

Abstract
Physical changes in skin are among the most visible signs of aging. We found that young dermal fibroblasts secrete high levels of extracellular matrix (ECM) constituents, including proteoglycans, glycoproteins and cartilage-linking proteins. The most abundantly secreted was HAPLN1, a hyaluronic and proteoglycan link protein. HAPLN1 was lost in aged fibroblasts, resulting in a more aligned ECM that promoted metastasis of melanoma cells. Reconstituting HAPLN1 inhibited metastasis in an aged microenvironment, in 3D skin reconstruction models, and in vivo. Intriguingly, aged fibroblast-derived matrices had the opposite effects on the migration of T-cells, inhibiting their motility. HAPLN1 treatment of aged fibroblasts restored motility of mononuclear immune cells, while impeding that of polymorphonuclear immune cells, which in turn affected Treg recruitment. These data suggest while age-related physical changes in the ECM can promote tumor cell motility, they may adversely impact the motility of some immune cells, resulting in an overall change in the immune microenvironment. Understanding the physical changes in aging skin may provide avenues for more effective therapy for older melanoma patients.
INTRODUCTION
Melanoma, the malignant transformation of epidermal melanocytes, is the leading global cause of skin cancer related deaths. Increasing age is a negative prognostic indicator, and elderly patients with melanoma have inferior disease-specific survival even when controlling for primary tumor factors (1). While age-related differences in tumor molecular pathways and host immune response may partly underlie these findings (2), the influence of age on the architectural changes that may govern immune and tumor cell trafficking through the skin have not been well studied. Previously, we reported that fibroblasts in the aged dermal microenvironment (age >55 years) contribute to melanoma tumor progression by secreting factors that promote metastasis and resistance to targeted therapy (3). In the present study, we performed a proteomics analysis of secreted factors from fibroblasts from young (<45 years) and aged (>55) human donors, and found striking changes specifically in a group of proteins associated with the integrity of the skin extracellular matrix (ECM).
Human skin is characterized by an epidermal layer comprised primarily of keratinocytes and a dermal layer comprising mostly of dense collagen-rich ECM largely secreted by dermal fibroblasts (4). Age-related changes in the physical properties of skin include decreases in collagen density (5, 6), ECM fiber area and thickness (7–9) as well as changes in the mechanical properties of the ECM such as stiffness (6). Collagen crosslinking with fibulin, fibrillin and elastin (10, 11) further enhances its structural stabilization (10, 12, 13). Changes in the turnover of these proteins are known to occur during natural aging (14). Specifically, collagen fibers in young skin are known to intersect in what is known as a “basketweave” pattern, where fibers cross each other at ~90° angles (15). This pattern breaks down during aging, giving way to a decreasingly dense matrix, that has larger gaps between collagen fibers. These changes further contribute to mechanical and structural alterations, often visible as wrinkles in the skin.
Changes in matrix stiffness and density have long been associated with invasion of tumor cells. We recently developed a mathematical fiber network model that simulates the deformation of collagen networks (16) induced by cellular forces such as those experienced during the invasion of cancer cells, which led us to re-evaluate and refine the current thinking that linear increases in the stiffness of the ECM promote metastasis. Instead, we hypothesized that stiffness may be relative, depending in which organ a tumor arises. For example, a breast cancer cell may arise in a “soft” environment that requires immense plasticity during lactation, and menstruation, and this may need to stiffen for optimal invasion. A melanoma however, arises in the skin, which by definition must form a strong, stiff barrier against external insults. Our data supported this, suggesting that when stiffness increases from a very soft “loose” ECM to a stiffer one, invasion increases; as elegantly reported in breast cancer studies (17). However, as fiber crosslinking and ECM stiffness increase further, a biphasic (e.g., as opposed to linear) tendency is evident in which cells under these conditions are no longer able to pass through tightly cross linked pores. Our published model takes into account discrete morphological alterations in the ECM, such as the realignment of the fibers and strain-stiffening, predicting a deformation zone around a contractile cell (18). This model was supported by our in vitro experiments showing that the fibrous nature and mechanical properties of the crosslinked ECM play key roles in the ability of the cells to invade (19). Hence our data, based on spheroid models, are more consistent with recent data showing that 3D cell invasion is enhanced by increasing matrix stiffness and alignment until pore size becomes constrained and restricts cellular motility (20). We confirmed our models in 3D spheroid assays and further showed that this effect was both proliferation and MMP-independent (18). In the present study we query the effect of aging on changes in the architecture of the ECM, and how those affect the migration of tumor and immune cells.
Our analysis of young vs. aged fibroblast secretomes demonstrated that aging results in marked decreases in proteins involved in ECM production and remodeling, including Fibulin, Agrin, and the Hyaluronan and Proteoglycan Link Protein 1 (HAPLN1). We focused this study on HAPLN1, because it was the most differentially expressed protein in our study, and was highly expressed by young fibroblasts. HAPLN1 is a crosslinking protein that stabilizes proteglycan monomer aggregates with hyaluronic acid (HA) (21). HA alterations have been shown to increase the ability of fibroblasts to contract collagen matrices, suggesting that changes in HAPLN1 could affect collagen crosslinking and ECM contractility. In the present study, we specifically assess the role of HAPLN1, its loss during aging, and its effects on tumor cell motility. Further, we queried the effect of HAPLN1 loss on immune cell infiltrates, since immune cells have been shown to rely on dense collagen networks to move in and out of sites such as wounds, and the tumor microenvironment (22). Overall, using a mixture of matrix deposition assays, novel 3D immune infiltration assays, and orthotopic mouse models, we show that increasing HAPLN1 in the aged microenvironment inhibits invasion and metastasis of tumor cells, while also promoting an active immune microenvironment. An understanding of ECM architecture in mediating age-related melanoma progression reveals a unique pathway to increase the effectiveness of current therapies and improve disease outcome in elderly patients.
Results
Aging affects the structural organization of the dermal extracellular matrix
We performed a proteomics study on the secretome of aged vs. young fibroblasts. Fibroblasts were obtained from young (<45 years) and aged (>55 years) healthy donors who participated in the Baltimore Longitudinal Study of Aging (23) and have been used previously to model the aging microenvironment (3, 24). To achieve robust and unbiased identification of factors that are differentially regulated in the secretome during normal aging, a SILAC quantitative proteomics experiment was performed on the secretomes of young/aged fibroblasts that had been cultured in “heavy” or “light” media with serum for nine cell doublings prior to incubation in the same media without serum for 8 h. This serum-free media was used for proteome analysis and 937 proteins were quantified with a protein and peptide false discovery rate of <1.0%. A total of 90 proteins showed significant changes and were used to identify the top pathways that change during fibroblast aging (Figure 1A, S1A). For quality control purposes, fibroblasts were assessed for viability in serum-free medium using calcein AM staining (Figure S1B), to ensure cells were still viable in the serum-free conditions used in the last phase of the SILAC experiment (Figure S1C-E). A list including all proteins exhibiting significant differences in abundance is shown in Supplemental Table 1. Notably, the most significantly changed proteins, between young and aged fibroblastic secretomes, were those involved in ECM remodeling (HAPLN 1, agrin, laminin β2, fibulin, tenascin).
Figure 1: The extracellular matrix is significantly altered during aging.

a) Secretome analysis from conditioned media from young and aged fibroblasts showing top overexpressed proteins in both and young and aged microenvironment. b) Dermal fibroblasts from healthy human donors were used to prepare skin reconstructs. Invasion and collagen deposition was assessed by H&E staining (upper panels; bar=100microns) as well as two-photon microscopy (bottom panels; bar=25microns). The color scale is indicative of the thickness of the collagen. c) C57/BL6 mouse skin was assessed for collagen composition using two-photon microscopy (bar=25 microns). d) Young and aged dermal fibroblasts were allowed to form matrices and color-coded for fiber alignment. More colors represent less alignment. Panel shown in black and white indicates no significant matrix. Results are also quantified along with controls. N/A indicates no significant matrix formed. e) Skin reconstructs were prepared using multiple young (GM01948, GM02674) and aged (AG11726, AG12988) fibroblasts and matrix alignment was measured. f) Young and aged C57/BL6 mice were injected with Yumm1.7 tumors and analyzed for matrix orientation at the skin/tumor interface. g) Normal human non-melanoma skin from young and aged donors was stained for expression of HAPLN1 and scored based on intensity (3-highest, 0-lowest/absent).
To test the effects of the biomechanical changes of the aged skin on melanoma metastasis, we used our in vitro models to emulate the structural changes observed in the skin during aging. We prepared artificial reconstructions of skin (3D skin reconstructs) using young and aged dermal fibroblasts, to recreate the dermis, along with epidermal keratinocytes and melanoma cells as previously described (3). We first confirmed reported increases in the invasive ability of melanoma cells in the aged microenvironment (3). Moreover, the collagen in reconstructs made with aged fibroblasts had a lacier and looser appearance by H&E staining, which corresponded to less dense collagen auto-fluorescent signal by two-photon microscopy (Figure 1B). Similar age-related changes in collagen could also be captured by two-photon microscopy in the normal mouse skin (in the absence of tumor), where aged mice demonstrated a loss of dermal collagen density with an increase in the dead space between more narrow collagen fibers (Figure 1C). To determine whether the observed changes in collagen fibrils during aging translated to altered matrix contractility, we embedded young or aged fibroblasts in collagen gels, and measured the ability of the fibroblasts to contract the gels. The percent of contraction was greater in aged fibroblasts (Figure S2A, B).
Our data suggested that aged fibroblasts organize their ECM in a manner different to that of young fibroblasts. To better analyze changes in the ECM during aging, we employed an established in vitro cell-derived matrix (CDM) technique used to recreate ECM production by fibroblasts alone, in the absence of tumor cells (25, 26). In this model, fibroblasts are treated with ascorbic acid to stabilize collagen secretion and assure its incorporation into CDM fibrils. The manner by which the matrix is deposited (e.g., aligned/anisotropic vs. unaligned/isotropic) is reflective of the activation state of fibroblasts (27). Activated fibroblasts are determined by their ability to align CDMs to a greater extent (anistropic); >55% of fibers distributed 15 degrees from the mode angle. The aged matrices were characterized as producing anisotropic CDMs, (Figure 1D), and relatively decreased ECM thickness (Figure S2C). Young matrices, on the other hand, were isotropic and appeared to be denser than aged CDMs (Figure 1D). Fibronectin levels did not change during aging (Figure S2D). Transforming growth factor beta (TGFβ), which is known to promote ECM remodeling, is often used to activate dermal fibroblasts, driving them towards a cancer-associated fibroblast (CAF) phenotype (28). We find that young fibroblasts are responsive to TGFβ, becoming activated, and modify their CDMs to become anisotropic (Figure 1D); The matrices laid down by aged fibroblasts are already aligned, and TGFβ serves to further increase their anisotropy (Figure 1D). Interestingly, treatment of young fibroblasts with a TGFβ inhibitor disrupts their matrix production, whereas aged fibroblasts are less responsive to the TGFβ inhibitor (Figure 1D).
Since these observations were made using only fibroblasts, we wanted to explore whether they remained consistent in a 3D setting that includes dermal and epidermal components. First, we explored the matrix alignment of the ECM in our skin reconstructs built using young or aged fibroblasts, and found that the CDM anisotropy seen using aged fibroblasts alone was recapitulated in the skin reconstructs (Figure 1E). To extend these data to an in vivo model, we assessed the ECM in the skin of young and aged C57/BL6 mice. Aged mice (>52 weeks of age) showed an increased matrix alignment as compared to the skin of young mice that were 6 weeks of age (Figure 1F). Overall these data suggest that aged fibroblasts can significantly change the architecture of the ECM, in manner consistent with that seen in activated, tumor-permissive fibroblasts. Conversely, young fibroblasts lay down a dense and isotropic matrix that is more consistent with the reported “basket weave” pattern (15).
Of all proteins measured (Figure 1A, Supplemental Table 1), the most significantly changed was HAPLN1. We observed a 35-fold decrease in HAPLN1 in aged fibroblasts compared to young. We confirmed that HAPLN1 was increased in young fibroblasts compared to aged by performing ELISA on the conditioned media of 9 young and 9 aged fibroblast lines (Figure S3A). This was also true for normal young (<45 years) vs. aged (>55 years) human skin (Figure 1G). To determine whether TGFb was able to directly affect HAPLN1 levels in young and aged fibroblasts, we treated fibroblasts directly with TGFb1 and 2, and found no significant difference in the levels of HAPLN1 (Fig S3B, C). Next we queried the correlation of HAPLN1 gene expression with age using the TCGA databases of cutaneous melanoma, and found that HAPLN1 is decreased during aging in melanoma patient samples (Figure S3D). Because HAPLN1 was decreased in the melanoma samples, we then asked whether HAPLN1 secretion from dermal fibroblasts affects the HAPLN1 levels in melanoma cells. We found that indeed, melanoma cells exposed to young fibroblast conditioned media increase HAPLN1 production (Figure S3E, F). Overall our results suggest that HAPLN1 is an abundant dermal protein secreted by fibroblasts, which is decreased during aging.
HAPLN1 regulates the structural organization of the dermal microenvironment.
A dense network composed mostly of collagen and elastin fibers along with large “space-filling” proteoglycans maintains the firmness of young skin. These discrete proteoglycans are bound to collagen with hyaluronic acid. HAPLN1 is purported to play a role in the crosslinking of hyaluronic acid and proteoglycans (21). Given the significant decrease of HAPLN1 in aged fibroblasts, we asked whether low levels of HAPLN1 contributed to the alteration of the young collagen basket weave structure rendering the observed increased alignment of dermal ECM fibers associated with aging. Using the ECM alignment assays described above, we tested CDM phenotypes in vitro using aged fibroblasts (in the absence of tumor cells) treated with increasing doses of HAPLN1 to achieve the physiological range that was detected via ELISA in the young fibroblasts as measured in Figure S3A. Aged fibroblasts showed increases in collagen basket weave structure with increasing concentrations of HAPLN1 (Figure 2A, a second fibroblast line is shown in S4A). We analyzed the percentage of fibers oriented within the 30° range and observed a rHAPLN1 dose-dependent decrease in the anisotropic levels of aged CDM fibers, suggesting that HAPLN1 decreases matrix activation (Figure 2A, S4B). Denatured HAPLN1 did not affect CDM fibers (Figure 2B). Conversely, we knocked down HAPLN1 in young fibroblasts (Figure S4C) and questioned how this affects CDM architecture. As expected, the high degree of CDM isotropy seen in the young fibroblasts was lost with HAPLN1 knockdown (Figure 2C). This could be rescued by simply adding back HAPLN1 (Figure 2D, S4D,E), emphasizing the importance of HAPLN1 in mediating fibroblastic CDM organization. The corresponding CDMs also showed HAPLN1 mediated changes in the thickness of the matrix, where increased HAPLN1 promoted thicker matrix deposition (Figure S4F), and knockdown of HAPLN1 resulted in decreased matrix deposition (Figure S4G) and changes in SMA indicative of fibroblast activation (Figure S4H).
Figure 2: HAPLN1 loss in aging microenvironment promotes extracellular matrix remodeling.

a) Matrix production by aged fibroblasts was carried out in presence of various concentrations of recombinant HAPLN1, and alignment of the matrix was measured. b) Aged fibroblasts were treated with active or denatured HAPLN1 (25ng/ml), allowed to form matrices and matrix alignment was measured. c) Young fibroblasts with HAPLN1 knockdown were allowed to produce matrix and assessed for matrix production. d) Young fibroblasts with HAPLN1 knockdown were allowed to form matrices with or without presence of rHAPLN1 (25ng/ml) and assessed for matrix alignment. e) Matrices were produced by young fibroblasts with HAPLN1 knockdown and aged fibroblasts with rHAPLN1 treatment (25ng/ml). Matrices were assessed for Collagen I by immunofluorescence (bar=25microns). f) Skin reconstructs were prepared with young fibroblasts with HAPLN1 knockdown and aged fibroblasts with rHAPLN1 treatment (25ng/ml). Skin reconstructs were embedded in paraffin and imaged for collagen bundles using Differential Interference Contrast (DIC) microscopy. g) Aged C57/BL6 mice were treated intradermally with 100ng (50ng/mL) rHAPLN1 for 7 days, followed by two photon imaging for dermal collagen (bar=25microns). h) Collagen was embedded with aged fibroblasts treated with varying concentrations of rHAPLN1 and young fibroblasts with HAPLN1 knockdown and layered in 48 well plates and assessed for contractility over 3 days (ANOVA (young+ shHAPLN1) 0.0022, (aged+rHAPLN1) 0.0031). For all experiments, ANOVA post-hoc tests were performed using Bonferroni correction and the significance values are shown in the figures.
Since CDM assays are conducted largely by staining for fibronectin, we also wanted to stain and look for other changes in the ECM. First, we stained the CDMs for collagen, by immunofluorescence, and noted a discernible change in the distribution of collagen that occurs during aging (Figure 2E, top panels). This can be mimicked in young fibroblasts by knocking down HAPLN1, or the collagen loss reversed in aged fibroblasts by adding in recombinant HAPLN1 (Figure 2E, lower panels). DIC imaging of the CDMs also shows age-related changes in collagen that can be reversed by HAPLN1 (Figure 2F). We also measured changes in the expression of alpha smooth muscle actin (ɑ-SMA), known to be upregulated during desmoplasia, and in naïve fibroblastic cells in response to CAF CDMs (7, 27, 29). Consistent with these reports, we observed activated levels of ɑ-SMA in aged fibroblasts, which decreased in a dose-dependent manner to rHAPLN1 treatment (Figure S4H). Conversely, knockdown of HAPLN1 in young fibroblasts, assessed during ECM production, showed an upregulation in ɑ-SMA levels (Figure S4H).
To extend these results to in vivo models, we injected rHAPLN1 (100ng) in aged (>52 weeks) C57/BL6 mice and observed a loss of collagen fiber anistropy (Figure 2G, Figure S4I,J). To determine whether this correlated to the ability of the fibroblasts to contract collagen, a feature of activated fibroblasts and of CAFs, we treated aged fibroblasts with increasing doses of rHAPLN1, and measured their ability to contract collagen. With increased HAPLN1, collagen contractility decreased, consistent with the results observed in young fibroblasts (Figure 2H). Similarly, knocking down HAPLN1 in young fibroblasts increased their ability to contract collagen gels (Figure 2H). Overall these data suggest that HAPLN1 plays a major role in maintaining the dense basket weave structure of the collagenous ECM in young skin, while maintaining relatively low ɑ-SMA levels, and that its loss in aged skin can promote increased ɑ-SMA and matrix alignment.
Mathematical modeling suggests that HAPLN1 alters matrix organization, and inhibits motility.
The increase in matrix alignment and the loss of a tight basketweave structure suggested that the loss of HAPLN1 might promote cell invasion, given that an increase in porosity of the matrix might lead to a more invasion-permissive matrix. To predict how HAPLN1 impacts invasion of cancer cells, we used a mathematical model that takes into account the effect of inter-fiber crosslinks on the deformation of collagenous ECMs and the two-way feedback between cellular contractility and the strain-stiffening behavior of fibrous ECMs. We modeled the ECM as a network of crosslinked filaments that can deform by both bending and stretching. The network model reproduced the experimentally observed mechanical behavior of collagen networks in stretch. An initial linear stress-strain behavior was observed at small strains (smaller than ~5%) followed by strain-stiffening of at large strains (greater than ~5%, Figure 3A). The network model showed that the presence of HAPLN1 constrains the deformation of the network, leading to an increase in their stiffness (Figure 3A). Furthermore, the additional crosslinks constrained the lateral bending and buckling of fibers and hindered the reorganization and reorientation of the stretched fibers. Therefore, less fiber alignment was observed as the networks were stretched in the presence of HAPLN1.
Figure 3. Chemo-Mechanical model for HAPLN1 restriction of tumor invasion into the extracellular matrix.
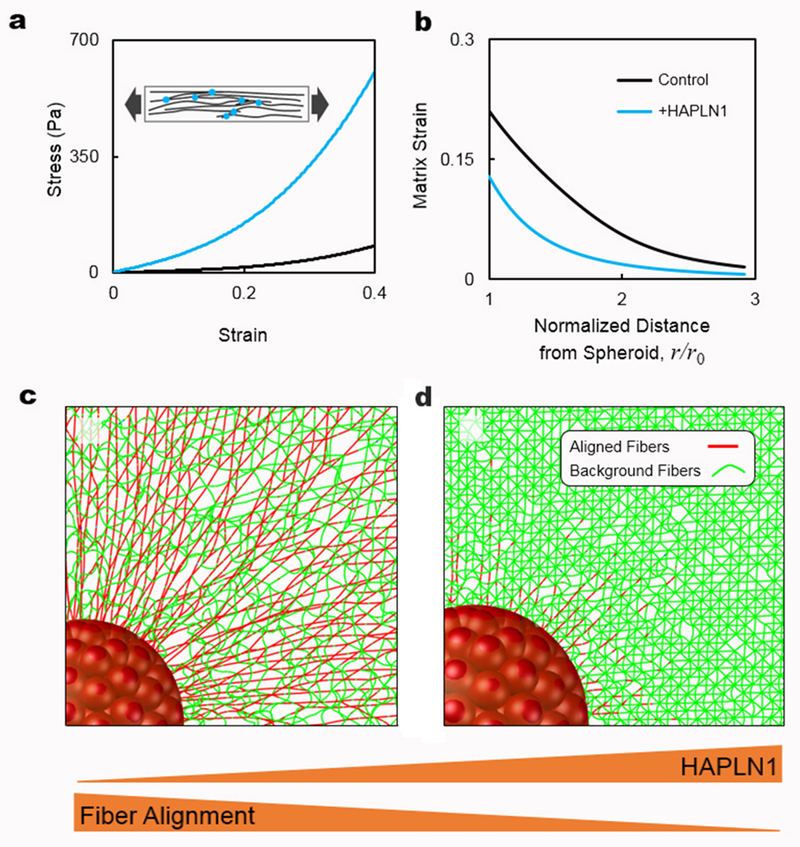
a) Stress-strain curves of fibrous networks with and without HAPLN1 obtained using stretch tests. The addition of HAPLN1 is modeled by the formation of crosslinks between nearby fibers. The inset shows a schematic of the fibrous networks in the stretch tests. The fibers and crosslinks are shown in black and red, respectively. b) The results from panel (a) are then used to inform our chemo-mechanical model about the mechanical behavior of fibrous matrices with and without HAPLN1. Strain in the matrix is plotted as a function of distance from the tumor spheroid. c) Fibers are aligned by the contractility of a tumor spheroid in the control case. The aligned and background fibers are displayed in red and green, respectively. Fibers are colored based on the stretching force present in the fibers. d) Reduced matrix deformation and fiber alignment are observed in the case where more fibers are crosslinked by HAPLN1. At the same level of strain, fewer fibers buckle in the matrix with HAPLN1 because crosslinks constrain the lateral bending of individual fibers, preventing the alignment of fibers due to cell contractility.
By incorporating the results of our network simulations, we next determined the influence of HAPLN1 on the structural organization of the ECMs in the presence of tumor spheroids. We modeled a spherical cluster of cells (with radius ~200 μm) embedded in ECMs containing or lacking HAPLN1. Following our previous work (18, 30), cells within the spheroid were modeled using a combination of a passive elastic element placed in parallel with an active contractile element. The passive element accounts for the stiffness of the cytoskeleton in response to external forces whereas the active element represents the contractility of the cells (e.g., as suggested by the observed changes in ɑ-SMA levels). The details of the mathematical model for the dependence of the contractility of the cells on the external stresses are presented in the Materials and Methods Section.
Our model demonstrates that the addition of HAPLN1 substantially hinders the realignment of fibers by the tumor spheroid. As the tumor cells exert contractile forces on the surrounding ECM, they realign collagen fibers in the direction perpendicular to the boundary of the spheroid (31). Our model showed that the tumor spheroid induces more strain in the matrix that lacks HAPLN1 (Figure 3B). The presence of large strains in the absence of HAPLN1 leads to the substantial alignment of fibers that radiate from the tumor spheroid (Figure 3C). In contrast, in the presence of HAPLN1, the higher stiffness of the matrix hinders contraction of the cells and imposes high stresses on the cells. In this case, our model shows that the spheroid induces less strain and fiber alignment in the matrix since the crosslinking of fibers due to HAPLN1 constrains the lateral bending and buckling of fibers; Individual fibers buckle more easily if they are free to bend in the lateral direction. Taken together, our model suggests that in the presence of HAPLN1, cellular forces cannot reorganize the matrix, and the fibers remain randomly oriented, crosslinked, and unaligned/isotropic (Figure 3D).
Structural ECM organization influences melanoma cell invasion
Cell motility is thought to be a function of stiffness of the matrix as well as intrinsic invasiveness of the cells (32–34). We hypothesized that the aging microenvironment influences the intrinsic invasiveness by providing external cues to the cells to either limit or increase their motility. First, we assessed the ability of the ECMs to affect non-directional motility of melanoma cells by obtaining cell-free CDMs from young and aged fibroblasts and introducing melanoma cells into these matrices. We observed changes in cell shape, where cells seeded on aged, desmoplastic matrices were increasingly elongated and motile, as assessed by time-lapse acquisition analysis (Figure S5A). Next, to study the implications of HAPLN1 changes in CDMs, we recreated the matrix microenvironment to study the effect of HAPLN1 on the motility of melanoma cells. We found that melanoma cells plated on CDMs deposited by aged fibroblasts decrease their velocity if the fibroblasts were treated with rHAPLN1 during ECM production (Figure S5B, Supplemental Movies S1–4). Conversely, melanoma cells plated into young HAPLN1 knockdown fibroblasts CDMs increased their velocity of movement (Figure S5C, Supplemental Movies S5–7). Using 3D skin reconstructs we also showed that increasing HAPLN1 added together with aged fibroblasts altered the provided collagen, resulting in a decrease in melanoma invasion (Figure 4A), while loss of HAPLN1 in young fibroblasts led to alterations that resulted in increased melanoma cell invasion (Figure 4B). Results were supported by decreased invasion in melanoma spheroids embedded in collagen gels, embedded with aged fibroblasts, that were polymerized in the presence of increasing concentrations of HAPLN1 (Figure S5D), as well as by the increased invasion of melanoma spheroids that were co-cultured with HAPLN1 knockdown fibroblasts (Figure S5E). HAPLN1 also affects the motility of melanoma cells in the absence of fibroblasts (Figure S5F).
Figure 4: HAPLN1 decreases invasion and metastasis of melanoma cells.

a) Skin reconstructs were prepared with aged fibroblasts treated with varying concentrations of rHAPLN1 and invasion was calculated as a percent of reconstruct thickness (ANOVA p<0.0001, bar=100microns). b) Skin reconstructs prepared with young fibroblasts with HAPLN1 knockdown and 1205lu melanoma cells. Invasion of melanoma cells into the collagen layer was calculated as a percentage of reconstruct thickness (ANOVA, p<0.0001, bar=100microns). c) Aged C57/BL6 mice (>300 days old) were injected with Yumm1.7 melanoma cells overexpressing mCherry and treated with rHAPLN1 intra-dermally. Tumor growth was followed for 6 weeks (ANOVA p<0.0001). d) Lungs from mice injected with mCherry overexpressing Yumm 1.7 cells were assessed for metastatic burden by IHC (bar=100microns). e) total metastatic burden in mice from (d).
Finally, our data have shown that tumors in aged mice metastasize more effectively to the lung (3). Given the effects of HAPLN1 on in vitro invasion, we asked whether HAPLN1 treatment of aged mouse skin could inhibit melanoma invasion. We implanted invasive Yumm1.7 melanoma cells into aged (>52 week) C57/BL6 mice and treated the mice with 50ng/mL rHAPLN1 intra-dermally. Tumors in mice treated with rHAPLN1 were much smaller (Figure 4C) and had decreased lung metastatic burden rates (Figure 4D,E), compared to vehicle-treated controls. These data confirmed the predictions made by the mathematical models, and suggested that loss of HAPLN1 facilitates invasion of melanoma cells in aged skin.
Immune cell infiltration is also dependent on crosslinking of the tumor-associated ECM
The observed effects of HAPLN1 on tumor size were intriguing, and we asked whether HAPLN1 affected tumor cell proliferation. In vitro assays confirmed that HAPLN1 alone did not affect apoptosis of melanoma cells (Figure S6A, B), nor cellular proliferation in skin reconstructs (Figure S6C), nor did tumors in HAPLN1 treated mice show any loss of Ki67 activity (Figure S6D, E). These data suggested that perhaps decreased growth in the tumors in the HAPLN1 treated mice was reflective of an immunogenic response, such as increased T-cell infiltration. While the pore size of the basket weave is a restrictive factor for tumor cells that have a relatively large nucleus, we predicted that it would be less so for immune cells, which have much smaller nuclei, as suggested by the existing literature (35). Further, T cells have been known to travel along interstitial collagen fibers in an MMP-independent manner, to target the tumor (36–38). Hence, we hypothesized that T cell infiltration might be affected by the HAPLN1 levels in the tumor microenvironment. To test this in vitro, we embedded melanoma spheroids in collagen along with T cells that have been clonally expanded and demonstrated to target the melanoma cell line in vitro (39). Melanoma spheroids in the presence of aged fibroblast media were embedded into collagen plugs containing autologous T-cells, and varying concentrations of rHAPLN1. As HAPLN1 concentrations increased, the T-cells showed a dose dependent increase in the velocity of the T cell infiltrates targeting the melanoma cells that sprouted from tumor spheroids (Figure 5A). Interestingly, T cells were not able to penetrate into the spheroids in the presence of aged conditioned media; however, adding rHAPLN1 induced an increase in T cells spheroid infiltration (Figure 5B). Next we embedded melanoma spheroids and T-cells into collagen plugs containing young fibroblasts in which HAPLN1 had been knocked down. Loss of HAPLN1 in the young microenvironment decreased the velocity of T cells (Figure 5C), and inhibited the infiltration of the T cells into the melanoma spheroid as compared to the empty vector controls (Figure 5D). Loss of HAPLN1 decreased collagen density and affected the accumulation of the T cells along collagen fibrils (Figure S7A). This phenotype was rescued by the addition of recombinant HAPLN1, such that the T-cells reacquired velocity, and penetrated the spheroid once again (Figure 5E).
Figure 5: T cell motility is affected by HAPLN1 in the ECM.

a) Melanoma spheroids expressing mCherry were allowed to form spheroids, mixed with T cells stained with CalceinAM and embedded in a collagen plug. The collagen plug was treated with varying concentrations of rHAPLN1. Time lapse microscopy was used to image the movement of the T cells and their velocity was quantified (ANOVA, p<0.0001). b) Snapshots of the melanoma spheroids interacting with T cells in the presence of 25ng/ml rHAPLN1 (bar=25microns). c) Melanoma cells expressing mCherry were allowed to form spheroids and embedded in collagen plug mixed with T cells stained with CalceinAM and young fibroblasts with HAPLN1 knockdown. Time lapse microscopy was used to quantify the velocity of the T cells (ANOVA, p<0.0001). d) Snapshots of the melanoma spheroids interacting with T cells with HAPLN1 knockdown (bar=25microns). e) Melanoma cells expressing mCherry were allowed to form spheroids and embedded in collagen plugs prepared with young fibroblasts with HAPLN1 knockdown and reconstituted with rHAPLN1 (25ng/ml). Time lapse microscopy was used to assess velocity of T cells. Snapshots of the interaction between T cells and melanoma spheroids is also shown (bar=25microns). f) Melanoma cells expressing mCherry were layered at the bottom of the well followed by additional layer of Calcein-AM labeled T cells (green) and aged fibroblasts treated with rHAPLN1 in a 3D reconstruct model. g) Melanoma cells expressing mCherry were mixed in a collagen matrix and layered with a mix of young fibroblasts with HAPLN1 knockdown and Calcein-AM labeled T cells (green) in a separate layer of collagen. Imaging was performed within 24 hours of preparing the reconstruct (bar=100microns).
We next confirmed these data using a modified skin reconstruct model that we call a “sandwich reconstruct”, into which we embedded the assorted fibroblasts together with melanoma cells in collagen and added autologous T cells to question their ability to target the red pre-labeled melanoma cells (Figure S7B). Using a time-lapse imaging of the sandwich reconstruct, we observed striking increases in the motility of T cells within 6-8 hours in a HAPLN1 dependent manner (Figure 5F, Supplemental Movies 8,9). As with the spheroid assays, knockdown of HAPLN1 in young fibroblasts inhibited T cell movement towards the melanoma cells (Figure 5G, Supplemental Movie 10,11). Importantly, HAPLN1 treatment of a sandwich reconstruct containing melanoma cells, aged fibroblasts and autologous T cells showed a decrease in the number of melanoma cells, suggestive of the loss these cells due to the cytolytic activity of T cells (Figure S7C, arrows). Taken together these data suggest that HAPLN1 may play an important role in ECM guided T-cell infiltration into tumor sites.
ECM breakdown during aging differentially affects T-cell subpopulations.
To determine whether we could increase infiltration of T-cells into aged tumors by manipulating HAPLN1, we implanted Yumm1.7 cells in aged mice, and treated the tumors with rHAPLN1 (50ng/mL, twice weekly; control mice were injected with an equal volume of PBS as a control). Next, we proceeded to analyze these tumors for immune cell infiltration. Overall, likely due to the decrease in tumor size upon treatment with rHAPLN1, there was a decrease in overall (CD45+) immune cell infiltration (Figure S8A). However, when we analyzed the percentage of CD45+ cells that also expressed CD3+, we observed a clear increase in this T lymphocyte cell population in HAPLN1 treated mice (Figure 6A), and of these CD4+ and CD8+ cells were increased by HAPLN1 treatment (Figure S8B). We repeated the experiment, this time measuring immune cells as a percentage of all live cells. We observed that in response to rHAPLN1 treatment, CD8+ T-cells increased significantly (Figure 6B) as measured by flow cytometry. The change in CD4+ cells, while significant when taken as part of the CD45+ population (Figure S8B), was less so when calculated as % live, but still trended towards an increase (Figure S8C). We next measured dendritic cells, macrophages and M-MDSCs, none of which changed (Figure 6C-E). However, we found that HAPLN1 treatment significantly decreased the infiltration of PMN-MDSCs and Tregs into the tumor (Figure 6F,G), although PMN-MDSC populations are not changed in young vs. aged mice (Figure S8D). This resulted in a significant increase in the ratio of CD8+:Tregs (Figure 6H). Finally, to determine if this increase in the CD8+:Treg ratio correlated to increased cytolytic activity in the tumor, we stained the HAPLN1 vs. control tumors for cleaved caspase 3. HAPLN1 treated tumors had higher levels of cleaved caspase 3 than control (Figure 6I), suggesting that tumor cells were being lysed. Again, HAPLN1 does not affect tumor cell apoptosis in the absence of a microenvironment (Figure S6A, B), so these data suggest that the tumor microenvironment is required for the deleterious effects of HAPLN1. Overall, our data suggest that tumor cell invasion is inhibited by increased HAPLN1, and the immune tumor microenvironment is enhanced. Manipulating these mechanical changes could promote immune cell infiltration while inhibiting tumor cell extravasation to increase effectiveness of current therapies.
Figure 6: HAPLN1 affects immune cell infiltration in vivo.

a) Aged C57/BL6 mice were injected with Yumm1.7 allografts and treated intra-dermally with rHAPLN1. Tumors in mice were analyzed for CD3+ cells. b) Aged mice with Yumm1.7 allografts were treated with rHAPLN1 and assessed for infiltration of CD8 positive cells by flow cytometry. Yumm1.7 allografts were also assessed for infiltration of c) M-MDSC (CD11b+Ly6GnegLy6Chigh) infiltration, d) dendritic cells (CD11c+F4/80-/low) and e) macrophages (CD11b+F4/80+). f) Aged mice injected with Yumm1.7 allografts were treated intra-dermally with rHAPLN1 and assessed for PMN-MDSC cell (CD11b+, Ly6Ghi, Ly6Clo) infiltration. g) Aged mice with Yumm1.7 allografts were treated with rHAPLN1 and assessed for infiltration of T regulatory cells (CD4+, Foxp3+) by flow cytometry. h) Tumors from mice were compared for expression of CD8:Foxp3 per tumor. i) Tumors were also assessed for activity of cleaved caspase-3 using immunohistochemistry. Cleaved caspase-3 staining was quantified by counting positive cells across 20 sampled areas and dichotomized to low (0-1+ staining) and high (2-3+ staining) in mice (n=15) with or without rHAPLN1 treatment.
Discussion
We have presented here an analysis of ECM changes in the physical makeup of the aging skin, and the subsequent effects on tumor cell invasion and immune cell infiltration. It is well accepted that changes in matrix stiffness, such as loss of pliability, affect the metastatic properties of tumor cells (17). This occurs not only by providing optimal contractile forces for the migration of tumor cells, but also by affecting signaling, which can alter growth and even responses to drugs (40). Further, ECM alignment associated with assorted cancers effectively predicts patient outcomes and metastasis (41–43). We show here that age-related changes in the ECM facilitate the migration of tumor cells but may also hamper immune cell infiltration. We identify a novel role for HAPLN1, showing that it can suppress invasion of melanoma cells in young skin, and that it is lost during aging, creating an invasion-permissive microenvironment. We hypothesize that this is due to the fact that the nuclei of tumor cells can no longer pass through the small pores created by a highly crosslinked matrix. While our studies have focused solely on HAPLN1, given its large change in aged fibroblasts, we have a deep appreciation of the breadth of studies that can ensue from this work. Other factors secreted by our young fibroblasts are also known to be involved in crosslinking the ECM, such as aggrecan, and LOXL2. Intriguingly, increases in tumor cell associated LOXL2 have been associated with increased invasion (44). However, it may be that LOXL2 loss in the stromal cells, as suggested by our proteomic signature during aging, is perhaps more reflective of the loss of integrity in dermal collagen.
Our studies suggest that aged fibroblasts bear great similarity to cancer-associated fibroblasts (CAFs) in terms of myofibroblastic activation (e.g., upregulation of α-SMA), the alignment of CDMs, and the ability to increase tumor cell metastasis. Interestingly, targeting CAFs either with CAR-T cells designed to eliminate them, or with drugs such as Perfidine, have been shown to decrease metastatic breast cancer spread (45). Targeting aged fibroblasts might have similarly beneficial effects for melanoma patients. However, rather than eradicating them, reversing their phenotype to one where they produce “youthful” ECMs may be the most useful, as our data indicate that the matrix the fibroblasts produce is likely required for anti-tumor immune infiltration. T cells first use the vasculature to infiltrate the tumor (38, 46) and proceed to migrate along the ECM fibers of the tumor microenvironment (46), particularly the collagen fibers (36, 37). Our results show a decreased infiltration of CD4+ and CD8+ T cells on an age-impaired matrix, which could be improved by changing the matrix dynamics with HAPLN1. The maximum motility of a cell is a function of matrix porosity and cell deformation that can be achieved by that cell type, with the cell deformation being dependent on the nuclear size, rigidity and shape (35). The shape of the T cells, along with their polarity is eventually important in predicting the response of the T cells towards antigen presentation. Other factors also need to be considered- for example, the chemokine receptor and adhesion molecule makeup of CD4+ and CD8+ T-cells differ, and may affect their ability to migrate into the tumor microenvironment. Normal CD4+ T-cells have been shown to have a great propensity to infiltrate collagen, and this was initially thought to be due to their ability to express MMPs (47). However the concept of MMPs as an important factor in T-cell migration has been challenged, as the movement of T-cells through collagen has been shown to be MMP independent (35). This is in keeping with our own data on the effects of increasingly stiff matrices on tumor cell migration, as we have shown that to be partially MMP independent as well (18). More recently, the ability of CD4+ and CD8+ cells to differentially express chemokine receptors, integrins and selectins during homing to different organ sites, has been explored. Both CD4+ and CD8+ T-cells express P and E-selectin when homing to the skin (48), but in a gene expression analysis study of activated immune cells, CD4+ cells were shown to also express CCR5, CX3CR1, where CD8+ cells do not (49). On the other hand, the IL7R was more highly expressed in CD8+ cells. While the results from these studies, due to the nature of the immune response elicited, cannot be directly correlated to our work, they do highlight the fact that CD4+ and CD8+ cells differentially express receptors that are known to depend on the ECM for their transduction. Therefore it is not unreasonable to expect that the age-related changes in the ECM may differentially affect the chemokine, integrin and adhesion molecule profiles in the different populations of immune cells in the TME. We surmise that the differences in tumor volumes in mice with HAPLN1 treatment was based on both restriction of tumor spreading by efficiently cross-linked matrix as well as improved immune cell kinetics, however, additional studies are needed to further study these factors. What is intriguing is that increasing HAPLN1 appears to selectively inhibit the movement of PMN-MDSCs, which have a larger nuclear volume, and this may contribute to the inhibition of Tregs in the HAPLN1 treated tumors.
Our studies imply that changes in the immune microenvironment of aged tumors cannot be purely ascribed to age-related immunosenescence. It is known that during aging, there is a decrease in the ability of T-cells to traffic from lymph nodes and other sites to sites of injury, infection or cancer. Studies assessing age-related decline in immune cell trafficking tend to focus on response to chemokines, or immunosenescence of the T-cells, but the role of the ECM as modulator of T-cell motility is beginning to emerge. For example, the role of advanced end glycation products found in aging may affect T-cell activity (50) and T-cell expression of proteins that can interact with ECM has been identified (51). However, the effects of age-related ECM changes on either tissue resident lymphocytes found in the skin, or trafficking of lymphocytes to tumor sites have not been explored (52). Our results highlight the importance of physical changes in the ECM as a mediator of immune and tumor cell trafficking that transcend signaling or other T-cell intrinsic changes such as immunosenescence, since simply reconstituting HAPLN1 in the ECM affects immune cell infiltration. Studies of aging and immunosenescence are largely performed on peripheral blood samples, just as studies of intratumoral T-cell populations have largely been conducted in young mice. The first excludes the role of the ECM on T-cells, and the latter excludes the effects of age on T-cell infiltration, which we attempt to address in this study. Further, with the identification of tissue resident memory T-cells (TRM) (53), which reside in tissues such as the skin, lung and gut during the lifetime of an organism, understanding the role of the ECM in regulating TRM becomes increasingly important. It will be interesting to explore the maintenance vs. egress of these cells during aging in organ sites such as the skin.
We have previously examined the effects of immunotherapy in young vs. aged microenvironments (54). Surprisingly, in patients under the age of 50, there is a much lower ratio of CD8+: Tregs, rendering these patients less responsive to anti-PD1 than older patients. Depleting Tregs in young mice increases sensitivity to anti-PD1 therapy. We were therefore somewhat surprised that HAPLN1 did not also lower the CD8:Treg ratio, but this is likely due to the fact that HAPLN1 may alter the ability of PMN-MDSCs to get to the tumor site. This is further supported by the fact that in tumors in young vs aged mice, we see no difference in the infiltration of PMN-MDSCs, M-MDSCs, or macrophages (Figure S8D) although we do see a shift in CD4s, CD8s and Tregs as we have previously published (54). Additionally, there are likely numerous secreted and other changes that occur during aging, which cannot be completely reversed simply by HAPLN1 addition, but this re-emphasizes the critical role that mechanical changes play. Since immunotherapy has had such remarkable success in melanoma, enhancing the immune microenvironment by promoting a favorable CD8+:Treg ratio may increase response to immune checkpoint inhibitors. These analyses help to provide an outlook whereby understanding the mechanical structure of the tumor microenvironment, particularly during aging can provide means by which to increase effectiveness of current therapies.
Materials and methods:
Cell Culture
1205lu and WM3918 cells were maintained in DMEM (Invitrogen, Carlsbad, CA), supplemented with 5% FBS and 4mM L-glutamine. WM35 cells were maintained in MCDB153 (University of Pennsylvania Cell Center Services)/ L-15 (Cellgro, Manassas, VA) (4:1 ratio) supplemented with 2% FBS and 1.6 mM CaCl2 (Tumor growth media). YUMM1.7 cells were maintained in DMEM supplemented with 10% FBS and 4mM L-glutamine. Fibroblasts were maintained in DMEM, supplemented with 10% FBS and 4mM L-glutamine. Keratinocytes were maintained in keratinocyte SFM supplemented with human recombinant Epidermal Growth Factor 1-53 (EGF 1-53) and Bovine Pituitary Extract (BPE) (Invitrogen).
Cell lines were cultured at 37°C in 5% CO2 and the medium was replaced as required. Cell stocks were fingerprinted using AmpFLSTR® Identifiler® PCR Amplification Kit from Life Technologies TM at The Wistar Institute Genomics Facility. Although it is desirable to compare the profile to the tissue or patient of origin, our cell lines were established over the course of 40 years, long before acquisition of normal control DNA was routinely performed. However, each STR profile is compared to our internal database of over 200 melanoma cell lines, as well as control lines, such as HeLa and 293T. STR profiles are available upon request. Cell culture supernatants were mycoplasma tested using Lonza MycoAlert assay at the University of Pennsylvania Cell Center Services.
Proteomics
Fibroblasts were incubated for 9 doublings in SILAC-labeled media (MS 10030, Life Technologies). The young fibroblasts were labeled with heavy [13C6,15N4]-L-arginine and 13C6-L-lysine while the aged fibroblasts were labeled with light arginine and lysine. Cells were then seeded at 2.2×106/150cm2 dish, incubated overnight, washed with HBSS and incubated for 8 h in appropriate SILAC media. Conditioned media was then centrifuged at 10,000 g for 30 min, filtered through a 0.2 μm filter and concentrated using an Amicon Ultra 10K filter (Millipore) to a protein concentration of approximately 1 μg/μl. Samples were then combined and proteins separated on an SDS-PAGE gel for 3.5 cm. The gel lane was sliced into 35 fractions, digested with trypsin and analyzed by LC-MS/MS on an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) as described previously (55).
Data analysis was performed using MaxQuant version 1.3.0.5 (56). MS/MS data were searched against the human UniRef 100 protein database (July 2012, Protein Information Resource, Georgetown University) using full trypsin specificity with up to two missed cleavages, 6 ppm precursor mass tolerance, 0.5 Da fragment ion mass tolerance, static carboxamidomethylation of Cys, and variable oxidation of Met and protein N-terminal acetylation. Consensus protein lists were generated with false discovery rates of 1% at both peptide and protein levels. Proteins were also required to be identified by at least two razor plus unique peptides, and a minimum ratio count of three. Protein fold changes were calculated from the normalized H/L ratio. A 3 standard deviation (SD) cutoff was determined from a control heavy/light labeled young fibroblasts sample, and was used to identify proteins with significant change in expression.
Organotypic 3D Skin Reconstructs
Organotypic 3D skin reconstructs were generated as previously described (57). Briefly, an acellular layer of collagen was prepared in transwells for 6 well dishes (BD #355467 and Falcon #353092) and allowed to solidify for 1 hour at room temperature. Next, 6.4 × 104 fibroblasts were mixed with collagen and plated on the acellular layer and incubated for 45 min at 37°C in a 5% CO2 tissue culture incubator. DMEM containing 10% FBS was added to each well of the tissue culture tray and incubated for 4 days. Next, reconstructs were pre-incubated for 1 h at 37°C in HBSS containing 1% dialyzed FBS to wash off DMEM with 10%FBS and replaced with reconstruct media I. Keratinocytes (4.17 × 105) and melanoma cells (8.3 × 104) were mixed in 1:5 ratio and added to the inside of each insert. Media was changed to reconstruct media III and replaced every other day until day 18 when the reconstructs were harvested. For conditions requiring treatment with recombinant protein, fibroblasts were pre-treated with rHAPLN1 for 72 hours, and rHAPLN1 (varied doses) was freshly added to the media during media replacement. After the reconstructs were harvested, they were fixed in 10% formalin, paraffin embedded, sectioned and stained. Quantification of the invasion was performed using ImageJ software (available at http://imagej.nih.gov/ij/; developed by Wayne Rasband, National Institutes of Health, Bethesda, MD)
3D Spheroid Assays
Tissue culture-treated 96-well plates were coated with 50μl 1.5% Difco Agar Noble (Becton Dickinson). Melanoma cells were seeded at 5 × 103 cells/well and allowed to form spheroids over 72-96 h. Spheroids were harvested and embedded using collagen type I (GIBCO, #A1048301). The collagen plug was prepared as 300μl mix per layer and two layers were added into each well (1× EMEM [12-684, Lonza]; 10% FCS; 1× L-Glutamine; 1.0 mg/ml Collagen I; NaHCO3 [17-613E, Lonza], diluted in PBS as required). The first layer was added to each well and allowed to solidify. After 5-10 minutes, spheroids were mixed with the remaining 300μl mix and added to the well to solidify. Once the plug was solidified, media was added to the well and incubated at 37°C at 5% CO2 and imaged daily until invasion had surpassed the field of view. For treatment with recombinant protein, rHAPLN1 (#2608-HP, R&D Systems) was added to both the top and bottom layer while preparing the collagen plug. For experiments with shHAPLN1 fibroblasts, melanoma cells were pre-labeled with mCherry before spheroid formation, while 6,000 fibroblasts labeled with GFP were added to mixed in each layer of the collagen plug. Spheroids were imaged in mCherry fluorescence channel to quantify invasive area. For spheroids embedded with T cells, melanoma cells pre-labeled with mCherry were used. A CD4+, HLA class I-restricted CTL clone was isolated from the peripheral blood of a patient with primary melanoma. This clone was then shown to be able to stably lyse autologously matched melanoma cells for up to 9 months in culture. T cells were obtained by selection of immunoreactive T cells against target melanoma cells (WM793/1205lu) as previously described (39). T cells were labeled with Calcein AM (1:10,000; L3224, Invitrogen) for 1 hour at 37°C, 5% CO2. Cells were washed with 1×PBS before being added to the collagen plug mix. The collagen plug mix was used as described above. Spheroid images were acquired on Nikon TE2000 inverted microscope and time lapse imaging of spheroids was performed on Leica TCS SP8 X WLL laser scanning spectral confocal microscope. Quantitation of invasive surface area as well as tracking of T cells was performed using NIS Elements Advanced Research software.
Denaturation of rHAPLN1
Recombinant HAPLN1 was purchased from R&D Systems (#2608-HP) and reconstituted in sterile PBS at 100μg/ml. For denaturation, reconstituted rHAPLN1 was mixed in 9M urea and boiled for 10 minutes at 95°C followed by sterilization through 0.22micron filter. Denatured HAPLN1 was stored at room temperature until use. 9M urea was used as control as the same concentration as denatured HAPLN1.
Production of fibroblastic CDMs
Fibroblast CDMs were prepared as previously described (25, 26). Briefly, 12mm coverslips (No.1) were added to 24 well plates and coated with 0.2% gelatin solution for 1 hour. Coverslips were washed with DPBS and treated with 1% glutaraldehyde solution for 30 minutes at room temperature. After washing with DPBS, coverslips were incubated with 1M ethanolamine for 30 minutes at room temperature. After 5 washes with DPBS, 1.0 × 105 fibroblasts were plated onto the coverslips and incubated overnight at 37°C, 5% CO2. Following day, media was replaced with fresh media containing 50μg/ml L-ascorbic acid. L-ascorbic acid was added daily to the wells to a final concentration of 50μg/ml with fresh media replacement every other day. After 5 treatments, CDMs and fibroblasts were analyzed or fibroblasts were and the remaining matrices were used as described below.
Extraction of fibroblast from CDMs and reconstitution of melanoma cells
Fibroblast CDMs were prepared as stated above. After the matrix production was complete, fibroblasts were removed from the matrices to allow seeding of other cells for functional assays. Wells were washed with Ca2+ and Mg2+ free DPBS twice. This was followed by treatment with extraction buffer (0.05% TritonX-100, 20mM NH4OH in DPBS lacking Ca2+ and Mg2+) for 10 minutes, followed by 1:1 dilution with DPBS and incubated overnight at 4°C. Next day, DPBS was used to wash the wells multiple times to remove cellular debris. The extracted matrices were stored at 4°C until used for no longer than 2 weeks. 3000 melanoma cells (1205lu melanoma cells expressing mCherry) were seeded in a 24 well plate and time lapse imaging was performed on Nikon TE300 inverted microscope mounted in an incubation chamber. Quantification of melanoma cell velocity was performed using NIS Elements advanced research software and graphed in graphpad/Prism6.
Immunofluorescence (IF)
Samples were fixed with 4% paraformaldehyde, containing 0.5% TritonX, for 5 minutes at room temperature followed by treatment with 4% paraformaldehyde at room temperature for 20 minutes. Samples were blocked using “IF blocking buffer” comprising of PBS complemented with 0.2%Triton-X100, 0.2%BSA, 0.2% Casein, 0.2% gelatin and 0.02% sodium azide and filtered prior to use. Primary antibodies were incubated overnight at 4°C using the following concentrations: αSMA (1:100, A2547, Sigma), Fibronectin (1:200, F3648, Sigma), Collagen I (1:100, ab34710, abcam), diluted using “IF blocking buffer” as above. After washing with PBS, samples were incubated with the appropriate secondary antibodies (1:2000, Invitrogen) for 1 h at room temperature, followed by additional PBS wash and mounted in Prolong Gold anti-fade reagent containing DAPI (Invitrogen). Images were captured on a Leica TCS SP5 II scanning laser confocal system.
Anisotropy levels and thickness measurements of ECMs
ECMs were stained for fibronectin and imaged using Leica SP5 II Confocal System. Samples were imaged using 63X objective with 2X zoom power and each image composing the resultant z-stacks was set to step 0.5μM. Images composing these z-stacks were analyzed and counted to calculate the overall matrix thickness, using a minimum of 9 stacks for averaging the matrix thickness per condition. In addition, images were analyzed using ImageJ Plugin OrientationJ (available for download at http://bigwww.epfl.ch/demo/orientation/ (58)). Maximum stack 3D reconstruction images of the ECM fibers were normalized for orientation measurements using R and graphed. Anisotropic measurements in images were normalized using Adobe Photoshop. Mode value for each image(x) obtained from OrientationJ was used to calculate −2× values and this value was incorporated in hue settings to normalize each image to mode value. Source code for R is provided in supplemental files.
Immunohistochemistry (IHC)
Patient samples were collected under IRB exemption approval for protocol #EX21205258-1. Skin reconstructs were paraffin embedded and sectioned. Paraffin embedded sections were rehydrated through a xylene and alcohol series, rinsed in H2O and washed in PBS. Antigen retrieval was performed using target retrieval buffer (#H3300, Vector Labs, Burlingame, CA) and steamed for 20 min. Samples were blocked in a peroxidase blocking buffer (#TA060H2O2Q, Thermo Scientific) for 15 min, followed by Protein block (#TA-060-UB, Thermo Scientific) for 5 min, and incubated in appropriate primary antibody diluted in antibody diluent (S0809, Dako) at 4°C overnight in a humidified chamber. Following washing in PBS, samples were incubated in biotinylated anti-rabbit (abcam) followed by streptavidin-HRP solution at room temperature for 20 min. Samples were then washed in PBS and incubated in 3-Amino-9-Ethyl-l-Carboazole (AEC) chromogen for 15 min (#TA060SA, Thermo Scientific). Finally, samples were washed in H2O, incubated in Mayer’s hematoxylin (MHS1, Sigma) for 1 min, rinsed in cold H2O, and mounted in Aquamount (#143905, Thermo Scientific). Primary antibodies used were purchased as described: HAPLN1 (TA325115, Origene), Ki67 (CloneSP6, ThermoScientific), mCherry (NBP2-25157, Novus Biologicals), CD4 (14-9766-82, ThermoScientific), CD8a (14-0808-82, ThermoScientific), cleaved caspase 3 (9661S, Cell Signaling).
T cell migration in organotypic culture (reconstruct)
T-cell cultures were prepared using a modified approach as previously described (39). Briefly, a CD4+, HLA class I-restricted CTL clone was isolated from the peripheral blood of a patient with primary melanoma. This clone was then shown to be able to stably lyse autologously matched melanoma cells for up to 9 months in culture. Organotypic cultures were prepared in 4 well 35mm glass bottom dishes for optimal imaging (Greiner cellview #50590467, Thermofisher Scientific). An acellular bottom layer was prepared with collagen matrix (1.6ml 10×EMEM [12-684F, Lonza], 0.16ml L-glutamine, 1.82ml heat inactivated FCS, 0.2ml NaHCO3 [17-613E, Lonza], 14.8ml Rat Tail Collagen I [final concentration 2.0mg/ml, #354249, Corning] and plated 200μl of this mix in each well and allowed to solidify for 1 hour. 1205lu melanoma cells labeled with mCherry were harvested and plated on the acellular layer at 6 × 105 cells per well in the Tu2% media. On the following day, fibroblasts (6×104 cells) were harvested and mixed with T cells (1-3×106 cells), mixed with 250μl collagen matrix and layered on the melanoma cells. The layers were allowed to solidify and imaging was started after 3-4 hours of addition of T cells at 37°C, 5% CO2. After 5 days, reconstructs were harvested and embedded in OCT and sectioned for histological evaluation. H&E stained slides were imaged on EVOS XL Core Cell Imaging System. Time-lapse imaging was performed on Leica TCS SP8 X WLL laser scanning spectral confocal microscope for 24 hours.
2-Photon Microscopy
2.5 × 2.5 cm samples of skin were shaved and collected from C57/BL6 mice, held in buffer solution under nylon mesh and imaged with a Leica TCS SP8 MP 2-photon intravital microscope (Leica Microsystems, Inc, Buffalo Grove, IL). The specific region of interest was in the dermis. Alternatively, for skin reconstructs, 15 um thick sections were collected on glass slides, deparaffinized and passed through a series of alcohol washes, followed by mounting in Aquamount (#143905, Thermo Scientific). Collagen was visualized using second harmonic generation (SHG) from 900nm excitation in a Chameleon XR Ti:Saphire laser (Coherent, Inc., Santa Clara, CA). SHG emission was captured in 12 bits, at 700 Hz, through a 25×/1.00 water immersion objective in reflected mode using a HyD detector with a standard DAPI filter set. Mouse tissue images shown are composites of 15 z-stacks and skin reconstruct images are composites from 21 stacks, with 10mm step size. The images were further processed using Huygens Professional Deconvolution software (Scientific Volume Imaging, B.V., The Netherlands).
In Vivo rHAPLN1 assay
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) (IACUC #112503X_0) and were performed in an Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited facility. Yumm1.7 cells were injected into young (6-8 weeks old) or aged (>300days old) mice (Charles River, Frederick). For experiments with rHAPLN1 treatments, aged mice (>300 days old) were purchased from Taconic. YUMM1.7 (1 × 105 cells) overexpressing mCherry were injected intradermally into mice that were treated with 100ng (total) rHAPLN1 (#2608-HP, R&D Systems) or PBS as control at the time of allograft injection. Tumor sizes were measured every 3-4 days using digital calipers, and tumor volumes were calculated using the following formula: volume = 0.5 × (length × width2). Time-to-event (survival) was determined by a 5-fold increase in baseline volume (~1000 mm3) and was limited by the development of skin necrosis. Mice were euthanized, lungs were harvested and metastases counted. Half of the tissue was embedded in paraffin and half in optimal cutting temperature compound (O.C.T, Sakura, Japan City) and flash frozen for sectioning. Lungs were sectioned and stained with mCherry antibody (NBP2-25157, Novus Biologicals) to determine melanoma metastasis. All reagents injected in live mice were tested for endotoxin levels at University of Pennsylvania Cell Center Services using The Associates of Cape Cod LAL test and results are available upon request.
Flow cytometry analysis
Tumors were harvested from mice immediately after euthanasia and were processed through tumor dissociation kit (#130-096-730, Miltenyi Biotec, CA) using manufacturer’s protocol. Single cells were incubated with primary antibodies for 30minutes at room temperature. Cells were washed once with PBS and analyzed using LSRII (18-color), Becton-Dickinson. Primary antibodies were purchased from Biolegend, CA [CD4-BV510 (100553); CD8b-PerCP/Cy5.5 (100733); CD45-APC (103112); CD3-AF700 (100216); Zombie-yellow (423103) CD11b-PE/CY7 (301321) CD11c-APC/CY7 (337217) F4/80-PE (123109) Foxp3-PacBlue (126409)] and BD Bioscience [Ly6G-PE (561104) Ly6C-FITC (561085). Results were analyzed on FlowJo and graphed in graphpad/Prism6.
Collagen contractility assay
3000 fibroblasts were mixed with collagen matrix (1× EMEM [12-684, Lonza]; 10% FCS; 1× L-Glutamine; 1.0 mg/ml Collagen I; NaHCO3 [17-613E, Lonza], diluted in PBS as required) and 300μl were plated in a 48 well plate and allowed to set. Images were taken at time 0 and used as control, and the collagen plug was separated from the well using a fine needle. After three days, wells were imaged again and contractility was measured based on the area of the collagen plug at day 3 normalized to area on day 0. Images were acquired on UVP Gel Doc-It Imager.
SILAC Proteomics Analysis
Fibroblasts were incubated for nine doublings in SILAC-labeled media (MS 10030, Life Technologies). The young fibroblasts were labeled with heavy [13C6,15N4]-L-arginine and 13C6-L-lysine while the aged fibroblasts were labeled with light arginine and lysine. Cells were then seeded at 2.2×106/150cm2 dish, incubated overnight, washed with HBSS and incubated for 8 h in appropriate serum-free SILAC media (conditioned media). Conditioned media was then centrifuged at 10,000 g for 30 min, filtered through a 0.2 μm filter and concentrated using an Amicon Ultra 10K filter (Millipore) to a protein concentration of approximately 1 μg/μl. The heavy (young) and light (aged)-labeled samples were then combined at a 1:1 ratio based on total protein followed by separation on an SDS-PAGE gel for 3.5 cm. A control consisting of a 1:1 mixture of heavy (young) and light (young) samples were processed in the same manner. The gel lanes were sliced into 35 fractions, digested with trypsin and analyzed by LC-MS/MS on an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) as described previously (55). Data analysis was performed using MaxQuant version 1.3.0.5 (56). MS/MS data were searched against the human UniRef 100 protein database (July 2012, Protein Information Resource, Georgetown University) using full trypsin specificity with up to two missed cleavages, 6 ppm precursor mass tolerance, 0.5 Da fragment ion mass tolerance, static carboxamidomethylation of Cys, and variable oxidation of Met and protein N-terminal acetylation. Consensus protein lists were generated with false discovery rates of 1% at both peptide and protein levels. Proteins were also required to be identified by at least two razor plus unique peptides, and quantitated by a minimum ratio count of three. Protein fold changes were calculated from the normalized H/L ratio. A 3 standard deviation (SD) fold change cutoff was determined from the control heavy/light labeled young fibroblasts sample, and was used to identify significantly changed proteins in the young/old fibroblast secretome.
ELISA
Nunc™ MaxiSorp™ ELISA plates (ebiosciences, CA) were coated with 50μl of 3μg/ml HAPLN1 (1:500, #P51999PU-N, Acris antibodies, CA) overnight at 4°C. Plates were washed in PBS containing 0.1% Tween20 and blocked in ELISA diluent (00-4202-56, ebiosciences, CA) for 2 hours. Conditioned media was harvested after 72 hours and used without any dilution at 50μl per well and incubated overnight at 4°C. Next day, the plates were washed in PBS containing 0.1% Tween20 and incubated with detection antibody, for 1 hour at room temperature. Plates were washed and incubated with secondary antibody HAPLN1 (MAB2608, R&D systems) diluted 1:1000 for 1 hour. After washing, 100μl of 1× TMB (00-4201-56, ebiosciences, CA) solution was added to the plates and incubated for 15 minutes. The reaction was stopped using 50μl of 2N H2SO4 and absorbance was measured at 450nm.
Lentiviral production and infection
HAPLN1 shRNA was obtained from the TRC shRNA library available through the Molecular Screening Facility at The Wistar Institute. Plasmids without ORF expression were used as control plasmids. pLU-EF1α-mCherry was used for mCherry overexpression in multiple cell lines. PLX304 –GFP overexpression plasmid was obtained as a gift from lab of Dr. Meenhard Herlyn. Lentiviral production was carried out as described in the protocol developed by the TRC library (Broad Institute). Briefly, 293T cells were co-transfected with shRNA/ ORF vector and lentiviral packaging plasmids (pCMV-dR8.74psPAX2, pMD2.G). The supernatant containing virus was harvested at 36 and 60 hours, combined and filtered through a 0.45μm filter. For transduction, the cells were layered overnight with lentivirus containing 8μg/ml polybrene. The cells were allowed to recover for 24 hours and then selected using puromycin selection marker.
Western blotting
Melanoma samples were harvested in appropriate amounts of whole cell lysis buffer (6% SDS, 30% glycerol, 0.18M Tris pH6.8, 5% DTT, bromophenol blue). Protein was quantified using Qubit assay kit (#Q33212, ThermoScientific) and 50μg protein was boiled for 10 minutes at 95°C and loaded on 4-20% Bis-Tris gels. Samples were run at 165V and transferred on PVDF membranes. Membranes were blocked for 1 hour in 5% milk and incubated with appropriate antibody overnight at 4°C. Next day, membranes were washed in TBS with 0.1% Tween and incubated with secondary antibody for 1 hour at room temperature. Membranes were washed and developed using chemiluminescence based detection reagents (#PI80196, ThermoScientific). Chemiluminescence was detected using ImageQuant™ LAS 4000 (GE Healthcare Life Sciences, Pittsburgh, PA). Primary antibody to HSP90 was purchased from Cell Signaling (1:4000, 4877S), HAPLN1 (1:500, TA325115, Origene) and anti-rabbit secondary antibody was purchased from Jackson Immunoresearch (#211-032-171).
Quantitative PCR
Cells were plates as required for every experimental procedure. After the treatment had been complete, cells were harvested using Trizol (Invitrogen, #15596026) and subjected to phenol: chloroform extraction. Approximately 500μl trizol was used for a T25 flask and 5:1 Phenol: chloroform ratio was used. After addition of 100μl chloroform, samples were centrifuged for separation of different layers for 15 minutes at 4°C and RNA layer was collected and mixed with fresh 70% ethanol in a 1:1 ratio and subjected to cleanup using RNeasy Mini kit (# 74106, Qiagen) as per manufacturer’s protocol. 1μg RNA was used to prepare cDNA using iscript DNA synthesis kit (#1708891, Bio-Rad, CA). cDNA was diluted 1:5 before use for further reactions. Each 20μl well reaction comprised of 10μl Power SYBR Green Master mix (4367659, Invitrogen), 1μl primer mix (Final concentration 0.5μM) and 1μl cDNA. Standard curves were generated for all primers and each set of primers were normalized to 18 Primer pair (#AM1718, Ambion, Invitrogen). Primer sequences are described here, HAPLN1 (forward – CAGACCTCACTCTGGAAGATTATG, reverse – GGGAATACCAGACCTTGTAAGT), Fibronectin (forward – TTGCTCCTGCACATGCTTTG, reverse – CATGAAGCACTCAATTGGGCA)
Chemomechanical model for the stress-dependent contractility of the cells.
Following our previous work (18, 30), the chemomechanical behavior of the cells was characterized by introducing a spatially varying strain εij and a contractility tensor ρij. In the coarse-grained system, the trace of the contractility tensor (ρ11 + ρ22 + ρ33) represented the density of the recruited myosins in the actomyosin system. Here we adopted an energy-based approach and obtained the total energy of the cells, accounting for mechanical deformations, motor binding energy and mechano-chemical feedback as follows:
The mechanical energy accounts for the strain energy stored in the cytoskeleton (Cijkl being the elastic constants) and the mechanical work done by the myosin motors as they generate the cellular contractions. ρ0 is the density of the motors attached to the cytoskeleton in the quiescent state. The motor binding energy, proportional to the to the “chemical stiffness β” ensured that perturbations of the motor density from ρ0 leads to an increase in the energy. And finally, the mechano-chemical feedback part accounts for the activity of the molecular pathways such as Rho-pathway and the increase in the cell contractility in response to external tensile stresses. The mechano-chemical coupling parameter αv is responsible for coupling of the overall density of the motors to the external stress while αd determines the polarization in motor recruitment.
By minimizing this total free energy function with respect to the strains (εij) and cell polarization (ρij), we determined the strain and the contractility of the cells and finally the force exerted by the cell cluster on the surrounding ECM. The parameters used in the model are presented in Table 1.
Table 1.
The parameters used in the chemo-mechanical model.
| Parameter | Symbol | Value | Reference |
|---|---|---|---|
| E | Cell elastic modulus | 0.3 kPa | (18) |
| ν | Cell Poisson’s ratio | 0.3 | (18) |
| ρ0 | Cell initial motor density | 0.15 kPa | (18) |
| αv | Chemo-mechanical feedback parameter | 15 kPa−1 | (18) |
| αd | Chemo-mechanical feedback parameter | 25 kPa−1 | (18) |
| β | motor turnover parameter | 30 kPa−1 | (18) |
| r0 | Cluster initial radius | 200 μm | (18) |
Statistical Analysis
For in vitro studies, a Student’s t test or Wilcoxon rank-sum test (Mann Whitney) was performed for two-group comparison. Estimate of variance was performed and unequal variances for the t test were adjusted accordingly using Welch’s correction. ANOVA or Kruskal-Wallis test with post-hoc Holm-Sidak’s adjusted p-values was used for multiple comparisons. For in vivo studies, the indicated sample size for each experiment was designed to have 80% power at a two-sided alpha of 0.05 to detect a difference of large effect size about 1.51 between two groups on a continuous measurement. The change in tumor volume at each time point after treatment relative to baseline was calculated and then the change in treatment group relative to age matched control group was compared using a mixed-effect model to evaluate the treatment effect between experimental groups. Graphpad/Prism6 and Stata14 were used for plotting graphs and statistical analysis. Significance was designated as follows: *, p<0.05; **, p<0.01; ***, p<0.001.
Supplementary Material
Significance.
These data shed light on the mechanochemical interactions that occur between aged skin, tumor and immune cell populations, which may affect tumor metastasis and immune cell infiltration, with implications for the efficacy of current therapies for melanoma.
Acknowledgements:
We thank Dr. Katie Marchbank for technical assistance with the preparation of fibroblasts for proteomics and Dr. Chi Van Dang for critical reading of the manuscript.
Financial Support: NIH Funding (NCI): We thank the outstanding Core Facilities of the Wistar Institute supported by P30CA010815; A.T. W, S.M.D, and Q.L. are supported by R01CA174746 and R01CA207935. A.K. is supported by F99CA212437. X X, X Y, Q L, M H, D S., M.F., and A.T. W. are also supported by P01 CA114046 and P50CA174523. CHK III is supported by T32CA009171. MRW is supported by K99CA208012. DS is supported by RO1CA131582. EC is supported by R01 CA113451 and P30CA06927. Other funding: ATW is also supported by the Melanoma Research Fund, Melanoma Research Alliance/ L’Oréal Paris-USA Women in Science Team Science Award, the Ira Brind Endowment, and the Wistar Science Discovery Fund. MH is supported by a gift from the Adelson Medical Research Foundation. EC is supported by DOD W81XH-15-1-0170 and a gift from Mrs. Concetta Greenberg in memory of Dr. Marvin Greenberg. VS is supported by the NSF Center for Engineering Mechanobiology (CMMI-154857).
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest
References.
- 1.Balch CM, Thompson JF, Gershenwald JE, Soong SJ, Ding S, McMasters KM, et al. Age as a predictor of sentinel node metastasis among patients with localized melanoma: an inverse correlation of melanoma mortality and incidence of sentinel node metastasis among young and old patients. Ann Surg Oncol 2014;21:1075–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsai S, Balch C, Lange J. Epidemiology and treatment of melanoma in elderly patients. Nat Rev Clin Oncol 2010;7:148–52. [DOI] [PubMed] [Google Scholar]
- 3.Kaur A, Webster MR, Marchbank K, Behera R, Ndoye A, Kugel CH, et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Situm M, Buljan M, Cavka V, Bulat V, Krolo I, Mihic LL. Skin changes in the elderly people--how strong is the influence of the UV radiation on skin aging? Coll Antropol. 2010;34 Suppl 2:9–13. [PubMed] [Google Scholar]
- 5.Diridollou S, Vabre V, Berson M, Vaillant L, Black D, Lagarde JM, et al. Skin ageing: changes of physical properties of human skin in vivo. International journal of cosmetic science. 2001;23:353–62. [DOI] [PubMed] [Google Scholar]
- 6.Panwar P, Lamour G, Mackenzie NC, Yang H, Ko F, Li H, et al. Changes in Structural-Mechanical Properties and Degradability of Collagen during Aging-associated Modifications. The Journal of biological chemistry. 2015;290:23291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee HO, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E, Cheng JD. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer. 2011;11:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marcos-Garces V, Molina Aguilar P, Bea Serrano C, Garcia Bustos V, Benavent Segui J, Ferrandez Izquierdo A, et al. Age-related dermal collagen changes during development, maturation and ageing - a morphometric and comparative study. Journal of anatomy. 2014;225:98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oh JH, Kim YK, Jung JY, Shin JE, Kim KH, Cho KH, et al. Intrinsic aging- and photoaging-dependent level changes of glycosaminoglycans and their correlation with water content in human skin. Journal of dermatological science. 2011;62:192–201. [DOI] [PubMed] [Google Scholar]
- 10.Roark EF, Keene DR, Haudenschild CC, Godyna S, Little CD, Argraves WS. The association of human fibulin-1 with elastic fibers: an immunohistological, ultrastructural, and RNA study. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 1995;43:401–11. [DOI] [PubMed] [Google Scholar]
- 11.Sephel GC, Davidson JM. Elastin production in human skin fibroblast cultures and its decline with age. The Journal of investigative dermatology. 1986;86:279–85. [DOI] [PubMed] [Google Scholar]
- 12.Neame PJ, Barry FP. The link proteins. EXS. 1994;70:53–72. [DOI] [PubMed] [Google Scholar]
- 13.Yeowell HN, Marshall MK, Walker LC, Ha V, Pinnell SR. Regulation of lysyl oxidase mRNA in dermal fibroblasts from normal donors and patients with inherited connective tissue disorders. Archives of Biochemistry and Biophysics. 1994;308:299–305. [DOI] [PubMed] [Google Scholar]
- 14.Martin H, Dean M. An N-terminal peptide from link protein is rapidly degraded by chondrocytes, monocytes and B cells. European journal of biochemistry / FEBS. 1993;212:87–94. [DOI] [PubMed] [Google Scholar]
- 15.Berthod F, Germain L, Li H, Xu W, Damour O, Auger FA. Collagen fibril network and elastic system remodeling in a reconstructed skin transplanted on nude mice. Matrix Biol 2001;20:463–73. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Abhilash AS, Chen CS, Wells RG, Shenoy VB. Long-range force transmission in fibrous matrices enabled by tension-driven alignment of fibers. Biophys J 2014;107:2592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmadzadeh H, Webster MR, Behera R, Jimenez Valencia AM, Wirtz D, Weeraratna AT, et al. Modeling the two-way feedback between contractility and matrix realignment reveals a nonlinear mode of cancer cell invasion. Proc Natl Acad Sci U S A. 2017;114:E1617–E26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao X, Lin Y, Driscoll TP, Franco-Barraza J, Cukierman E, Mauck RL, et al. A Chemomechanical Model of Matrix and Nuclear Rigidity Regulation of Focal Adhesion Size. Biophys J 2015;109:1807–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lang NR, Skodzek K, Hurst S, Mainka A, Steinwachs J, Schneider J, et al. Biphasic response of cell invasion to matrix stiffness in three-dimensional biopolymer networks. Acta biomaterialia. 2015;13:61–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang JS, Kawakami Y, Bekku Y, Ninomiya Y, Izpisua Belmonte JC, Oohashi T. Molecular cloning and developmental expression of a hyaluronan and proteoglycan link protein gene, crtl1/hapln1, in zebrafish. Zoological science. 2008;25:912–8. [DOI] [PubMed] [Google Scholar]
- 22.Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, et al. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol 2003;160:267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park HW. Biological aging and social characteristics: gerontology, the Baltimore city hospitals, and the national institutes of health. J Hist Med Allied Sci 2013;68:49–86. [DOI] [PubMed] [Google Scholar]
- 24.Behera R, Kaur A, Webster MR, Kim S, Ndoye A, Kugel CH 3rd, et al. Inhibition of Age-Related Therapy Resistance in Melanoma by Rosiglitazone-Mediated Induction of Klotho. Clin Cancer Res 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franco-Barraza J, Beacham DA, Amatangelo MD, Cukierman E. Preparation of Extracellular Matrices Produced by Cultured and Primary Fibroblasts. Curr Protoc Cell Biol 2016;71:1091–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franco-Barraza J, Francescone R, Luong T, Shah N, Madhani R, Cukierman G, et al. Matrix-regulated integrin alphavbeta5 maintains alpha5beta1-dependent desmoplastic traits prognostic of neoplastic recurrence. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amatangelo MD, Bassi DE, Klein-Szanto AJ, Cukierman E. Stroma-derived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. The American journal of pathology. 2005;167:475–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papageorgis P, Stylianopoulos T. Role of TGFbeta in regulation of the tumor microenvironment and drug delivery (review). International journal of oncology. 2015;46:933–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dugina V, Alexandrova A, Chaponnier C, Vasiliev J, Gabbiani G. Rat fibroblasts cultured from various organs exhibit differences in alpha-smooth muscle actin expression, cytoskeletal pattern, and adhesive structure organization. Experimental cell research. 1998;238:481–90. [DOI] [PubMed] [Google Scholar]
- 30.Shenoy VB, Wang H, Wang X. A chemo-mechanical free-energy-based approach to model durotaxis and extracellular stiffness-dependent contraction and polarization of cells. Interface Focus. 2016;6:20150067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC medicine. 2006;4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aladowicz E, Ferro L, Vitali GC, Venditti E, Fornasari L, Lanfrancone L. Molecular networks in melanoma invasion and metastasis. Future oncology (London, England). 2013;9:713–26. [DOI] [PubMed] [Google Scholar]
- 33.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–400. [DOI] [PubMed] [Google Scholar]
- 34.Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–40. [DOI] [PubMed] [Google Scholar]
- 35.Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, et al. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol 2013;201:1069–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arencibia I, Sundqvist KG. Collagen receptor on T lymphocytes and the control of lymphocyte motility. European journal of immunology. 1989;19:929–34. [DOI] [PubMed] [Google Scholar]
- 37.Friedl P, Noble PB, Zanker KS. Lymphocyte locomotion in three-dimensional collagen gels. Comparison of three quantitative methods for analysing cell trajectories. Journal of immunological methods. 1993;165:157–65. [DOI] [PubMed] [Google Scholar]
- 38.Peske JD, Woods AB, Engelhard VH. Chapter Eight - Control of CD8 T-Cell Infiltration into Tumors by Vasculature and Microenvironment. Immunotherapy of Cancer. 2015;128:263–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Somasundaram R, Robbins P, Moonka D, Loh E, Marincola F, Patel A, et al. CD4(+), HLA class I-restricted, cytolytic T-lymphocyte clone against primary malignant melanoma cells. Int J Cancer. 2000;85:253–9. [DOI] [PubMed] [Google Scholar]
- 40.Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gehler S, Ponik SM, Riching KM, Keely PJ. Bi-directional signaling: extracellular matrix and integrin regulation of breast tumor progression. Crit Rev Eukaryot Gene Expr 2013;23:139–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol 2011;178:1221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oudin MJ, Jonas O, Kosciuk T, Broye LC, Guido BC, Wyckoff J, et al. Tumor Cell-Driven Extracellular Matrix Remodeling Drives Haptotaxis during Metastatic Progression. Cancer Discov 2016;6:516–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pickup MW, Laklai H, Acerbi I, Owens P, Gorska AE, Chytil A, et al. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-beta-deficient mouse mammary carcinomas. Cancer Res 2013;73:5336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lo A, Wang LC, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res 2015;75:2800–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madri JA, Graesser D. Cell migration in the immune system: the evolving inter-related roles of adhesion molecules and proteinases. Developmental immunology. 2000;7:103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ivanoff J, Talme T, Sundqvist KG. The role of chemokines and extracellular matrix components in the migration of T lymphocytes into three-dimensional substrata. Immunology. 2005;114:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferguson AR, Engelhard VH. CD8 T cells activated in distinct lymphoid organs differentially express adhesion proteins and coexpress multiple chemokine receptors. J Immunol 2010;184:4079–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cliff JM, Andrade IN, Mistry R, Clayton CL, Lennon MG, Lewis AP, et al. Differential gene expression identifies novel markers of CD4+ and CD8+ T cell activation following stimulation by Mycobacterium tuberculosis. J Immunol 2004;173:485–93. [DOI] [PubMed] [Google Scholar]
- 50.Han XQ, Gong ZJ, Xu SQ, Li X, Wang LK, Wu SM, et al. Advanced glycation end products promote differentiation of CD4(+) T helper cells toward pro-inflammatory response. J Huazhong Univ Sci Technolog Med Sci 2014;34:10–7. [DOI] [PubMed] [Google Scholar]
- 51.Moreau JF, Pradeu T, Grignolio A, Nardini C, Castiglione F, Tieri P, et al. The emerging role of ECM crosslinking in T cell mobility as a hallmark of immunosenescence in humans. Ageing Res Rev 2017;35:322–35. [DOI] [PubMed] [Google Scholar]
- 52.Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, et al. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood. 2014;123:239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med 2015;21:688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kugel CH 3rd, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X, et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin Cancer Res 2018. doi: 10.1158/1078-0432.CCR-18-1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rasanen K, Sriswasdi S, Valiga A, Tang HY, Zhang G, Perego M, et al. Comparative secretome analysis of epithelial and mesenchymal subpopulations of head and neck squamous cell carcinoma identifies S100A4 as a potential therapeutic target. Mol Cell Proteomics. 2013;12:3778–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26:1367–72. [DOI] [PubMed] [Google Scholar]
- 57.Li L, Fukunaga-Kalabis M, Herlyn M. The three-dimensional human skin reconstruct model: a tool to study normal skin and melanoma progression. J Vis Exp 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Puspoki Z, Storath M, Sage D, Unser M. Transforms and Operators for Directional Bioimage Analysis: A Survey. Adv Anat Embryol Cell Biol 2016;219:69–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.