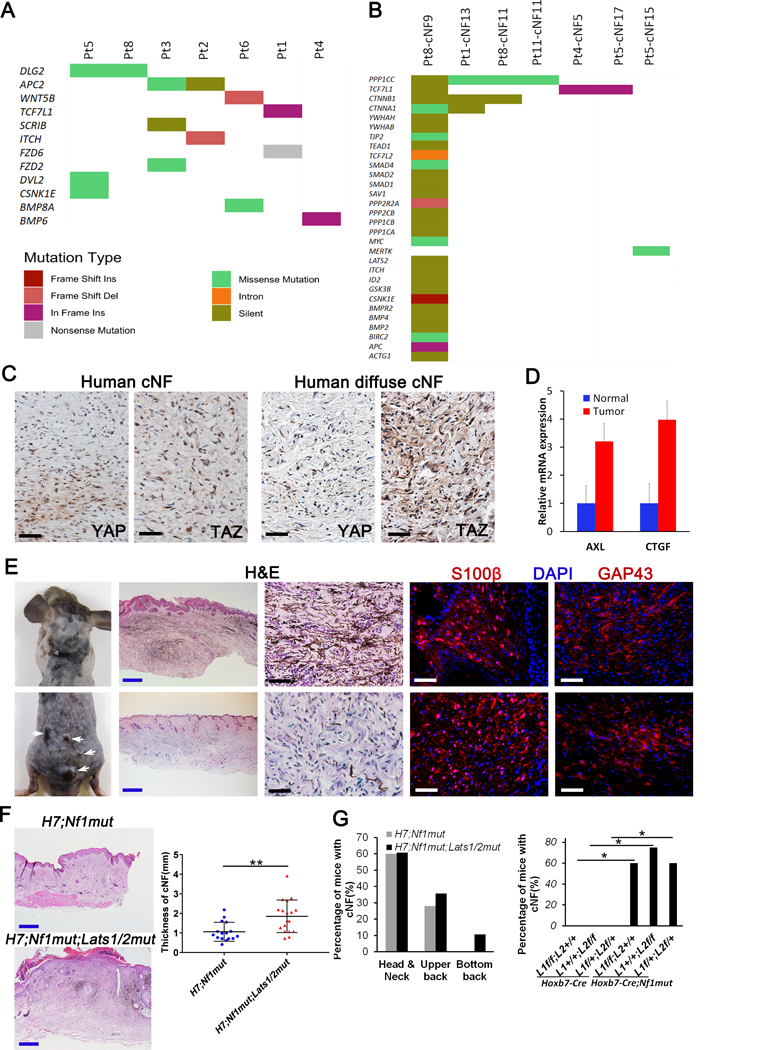
(A-B) Hippo pathway mutation analysis of human cNF. 165 genes known to play a role in the Hippo pathway (literature search and KEGG) was used to mine genomic alterations identified in a dataset of 33 cutaneous neurofibroma collected from 9 individual NF1 patients. Hippo pathway mutations were plotted for both (A) germline (7 out of 9 patients have mutations) and (B) somatic datasets (7 out of 33 cNFs have additional somatic mutations). Colors indicate the type of mutation observed as described in the figure legend. (C) Immunohistochemistry of human discrete (upper panels) and diffuse (lower panels) cNF using anti-YAP and anti-TAZ antibodies. (D) Relative mRNA expression of AXL and CTGF by real-time PCR on 5 cNF and their adjacent normal margin. (E) Representative picture of H7;Nf1mut;Lats1/2mut mice that develop diffuse cNF (upper panel) and discrete cNF nodule (lower panel). Histological characterization of H7;Nf1mut;Lats1/2mut mice that develop diffuse cNF (upper panels) and discrete cNF nodule (lower panels) by H&E and immunofluorescence with anti-S100β and anti-GAP43 (Schwann cell markers). (F) Representative H&E used to measure the skin thickness in H7;Nf1mut (upper left panel) and H7;Nf1mut;Lats1/2mut (lower left panel). Scattered plot representing the skin thickness in function of genotype (right panel) ** = p < 0.01. (G) Bar graph representing the percentage of mice developing diffuse cNF in specific body location (left panel). Bar graph of the percentage of mice developing diffuse cNF in function of genotype * = p < 0.05 (right panel). Black and white scale bar = 50μm. Blue scale bar = 500μm. L1=Lats1. L2=Lats2.
Consequently, we decided to test the impact of altering the Hippo pathway on the development of cNF in vivo. Since Lats1/2 regulate the intracellular localization of the key transcriptional effectors of the Hippo pathway (YAP and TAZ) through phosphorylation, ablating Lats1 and/or Lats2 leads to nuclear accumulation of YAP/TAZ and subsequent transcription of a cell growth gene expression program (19). We crossed the H7;Nf1mut mice to Lats1f/f or Lats2f/f mice to generate H7;Nf1mut mice harboring a combination of two flox allelles from Lats1 and/or Lats2 (H7;Nf1mut;Lats1f/f or H7;Nf1mut;Lats2f/f or H7;Nf1mut;Lats1f/+;Lats2f/+) and hereafter called H7;Nf1mut;Lats1/2mut mice. Strikingly, a fraction of H7;Nf1mut;Lats1/2mut mice (10.7%) develop a mixture of classic discrete nodular cNF as seen in NF1 patients and spreading to almost whole back skin (Figure 5E, far left panels). Histological evaluation of the diffuse (Figure 5E, upper panels) and nodular (Figure 5E, lower panels) mouse cNF confirms that the mouse lesions match human cNFs.
The impact of the modifier can also be appreciated in the diffuse cNF variant. The skin is thicker with infiltrating cNF in the H7;Nf1mut;Lats1/2mut mice (Figure 5F). cNFs also develop in broader areas of different body locations in the H7;Nf1mut;Lats1/2mut mice (Figure 5G, left panel; Supplemental Table 1). Since none of the control mice (Hoxb7-Cre;Nf1+/+;Lats1f/f;Lats2+/+ or Hoxb7-Cre;Nf1+/+;Lats1+/+;Lats2f/f or Hoxb7-Cre;Nf1+/+;Lats1f/+;Lats2f/+, hereafter called H7;Lats1/2mut mice) develop any type of cNF (Figure 5G, right panel), it indicates that the Hippo pathway acts as a modifier for cutaneous neurofibroma tumorigenesis.
Additionally, Nf1 heterozygosity has long been considered as an important supportive factor to sustain neurofibroma tumor microenvironment in Krox20-Cre mouse models (9). However, the requirement of Nf1+/− microenvironment was not fully emulated in another neurofibroma model system by using Dhh-Cre (25), suggesting that Nf1 haploinsufficiency might contribute to neurofibroma development in a context-dependent manner. Therefore, we also re-evaluated the contribution of Nf1 heterozygosity by comparing the tumor progression between Nf1f/f and Nf1f/- mice with Hoxb7-Cre mice. We found that both Hoxb7-Cre;Nf1f/- and Hoxb7-Cre;Nf1f/f from both H7;Nf1mut and H7;Nf1mut;Lats1/2mut mice in Supplemental Table 1 succumbed to neurofibroma development. However, mice in the Nf1f/- group have thicker skin with infiltrating cNF and they died from tumor development much faster than those in the Nf1f/f group (Supplemental Figure 2). These findings revealed that Nf1 heterozygosity is not absolutely required for neurofibroma development. However, inclusion of Nf1 heterozygosity in non-tumor cells significantly enhanced neurofibroma progression, suggesting that germline Nf1 heterozygosity is a modifying factor for neurofibroma development.
Hippo pathway also acts as a modifier of plexiform neurofibroma
Since alteration of the Hippo pathway modified the growth of cNF, we hypothesized that it may also modify the development of pNF. To determine pNF tumor burden, we further performed whole spinal cord dissection as described in Figure 4 to quantify pNF development in the mice cohort previously mentioned in Figure 5. Indeed, para-spinal neurofibromas developed at DRGs are substantially larger in H7;Nf1mut;Lats1/2mut compared to H7;Nf1mut (Figure 6A). Accordingly, histological evaluation revealed a higher cellularity, chaotic distribution of neuron bodies in H7;Nf1mut;Lats1/2mut DRGs compared to H7;Nf1mut (Figure 6B). Moreover, the number of para-spinal NF causing enlarged DRGs was significantly higher in H7;Nf1mut;Lats1/2mut (Figure 6C). Similarly, the diameter of large peripheral nerve such as the sciatic nerve was increased in H7;Nf1mut;Lats1/2mut mice (Figure 6D, F). Histological evaluation revealed that the nerve fiber high disorganization and hypercellularity found in H7;Nf1mut;Lats1/2mut was almost completely absent in H7;Nf1mut and H7;Lats1/2mut sciatic nerves (Figure 6E). Thus, we conclude that the Hippo pathway acts as a modifier to promote para-spinal and peripheral nerve pNF development.
Figure 6. Hippo pathway act as a modifier of plexiform neurofibroma.
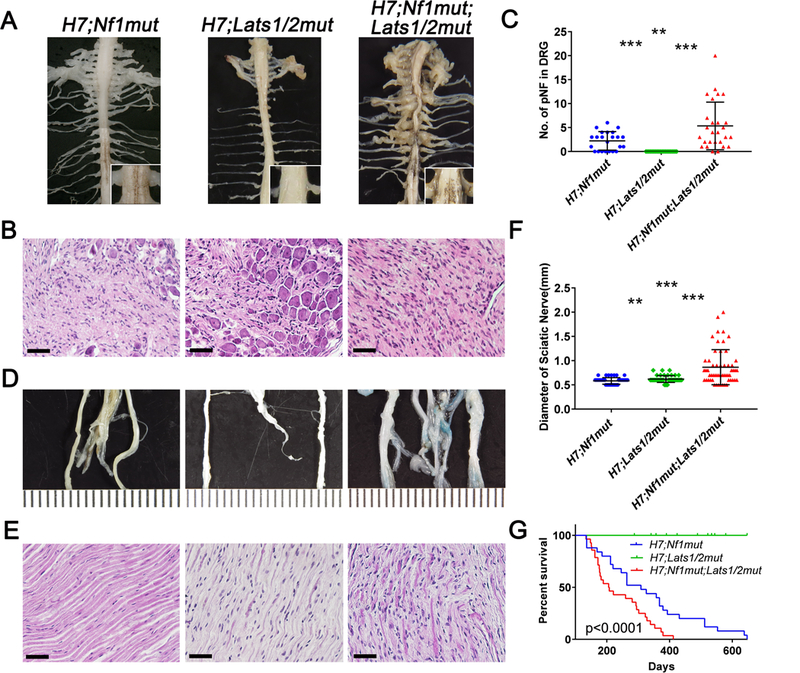
(A, D) Picture of the cervical and trunk spinal cord and peripheral nerves (A) and sciatic nerves (D) of H7;Nf1mut (left panels), H7;Lats1/2mut (middle panels) and H7;Nf1mut;Lats1/2mut (right panels). (B, E) Histological evaluation of DRGs (B) and sciatic nerves (E) in function of the genotype. (C, F) Scattered plot of (C) the number of enlarged DRGs (> 1mm diameter) and (F) diameter of sciatic nerves as a function of genotype. (G) Kaplan-Maier plot illustrating the percentage of mice surviving as a function of time and genotype. ** = p < 0.01; *** = p < 0.001. Scale bar = 50μm.
Finally, we investigated the impact of interfering with the Hippo pathway on the overall survival using a Kaplan-Meier plot. At 200 days, about 50% of H7;Nf1mut;Lats1/2mut already died because of extensive cutaneous tumors or pNF requiring sacrifice, whereas more than 80% and 100% of H7;Nf1mut and H7;Lats1/2mut survived, respectively (Figure 6G). Altogether, it indicates that the Hippo pathway is acting as a modifier of the plexiform in addition to cutaneous neurofibromagenesis and suggests that dampening the Hippo pathway could be an attractive therapeutic target.
Recent studies have shown that estrogen from females correlates with the prevalence of optic glioma, another NF1-associated neoplasm (26, 27). Therefore, in this study, we also utilized our survival data set from both H7;Nf1mut and H7;Nf1mut; Lats1/2mut mice in Supplemental Table 1 to further characterize the plexiform neurofibroma development in this new model and to compare the progression of neurofibromas in male and female mice. Although we observed that 100% of female mice developed plexiform neurofibroma compare to 71% of their male counterparts, both male and female mice developed similar numbers of plexiform neurofibroma in both cervical and thoracic dorsal root ganglia. Furthermore, although more male mice survived beyond 400 days than female mice, the Kaplan-Meier survival analysis between male and female mice is not statistically significant (Supplemental Figure 3).
Hippo pathway dysregulation enhances MAPK pathway activation induced by NF1 loss
To probe the molecular mechanistic link between the Hippo pathway deregulation and its effect on neurofibromagenesis which is driven by NF1 loss leading to MAPK pathway activation, we performed immunohistochemical analysis to monitor MAPK pathway activity using phosphorylated ERK (p-ERK1/2) as a surrogate marker. Indeed, p-ERK1/2 is increased in H7;Nf1mut;Lats1/2mut when normalized to total ERK and compared to H7;Nf1mut in both cNF and pNF (Figure 7A, B). As expected, histological evaluation of skin and DRG from H7;Lats1/2mut revealed minimal change in the normal tissue organization and confirmed the role of Hippo pathway as a modifier of neurofibroma (Figure 7A,B middle panels). We confirmed these results with western blot analysis by quantifying p-ERK1/2 in two sets of experiments. First, we compared mouse embryonic DRG/Nerve root Schwann cell precursors (the cell of origin for pNF) (16) with or without ablation of one or two copies of Lats2 and observed an increase in the level of p-ERK1/2 (Figure 7C). Second, we compared skin and DRG from wild type mice versus H7;Nf1mut, H7;Lats1/2mut and H7;Nf1mut;Lats1/2mut and observed again an increase in the level of p-ERK1/2 in tissues from H7;Nf1mut;Lats1/2mut mice (Figure 7D). Altogether, we reasoned that the Hippo pathway deregulation enhances the MAPK pathway activation by NF1 loss.
Figure 7. Hippo pathway inactivation enhances MAPK pathway activation induced by NF1 loss.
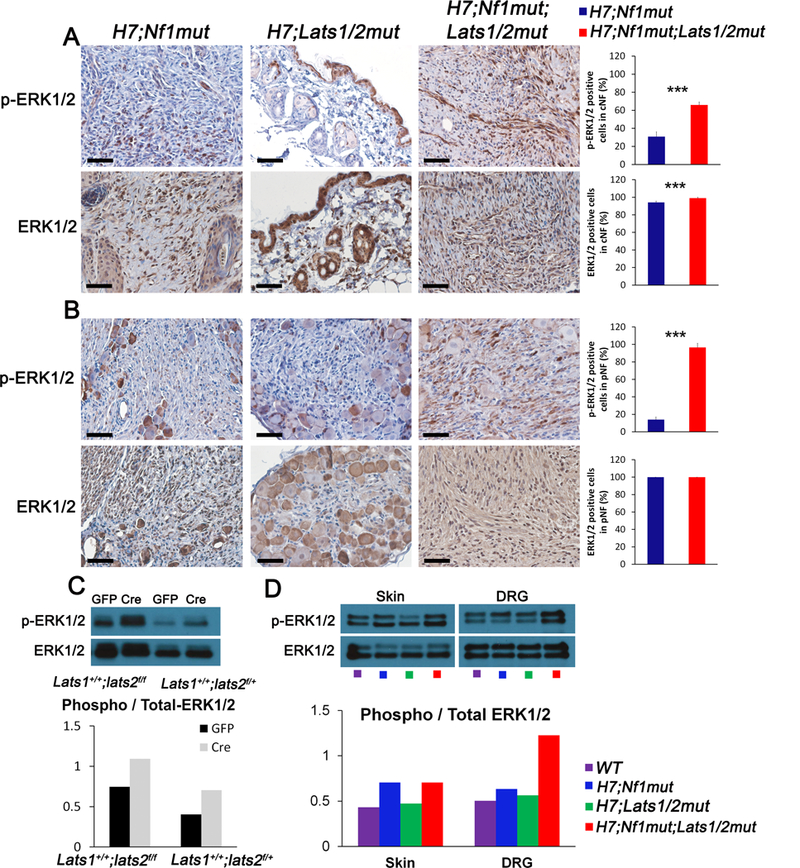
(A-B) Representative immunohistochemistry using anti p-ERK1/2 (upper panels) and anti-total ERK1/2 (lower panel) of (A) skin, (B) DRG of H7;Nf1mut (left panels), H7;Lats1/2mut (middle panels) and H7;Nf1mut;Lats1/2mut (right panels). Bar graphs (far right) indicate the quantification as percentage of positive cells found in A,B. (C) Western-Blot analysis using anti p-ERK1/2 and anti-total ERK1/2 in mouse embryonic DRG/Nerve root sphere cells from Lats1+/+;Lats2f/f and Lats1+/+;Lats2f/+ treated with adenovirus cre (Cre) or control adenovirus (GFP) (upper panel). Quantification as ratio of p-ERK1/2 over total ERK1/2 immunoblot (lower panel). (D) Western-Blot analysis using anti p-ERK1/2 and anti-total ERK1/2 in whole tissue extract from murine skin and DRG from wild type (WT), H7;Nf1mut, H7;Lats1/2mut and H7;Nf1mut;Lats1/2mut. Quantification as ratio of p-ERK1/2 over total ERK1/2 immunoblot (lower panel).
Discussion
NF1 patients present with a wide range of phenotypic manifestations, including tumors that develop at different time point during lifetime. Several NF1 mice faithfully model human NF1-associated tumor such as pNF, astrocytomas, MPNST and pheochromocytomas, both histologically and with regards to NF1 mutation status (28). However, the most prevalent pathological signs of NF1 occur in the skin in the form of cNFs and pigmentation. Ironically, these characteristic features are currently the most challenging to model in vivo. We discovered that deleting Nf1 in the Hoxb7 lineage derived cells generates both cNF and pNF spontaneously, faithfully recapitulating human neurofibromatosis. In addition to the histological signatures and NF1 status, this novel NF1 mouse model highlights three more hallmarks of human neurofibroma: 1) Neurofibroma exclusively affects the dorsal sensory nerve fibers; 2) Diffuse cNF is consistently present with pigmented skin over cNF; 3) Neurofibroma development is susceptible to modifiers.
Spatio temporal loss of NF1 control the types and subtypes of neurofibroma
From a developmental perspective, most of the tissues sensitive to NF1 gene dosage are derived from the neural crest. Neural crest generates an important number of tissues above the shoulder including the facial muscles and bones (29) as well as the peripheral nervous system (14). Indeed, Nf1 LOH in Schwann cell lineage lead to neurofibroma development (9, 30), Nf1-/- melanocytes lead to café-au-lait macules (CALM) (3) and Lisch nodules (iris hamartoma), NF1-/- osteoblast lead to pseudoarthrosis of the tibia (31), Nf1-/- in glial cells leads to astrocytomas (4). Attempts to study the development of neurofibromas through the use of a neural crest-specific Cre driver in these mice have failed because mice do not survive long enough to allow the development of phenotypic manifestations (12). To circumvent this issue, Schwann cell-specific Cre were successfully used (9, 17, 25, 32, 33). Since cNF and pNF have identical histological features including neoplastic Schwann cells, one can expect to find a mixture of cNF and pNF in these mouse models. Strikingly, none of these models develop cNF (9, 17, 25, 32, 33). This means that although both tumors contain Schwann cells, they may not come from the same cell of origin. In fact, the biology of these tumors is widely different. First, pNF involves internal nerve plexuses and can further progress to a malignant tumor while a malignant form of cNF has rarely if ever been reported. Second, the onset of pNFs can be as early as the embryonic stage of development whereas the cNFs typically occur around puberty and cNFs are exclusively in the skin. Thus, the presence of at least two cells of origin populations within the Schwann cell lineage may explain the different types of neurofibroma due to inactivation of NF1 in two different spatio-temporal locations.
As cells of origin for both cNF and pNF originate from the neural crest, one can then envision three scenarios (34): if the Nf1 LOH occurs before their bifurcation into distinct lineage, then it will lead to both cNF and pNF development (as in Hoxb7-Cre). If Nf1 LOH occurs only in the pNF cell of origin after the bifurcation, only pNF develop (as in PLP-Cre and Krox20-Cre). On the other hand, if Nf1 LOH only occurs in the cNF cell of origin, then this will lead to cNF development without pNF (as in topical induction with CMV-Cre-ERT2). At first sight, this model does not seem to reflect what is observed in clinic where most if not all NF1 patients that develop pNF also have cNF. One explanation for this may be the mechanism by which mutations or LOH are introduced in mouse models but occur spontaneously in humans. In order to develop pNF, biallelic Nf1 inactivation has to occur precisely at define embryonic stage, a narrow window of opportunity. In humans, this window spans only a short amount of time during embryonic development and the “2nd hit” happens stochastically. In contrast, in mice, the time and cell type where the Nf1 biallelic inactivation occurs is controlled and purposely made in the pNF cell of origin. Thus, a minority of NF1 patient develop pNF and even if they do, most of them will eventually also develop cNF because the window for developing solely pNF is very small. On the other hand, the cell of origin of cNF is present throughout adulthood, giving ample time to undergo Nf1 inactivation. This may explain why a near-totality of adult patients bear cNF. Thus, our model reconciles the apparent discrepancies between in vivo mouse experiment and human clinical scenarios.
Spatio temporal loss of NF1 control pigmentation
Modeling NF1 skin pigmentation in mice is not trivial because mice and human have different pigmentation mechanisms (35). Human skin gets pigmented because its epithelial cells uptake the melanin produced by melanocytes through Stem Cell Factor (SCF) dependent signaling, resulting in interfollicular pigmentation. In mice, the production of SCF in interfollicular epithelial cells drastically drops at about two weeks after birth. Therefore, melanocytes still produce melanin but only in the hair follicle. Consequently, murine skin color is dependent on the presence of hair and follows the temporal cycle of hair growth. In addition, it is technically challenging to formally demonstrate the origin of melanocytic cells due to the inherent nature of dark pigmentation overwhelming any co-localization signal. Therefore, and not surprisingly, the mechanism underlying pigmentation defect in the context of NF1 is still a matter of debate and remains elusive.
Nf1+/− mice exhibit a subtle darkening of the skin (36) and biallelic inactivation of Nf1 using melanocyte-specific Cre mice phenocopy this trait (36, 37). The hyperpigmentation phenotype in these models is spread across the body and accumulation of pigmented cells takes place in the dermis. Of note, the skin darkening can be easily overlooked by naked eyes. Since Nf1-/- melanocyte from the Tyr and Mitf lineage also originate from neural crest cells and HOXB7 expression coincides with the first wave of neural crest cell migration, it is possible that these melanocytic lineage cells are in the Hoxb7 lineage. Consistent with this, we observed more pronounced pigmentation of tails and paws in H7;Nf1mut mice suggesting that the melanocyte responsible for the global and subtle pigmentation in NF1 may be derived from the neural crest derived melanocyte within the Hoxb7 lineage.
The fact that in diffuse cNF, skin hyperpigmentation surface matches the tumor area in the dermis suggests that Nf1-/- Schwann cell and Nf1-/- melanocytes may share the same cell of origin (a neural crest cell progenitor). Indeed, very early in neural crest migration, SOX10+ Mitf+ stem cells (melanocyte precursor) migrate in a dorsolateral pathway to reach the skin dermis (38) whereas PLP+ and Krox20+ cells (Schwann cell precursors) start to appear at E10.5 (16). However, this hypothesis has never been demonstrated in mice because the Cre mice strains used up to now were not early enough in the neural crest lineage to encompass the melanocyte lineage. Strikingly, we observed that ablating Nf1 in the Hoxb7 lineage results in the concomitant generation of pigmented skin on top of cutaneous neurofibroma. As seen in NF1 patients, the location of the tumor is mostly restricted to the head & neck area. We conclude that the type of cells, their location and the timing of NF1 loss along the developmental course may dictate skin pigmentation in NF1 patients.
Modifiers of Neurofibromagenesis
Several lines of evidence indicate that modifiers may explain some of the high phenotypic variability observed within a sub-group of NF1 patients sharing the same germline mutation. A genome-wide high resolution array-comparative genomic hybridization strategy identified single nucleotide polymorphism in the gene ANRIL that is associated specifically with the genesis of pNF (39). Interestingly, type 1 microdeletions encompassing the NF1 gene is associated with a high number of cNF and an increased risk for malignant peripheral nerve sheath tumor (MPNST) in NF1 patients. In the context of MPNST, the modifier function has been genetically linked to SUZ12 (40) but it is currently unknown if SUZ12 or any other gene that is commonly inactivated with NF1 in the setting of these large gene deletions play a role in determining the number of cNF. Other interesting candidate genes were identified and need further validation (41, 42).
Robust cNF mouse model recapitulating the human cNF represents an important milestone. H7;Nf1mut mice develop a robust diffuse subtype of cNF rather than the discrete cNF found in many NF1 patients. Therefore, we were looking for factors beyond the cell type of origin and the timing of the Nf1 biallelic inactivation that could modify the development of cNF (body location, time of onset, gross morphology) to be more representative of the discrete human cNF. In the context of mouse pNF, the Nf1+/− microenvironment is known to promote the development of pNF (9). In fact, and depending on the context, the Nf1+/− microenvironment may be absolutely required (9); without significant effect (17); or anywhere between these two extremes. In the current study, we didn’t find any significant difference in the percentage of mice developing cNF between Nf1+/+ and Nf1+/− backgrounds, although mice with the Nf1+/− background develop both cNF and pNF and succumbed to death earlier than the Nf1+/+ background. Therefore, we looked for alternative modifiers of neurofibromagenesis.
The Hippo pathway, which regulates cell growth and has been implicated in a number of cancer types (19), may be altered in a subset of cNF based on whole exome sequencing analysis (20). We reported here a high expression level of the key effectors of the Hippo pathway (YAP and TAZ) in the nucleus of both human discrete and diffuse cNF. Our data supports the clinical fact that individuals with mutation in the Hippo regulator NF2 develop Neurofibromatosis type 2, where cNF can grow (21, 22). Moderately altering the Hippo pathway in addition to Nf1 deletion modifies the course of neurofibroma development. H7;Nf1mut;Lats1/2mut mice generates higher numbers of enlarged DRGs and peripheral nerves as well as much thicker skin lesions where cNFs developed. There is also a trend toward a faster tumor onset compared to H7;Nf1mut mice and a broader body location for tumor distribution. Strikingly, nodular cNFs similar to the classic discrete cNF found in NF1 patient were observed in a subset of H7;Nf1mut;Lats1/2mut mice that was not found in control littermates (H7;Nf1mut or H7;Lats1/2mut mice). In contrast to the diffuse cNF, these tumor nodules were not restricted to a particular body location, a characteristic shared with the human scenario. Furthermore, moderately affecting the Hippo pathway alone (H7;Lats1/2mut mice) does not produce any skin or nervous system tumor even after 1 year of age. It is also possible that we underestimated the number of mice developing discrete cNF as the high tumor burden of diffuse cNF and pNF required early mice sacrifice. Mechanistically, there are many ways NF1 could interact with the Hippo pathway. Since it is known that NF1 plays an inhibitory role in the MAP kinase pathway in cutaneous neurofibroma, and it is also known that there is cross-talk reported between the MAP kinase and the Hippo pathway in some cancer types (43–45), one possibility could be that the Hippo pathway is modulated indirectly due to NF1 loss of function-induced MAP kinase oversignaling. Alternatively, the Hippo pathway could be tempered in neurofibroma through genetic mutation in the Hippo pathway (20) and (Figure 5A). This suggests that dampening the Hippo pathway may serve as part of a comprehensive treatment approach for cNF and pNF, specifically using new inhibitors in development for human clinical trials that target the Hippo pathway (46).
In conclusion, we discovered that Hoxb7 serves as a lineage marker to trace the developmental origin of cNF neoplastic cells. Ablating Nf1 in the Hoxb7 lineage faithfully recapitulates both human cutaneous and plexiform neurofibroma. In addition, we discovered that the Hippo pathway acts as a modifier to promote neurofibroma tumorigenesis. This novel mouse model opens the doors for deciphering the evolution of cNF to identify effective therapies, where none exists today.
Methods
In vivo mouse studies and human cNF biopsy
The Nf1 knockout (8), Nf1 flox (9), Hoxb7-Cre (47), Lats1 flox (48), Lats2 flox (48) ROSA26-YFP (49), ROSA26-lacZ and athymic nude mice (Foxn1−/−) are available from Jackson Laboratory (Bar Harbor, ME). Genotyping was performed by PCR as previously reported (8, 9, 47–49). All mice were housed in the Animal Care Facility at The University of Texas Southwestern Medical Center (Dallas, TX), and all procedures were approved by Institutional Animal Care and Use Committee at University of Texas Southwestern Medical Center and conformed to NIH guidelines. Human subjects and cNF sample collection and use were approved by Institutional Review Board at University of Texas Southwestern Medical Center and conformed to NIH guidelines. Written informed consent was obtained from patients.
SKP isolation and cell culture
SKP isolation, culture and sciatic nerve implantation protocols were performed as previously described (50). Briefly, dorsal skin was cut to yield about 1 square inch of tissue and was dissected into small pieces. To isolate SKP cells, the suspension of tissue pieces was centrifuged (30 sec, 1200 rpm), supernatant was discarded and resuspended in a solution of PBS (Sigma, D8662) (8 mL) and collagenase I (2 mL) by inversion and let it moderately shake (37°C, 40 min). Next, tissue pieces were mechanically disrupted by up and down pipetting (10 times), were centrifuged (10 min, 2000 rpm, 4°C), supernatant was discarded and was resuspended in ice cold HBSS (10 mL). The procedure was repeated twice starting back at the mechanical pipetting disruption step. Finally, the resuspension was filtered on a 70 um sterile cell strainer and viable cells were counted (trypan blue). To enrich for SKPs, 1 × 106 cells per 10 cm dish were seeded on ultra-low culture dish (Corning, 3262). After 3–4 days, half of the supernatant (5 mL) was discarded and fresh SKP complete media (5 mL) was added.
SKP complete media
To make SKP base media: Mix and filter at 0.22um heparin 0.2% (1 mL), glucose 30% (1 mL), NaHCO3 7.5% (750 uL), HEPES 1M (250 uL), glutamine (5 mL), sodium pyruvate (5 mL), penicillin-streptomycin (5 mL), N2 supplement (Gibco, 1750248) (5 mL) into 500 mL of DMEM/F12. To make complete SKP media: Add basic fibroblast growth factor (Sigma, F0291) (0.16 ng), Epidermal growth factor (Gibco 13247–051) (0.8 ng), amphoterin B (160 uL) and B27 (800 uL) into SKP base media (40 mL). Use within 2 weeks.
Histology
Tissues were fixed in 10% formalin-buffered solution for at least 48 h. Then tissues were paraffin embedded (Rushabh Instruments LLC), were sectioned at 5 um using a microtome (Leica RM2135) and were allowed to dry on glass slide at room temperature. Tissue slides were deparaffinized, were progressively rehydrated and were stained with hematoxylin (2 min) followed by high definition (10 sec), bluing agent (10 sec) and eosin (H&E staining) or a solution of Sirius Red (0.5g of Direct Red80 dissolved in 500 mL of saturated picric acid) for 1h (collagen staining) or a solution of toluidine blue (0.05g of toluidine blue O dissolved in 70% ethanol (5 mL) and freshly further diluted in 1% sodium chloride (45 mL)) for 2 min (toluidine blue staining). Finally, tissue slides were progressively dehydrated and coverslipped.
X-Gal staining
Tissues were briefly incubated in 4% paraformaldehyde (10 min, r.t.), were rinsed twice with PBS 1X followed by incubation into X-Gal staining solution (X-Gal 1 mg/mL, KFerro 1X, KFerri 1X, MgCl2 50 mM) for 24–48h and tissues were finally fixed with 10% formalin-buffered solution.
Immunohistochemistry
Tissue slides were blocked with 10% donkey serum in PBS (1h) and were incubated with primary antibodies (Rabbit anti-YAP (Cell signaling; AB-2650491); Rabbit anti-TAZ (abclonal; AB-2721146); Rabbit anti-Hoxb7 (Novus; AB-2721144); Rabbit anti-S100β (Dako; AB-10013383); Rabbit anti-GAP43 (Abcam; AB-2247459); Rabbit anti-βIII-tubulin (Sigma-Aldrich AB-262133) diluted in 3% donkey serum (16h, 4°C). The next day, tissue slides were rinsed in PBS (3 × 5 min), were incubated with secondary antibodies coupled to biotin and diluted in 3% donkey serum (1h), were rinsed again in PBS (3 × 5 min), were incubated with a pre-mixture of avidin and biotin (following Vecta Stain Elite ABC kit procedure) (30 min), were rinsed again in PBS (3 × 5 min) and were visualized by adding the DAB substrate (following Vecta Stain Elite ABC kit procedure). Finally, reaction was quenched in distilled water and tissue slides were counterstained with hematoxylin, dehydrated and were cover slipped. A brown precipitate was deposited on positive cells.
Immunofluorescence
Tissue slides were blocked with 10% donkey serum in PBS (1h) and were incubated with primary antibodies (Rabbit anti-S100β (Novus; AB-2184423); Rabbit anti-GAP43 (Abcam; AB-2247459); Chicken anti-GFP (Aves Labs; AB-10000240) diluted in 3% donkey serum (16h, 4°C). The next day, tissue slides were rinsed in PBS (3 × 5 min), were incubated with secondary antibodies coupled to a fluorophore from Jackson Immunoresearch (Cy3-AffiniPure Donkey anti-Rabbit; AlexaFluor488, Donkey anti-Goat) and diluted in 3% donkey serum (1h), were rinsed again in PBS (3 × 5 min), Slides were visualized under fluorescence microscope.
Mouse Skin tumor dissection
Skin lesion body location was classified as follow: Head and neck is defined as diffuse cNF located at least above the shoulder; upper back tumor is defined as diffuse cNF located at least between the shoulder and the upper 50% of the dorsal skin and bottom back is defined as diffuse cNF where tumor located at least in the lower 50% of the dorsal skin. The number of mice with diffuse cNF in these 3 body locations were expressed as percentage over the total number of mice for each genotype. After euthanasia, a skin incision along the spinal cord was performed and the skin was lifted up to evaluate potential involvement of subcutaneous region. Finally, skin tumors of interest were excised, were rinsed with PBS 1X and either quickly immersed in 10% formalin-buffered solution or submitted to X-Gal staining procedure.
Mouse whole spinal cord dissection
Whole spinal cord dissection was performed as previously reported (33). To perform mouse anesthesia, a mixture of ketamine (10 mg/ml) and xylazine (1 mg/ml) solution (100 uL per 25g of mouse) was administered intraperitoneally. After 15–20 min, mouse was placed face up in a surgical field and the chest area was sprayed with 70% ethanol. The left thoracic cage was removed, a catheter was installed in the heart left ventricle and the mouse was perfused intracardiacally with 4% para-formaldehyde. Then, the mouse was prepared for microscopic dissection by removing gross tissue (cervical decapitation, whole skin removal and all internal organs). Next, muscle and other tissue were carefully removed and bones from the vertebrate column were broken one by one under dissection microscope to end up with intact spinal cord and peripheral nerves. Finally, whole spinal cord and peripheral nerves were rinsed with PBS 1X and either immersed in 10% formalin-buffered solution or submitted to X-Gal staining procedure.
Skin thickness measurement
To quantify the thickness of skin samples, digital H&E images were reviewed and a line was drawn between epidermis and the tumor area (perpendicular to the epidermis layer) using the cellsenstandard software. Then, the longest line (in mm) was used as a data point to generate the scatter plot in Figure 5 (one data point per mouse).
Sciatic nerve diameter measurement
To quantify the sciatic nerve diameter, sciatic nerve from dissected whole spinal cord were reviewed and the point at which L3, L4 and L5 merge was selected as the measurement location (51). Then, the left and right nerve diameters were measured using a vernier caliper and were used to generate the scatter plot in Figure 6F (two data points per mouse).
DRGs size measurement
To quantify the number of para-spinal neurofibromas, all DRGs from dissected whole spinal cord were measured using a vernier caliper. DRGs with a diameter of at least 1mm were considered neurofibromas (52). The numbers of neurofibromas were counted and were used to generate the scatter plot in Figure 6C (one data point per mouse).
Estimation of Hoxb7 positive SKPs
To estimate the fraction of SKPs derived from Hoxb7 lineage, SKPs from Hoxb7-Cre;Nf1f/f;YFP were passage and final resuspension was done in sterile PBS. SKPs were analyzed on a FACSAria instrument (BD Biosciences) equipped with a 488 nm solid-state laser where green fluorescence was detected using 490 LP and 510/20 filters. The fraction of YFP positive SKPs was estimated using FACSDiva software. Analyses were repeated on SKP cultures derived from at least 3 mice.
Estimation of Hoxb7 positive cells in neurofibroma
To estimate the fraction of cells in neurofibroma derived from Hoxb7 lineage, SKPs from Hoxb7-Cre;Nf1f/f;YFP were implanted into sciatic nerve of nude mice. Resulting neurofibromas were analyzed by immunohistochemistry using anti-GFP antibodies for the presence of GFP positive cells. Total cell number (nuclei) and GFP positive cells were counted in at least 3 random fields from at least 3 different neurofibroma-bearing mice.
Gene expression measurement
RNA was extracted from 30 mg of tissue using the Trizol reagent followed by on-column DNAse digestion as described elsewhere (53). Reverse transcription step was performed using iScript Select cDNA synthesis kit as per manufacturer recommended procedure using a random priming strategy. Real-time PCR assays validation for AXL (5’-TGGAAGGCCAGCTCAACCAG-3’ and 5’-TGCAGACCGCTTCACTCAGG-3’) and CTGF (5’-ACCTGTGCCTGCCATTACAA-3’ and 5’-GCTTCATGCCATGTCTCCGT-3’) were performed as described elsewhere (54) and normalized with GAPDH (5’- AGGGCTGCTTTTAACTCTGGT-3’ and 5’- CCCCACTTGATTTTGGAGGGA-3’).
Hippo pathway mutation plotting
To investigate hippo pathway mutations in a clinical dataset, somatic and germline variant calls were acquired from the CTF Cutaneous Neurofibroma Data Resource: http://doi.org/10.7303/syn4984604 (23). This database includes summarized germline mutation data from 9 patients and somatic mutation data from 33 patient-matched cutaneous neurofibroma samples. All analysis was performed in R 3.5 (https://r-project.org/). Hippo pathway genes were identified by taking the union of genes listed in the literature and in the KEGG database (hsa04390) (55). Somatic and germline mutations in the Hippo pathway were summarized and plotted using the R package GeneVisR (56). The scripts used to summarize the variants and generate this figure is available here: http://doi.org/10.5281/zenodo.1273864.
Quantification and Statistical Analysis
All data are displayed as the mean ± SEM unless specified otherwise. A two-tailed t test and Fisher exact test were applied as appropriate to evaluate statistical significance (* = p < 0.05; ** = p < 0.01; *** = p < 0.001).
Supplementary Material
Significance.
This study provides insights into the developmental origin of cNF, the most common tumor in NF1, and generates the first mouse model that faithfully recapitulates both human cutaneous and plexiform neurofibroma. The study also demonstrates that the Hippo pathway can modify neurofibromagenesis, suggesting that dampening the Hippo pathway could be an attractive therapeutic target.
Acknowledgments
We thank all members of the Le lab for helpful suggestions and discussions.
Grant Supports
J.P. Brosseau and C.P. Liao are recipients of the Young Investigator Award from Children’s Tumor Foundation. C.P. Liao also receives a Career Development Award from Dermatology Foundation. J.M. Cooper is funded by the Dermatology Research Training Program T32 Grant T32AR065969. L.Q. Le holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund and the Thomas L. Shield, M.D. Professorship in Dermatology. This work was supported by funding from the National Cancer Institute of the NIH grant number R01 CA166593, the US Department of Defense grant number W81XWH-17–1-0148, the Giorgio Foundation, the Neurofibromatosis Therapeutic Acceleration Program and the NF1 Research Consortium Fund to L.Q. Le.
Footnotes
The authors declare no potential conflicts of interest
References
- 1.Xu GF, O’Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R, et al. 1990. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62:599–608. [DOI] [PubMed] [Google Scholar]
- 2.Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, and Collins F 1990. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 63:851–859. [DOI] [PubMed] [Google Scholar]
- 3.Maertens O, De Schepper S, Vandesompele J, Brems H, Heyns I, Janssens S, Speleman F, Legius E, and Messiaen L 2007. Molecular dissection of isolated disease features in mosaic neurofibromatosis type 1. Am J Hum Genet 81:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, and Gutmann DH 2003. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res 63:8573–8577. [PubMed] [Google Scholar]
- 5.Lin AL, and Gutmann DH 2013. Advances in the treatment of neurofibromatosis-associated tumours. Nat Rev Clin Oncol 10:616–624. [DOI] [PubMed] [Google Scholar]
- 6.Chander V, Rao RS, Sekhar G, Raja A, and Sridevi M 2015. Recurrent Diffuse Neurofibroma of Nose Associated with Neurofibromatosis Type 1: A Rare Case Report with Review of Literature. Indian J Dermatol 60:573–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Megahed M 1994. Histopathological variants of neurofibroma. A study of 114 lesions. Am J Dermatopathol 16:486–495. [DOI] [PubMed] [Google Scholar]
- 8.Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, and Weinberg RA 1994. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet 7:353–361. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Y, Ghosh P, Charnay P, Burns DK, and Parada LF 2002. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science 296:920–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le LQ, Shipman T, Burns DK, and Parada LF 2009. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell 4:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toma JG, Akhavan M, Fernandes KJ, Barnabe-Heider F, Sadikot A, Kaplan DR, and Miller FD 2001. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol 3:778–784. [DOI] [PubMed] [Google Scholar]
- 12.Gitler AD, Zhu Y, Ismat FA, Lu MM, Yamauchi Y, Parada LF, and Epstein JA 2003. Nf1 has an essential role in endothelial cells. Nat Genet 33:75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacques-Fricke BT, Roffers-Agarwal J, and Gammill LS 2012. DNA methyltransferase 3b is dispensable for mouse neural crest development. PLoS One 7:e47794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jessen KR, and Mirsky R 2005. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci 6:671–682. [DOI] [PubMed] [Google Scholar]
- 15.Lopez SL, Dono R, Zeller R, and Carrasco AE 1995. Differential effects of retinoic acid and a retinoid antagonist on the spatial distribution of the homeoprotein Hoxb-7 in vertebrate embryos. Dev Dyn 204:457–471. [DOI] [PubMed] [Google Scholar]
- 16.Chen Z, Liu C, Patel AJ, Liao CP, Wang Y, and Le LQ 2014. Cells of origin in the embryonic nerve roots for NF1-associated plexiform neurofibroma. Cancer Cell 26:695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ribeiro S, Napoli I, White IJ, Parrinello S, Flanagan AM, Suter U, Parada LF, and Lloyd AC 2013. Injury signals cooperate with Nf1 loss to relieve the tumor-suppressive environment of adult peripheral nerve. Cell Rep 5:126–136. [DOI] [PubMed] [Google Scholar]
- 18.Liao CP, Pradhan S, Chen Z, Patel AJ, Booker RC, and Le LQ 2016. The role of nerve microenvironment for neurofibroma development. Oncotarget 7:61500–61508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harvey KF, Zhang X, and Thomas DM 2013. The Hippo pathway and human cancer. Nat Rev Cancer 13:246–257. [DOI] [PubMed] [Google Scholar]
- 20.Faden DL, Asthana S, Tihan T, DeRisi J, and Kliot M 2017. Whole Exome Sequencing of Growing and Non-Growing Cutaneous Neurofibromas from a Single Patient with Neurofibromatosis Type 1. PLoS One 12:e0170348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, Pikus A, and Patronas N 1994. Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet 52:450–461. [DOI] [PubMed] [Google Scholar]
- 22.Evans DG 2009. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis 4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gosline SJ, Weinberg H, Knight P, Yu T, Guo X, Prasad N, Jones A, Shrestha S, Boone B, Levy SE, et al. 2017. A high-throughput molecular data resource for cutaneous neurofibromas. Sci Data 4:170045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang SD, McCrudden CM, Yuen HF, Leung KL, Hong WJ, and Kwok HF 2016. Association between the expression levels of TAZ, AXL and CTGF and clinicopathological parameters in patients with colon cancer. Oncol Lett 11:1223–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, Stemmer-Rachamimov AO, Cancelas JA, and Ratner N 2008. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell 13:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toonen JA, Solga AC, Ma Y, and Gutmann DH 2017. Estrogen activation of microglia underlies the sexually dimorphic differences in Nf1 optic glioma-induced retinal pathology. J Exp Med 214:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diggs-Andrews KA, Brown JA, Gianino SM, Rubin JB, Wozniak DF, and Gutmann DH 2014. Sex Is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol 75:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brossier NM, and Carroll SL 2012. Genetically engineered mouse models shed new light on the pathogenesis of neurofibromatosis type I-related neoplasms of the peripheral nervous system. Brain Res Bull 88:58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santagati F, and Rijli FM 2003. Cranial neural crest and the building of the vertebrate head. Nat Rev Neurosci 4:806–818. [DOI] [PubMed] [Google Scholar]
- 30.Serra E, Rosenbaum T, Winner U, Aledo R, Ars E, Estivill X, Lenard HG, and Lazaro C. 2000. Schwann cells harbor the somatic NF1 mutation in neurofibromas: evidence of two different Schwann cell subpopulations. Hum Mol Genet 9:3055–3064. [DOI] [PubMed] [Google Scholar]
- 31.Kolanczyk M, Kuhnisch J, Kossler N, Osswald M, Stumpp S, Thurisch B, Kornak U, and Mundlos S. 2008. Modelling neurofibromatosis type 1 tibial dysplasia and its treatment with lovastatin. BMC Med 6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayes DA, Rizvi TA, Cancelas JA, Kolasinski NT, Ciraolo GM, Stemmer-Rachamimov AO, and Ratner N 2011. Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer Res 71:4675–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le LQ, Liu C, Shipman T, Chen Z, Suter U, and Parada LF 2011. Susceptible stages in Schwann cells for NF1-associated plexiform neurofibroma development. Cancer Res 71:4686–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown RM, Klesse LJ, and Le LQ 2010. Cutaneous features predict paraspinal neurofibromas in neurofibromatosis type 1. J Invest Dermatol 130:2167–2169. [DOI] [PubMed] [Google Scholar]
- 35.Fitch KR, McGowan KA, van Raamsdonk CD, Fuchs H, Lee D, Puech A, Herault Y, Threadgill DW, Hrabe de Angelis M, and Barsh GS 2003. Genetics of dark skin in mice. Genes Dev 17:214–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deo M, Huang JL, Fuchs H, de Angelis MH, and Van Raamsdonk CD 2013. Differential effects of neurofibromin gene dosage on melanocyte development. J Invest Dermatol 133:49–58. [DOI] [PubMed] [Google Scholar]
- 37.Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter S, Flaherty K, et al. 2013. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov 3:338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dupin E, and Sommer L 2012. Neural crest progenitors and stem cells: from early development to adulthood. Dev Biol 366:83–95. [DOI] [PubMed] [Google Scholar]
- 39.Pasmant E, Sabbagh A, Masliah-Planchon J, Ortonne N, Laurendeau I, Melin L, Ferkal S, Hernandez L, Leroy K, Valeyrie-Allanore L, et al. 2011. Role of noncoding RNA ANRIL in genesis of plexiform neurofibromas in neurofibromatosis type 1. J Natl Cancer Inst 103:1713–1722. [DOI] [PubMed] [Google Scholar]
- 40.De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, Helin K, Hornick JL, Mautner V, Kehrer-Sawatzki H, et al. 2014. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514:247–251. [DOI] [PubMed] [Google Scholar]
- 41.Titze S, Peters H, Wahrisch S, Harder T, Guse K, Buske A, Tinschert S, and Harder A 2010. Differential MSH2 promoter methylation in blood cells of Neurofibromatosis type 1 (NF1) patients. Eur J Hum Genet 18:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu J, Liu W, Williams JP, and Ratner N 2017. EGFR-Stat3 signalling in nerve glial cells modifies neurofibroma initiation. Oncogene 36:1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reddy BV, and Irvine KD 2013. Regulation of Hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Dev Cell 24:459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nussinov R, Tsai CJ, Jang H, Korcsmaros T, and Csermely P 2016. Oncogenic KRAS signaling and YAP1/beta-catenin: Similar cell cycle control in tumor initiation. Semin Cell Dev Biol 58:79–85. [DOI] [PubMed] [Google Scholar]
- 45.Feng R, Gong J, Wu L, Wang L, Zhang B, Liang G, Zheng H, and Xiao H 2017. MAPK and Hippo signaling pathways crosstalk via the RAF-1/MST-2 interaction in malignant melanoma. Oncol Rep 38:1199–1205. [DOI] [PubMed] [Google Scholar]
- 46.Woodard GA, Yang YL, You L, and Jablons DM 2017. Drug development against the hippo pathway in mesothelioma. Transl Lung Cancer Res 6:335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu J, Carroll TJ, and McMahon AP 2002. Sonic hedgehog regulates proliferation and differentiation of mesenchymal cells in the mouse metanephric kidney. Development 129:5301–5312. [DOI] [PubMed] [Google Scholar]
- 48.Yi J, Lu L, Yanger K, Wang W, Sohn BH, Stanger BZ, Zhang M, Martin JF, Ajani JA, Chen J, et al. 2016. Large tumor suppressor homologs 1 and 2 regulate mouse liver progenitor cell proliferation and maturation through antagonism of the coactivators YAP and TAZ. Hepatology 64:1757–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, and Costantini F 2001. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Z, Pradhan S, Liu C, and Le LQ 2012. Skin-derived precursors as a source of progenitors for cutaneous nerve regeneration. Stem Cells 30:2261–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rigaud M, Gemes G, Barabas ME, Chernoff DI, Abram SE, Stucky CL, and Hogan QH 2008. Species and strain differences in rodent sciatic nerve anatomy: implications for studies of neuropathic pain. Pain 136:188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu J, Keng VW, Patmore DM, Kendall JJ, Patel AV, Jousma E, Jessen WJ, Choi K, Tschida BR, Silverstein KA, et al. 2016. Insertional Mutagenesis Identifies a STAT3/Arid1b/beta-catenin Pathway Driving Neurofibroma Initiation. Cell Rep 14:1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Venables JP, Klinck R, Bramard A, Inkel L, Dufresne-Martin G, Koh C, Gervais-Bird J, Lapointe E, Froehlich U, Durand M, et al. 2008. Identification of alternative splicing markers for breast cancer. Cancer Res 68:9525–9531. [DOI] [PubMed] [Google Scholar]
- 54.Brosseau JP, Lucier JF, Lapointe E, Durand M, Gendron D, Gervais-Bird J, Tremblay K, Perreault JP, and Elela SA 2010. High-throughput quantification of splicing isoforms. RNA 16:442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanehisa M, Furumichi M, Tanabe M, Sato Y, and Morishima K 2017. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45:D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Skidmore ZL, Wagner AH, Lesurf R, Campbell KM, Kunisaki J, Griffith OL, and Griffith M 2016. GenVisR: Genomic Visualizations in R. Bioinformatics 32:3012–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.