

Abstract
Background:
Glycogen storage disease type Ia (GSD Ia) in dogs closely resembles human GSD Ia. Untreated patients with GSD Ia develop complications associated with glucose-6-phosphatase (G6Pase) deficiency. Survival of human patients on intensive nutritional management has improved; however, long-term complications persist including renal failure, nephrolithiasis, hepatocellular adenomas (HCA), and a high risk for hepatocellular carcinoma (HCC). Affected dogs fail to thrive with dietary therapy alone. Treatment with gene replacement therapy using adeno-associated viral vectors (AAV) expressing G6Pase has greatly prolonged life and prevented hypoglycemia in affected dogs. However, long-term complications have not been described to date.
Methods:
Five GSD Ia affected dogs treated with AAV-G6Pase were evaluated. Dogs were euthanized due to reaching humane endpoints related to liver and/or kidney involvement, at 4 to 8 years of life. Necropsies were performed and tissues were analyzed.
Results:
Four dogs had liver tumors consistent with HCA and HCC. Three dogs developed renal failure, but all dogs exhibited progressive kidney disease histologically. Urolithiasis was detected in two dogs; uroliths were composed of calcium oxalate and calcium phosphate. One affected and one carrier dog had polycystic ovarian disease. Bone mineral density was not significantly affected.
Conclusions:
Here we show that the canine GSD Ia model demonstrates similar long-term complications as GSD Ia patients in spite of gene replacement therapy. Further development of gene therapy is needed to develop a more effective treatment to prevent long-term complications of GSD Ia.
Introduction
Glycogen storage disease type Ia (GSD Ia) (OMIM #232200) is caused by a glucose-6-phosphatase (G6Pase) deficiency that leads to several metabolic complications early in life of patients affected, which include life-threatening hypoglycemia, hyperlipidemia, lactic acidosis, hyperuricemia, growth retardation, and early death (Koeberl et al 2007). For the past 3 decades, survival of human patients that are placed under intensive nutritional management with uncooked corn starch has improved; however, long-term complications persist including renal failure, nephrolithiasis, gout, pulmonary hypertension, hepatocellular adenomas (HCA), and a high risk for hepatocellular carcinoma (HCC) (Koeberl et al 2007). There is currently no cure for these long-term complications, but liver and kidney transplantation has been shown effective. This has led to numerous studies in animal models of GSD Ia to find a cure for the disease.
A G6Pase knockout mouse model has been described to demonstrate similar life-threatening hypoglycemia, early morbidity and mortality. More recently, liver, kidney and intestine specific G6Pase knock-out mouse models have been described to prolong life and develop complications related to the specific organ; for example, hepatocellular adenomas were noted in all liver specific G6Pase knock-out mice analyzed at 18 months of age (Mutel et al 2011; Clar et al 2014; Penhoat et al 2014). While murine GSD Ia models have some advantages relating to shorter lifespan and smaller costs, the canine GSD Ia canine model (OMIA 000418–9615) has been shown to simulate the human disease more accurately than the mouse models by also demonstrating lactic acidosis (Kishnani et al 2001). Both murine and canine models have demonstrated growth deficiencies likely due to growth hormone resistance in the diseased liver, but effects appear to be more severe in dogs (Brooks et al 2013).
Gene therapy has been shown quite effective at preventing hypoglycemia and greatly prolonging lifespan in murine and canine GSD Ia models, albeit with limitations. Episomal AAV vector genomes expressing G6Pase were lost over time requiring retreatment with an AAV vector of a new serotype (Demaster et al 2012). Gene therapy treated dogs continue to display growth deficiencies in spite of greatly prolonging life and preventing hypoglycemia (Brooks et al 2013). Prevention of some other long-term complications, such as HCA has been described in G6Pase−/− mice treated with AAV-G6Pase followed for 70–90 weeks (Lee et al 2012), but has not been described to date in gene therapy treated dogs. Some long-term complications, such as HCA, cytoplasmic vacuolation, glomerulosclerosis and cortical tubule nephrosis with renal failure have been previously described in dogs treated with helper-dependent adenovirus gene therapy (Crane et al 2012), but the oldest of these dogs was 3 years of age. The current study further provides follow-up and further description of the long-term complications in previously described GSD Ia dogs treated with AAV-G6Pase therapy living up to 8 years of age (Demaster et al 2012; Brooks et al 2013).
Materials and Methods
Animal use
Five GSD Ia affected dogs treated with AAV-G6Pase were followed up to eight years of life. All required readministration of AAV vector(s), pseudotyped as a new serotype to avoid anti-AAV antibodies, due to decreased ability to maintain normoglycemia during fasting (Table 1). All dogs were maintained on diets formulated for the growth and stage of each dog. Occasionally appetite was suppressed in the dogs, requiring a variety of dog food options available throughout each dog’s life. However, dogs remained largely symptom free throughout life, and recurrent hypoglycemia was managed by vector readministration (Demaster et al 2012). In general, most dogs were able to thrive on 2–3 feedings/day similar to unaffected dogs. All dogs were euthanized due to reaching humane endpoints related to liver and/or kidney involvement at 4 to 8 years of age. Complete necropsies were performed on all dogs immediately and tissues were fixed in 10% neutral-buffered formalin and stored at 4° C until embedded in paraffin and sectioned at 5μm. Hepatic DNA and G6Pase were not able to be detected in Dog De due to improper liver storage at necropsy by first placing entire liver in formalin overnight. Multiple sections of liver and kidney were examined from all dogs as well as sections from all organs that had macroscopic lesions or that were related to clinical findings in life. Histologic stains included hematoxylin and eosin (H&E), as well as Masson’s trichrome and Periodic acid-Schiff (PAS) on selected sections.
Table 1.
GSD Ia dogs described in current study.
| Dog | Sex | AAV vector treatments | Age at euthanasia or death | Reason for euthanasia/death | Health abnormalities during lifetime | Previously published data |
|---|---|---|---|---|---|---|
| L | F | Birth: AAV2/8 (1×1013vp/kg) 36 mo: AAV2/9 (3×1012vp/kg) |
8 years | Renal failure | Renal disease, elevated serum liver enzymes and cholesterol, HCA and HCC, polycystic ovarian syndrome, urolithiasis | (Koeberl et al 2008; Demaster et al 2012; Farah et al 2016) |
| R | M | Birth: AAV2/8 (1×1013vp/kg) 34 mo: AAV2/9 (3×1012vp/kg) 43 mo: AAV2/7 (1×1013vp/kg) 58 mo: AAV2/9 (3×1012vp/kg) |
5.7 years | Renal failure | Elevated serum liver enzymes and cholesterol, severe end stage renal disease | (Koeberl et al 2008; Demaster et al 2012) |
| W | F | Birth: AAV2/9 (2×1013vp/kg) 15 mo: AAV2/8 (1×1012vp/kg) |
7.4 years | Rapid decline in health from hemorrhage from HCC | Elevated serum liver enzymes and cholesterol, one pancreatitis event that resolved with vector therapy, HCC | (Demaster et al 2012; Brooks et al 2013; Farah et al 2016) |
| H | F | Birth: AAV2/9 (4×1013vp/kg) 3 mo: AAV2/7 (2×1012vp/kg) 5 mo: AAV2/8 (1×1013vp/kg) |
6.9 years | Acute renal failure, pancreatitis and HCC | Elevated liver enzymes, triglycerides and cholesterol, HCA and HCC, acute renal failure and pancreatitis | (Demaster et al 2012; Brooks et al 2013; Farah et al 2016) |
| De | M | Birth: AAV2/9 (4×1013vp/kg) 2 mo: AAV2/7 (2×1012vp/kg) 4 mo: AAV2/8 (1×1013vp/kg) |
4.1 years | Rapid decline in health likely from hemorrhage from HCC | Urolithiasis, elevated serum liver enzymes and cholesterol, HCC | (Demaster et al 2012; Brooks et al 2013) |
| A | F | GSD Ia carrier- not treated | Adopted | NA | Unable to breed successfully beginning at about 5 years of age, noted polycystic ovaries during ovariohysterectomy | |
| Li | M | GSD Ia carrier-not treated | 11 years | Intervertebral Disk Disease | Intervertebral Disk Disease, BUN elevated later in life |
Vector Therapy
AAV vectors were prepared using a minimal human G6Pase promoter and human G6Pase cDNA as described previously (Koeberl et al 2008). Briefly, vectors were purified by cesium chloride gradient centrifugation to remove empty capsids, and quantified by Southern blotting to detect only full length vector genomes. Vector bioactivity was evaluated in mice with GSD Ia as described to confirm potency (Luo et al 2011). Vector administration was as reported (Demaster et al 2012), with the exception of dog R, who received an additional vector due to renal disease (Table 1). Dog R was retreated with AAV2/9 due to data showing very low serum anti-AAV9 antibodies (titer < 1:20) after his first treatment with AAV2/9 (Demaster et al 2012). Data from Dog T in Demaster et al was not included due to ineffective treatment effect from multiple vector treatments; he ultimately died of acute hypoglycemia at 4 years of age (Supplementary Fig. 1). Dog T had elevated anti-AAV8 antibodies despite the lack of previous exposures, prior to AAV8 vector administration, raising the possibility that cross-reacting anti- AAV antibodies reduced efficacy from gene therapy in that dog but not in others that had low baseline anti-AAV antibody titers (Demaster et al 2012).
Plasma and serum chemistry
Complete blood counts, blood chemistries and urinalyses analyzed prior to 2009 were submitted to the North Carolina State University College of Veterinary Medicine clinical pathology lab. After 2009, blood analysis was either performed in house at the Duke University Division of Laboratory Animal Services clinical pathology lab or sent out to a commercial laboratory, Antech Diagnostics (Antech, Diagnostic Laboratories, Cary, NC). Lactate concentration was determined on serum or plasma samples saved at - 80°C using the Lactate Assay Kit II (Sigma-Aldrich) according to manufacturer directions; performed controls with stored plasma from 3 unaffected dogs and results were within normal dog range.
Glucose curves
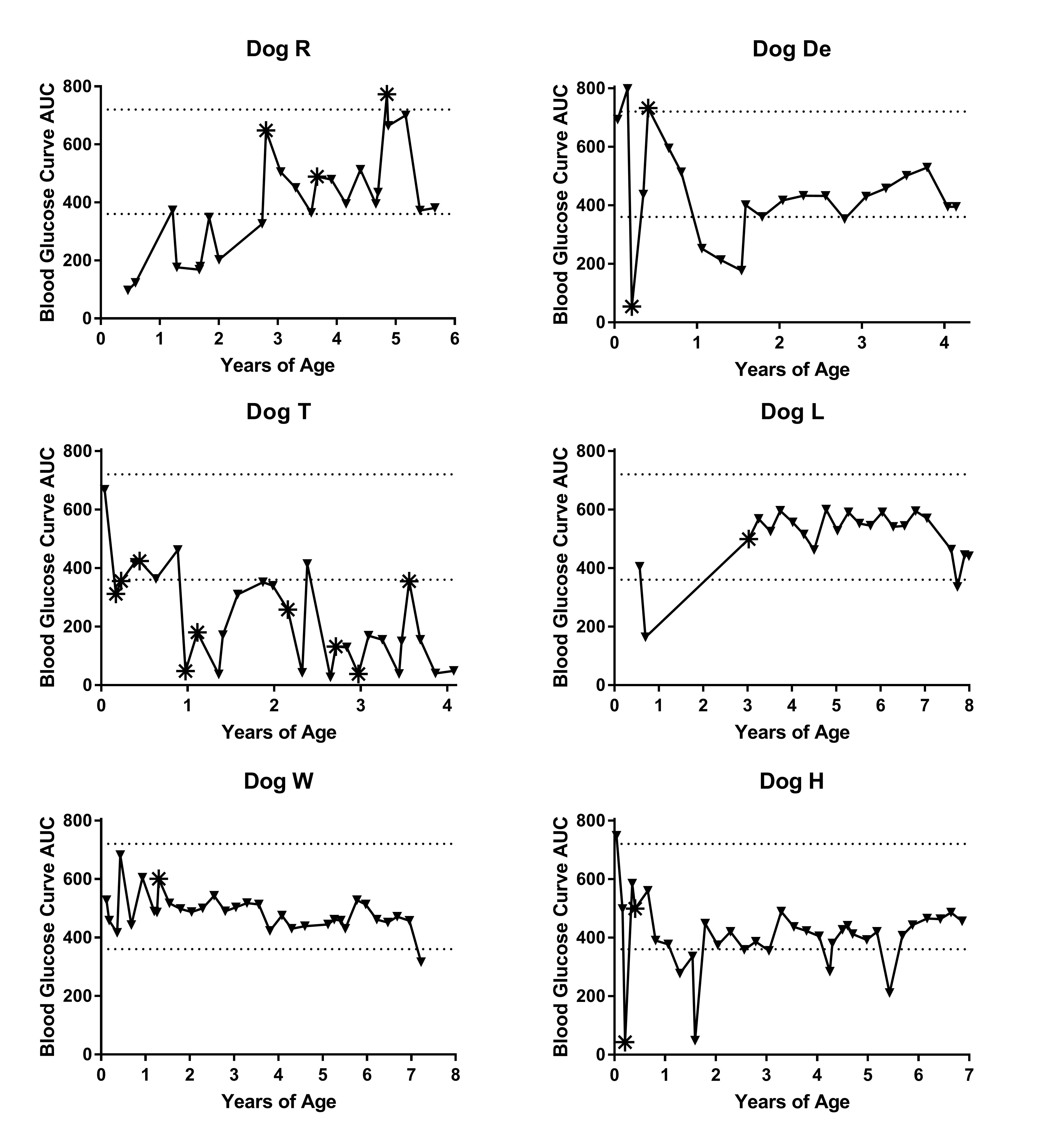
Glucose curves for monitoring hypoglycemia were performed by fasting the dogs for up to 8 hours and monitoring blood glucose every 2 hours. If blood glucose dropped below 50–60 mg/dL or clinical signs of hypoglycemia occurred, the curve was stopped, and dogs were given dextrose therapy as needed and fed. Blood glucose was measured by a point of care glucometer, either the AlphaTRAK or AlphaTRAK 2 (Zoetis). The area under the curve (AUC) was determined for each glucose curve for each dog over time using GraphPad Prism 7.03. For example, the low and high normal AUC was determined using 60 mg/dl for 4 time points and 120 mg/dl for 4 time points, resulting in total area of 360 and 720, respectively (Fig. 1f, supplementary Fig. 1). If a dog had to stop the curve before 8 hours of fasting due to hypoglycemia, the value for subsequent time points was reported as 0.
Fig. 1.
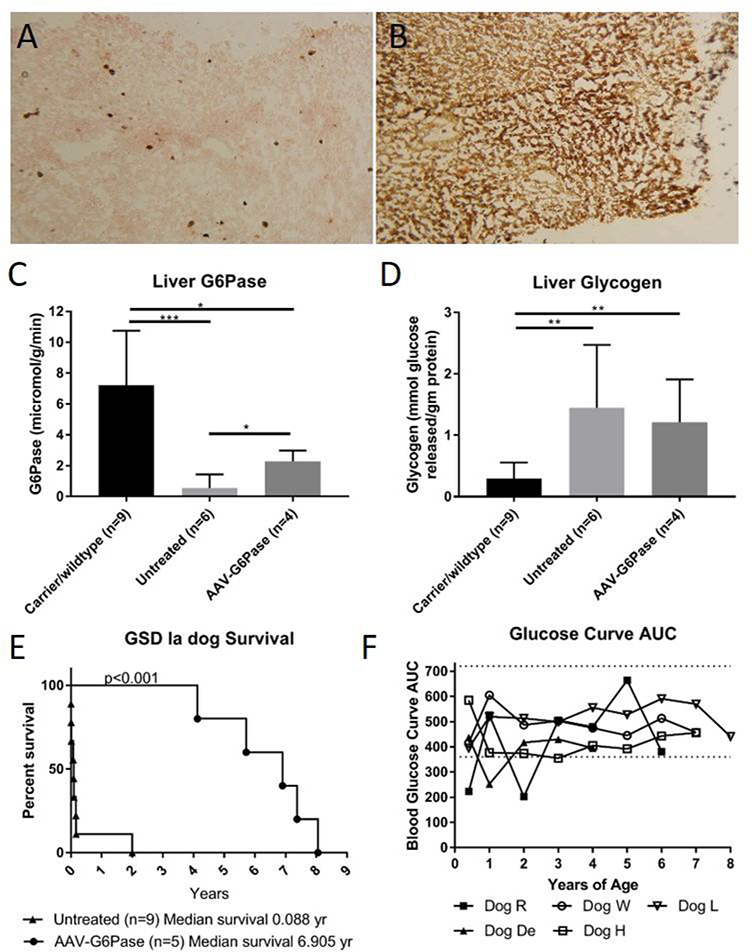
Liver G6Pase persists throughout lifetime of AAV-G6Pase treated GSD Ia dogs increasing survival and ability to maintain normoglycemia during fasting. a. Hepatic G6Pase staining of AAV-G6Pase treated GSD Ia, Dog R. G6Pase is shown with brown staining. 100x b. Hepatic G6Pase staining of a GSD Ia carrier shown for comparison. 100x c. Mean ±SD of G6Pase activity detected in livers of GSD Ia dogs at end of life, AAV-G6Pase treated dogs have significantly higher G6Pase activity than untreated GSD Ia dogs , but both treated and untreated GSD Ia dogs do not achieve G6Pase activity as high as that found in carrier/wild type animals as shown through Student’s t test. Untreated, affected puppies had a low residual G6Pase activity in liver as described (Kishnani et al 2001). d. Mean ±SD of glycogen detected in GSD Ia dogs at end of life. Both treated and untreated GSD Ia dogs have increased glycogen compared with carriers and wild type through student’s t test. e. AAV-G6Pase dogs have a greatly increased survival over untreated GSD Ia dogs; as shown using a Log-rank (Mantel- Cox) test, p=0.008. f. Using the Area Under Curve (AUC), glucose curves throughout the lifetime of AAV-G6Pase treated dogs demonstrate abilities to fast and maintain normoglycemia.
G6Pase and Glycogen Biochemical and Histochemical Detection
G6Pase enzyme and glycogen analysis were performed as previously described on kidney and liver tissues collected at necropsy for affected, control and experimental samples (Fig. 1) (Kishnani et al 2001). Tissues were frozen and stored at −80°C. G6Pase activity was quantified by using glucose-6-phosphate (Sigma) as substrate after subtraction of nonspecific phosphatase activity as estimated by glycerol 2-phosphate disodium salt hydrate (Sigma).
G6Pase was assessed qualitatively in flash frozen sections of dog liver by an optimized cerium-diaminobenzidine method as described (Jonges et al 1990; Luo et al 2011). Briefly, 6 μm liver sections were first incubated in a medium consisting of 5 mmol/l cerium chloride heptahydrate (Aldrich), 10 mmol/l glucose-6-phosphate (Sigma), 50 mmol/l tris-maleate buffer (pH 6.5), at room temperature for 5 minutes. The incubation was stopped by rinsing with 50 mmol/l tris-maleate buffer (pH 8.0). G6Pase was visualized by a second incubation in 10 mmol/l glucose-6-phosphate, 5 mmol/l calcium chloride dihydrate (Sigma), 10 mmol/l sodium azide, 40 mmol/l tris-maleate (pH 8.0), 10 mmol/l diaminobenzidine, 10 mmol/l tris-maleate (pH 6.5), 4% hydrogen peroxide for 10 minutes at room temperature. Slides were then rinsed with distilled water, counter-stained with nuclear fast red (Sigma- Aldrich), and mounted with Shur/Mount medium (Triangle Biomedical Sciences).
Femur Bone Mineral Density Calculations
GSD Ia dog legs were wrapped in saline-coated paper towels at necropsy and stored at −80°C. Average bone mineral densities (BMD) of were calculated using quantitative computed tomography (QCT) in the Shared Material Instruments Facility’s MicroCT lab at Duke University. CT scans were done using 2000 projections with uniform energy (95 kV, 300 uA). Grey scale values from the scans were calibrated by placing materials with known volumetric densities and linear attenuations in each scan.
Orthogonal slices of the mid-shaft, 5% and 10% distal (referred to as the −5% and −10% locations, respectively), and 5% and 10% proximal to the mid-shaft (referred to as the 5% and 10% locations, respectively) regions of each femur were obtained similar as that described previously (Schneider et al 2004). ImageJ software was used to calculate the mean total femur BMDs in each slice by dividing the grey scale integrated density of the femur by the total area to give a mean grey scale value for the location.
Results
Partial Correction of G6Pase deficiency throughout lifetime
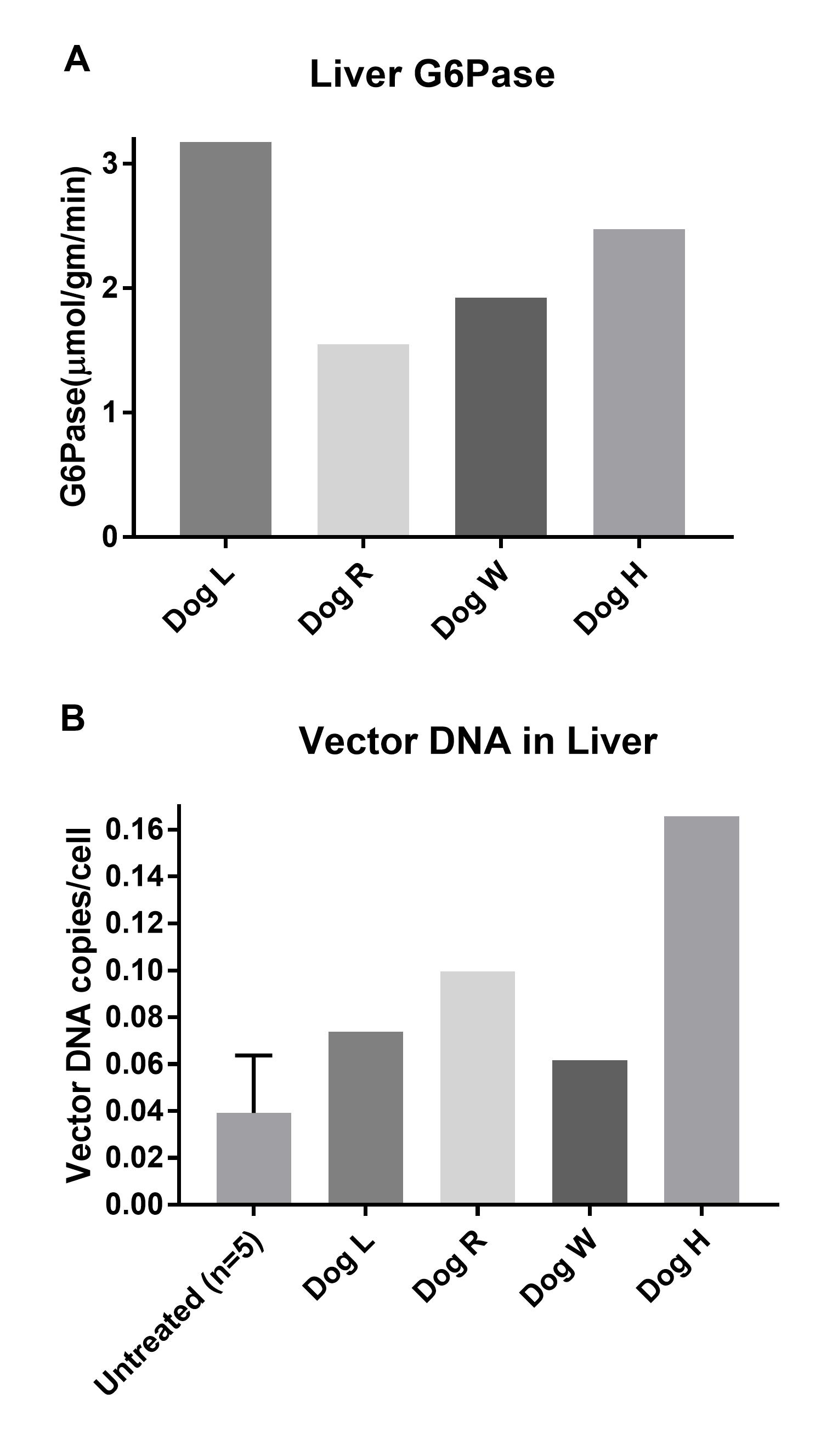
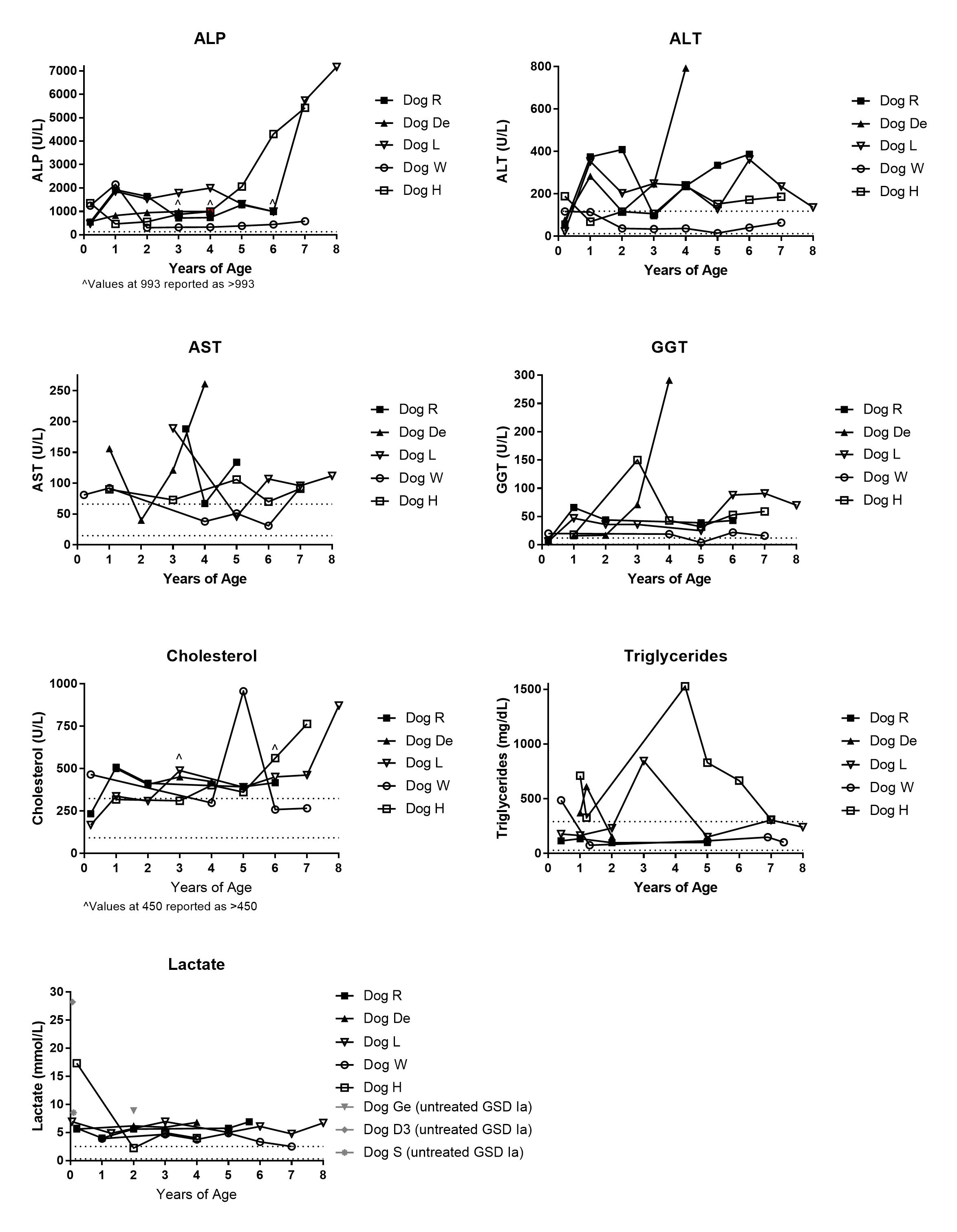
Through G6Pase histochemical staining, we noted persistent foci of G6Pase-producing hepatocytes in dogs at necropsy, years after receiving AAV-G6Pase vector therapy (Fig 1a). Furthermore, biochemical detection of G6Pase in liver revealed G6Pase activity (2.28 ± 0.71 μmol/min/g tissue) between 21–52% of unaffected dogs (Fig. 1c, Supplementary Fig. 2a). G6Pase activity for the untreated, affected dogs was low (0.57 ± 0.86 μmol/min/g tissue), in comparison with the activity measured in the unaffected dogs (7.21 ± 3.54 μmol/min/g tissue), thus confirming that this method appropriately monitored the response to gene therapy in GSD Ia dogs. Of note, the normal range of G6Pase activity in dogs was higher than the reported activity in normal human liver tissue, 4.37 ± 1.65 μmol/min/g tissue (Kishnani et al 2014). In contrast with survival liver biopsy data (Demaster et al 2012), liver glycogen was not significantly decreased in treated dogs compared to untreated dogs at necropsy (Fig. 1d). Detection of AAV-G6Pase DNA was low in all dogs examined at necropsy (Supplementary Fig. 2b). In spite of low G6Pase activity detected, survival vastly improved in treated dogs compared to untreated dogs (Fig. 1e), increased from a mean of 0.09 years in untreated dogs to a mean of 6.9 years in treated dogs. AAV-G6Pase treated dogs were able to maintain normoglycemia throughout their lifetime as demonstrated through fasting glucose curves (Fig. 1f, Supplementary Fig 1). Although dogs did not appear to have clinical signs of lactic acidosis, lactate was elevated higher than normal throughout their life, albeit lower than untreated GSD Ia dogs (Supplementary Fig 3).
Liver Disease
Histologically and biochemically, AAV-G6Pase treated dogs demonstrated changes consistent with GSD Ia. Beginning at birth, serum ALP is elevated in GSD Ia dogs much higher than normally seen in a healthy puppy, and serum AST, ALT, GGT, and cholesterol frequently are elevated (Supplementary Fig. 3). Over time, the serum transaminase concentrations remained elevated, as did triglycerides and cholesterol.
Hepatomegaly was noted throughout the lifetime of each dog through serial radiographic and ultrasonographic examinations (Fig. 2a). Liver tumors were first appreciable on ultrasound examination in dog H and W at 6–7 years of age, while other tumors were detected at necropsy. Upon necropsy, GSD Ia dog livers weighed an average of 12.1 ± 5.4% of body weight (n=4), while an average of 3.4% is normal for healthy dogs (Evans and Miller 2013). Livers appeared diffusely pale, enlarged with rounded lobar edges, friable, with multiple regions consistent macroscopically with focal nodular hyperplasia (FNH) (Fig. 2b). Areas of altered hepatic parenchyma compatible with FNH were noted multicentrically scattered in multiple lobes throughout the liver in all seven dogs.
Fig. 2.
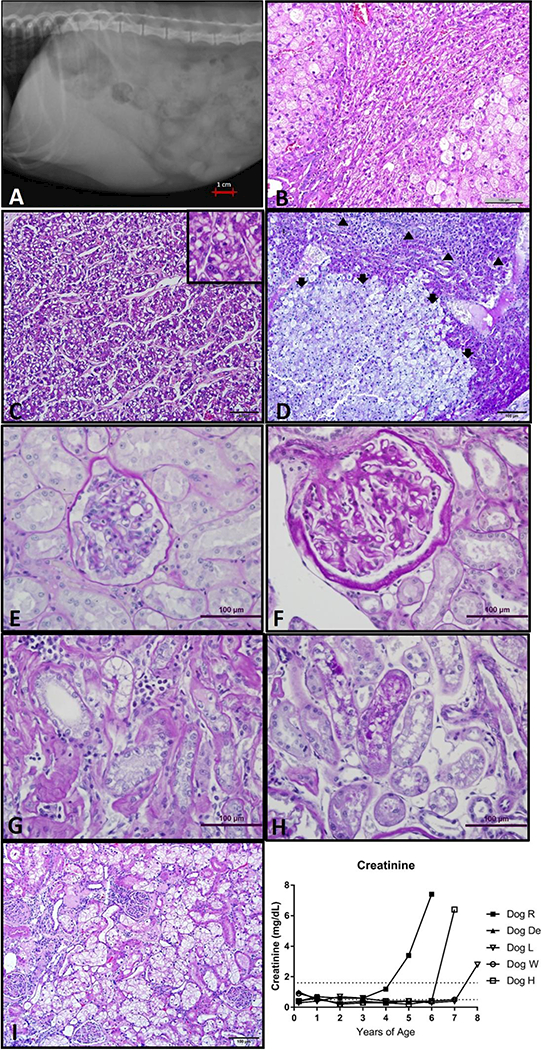
Liver and kidney disease in GSD Ia dogs. a. Hepatomegaly of Dog H noted on right lateral abdominal radiographs, liver far surpasses the border of the rib cage. b. Liver histology from Dog L with relatively normal parenchyma entrapped between two encroaching areas of focal nodular hyperplasia with marked hepatocyte enlargement and vacuolation (H&E 200x). c. Liver mass histology from dog De demonstrates nests and thickened trabeculae of neoplastic cells more prominent with absence of lobular architecture (H&E 200x). Inset box (400x) demonstrates a mitotic figure in center. d. Liver mass histology from dog L is composed of large, vacuolated neoplastic hepatocytes compresses and invades adjacent more normal hepatic parenchyma. Normal lobular architecture not present in neoplastic mass (adenoma, Arrows). There is a thin layer of compressed more normal hepatocytes above the adenoma. In upper most part of photomicrograph are nests and trabeculae thickened by neoplastic hepatocytes 2 or more cells wide (carcinoma, arrowheads). (PAS, 100x). e. Normal renal glomerulus (PAS, 200X). f. Glomerulus with changes of FSGS demonstrating thickened PAS positive glomerular basement membrane as well as thickened Bowman’s capsule (PAS 200x). g. Tubulo-interstitial changes as part of the spectrum of chronic renal lesions in dog H. Note interstitial mononuclear inflammatory infiltrate and thickened peritubular basement membrane. (PAS 200x) h. PAS positive droplets in proximal tubular epithelium (PAS 200x). i. Kidney histology from Dog R (100x) There is clustering of glomeruli with varying changes. In several glomeruli, there is moderate eosinophilic thickening of Bowman’s capsules (sclerosis). In other glomeruli, the sclerosis of the Bowman’s capsules is more severe and there is moderated to marked eosinophilic thickening of the mesangium of one or more segments of the glomeruli tufts. A sclerotic remnant of a glomerulus is also present. The proximal convoluted tubular epithelial cells are markedly swollen with clear, cytoplasmic vacuoles. j. Creatinine increased over time in dogs R, H and L.
Microscopically, livers from all dogs had variably sized multifocal to coalescing proliferative masses with irregular borders and relatively poor demarcation from more normal surrounding hepatic parenchyma. The histologic appearance of the proliferative regions in the GSD Ia dogs was somewhat unique to this series of animals. Unlike the typical microscopic appearance of canine FNH that is often compressive and relatively well demarcated from surrounding normal hepatic parenchyma, many of the lesions in the GSD Ia dog livers had irregular scalloped borders that merged without encapsulation into surrounding areas of more normal parenchyma that also had vacuolar change (glycogen and non-glycogen vacuoles) (Fig 2b). Canine FNH is most commonly characterized by vacuolar change within hepatocytes along with changes in cell size and number (Fabry et al 1982). These features were prominent within the GSD Ia proliferative masses that were also characterized by the maintenance of normal hepatic architecture, a feature that differentiates the hyperplastic/dysplastic lesions from hepatocellular neoplasms such as HCA and HCC (Fig 2).
An unusual feature of the GSD Ia dog livers was the number and size of the proliferative nodules that often seemed to merge into one another, often entrapping more normal hepatic parenchyma such as portal tracts. For most dogs, it was difficult to enumerate the true number of non-neoplastic proliferative masses due to the merging of masses and the “collision” of these lesions with the multiple large hepatocellular tumors (HCA and HCC) found in four of the five dogs. HCA was differentiated from non-neoplastic FNH by the loss of normal hepatic architecture (hepatic cord structure, portal tracts, central veins etc.) (Fig 2d). Four dogs had HCC that was predominantly of either a pelioid or trabecular histopathology. Both neoplastic and non-neoplastic proliferative masses contained hepatocytes with intracytoplasmic eosinophilic inclusions believed to represent Mallory bodies (Denk et al 2000).
Renal Disease
The oldest male and female dogs in this study were euthanized due to severe renal disease (Table 1). Dog R had elevated serum creatinine and blood urea nitrogen (BUN) beginning at 4 years of age (Figure 2j); Creatinine was 3.4 and 7.6 mg/dL (0.5–1.6 mg/dL normal range), while BUN was 118 and 140 mg/dL (6–31 mg/dL normal range), at 5 years of age and at last blood draw at 5.7 years of age, respectively. Dog R exhibited proteinuria demonstrated by elevated urine protein:creatinine ratio (UPC), ranging from 0.8 to 2.9 (normal ≤0.5) and microalbuminuria ranging from 1.2 to 6.9 mg/dL (normal <2.5) during the last year of life. Dog L had a rapid decline in renal function that required euthanasia; BUN and creatinine were 50 mg/dL and 1.4 mg/dL, respectively, at endpoint with granular casts present in the urine. Dog H similarly had severe acute renal failure requiring euthanasia at almost 7 years of age; BUN and creatinine were 25 mg/dL and 6.4 mg/dL, respectively with microalbuminuria (26.1 mg/dL, normal <2.5) and UPC of 7.1 (normal ≤0.5) present at endpoint. All three dogs had a decrease in appetite, activity and attitude at endpoints. All other dogs had occasional mild increases in BUN noted, but did not develop clinical signs of renal disease. Macroscopic lesions of chronic renal disease was evident at necropsy of dogs that had clinical evidence of renal dysfunction. Kidneys were pale, firm, and had irregular cortical surfaces sometimes containing scattered cysts (Supplementary Fig 4c).
Microscopic examination of kidneys from all five dogs revealed both glomerular and tubular changes that increased in severity with increasing age. Glomerular changes consisted of focal segmental glomerular sclerosis (FSGS) and included thickening and lamellation of the glomerular basement membrane and marked thickening of the Bowman’s capsule (Fig. 2f). Glomerular lesions were pronounced in dogs that had clinical renal failure. These dogs also had evidence of tubulo-interstitial disease including interstitial inflammatory infiltrates, thick peritubular basement membranes and interstitial fibrosis (Fig. 2f-i). All GSD Ia dogs had evidence of PAS positive vacuoles in the cortical proximal tubular epithelium (Fig. 2h), although the degree of tubular change varied from dog to dog.
Urolithiasis
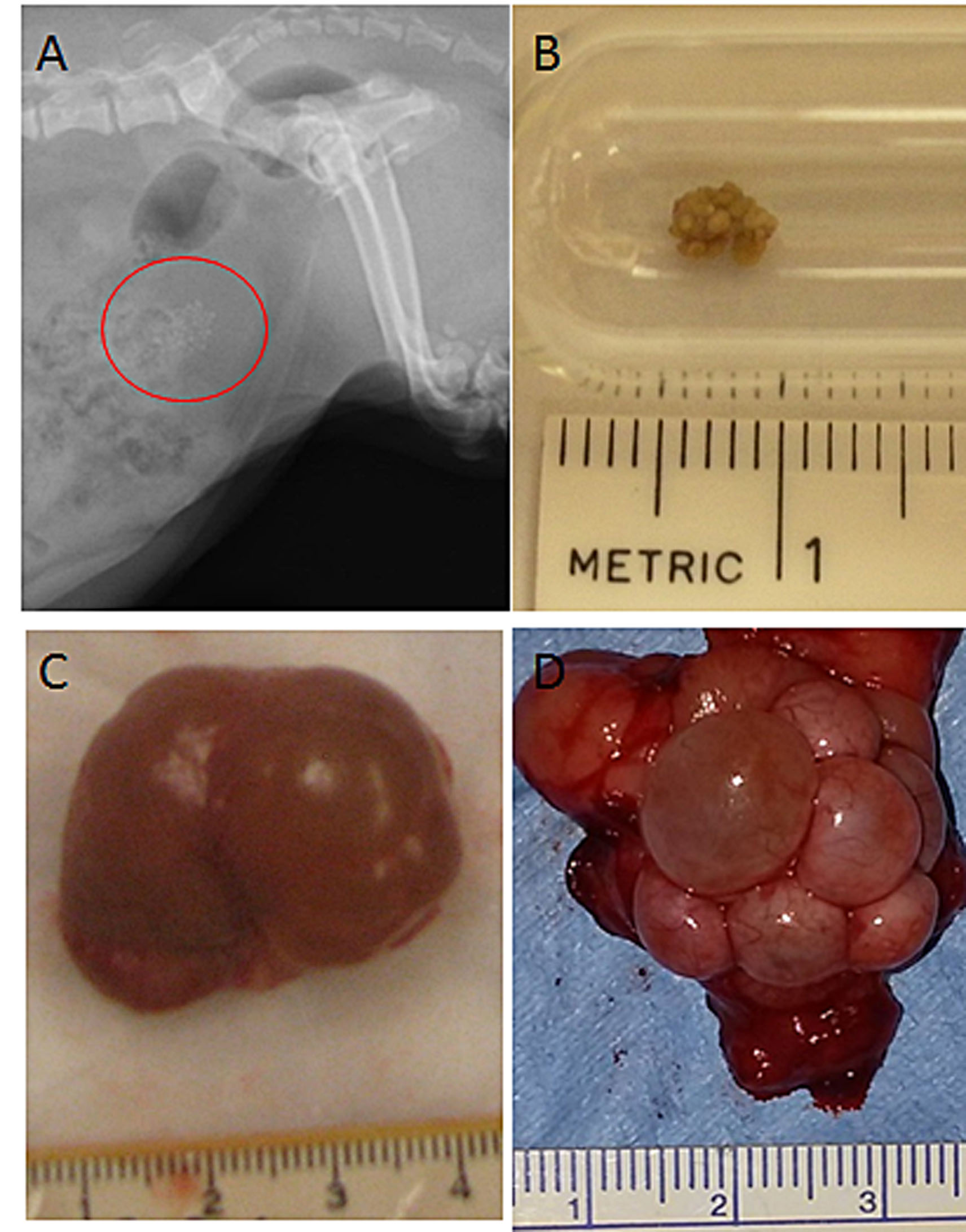
Two dogs were noted to have urolithiasis throughout their lifetime. Upon routine abdominal radiographic examination at 8 years of age, Dog L had evidence of radiopacities within her urinary bladder (Supplementary Fig. 4a). She was asymptomatic with no signs of stranguria or hematuria until she was begun on aggressive fluid therapy for concurrent renal disease, when she began to pass uroliths. Uroliths were removed and analyzed at necropsy and were determined to be 100% calcium oxalate monohydrate.
Dog D presented with hematuria and stranguria at 4 years of age; many oxalate crystals were detected in his urine sediment. Multiple uroliths ranging from small grains to a 0.5 cm stones were removed during cystotomy (Supplementary Fig. 4b). Microscopic examination of the bladder showed chronic inflammatory changes within the submucosa. Uroliths were determined to be 90% calcium oxalate and 10% calcium phosphate. Small nephroliths were also found upon necropsy and histopathologic examination of kidneys.
Polycystic Ovarian Syndrome
One GSD Ia dog, dog L, had irregularly prolonged and more frequent hemorrhagic heat cycles noted at 4 years of age. Due to this problem, she underwent an ovariohysterectomy, where she was noted to have polycystic ovaries. In addition to this, one GSD Ia carrier dog that was used for breeding developed infertility problems after 2–3 litters and was adopted out from the colony. Upon ovariohysterectomy, she had a thickened cervix with polycystic ovaries (Supplementary Fig. 4d). Microscopically the ovary contained numerous follicular cysts most of which were lined by granulosa cells.
Bone Malformations and Osteopenia
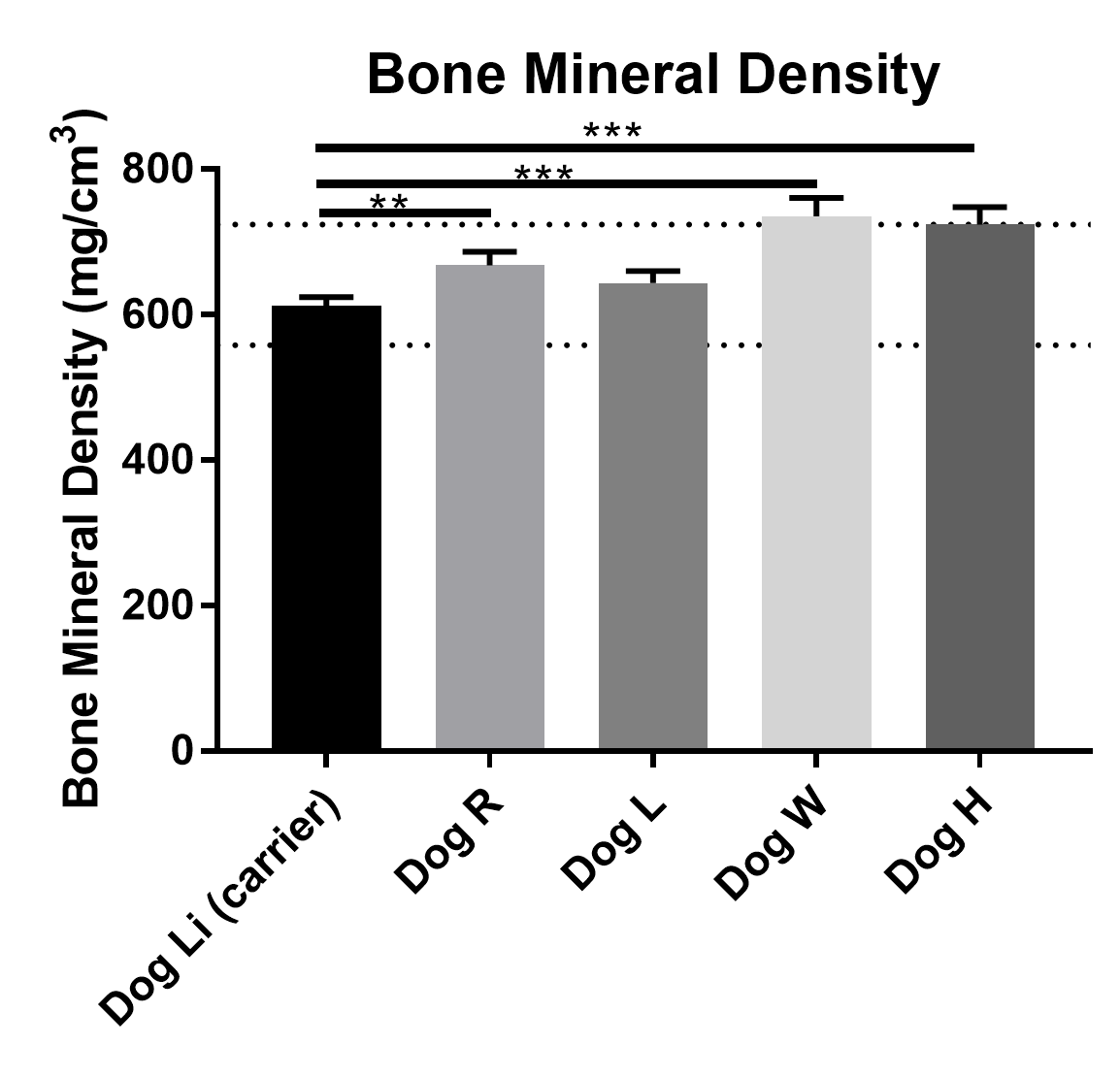
Growth deficiencies were previously reported in GSD Ia dogs likely due to delayed osteogenesis (Brooks et al 2013). Frequently adult dogs possess open fontanelles throughout their lifetime, angular limb deformities in a few dogs, as well as thinner, more brittle bones noted upon necropsy in dog R. A GSD Ia carrier, Dog Li also had signs of intervertebral disk disease at 11 years of age. QCT analysis revealed average bone mineral density (BMD) of the femur was significantly higher in dogs H, R, and W than found in Dog Li in five analyzed locations. Dog R also had decreased BMD compared to dogs H and W (Supplementary Table 1 and Supplementary Fig. 5).
Discussion
Mortality in GSD Ia dogs is greater than in human patients, but with the aid of AAV-mediated gene therapy, the lifespan of affected dogs has greatly increased (Demaster et al 2012) (Fig. 1e). GSD Ia dogs typically do not live more than two months with dietary therapy alone and show clinical signs almost immediately after birth; this is in contrast with human patients that sometimes are not diagnosed until 6 months of age or older (Rake et al 2002). This increased mortality in dogs may be due to the increased rate of growth comparatively; dogs reach sexual maturity and can be nearly full-grown at around 6 months of age vs 12–13 years as being the average age of onset of puberty in humans. This difference in the rate of growth and development underlies greater metabolic demands in dogs, in comparison with humans, which implies that GSD Ia might cause earlier, more severe complications in the canine model. This theory is supported by findings that dog metabolic rate is higher than people, consuming 0.6–1 g of nutrients per cubic centimeter of liver per day, vs 0.088–0.098 g in human liver (Shestopaloff 2014). Due to these metabolic differences, we presume that gene replacement therapy might be more effective in human GSD Ia patients than it has been in GSD Ia dogs. Which is highly applicable, as a clinical trial using AAV 8 gene therapy in GSD Ia human patients was recently approved by the FDA (NCT03517085).
As in the original report describing these dogs, biochemical correction demonstrated by low liver glycogen and absence of hypoglycemia during controlled fasting persisted long after AAV vector genomes decreased to lower numbers (Demaster et al 2012). The emergence of long-term complications of GSD Ia described herein further demonstrated that the canine GSD Ia model represents an accurate model for the development of novel therapies of GSD Ia. One benefit of the model is that long-term complications developed on a much more rapid time course, consistent with the concept of “dog years”. The dogs developed HCA and HCC, progressive kidney disease, urolithiasis, and polycystic ovarian disease within a few years, while these conditions occur during mid-life in humans with GSD Ia (Koeberl et al 2009).
The development of such complications in human patients is correlated with poor metabolic control (Kishnani et al 2014). The dogs on this study appeared to have well-regulated metabolic control without the need of rigorous dietary therapy and in fact, GSD Ia dogs not respond similarly to cornstarch therapy (Weinstein et al 2010). However, monitoring glucose metabolism in response to therapy is limited, longitudinal studies in dogs using isotopically labeled glucose following gene therapy would be another aid in determining metabolic control (van Dijk et al 2013). In this study, the mildly elevated lactate levels noted in the dogs over time (Supplementary Fig. 3) suggests that gene therapy did not achieve complete metabolic control and likely precipitated the long-term complications, in spite of greatly prolonging life.
The findings of such a high incidence (4/5) of hepatocellular neoplasms (HCA and HCC) in this series of dogs is quite striking, as these tumors are rare spontaneous findings in dogs. The incidence of HCA and HCC is under 1% of the canine population and usually occurs in dogs that are older than 10 years of age (Patnaik et al. 1981). Tumor multiplicity is also an unusual finding in spontaneous canine hepatocellular tumors. In several animals, development of HCC appeared to arise within hepatic lobes in conjunction with benign proliferative masses such as HCA, making it plausible that GSD Ia dogs may prove useful as a high incidence model to further our understanding of the molecular genetic alterations that occur in multistep hepatocellular neoplasia. Tumor formation correlated with the increase in glycogen content observed (Fig. 1d), in comparison with the low liver glycogen content reported in the first months following vector administration (Demaster et al 2012). It is also of note that the dogs in this study with hepatic tumors achieved between 27–52% of normal G6Pase activity (Supplementary Fig. 2). This is in contrast to reports of mice that showed achievement of >3% G6Pase correction prevented tumor formation (Lee et al 2012). It is likely that the long duration of this study with a large animal model left many uncorrected hepatic cells over years of time, in contrast to the shorter lifespan of mice, which were followed to 90 months of age (Lee et al 2012).
All five dogs in this series developed renal lesions that increased in severity with age. All dogs had lesions consistent with FSGS. Several of the dogs had significant tubule-interstitial changes including tubular atrophy and basement membrane changes as well as interstitial inflammatory infiltrates and fibrosis. FSGS in the canine model is identical to the glomerulopathy of GSD I patients. It is believed that GSD I tubulopathy responds to metabolic control but that the glomerular lesions in GSD I patients progresses despite metabolic control (Okechuku et al 2017). It is likely that the canine model will be useful for helping to understand the long-term management of GSD I renal disease, but additional studies to elucidate the pathophysiology of renal changes are warranted.
Bone mineral densities measured in GSD Ia dogs were comparable to those previously reported in dog femurs and talus (Schneider et al 2004; Dingemanse et al 2017). The slightly lower values of BMD and clinical disease observed in Dog Li, L and Dog R may be indicative of mild osteopenia, but a more complete analysis is needed.
GSD Ia dogs that have received gene replacement therapy have overall followed a closer course of disease to that seen in human patients than untreated GSD Ia dogs (Table 2). However, gene replacement therapy has failed to fully reverse disease within the liver of GSD Ia dogs and prevent the long-term complications such as HCC, as well as renal disease. The reasons for this are likely multifactorial, but all point to failure of the current vector strategies to fully correct all liver cells. One report found that the minimal G6PC promoter in AAV-G6Pase evaluated here had lower activity and conferred lower efficacy than a longer promoter (Lee et al 2013), but other studies showed high efficacy from AAV-G6Pase containing this minimal promoter (Koeberl et al 2008; Luo et al 2011). G6Pase histochemical staining in this (Fig. 1a) and previous studies show a small percentage of G6Pase- expressing cells within the liver (Weinstein et al 2010). While G6Pase activity is present throughout the lifetime of these dogs and sufficient in preventing hypoglycemia, it is significantly decreased from normal and may not quite achieve metabolic control, There are likely >75% uncorrected hepatocytes still diseased with glycogen and lipid accumulation. Studies have shown that metabolic syndrome and steatohepatitis are correlated with the occurrence of HCA (Chang et al 2013).
Table 2.
Comparison of clinical findings in GSD Ia dogs and human patients.
| Clinical findings in GSD Ia dogs | Clinical findings in GSD Ia human patients |
|---|---|
| Increased liver enzymes ALP, ALT, AST and GGT (Supplementary Fig. 3) | Increased ALP, ALT, AST and GGT (Moraru et al 2007) |
| Increased serum triglycerides and cholesterol (Supplementary Fig. 3) | Increased serum triglycerides and cholesterol- (Bali et al 1993; Rake et al 2002; Moraru et al 2007; Derks and van Rijn 2015) |
| Hepatomegaly (Fig. 2) | Hepatomegaly (Bali et al 1993; Rake et al 2002) |
| Fatty infiltration throughout all GSD Ia dog livers | Focal fatty infiltration, focal fatty sparing (Bali et al 1993; Rake et al 2002) |
| Glycogen accumulation with hepatomegaly in all dogs (Fig. 2) | Glycogen accumulation with hepatomegaly (Bali et al 1993; Moraru et al 2007) |
| Hepatocellular adenoma(s) with Mallory hyaline in 5/7 dogs (Fig. 2) | Hepatocellular adenoma with Mallory hyaline (Labrune et al 1997; Kelly and Poon 2001; Volmar et al 2003; Carreiro et al 2007; Reddy et al 2007; Di Rocco et al 2008; Kishnani et al 2009; Sakellariou et al 2012; Sever et al 2012; Calderaro et al 2013) |
| Hepatocellular carcinoma in 5/7 dogs (Fig. 2) | Hepatocellular carcinoma (Franco et al 2005; Manzia et al 2011; Baheti et al 2015) |
| Focal nodular hepatic hyperplasia in 7/7 dogs (Fig. 2) | Focal nodular hepatic hyperplasia (Carreiro et al 2007) |
| Impaired hepatocellular autophagy (Farah et al 2016) |
Not described |
| Moderate to severe biliary sludge in 7/7 dogs (unpublished observation) | Gallstones and biliary sludge (Kelly and Poon 2001; Rake et al 2002) |
| Splenomegaly in 1/7 dogs (unpublished observation) | Splenomegaly- (Rake et al 2002) |
| Pancreatitis in 2/7 dogs; dog W reported in (Demaster et al 2012) and dog H in current study based on elevated amylase and PrecisionPSL | Pancreatitis- (Bali et al 1993; Rake et al 2002) |
| Urolithiasis in 2/7 dogs (Supplementary Fig. 4a,b) | 2/5 patients with kidney stones(Talente et al 1994) (Rake et al 2002) 2 patients with recurrent nephrourolithiasis in Duke GSD Clinic |
| Proteinuria and microalbuminuria reported in dogs R and H |
Proteinuria and microalbuminuria (Bali et al 1993; Rake et al 2002; Gjorgjieva et al 2016) |
| Azotemia in 3/7 dogs (Fig. 2) | Azotemia (Rake et al 2002) |
| Kidney disease in 7/7 dogs plus those reported previously (Crane et al 2012; Demaster et al 2012) (Fig. 2) | Progressive Renal disease (Rake et al 2002; Gjorgjieva et al 2016) |
| Renal cysts in 1/7 dogs (Supplementary Fig. 4c) | Renal cysts (Gjorgjieva et al 2016) |
| Hypoglycemia key feature of disease (Kishnani et al 2001; Crane et al 2012; Demaster et al 2012) | Hypoglycemia key feature of disease (Bali et al 1993) |
| Elevated lactate throughout lifetime of treated dogs and lactic acidosis in untreated GSD Ia dogs (Kishnani et al 2001)(Supplementary Fig. 3) | Lactic acidosis (Bali et al 1993; Moraru et al 2007) |
| Polycystic ovaries in one GSD Ia affected dog and one GSD Ia carrier dog (Supplementary Fig. 4d) | Polycystic ovaries in GSD I and III patients associated with hyperinsulinemia (Lee et al 1995; Sechi et al 2013) |
| Increased bleeding and prolonged estrus in dog L (unpublished observation) | Menorrhagia (Austin et al 2013) |
| Not described | Bleeding Tendency (Rake et al 2002) |
| Brittle bones noted on necropsy of Dog R | Osteopenia/Osteoporosis (Bali et al 1993; Rake et al 2002; Minarich et al 2012) |
| Growth failure (Brooks et al 2013) | Growth failure (Rake et al 2002; Moraru et al 2007) |
| Intermittent diarrhea noted in many dogs (unpublished observation) | IBD (Visser et al 2002; Lawrence et al 2015) |
| Obesity observed in some dogs, but mostly controlled with diet (unpublished observation) | Obesity (Korljan Jelaska et al 2013) |
| Hyperuricemia in untreated GSD Ia dogs (Kishnani et al 2001) | Gout or increased uric acid (Bali et al 1993; Rake et al 2002) |
| Not described | Increased urinary biomarker Glc4(Lennartson et al 1976; Oberholzer and Sewell 1990; Sluiter et al 2012) |
| Not described | Anemia (Wang et al 2012) |
| Occasionally elevated blood glucose in dog R (unpublished observation) | Metabolic syndrome (Melis et al 2015) |
This study demonstrated the loss of AAV vector transduction and hepatic G6Pase at necropsy in the canine model for GSD, which correlated with the onset of long-term complications of GSD Ia (Supplementary Figure 2). AAV vector administration to young mice accomplished a high level of liver transduction followed by declining numbers of vector genomes over the ensuing months (Koeberl et al 2006; Cunningham et al 2008; Weinstein et al 2010; Yiu et al 2010). For example, an AAV2/8 vector decreased from >2 copies per liver cell at 1 month of age to 0.3 copies at 7 months of age in G6Pase- KO mice (Koeberl et al 2006). Similarly, an AAV2/8 vector was administered to a GSD Ia puppy at one day of age, and prevented hypoglycemia for three hours at one month of age; however, by two months of age the dog became hypoglycemic after one hour of fasting (Weinstein et al 2010). Chou and colleagues highlighted the transience of AAV vector-mediated expression by measuring a 12-fold decline in G6Pase expression in mice with GSD Ia between age 2 to 6 weeks (Yiu et al 2010).
Human patients have been treated with corticosteroids to suppress T cell responses in response to elevated transaminases following AAV vector administration, which preserved transgene expression (Nathwani et al 2011). This approach would be detrimental in GSD Ia, because glucocorticoid treatment prevents the uptake of glucose by cells and reduces intracellular glucose (Chen 2001). It is unlikely that a cellular immune response was the cause for the loss of AAV vector transduction over time in these dogs. We have demonstrated long-term biochemical correction of the liver following AAV vector administration in these dogs, with the exception of one of the dogs previously reported (Demaster et al 2012). At least one of these dogs had low titers of anti-AAV antibodies prior to vector exposure and clearly represented an outlier (Methods, dog T). However, no lymphocytic infiltrates were demonstrated in survival liver biopsy of that dog within 6 weeks of vector administration, reducing the likelihood of a cytotoxic T cell response against the vector (Demaster et al 2012). Finally, the lack of recurrent hypoglycemia is not consistent with elimination of transgene expression that would be expected following cytotoxic T lymphocyte responses.
It is likely that genome editing would increase the viability of current gene replacement techniques by allowing more cells to be corrected over time (Landau et al 2016). Currently transgene expression from AAV-G6Pase is gradually lost (Supplementary Fig. 2b), most likely due to dilution of episomal DNA in the GSD Ia liver, which exhibits elevated apoptosis, in comparison with normal liver (Sun et al 2009). However, other modifications to improve vector efficiency including codon optimization and advanced targeting of AAV to both liver and kidneys may lead to a stronger treatment effect. Work to improve AAV vectors is ongoing and will likely contribute to a greater treatment effect in the future.
Small molecule therapies in combination with gene therapies would likely be beneficial in treating the diseased hepatocytes and increase transduction of AAV vectors. We have demonstrated a beneficial effect in using rapamycin to increase hepatic autophagy and decrease hepatomegaly in dogs with GSD Ia (Farah et al 2016). Since gene replacement therapy has thus far stably corrected only a small number of cells, we expect that combination therapy with a pro-autophagy drug could improve liver function and decrease apoptosis leading to a better treatment effect from gene therapy.
In conclusion, the GSD Ia dog model follows a similar clinical outcome as human GSD la patients and is an important resource for developing novel therapies for GSD Ia. AAV gene therapy has greatly increased lifespan in dogs and has promising potential to treating hypoglycemia in patients decreasing the need for strict dietary regimens, but patients may still require conventional treatment for liver and renal disease. Our current data reveals limitations in AAV gene therapy preventing long-term complications, most importantly renal disease and hepatic tumors. Modification of current gene therapies to increase liver and renal transduction efficiency, as well as persistence of treatment effect is needed.
Supplementary Material
Supplementary Figure 1. Area under the curve (AUC) of 8-hour fasted glucose curves over the lifetime of GSD Ia AAV-G6Pase treated dogs. *indicates glucose curve 2–4 weeks after last vector treatment. See table 1 for vector therapy summary, excluding dog T, which had poor response to all vector therapies given and data is not included in main document.
{kind=link}
Supplementary Figure 2. a. Hepatic G6Pase from individual dogs at necropsy. b. Vector DNA copies/cell in dog liver at necropsy; mean ± SD for untreated dogs.
{kind=link}
Supplementary Figure 3. Serum enzyme concentrations of GSD Ia dogs throughout lifetime; Open symbols denote female dogs. Dotted lines denote enzyme concentrations for normal dogs. ^ denote that some values were higher than lab was able to detect. Dog De was >993 for ALP at 3 years of age, dogs De and H were >993 for ALP at 4 years of age, and dogs R and L were >993 for ALP at 6 years of age. Dog De was >450 for cholesterol at 3 years of age and dog L was >450 for cholesterol at 6 years of age.
{kind=link}
Supplementary Figure 4. a. Urolithiasis detected in Dog L on Radiographs at 8 years of age, urinary bladder denoted by circle. b. Urolith surgically removed from Dog De at 4 years of age. c. Renal cysts from Dog R at 5.7 years of age. d. Polycystic Ovaries in GSD Ia carrier, Dog A at 5 years of age.
{kind=link}
Supplementary Figure 5. Bone mineral densities of the femurs of GSD Ia dogs compared to a GSD Ia carrier. Mean ± SD of 5 different measurements through QCT. Dotted lines represent BMD range obtained from Schneider et al 2006 in normal dog femurs.
{kind=link}
Synopsis:
Gene therapy with AAV in GSD Ia dogs improves survival and prevents hypoglycemia; however, similar long-term complications as seen in human GSD Ia patients persist.
Acknowledgements
We acknowledge funding for this work provided by the Children’s Fund for GSD Research, Children’s Miracle Network, Association for Glycogen Storage Disease, For the Love of Christopher, the Alice and YT Chen Center for Pediatric Genetics and Genomics, and grant R01DK105434–01A1 from the National Institute of Diabetes and Digestive and Kidney Diseases. We would also like to acknowledge inspiration and support from Dr. Emory and Mrs. Mary Chapman and their son Christopher, and from Dr. John and Mrs. Michelle Kelly. We appreciate the care that these dogs received from the Duke Division of Laboratory Animal Resources staff, namely Ms. Laura Jordan and the veterinary support from Drs. Francis Sun, Amanda DeMaster, Sarah Faircloth, Shannon Smith, Feli Smith, Angela Garner and Jai Tubbs and veterinary technicians, namely Christian Marsini, Kelly Franke, Diego Zapata and Jeff Lee. The collective support from the multiple students and staff in the dog feeding team over these years ensured a long, comfortable life for the GSD Ia dogs.
Footnotes
Compliance with Ethics Guidelines
Conflict of Interest:
Elizabeth D. Brooks, Dustin J. Landau, Jeffrey I. Everitt, Talmage T. Brown, Kylie M. Grady, Lauren Waskowicz, Cameron R. Bass, John D’Angelo, Yohannes G. Asfaw, Kyha Williams, Priya Kishnani, and Dwight D. Koeberl have no conflicts of interest to report.
Animal Rights:
All institutional and national guidelines for the care and use of laboratory animals were followed.
Contributions of Authors:
Elizabeth Brooks wrote the manuscript, supervised the colony, and collected and compiled data from the canine model.
Dustin Landau assisted with manuscript preparation, made vector therapies and collected data from the canine model.
Jeffrey Everitt and Talmage Brown performed histologic analysis of tissues and prepared photomicrographs.
Kylie Grady performed biochemical and histochemical analysis of liver and assisted with manuscript preparation.
Lauren Waskowicz performed biochemical analysis of liver.
Cameron Bass and John D’Angelo performed and analyzed skeletal imaging.
Yohannes Asfaw contributed necropsies and assisted with preparation of manuscript and images.
Kyha Williams provided clinical veterinary care and aided in clinical care of the canine model.
Priya Kishnani helped with manuscript preparation and provided expert opinion.
Dwight Koeberl designed experiments, assisted with manuscript preparation and provided expert opinion.
References
- Austin SL, El-Gharbawy AH, Kasturi VG, James A, Kishnani PS (2013) Menorrhagia in patients with type I glycogen storage disease. Obstet Gynecol 122: 1246–1254. [DOI] [PubMed] [Google Scholar]
- Baheti AD, Yeh MM, O’Malley R, Lalwani N (2015) Malignant Transformation of Hepatic Adenoma in Glycogen Storage Disease Type-1a: Report of an Exceptional Case Diagnosed on Surveillance Imaging. J Clin Imaging Sci 5: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali DS, Chen YT, Austin S, Goldstein JL (1993) Glycogen Storage Disease Type I In Adam MP, Ardinger HH, Pagon RA et al. eds. GeneReviews((R)) Seattle (WA). [Google Scholar]
- Brooks ED, Little D, Arumugam R, et al. (2013) Pathogenesis of growth failure and partial reversal with gene therapy in murine and canine Glycogen Storage Disease type Ia. Mol Genet Metab 109: 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderaro J, Labrune P, Morcrette G, et al. (2013) Molecular characterization of hepatocellular adenomas developed in patients with glycogen storage disease type I. J Hepatol 58: 350–357. [DOI] [PubMed] [Google Scholar]
- Carreiro G, Villela-Nogueira CA, Coelho H, et al. (2007) Orthotopic liver transplantation in glucose-6-phosphatase deficiency--Von Gierke disease--with multiple hepatic adenomas and concomitant focal nodular hyperplasia. J Pediatr Endocrinol Metab 20: 545–549. [DOI] [PubMed] [Google Scholar]
- Chang CY, Hernandez-Prera JC, Roayaie S, Schwartz M, Thung SN (2013) Changing epidemiology of hepatocellular adenoma in the United States: review of the literature. Int J Hepatol 2013: 604860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YT (2001) Glycogen Storage Diseases In Scriver CR, Beaudet AL, Sly WS, Valle D eds. The Metabolic and Molecular Bases of Inherited Disease New York: McGraw-Hill, 1521–1551. [Google Scholar]
- Clar J, Gri B, Calderaro J, et al. (2014) Targeted deletion of kidney glucose-6 phosphatase leads to nephropathy. Kidney Int 86: 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane B, Luo X, Demaster A, et al. (2012) Rescue administration of a helper-dependent adenovirus vector with long-term efficacy in dogs with glycogen storage disease type Ia. Gene Ther 19: 443–452. [DOI] [PubMed] [Google Scholar]
- Cunningham SC, Dane AP, Spinoulas A, Logan GJ, Alexander IE (2008) Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol Ther 16: 1081–1088. [DOI] [PubMed] [Google Scholar]
- Demaster A, Luo X, Curtis S, et al. (2012) Long-term efficacy following readministration of an adeno-associated virus vector in dogs with glycogen storage disease type Ia. Hum Gene Ther 23: 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk H, Stumptner C, Zatloukal K (2000) Mallory bodies revisited. J Hepatol 32: 689–702. [DOI] [PubMed] [Google Scholar]
- Derks TG, van Rijn M (2015) Lipids in hepatic glycogen storage diseases: pathophysiology, monitoring of dietary management and future directions. J Inherit Metab Dis 38: 537–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemanse W, Muller-Gerbl M, Jonkers I, Sloten JV, van Bree H, Gielen I (2017) A prospective follow up of age related changes in the subchondral bone density of the talus of healthy Labrador Retrievers. BMC Vet Res 13: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rocco M, Calevo MG, Taro M, Melis D, Allegri AE, Parenti G (2008) Hepatocellular adenoma and metabolic balance in patients with type Ia glycogen storage disease. Mol Genet Metab 93: 398–402. [DOI] [PubMed] [Google Scholar]
- Evans HE, Miller ME (2013) Miller’s anatomy of the dog, St. Louis, Missouri: Elsevier. [Google Scholar]
- Fabry A, Benjamin SA, Angleton GM (1982) Nodular hyperplasia of the liver in the beagle dog. Vet Pathol 19: 109–119. [DOI] [PubMed] [Google Scholar]
- Farah BL, Landau DJ, Sinha RA, et al. (2016) Induction of autophagy improves hepatic lipid metabolism in glucose-6-phosphatase deficiency. J Hepatol 64: 370–379. [DOI] [PubMed] [Google Scholar]
- Franco LM, Krishnamurthy V, Bali D, et al. (2005) Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J Inherit Metab Dis 28: 153–162. [DOI] [PubMed] [Google Scholar]
- Gjorgjieva M, Raffin M, Duchampt A, et al. (2016) Progressive development of renal cysts in glycogen storage disease type I. Hum Mol Genet 25: 3784–3797. [DOI] [PubMed] [Google Scholar]
- Jonges GN, Van Noorden CJ, Gossrau R (1990) Quantitative histochemical analysis of glucose-6- phosphatase activity in rat liver using an optimized cerium-diaminobenzidine method. J Histochem Cytochem 38: 1413–1419. [DOI] [PubMed] [Google Scholar]
- Kelly PM, Poon FW (2001) Hepatic tumours in glycogen storage disease type 1 (von Gierke’s disease). Clin Radiol 56: 505–508. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Austin SL, Abdenur JE, et al. (2014) Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med 16: e1. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Chuang TP, Bali D, et al. (2009) Chromosomal and genetic alterations in human hepatocellular adenomas associated with type Ia glycogen storage disease. Hum Mol Genet 18: 4781–4790. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Faulkner E, VanCamp S, et al. (2001) Canine model and genomic structural organization of glycogen storage disease type Ia (GSD Ia). Vet Pathol 38: 83–91. [DOI] [PubMed] [Google Scholar]
- Koeberl DD, Kishnani PS, Bali D, Chen YT (2009) Emerging therapies for glycogen storage disease type I. Trends EndocrinolMetab 20: 252–258. [DOI] [PubMed] [Google Scholar]
- Koeberl DD, Kishnani PS, Chen YT (2007) Glycogen storage disease types I and II: Treatment updates. Journal of Inherited Metabolic Disease 30: 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberl DD, Pinto C, Sun B, et al. (2008) AAV vector-mediated reversal of hypoglycemia in canine and murine glycogen storage disease type Ia. MolTher 16: 665–672. [DOI] [PubMed] [Google Scholar]
- Koeberl DD, Sun BD, Damodaran TV, et al. (2006) Early, sustained efficacy of adeno-associated virus vector-mediated gene therapy in glycogen storage disease type Ia. Gene Ther 13: 1281–1289. [DOI] [PubMed] [Google Scholar]
- Korljan Jelaska B, Ostojic SB, Berovic N, Kokic V (2013) Continuous glucose monitoring in the treatment of obesity in patients with glycogen storage disease type Ia. Endocrinol Diabetes Metab Case Rep 2013: 130056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrune P, Trioche P, Duvaltier I, Chevalier P, Odievre M (1997) Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. J Pediatr Gastroenterol Nutr 24: 276–279. [DOI] [PubMed] [Google Scholar]
- Landau DJ, Brooks ED, Perez-Pinera P, et al. (2016) In Vivo Zinc Finger Nuclease-mediated Targeted Integration of a Glucose-6-phosphatase Transgene Promotes Survival in Mice With Glycogen Storage Disease Type IA. Mol Ther 24: 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence NT, Chengsupanimit T, Brown LM, Weinstein DA (2015) High Incidence of Serologic Markers of Inflammatory Bowel Disease in Asymptomatic Patients with Glycogen Storage Disease Type Ia. JIMD Rep 24: 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PJ, Patel A, Hindmarsh PC, Mowat AP, Leonard JV (1995) The prevalence of polycystic ovaries in the hepatic glycogen storage diseases: its association with hyperinsulinism. Clinical endocrinology 42: 601–606. [DOI] [PubMed] [Google Scholar]
- Lee YM, Jun HS, Pan CJ, et al. (2012) Prevention of hepatocellular adenoma and correction of metabolic abnormalities in murine glycogen storage disease type Ia by gene therapy. Hepatology 56: 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YM, Pan CJ, Koeberl DD, Mansfield BC, Chou JY (2013) The upstream enhancer elements of the G6PC promoter are critical for optimal G6PC expression in murine glycogen storage disease type Ia. Mol Genet Metab 110: 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennartson G, Lundblad A, Sjoblad S, Svensson S, Ockerman PA (1976) Quantitation of a urinary tetrasaccharide by gas chromatography and mass spectrometry. Biomedical mass spectrometry 3: 51–54. [DOI] [PubMed] [Google Scholar]
- Luo X, Hall G, Li S, et al. (2011) Hepatorenal correction in murine glycogen storage disease type I with a double-stranded adeno-associated virus vector. Mol Ther 19: 1961–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzia TM, Angelico R, Toti L, et al. (2011) Glycogen storage disease type Ia and VI associated with hepatocellular carcinoma: two case reports. Transplant Proc 43: 1181–1183. [DOI] [PubMed] [Google Scholar]
- Melis D, Rossi A, Pivonello R, et al. (2015) Glycogen storage disease type Ia (GSDIa) but not Glycogen storage disease type Ib (GSDIb) is associated to an increased risk of metabolic syndrome: possible role of microsomal glucose 6-phosphate accumulation. Orphanet J Rare Dis 10: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minarich LA, Kirpich A, Fiske LM, Weinstein DA (2012) Bone mineral density in glycogen storage disease type Ia and Ib. Genet Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraru E, Cuvinciuc O, Antonesei L, et al. (2007) Glycogen storage disease type I--between chronic ambulatory follow-up and pediatric emergency. J Gastrointestin Liver Dis 16: 47–51. [DOI] [PubMed] [Google Scholar]
- Mutel E, Abdul-Wahed A, Ramamonjisoa N, et al. (2011) Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J Hepatol 54: 529–537. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, et al. (2011) Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365: 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberholzer K, Sewell AC (1990) Unique oligosaccharide (apparently glucotetrasaccharide) in urine of patients with glycogen storage diseases. Clinical chemistry 36: 1381. [PubMed] [Google Scholar]
- Okechuku GO, Shoemaker LR, Dambska M, Brown LM, Mathew J, Weinstein DA (2017) Tight metabolic control plus ACE inhibitor therapy improves GSD I nephropathy. J Inherit Metab Dis 40: 703–708. [DOI] [PubMed] [Google Scholar]
- Patnaik AK, Hurvitz AI, Lieberman PH, Johnson GF (1981) Canine hepatocellular carcinoma. Vet Pathol 18: 427–438. [DOI] [PubMed] [Google Scholar]
- Penhoat A, Fayard L, Stefanutti A, Mithieux G, Rajas F (2014) Intestinal gluconeogenesis is crucial to maintain a physiological fasting glycemia in the absence of hepatic glucose production in mice. Metabolism 63: 104–111. [DOI] [PubMed] [Google Scholar]
- Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP (2002) Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 161 Suppl 1: S20–34. [DOI] [PubMed] [Google Scholar]
- Reddy SK, Kishnani PS, Sullivan JA, et al. (2007) Resection of hepatocellular adenoma in patients with glycogen storage disease type Ia. J Hepatol 47: 658–663. [DOI] [PubMed] [Google Scholar]
- Sakellariou S, Al-Hussaini H, Scalori A, et al. (2012) Hepatocellular adenoma in glycogen storage disorder type I: a clinicopathological and molecular study. Histopathology 60: E58–65. [DOI] [PubMed] [Google Scholar]
- Schneider S, Breit SM, Grampp S, et al. (2004) Comparative assessment of bone mineral measurements obtained by use of dual-energy x-ray absorptiometry, peripheral quantitative computed tomography, and chemical-physical analyses in femurs of juvenile and adult dogs. Am J Vet Res 65: 891–900. [DOI] [PubMed] [Google Scholar]
- Sechi A, Deroma L, Lapolla A, et al. (2013) Fertility and pregnancy in women affected by glycogen storage disease type I, results of a multicenter Italian study. J Inherit Metab Dis 36: 83–89. [DOI] [PubMed] [Google Scholar]
- Sever S, Weinstein DA, Wolfsdorf JI, Gedik R, Schaefer EJ (2012) Glycogen storage disease type Ia: linkage of glucose, glycogen, lactic acid, triglyceride, and uric acid metabolism. J Clin Lipidol 6: 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestopaloff YK (2014) Method for finding metabolic properties based on the general growth law. Liver examples. A general framework for biological modeling. PLoS One 9: e99836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluiter W, van den Bosch JC, Goudriaan DA, et al. (2012) Rapid ultraperformance liquid chromatography-tandem mass spectrometry assay for a characteristic glycogen-derived tetrasaccharide in Pompe disease and other glycogen storage diseases. Clinical chemistry 58: 1139–1147. [DOI] [PubMed] [Google Scholar]
- Sun B, Li S, Yang L, et al. (2009) Activation of glycolysis and apoptosis in glycogen storage disease type Ia. Mol Genet Metab 97: 267–271. [DOI] [PubMed] [Google Scholar]
- Talente GM, Coleman RA, Alter C, et al. (1994) Glycogen storage disease in adults. Annals of internal medicine 120: 218–226. [DOI] [PubMed] [Google Scholar]
- van Dijk TH, Laskewitz AJ, Grefhorst A, et al. (2013) A novel approach to monitor glucose metabolism using stable isotopically labelled glucose in longitudinal studies in mice. Lab Anim 47: 79–88. [DOI] [PubMed] [Google Scholar]
- Visser G, Rake JP, Kokke FT, Nikkels PG, Sauer PJ, Smit GP (2002) Intestinal function in glycogen storage disease type I. J Inherit Metab Dis 25: 261–267. [DOI] [PubMed] [Google Scholar]
- Volmar KE, Burchette JL, Creager AJ (2003) Hepatic adenomatosis in glycogen storage disease type Ia: report of a case with unusual histology. Arch Pathol Lab Med 127: e402–405. [DOI] [PubMed] [Google Scholar]
- Wang DQ, Carreras CT, Fiske LM, et al. (2012) Characterization and pathogenesis of anemia in glycogen storage disease type Ia and Ib. Genet Med 14: 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein DA, Correia CE, Conlon T, et al. (2010) Adeno-associated virus-mediated correction of a canine model of glycogen storage disease type Ia. Hum Gene Ther 21: 903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu WH, Lee YM, Peng WT, et al. (2010) Complete normalization of hepatic G6PC deficiency in murine glycogen storage disease type Ia using gene therapy. Mol Ther 18: 1076–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Area under the curve (AUC) of 8-hour fasted glucose curves over the lifetime of GSD Ia AAV-G6Pase treated dogs. *indicates glucose curve 2–4 weeks after last vector treatment. See table 1 for vector therapy summary, excluding dog T, which had poor response to all vector therapies given and data is not included in main document.
Supplementary Figure 2. a. Hepatic G6Pase from individual dogs at necropsy. b. Vector DNA copies/cell in dog liver at necropsy; mean ± SD for untreated dogs.
Supplementary Figure 3. Serum enzyme concentrations of GSD Ia dogs throughout lifetime; Open symbols denote female dogs. Dotted lines denote enzyme concentrations for normal dogs. ^ denote that some values were higher than lab was able to detect. Dog De was >993 for ALP at 3 years of age, dogs De and H were >993 for ALP at 4 years of age, and dogs R and L were >993 for ALP at 6 years of age. Dog De was >450 for cholesterol at 3 years of age and dog L was >450 for cholesterol at 6 years of age.
Supplementary Figure 4. a. Urolithiasis detected in Dog L on Radiographs at 8 years of age, urinary bladder denoted by circle. b. Urolith surgically removed from Dog De at 4 years of age. c. Renal cysts from Dog R at 5.7 years of age. d. Polycystic Ovaries in GSD Ia carrier, Dog A at 5 years of age.
Supplementary Figure 5. Bone mineral densities of the femurs of GSD Ia dogs compared to a GSD Ia carrier. Mean ± SD of 5 different measurements through QCT. Dotted lines represent BMD range obtained from Schneider et al 2006 in normal dog femurs.