

Abstract
Objective Alveolar macrophages play a key role in the development and resolution of acute respiratory distress syndrome (ARDS), modulating the inflammatory response and the coagulation cascade in lungs. Anti-coagulants may be helpful in the treatment of ARDS. This study investigated the effects of nebulized heparin on the role of alveolar macrophages in limiting lung coagulation and inflammatory response in an animal model of acute lung injury (ALI).
Methods Rats were randomized to four experimental groups. In three groups, ALI was induced by intratracheal instillation of lipopolysaccharide (LPS) and heparin was nebulized at constant oxygen flow: the LPS/Hep group received nebulized heparin 4 and 8 hours after injury; the Hep/LPS/Hep group received nebulized heparin 30 minutes before and 4 and 8 hours after LPS-induced injury; the LPS/Sal group received nebulized saline 4 and 8 hours after injury. The control group received only saline. Animals were exsanguinated 24 hours after LPS instillation. Lung tissue, bronchoalveolar lavage fluid (BALF) and alveolar macrophages isolated from BALF were analysed.
Results LPS increased protein concentration, oedema and neutrophils in BALF as well as procoagulant and proinflammatory mediators in lung tissue and alveolar macrophages. In lung tissue, nebulized heparin attenuated ALI through decreasing procoagulant (tissue factor, thrombin–anti-thrombin complexes, fibrin degradation products) and proinflammatory (interleukin 6, tumour necrosis factor alpha) pathways. In alveolar macrophages, nebulized heparin reduced expression of procoagulant genes and the effectors of transforming growth factor beta (Smad 2, Smad 3) and nuclear factor kappa B (p-selectin, CCL-2). Pre-treatment resulted in more pronounced attenuation.
Conclusion Nebulized heparin reduced pulmonary coagulopathy and inflammation without producing systemic bleeding, partly by modulating alveolar macrophages.
Keywords: acute respiratory distress syndrome, acute lung injury, anti-coagulants, heparin, alveolar macrophages
Introduction
Acute respiratory distress syndrome (ARDS) is a major cause of morbidity and mortality (30–40%) in critically ill patients. 1 2 Defined by non–heart-failure-related acute respiratory failure and inflammation, ARDS can arise from various local and systemic insults. 3 It is characterized by bilateral pulmonary infiltrates, increased endothelial permeability and oedema. 4 Although lung protective ventilation strategy and prone position have produced a major breakthrough in supportive care of ARDS patients, an effective pharmacological therapy for ARDS is not available yet.
Even though neutrophil influx and activation within the lungs contribute to the induction of ARDS, increasing evidence suggests that alveolar macrophages are critical to the initiation and maintenance of the inflammatory response and to the resolution phase. 5 6 More specifically, lung inflammation during ARDS is deeply correlated to the alveolar macrophages phenotype, function and cell–cell interactions. 6 7 Due to their plasticity, macrophages can be proinflammatory (M1) or anti-inflammatory activated (M2) depending on environmental signals. At the initial acute phase of ARDS, classically activated macrophages (M1) release early response cytokines such as tumour necrosis factor alpha (TNF-α) or inducible nitric oxide synthase (iNOS), stimulating the cells of the alveoli and recruiting neutrophils to the alveolar space, amplifying the inflammatory response and promoting the elimination of pathogens. At the resolution phase, alternative macrophages (M2) are activated releasing anti-inflammatory cytokines such as interleukin 10 (IL-10) or arginase 1 and promoting tissue remodeling. 5 7
ARDS is also associated with pulmonary activation of coagulation mediated by the tissue factor (TF) pathway. Exposure to proinflammatory cytokines causes alveolar macrophages and alveolar epithelial cells to produce TF, the key mediator of coagulation in severe infections. 8 Pulmonary coagulation is evident in increased markers of thrombin generation, soluble TF and factor VIIa activity found in bronchoalveolar lavage fluid (BALF) from ARDS patients, together with increased release of plasminogen activator inhibitor-1 (PAI-1) resulting in decreased fibrinolytic activity. 9 10 11
Previous findings indicate that anti-coagulants may help restore the coagulation cascade and treat ARDS; however, in experimental models of acute lung injury (ALI) and in ARDS patients, the beneficial effects of systemic anti-coagulants were outweighed by systemic bleeding. 12 13 14 15 16 17 18 Local administration of nebulized anti-coagulants to the lungs might reduce the risk of systemic bleeding and might be more effective than intravenous administration. 19 Preclinical studies with animal models of direct and indirect ALI have found that local administration of nebulized heparin improved pulmonary coagulopathy. 20 Intravenous anti-thrombin combined with nebulized heparin and tissue plasminogen activator restored gas exchange but not inflammation in a model of burn and smoke inhalation injured sheep. 21 In a phase I trial of nebulized heparin to ARDS patients, the activation of pulmonary coagulation was reduced without producing systemic bleeding. 22 23 In addition, nebulized heparin decreased the duration of mechanical ventilation in burn inhalation injured patients. 24
Besides its anti-coagulant effects, intravenously administered heparin showed anti-inflammatory effects, ameliorating lipopolysaccharide (LPS)-induced lung injury in rats via the inhibition of the nitric oxide synthase expression and transforming growth factor beta (TGF-β)/Smad pathway. 25 Heparin was also found to inhibit the nuclear factor kappa B (NF-κ≡) pathway in monocytes treated with LPS. 26 27 Furthermore, data recently obtained by our group showed that after LPS injury in human alveolar macrophages heparin limited the expression of TNF-α and IL-6, while in human alveolar type II cells heparin inhibited the NF-κ≡ pathway and their effectors IL-6, MCP-1 and IL-8. 28
Since alveolar macrophages play an important role in the development and resolution of ARDS, modulating the inflammatory response and the coagulation cascade in lungs, and heparin exhibits both anti-inflammatory and anti-coagulant properties, the hypothesis of this work was that nebulized heparin could attenuate ARDS through the involvement of alveolar macrophages. Accordingly, the current study aimed to assess the effects of nebulized heparin in a rat model of ALI induced by intratracheal instillation of LPS with special regard to the role that alveolar macrophages might have in limiting the coagulation and the inflammatory response. More in details, we postulated that: (1) in lungs, nebulized heparin decreases procoagulant markers and inflammation in terms of lung neutrophil influx, oedema, proinflammatory cytokines and histopathology; (2) in alveolar macrophages, nebulized heparin reduces the effectors of TGF-β and NF-κ≡ pathways and the expression of procoagulant genes.
Materials and Methods
Animals
We studied 64 pathogen-free male Sprague-Dawley rats (8 weeks old; 250–300 g; Charles River, Chatillon-sur-Chalaronne, France) housed in 12-hour light–dark-cycle, air-conditioned (23°C and 60% relative humidity) quarters with free access to standard food pellets (A04; Panlab, Barcelona, Spain) and tap water. The Animal Research Ethics Committee of the Autonomous University of Barcelona (UAB) approved the study.
Experimental Design
Rats were sedated with sevoflurane and randomized to four experimental groups (16 animals/group). Fig. 1 illustrates the experimental design. ALI was induced by intratracheal instillation of LPS ( Escherichia coli 055: B5, 10 µg/g body weight) (Sigma Chemical, St. Louis, MO). 29 Heparin (Vister, Parke-Davis, Linate, Milan, Italy) was nebulized through Aeroneb system (Philips Healthcare) at constant oxygen flow (2 L/min). Rats in the LPS/Hep group received two doses of 1,000 IU/kg nebulized heparin, administered 4 and 8 hours after LPS instillation. Rats in the Hep/LPS/Hep group received three doses of nebulized heparin: one dose 30 minutes before LPS instillation and one dose 4 and 8 hours after LPS instillation. Rats in the LPS/saline group received nebulized saline solution (0.9% NaCl) 4 and 8 hours after LPS instillation. Rats in the control group received only saline solution, by tracheal instillation at the time of ALI induction in the other animals and by nebulizer 4 and 8 hours afterwards. Animals were anesthetized (90 mg/kg ketamine and 10 mg/kg xylazine) and exsanguinated 24 hours after LPS instillation. BALF, lung tissue and blood were collected for further analyses; BALF and lung homogenate were obtained from eight animals in each group, and the lungs for histological examination and lung wet/dry weight ratio were obtained from the remaining eight animals in each group.
Fig.1.
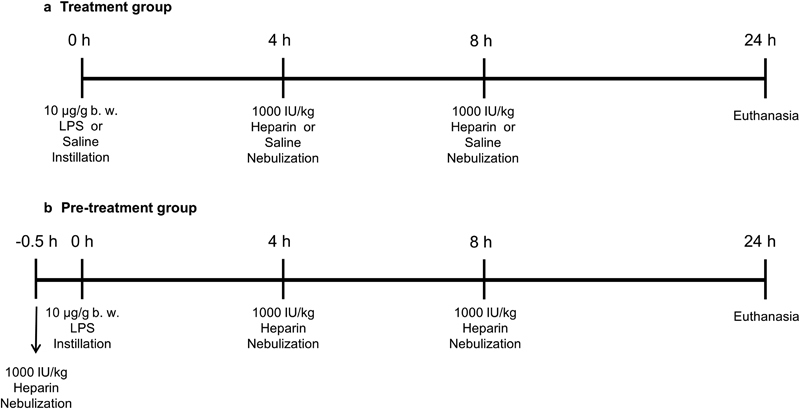
Experimental design. ( a ) Treatment group and ( b ) pre-treatment group.
Obtaining and Processing Bronchoalveolar Lavage Fluid
The left main bronchus was tied with a string at the hilum. BALF from all animal groups was obtained from the right lung by connecting a syringe to the cannula placed in the trachea and then gently flushing through it 5 mL sterile 0.9% NaCl solution with 1-mM Ethylenediaminetetraacetic acid (EDTA) five times. BALF volume recovery was always greater than 85%. BALF was spun at 800 × g for 10 minutes and the supernatant was stored at –80°C for subsequent analysis. Cells were counted using a haemocytometer (Neubauer, Marienfeld, Lauda-Königshofen, Germany), and slides were prepared by cytocentrifugation (Shandon Cytospin 4, Thermo Electron Corporation, Marietta, OH) and Diff-Quick staining (Panreac Quimica SAU; Castellar del Vallès, Spain). For each rat, approximately 500 cells were counted.
Lung Wet/Dry Weight Ratio
To assess oedema, the left lung was dissected immediately after exsanguination and the wet weight was recorded. The lung was then placed in an incubator at 80°C for 48 hours, the dry weight recorded and the lung wet-to-dry weight ratio was measured.
Histological Examination
The right lungs were removed and fixed in 4% paraformaldehyde. Two independent experts blinded to treatment analysed 4-µm sections excised of lung stained with haematoxylin and eosin (H&E). The entire surface of the lung was analysed for inflammation and damage, and was scored as follows: normal lung (0), haemorrhage (on a 0–1 scale), peribronchial infiltration (on a 0–1 scale), interstitial oedema (on a 0–2 scale), pneumocyte hyperplasia (on a 0–3 scale) and intra-alveolar infiltration (on a 0–3 scale).
Cytokine and Protein Measurements
Total protein concentration in BALF was quantified using the Micro BCA Protein Assay Kit (Pierce, Rockford, IL). IL-6, GRO-κC, TNF-α and IL-10 in lung homogenate were determined by multiplex assay following the manufacturer's protocol (Luminex, Merck Millipore, Darmstadt, Germany). PAI-1 in lung homogenate was determined by uniplex assay (Luminex, Merck Millipore, Darmstadt, Germany). Levels of TF, fibrin degradation products (FDPs), and thrombin-anti-thrombin complexes (TATc) in lung homogenate were measured by enzyme-linked immunosorbent assay (USCN Life Sciences, Hubei, China).
Activated Partial Thromboplastin Time
Blood (0.3 mL) was collected from the abdominal aorta in tubes containing 11-mM sodium citrate 24 hours after LPS instillation. Activated partial thromboplastin time (aPTT) was measured according to standard protocols (Echevarne Laboratories, Spain).
Isolation of Alveolar Macrophages
BALF pellet from all animal groups was seeded in Petri dishes with RPMI 1640 Medium supplemented with 10% foetal bovine serum (FBS), 100 IU/mL penicillin and 100 µg/mL streptomycin (Gibco, Langley, OK), for 1 hour at 37°C. The supernatant was discarded and purified attached alveolar macrophages were cryopreserved in 500 µL of TRIzol reagent (Ambion, Thermo Fisher Scientific, Madrid, Spain).
Alveolar macrophages' purity was assessed by Diff-Quick staining and immunofluorescence. To perform immunofluorescence, cells were fixed in 4% paraformaldehyde and incubated for 2 hours in a blocking solution (3% FBS and 1% bovine serum albumin in phosphate buffered saline [PBS]). Cells were then incubated overnight with a mouse anti-rat CD68 antibody (1:100) (Acris Antibodies, Rockville, MD), washed with PBS1X and incubated at 37°C for 1 hour with a goat anti-mouse IgG-FITC antibody (1:500) (Santa Cruz Biotechnology, Dallas, TX). Cells were finally washed with PBS 1X and incubated 5 minutes with HOECHST (1:1,000) (Invitrogen, Waltham, MA).
RNA Extraction and Real-Time PCR
Total RNA was extracted from isolated alveolar macrophages using chloroform, isopropanol and ethanol. The optical density at 260 nm and the ratio 260 nm/280 nm were measured to determine the RNA purity (spectrophotometer ND-1000, Nanodrop, Thermo Fisher Scientific, Wilmington, DE). Total RNA was reverse-transcribed into cDNA with a reverse transcriptase core kit (Eurogentec, Seraing, Belgium), using an Alpho-SC (Analytik Jena AG, Jena, Germany) thermocycler. DNA was amplified in a real-time polymerase chain reaction (PCR) system (7500 Real-Time PCR System, Applied Biosystems, Thermo Fisher Scientific, Madrid, Spain) using SYBR green (Kapa Biosystems, Cultek, Mataró, Spain) and the corresponding rat primers ( Table 1 ). Data were corrected by ΔΔCt method and shown as target gene expression relative to Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and fold over saline group.
Table 1. Rat primers.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| GAPDH | 5′ CTGTGCTTTCCGCTGTTTTC 3′ | 5′ TGTGCTGTGCTTATGGTCTCA 3′ |
| TNF-α | 5′ AACTCCCAGAAAAGCAAGCA 3′ | 5′ CGAGCAGGAATGAGAAGAGG 3′ |
| iNOS | 5′ CTTGGAGCGAGTTGTGGATT 3′ | 5′ GGTGGGAGGGGTAGTGATG 3′ |
| IL-10 | 5′ CATCCGGGGTGACAATAA 3′ | 5′ TGTCCAGCTGGTCCTTCT 3′ |
| Arginase-1 | 5′ GGGAAGACACCAGAGGAGGT 3′ | 5′ TGATGCCCCAGATGACTTTT 3′ |
| TGF-β | 5′ TGCTTCAGCTCCACAGAGAA 3′ | 5′ TGGTTGTAGAGGGCAAGGAC 3′ |
| Smad 2 | 5′ ACTCGTGGGGGAAGAAAAGT 3′ | 5′ CATGCTGCACTGCTTTGAAT 3′ |
| Smad 3 | 5′ GACCAGGCATTTTGAGGAAA 3′ | 5′ AGACCACAGCACCCCATAAG 3′ |
| IRAK1 | 5′ TACAAAGTGATGGACGCCCT 3′ | 5′ GGTGCCAGGCTGTAATGATG 3′ |
| P-Selectin | 5′ AGGTTGGCAATGGTTCACTC 3′ | 5′ ACCATTGGGAGCTACACCTG 3′ |
| CCL-2 | 5′ GCTGCTACTCATTCACTGGC 3′ | 5′ GGTGCTGAAGTCCTTAGGGT 3′ |
| TF | 5′ ACAATCTTGGAGTGGCAACC 3′ | 5′ TGGGACAGATAGGACCCTTG 3′ |
| PAI-1 | 5′ AGGGGCAGCAGATAGACAGA 3′ | 5′ CACAGGGAGACCCAGGATAA 3′ |
| Plasminogen | 5′ AAACGAAAGGGACTCCAGGT 3′ | 5′ TCTCGAAGCAAACCAGAGGT 3′ |
Note: Table of the primers used for the real-time polymerase chain reaction.
Statistical Analysis
Before starting the experiments, a power analysis was performed using the Gpower 3 program. The analysis indicated that 44 animals were needed to detect large effects (0.5) with 85% power using an analysis of variance (ANOVA) test between factors with α at 0.05. We used 20 extra animals to perform further investigational assessments. One-way ANOVA with Fisher's protected least-squares differences (PLSD) test as post hoc analysis was used for multigroup comparisons (StatView 5.0.1; Abacus Concept, Berkeley, CA). Results are reported as mean ± SEM. Statistical significance was set at p < 0.05.
Results
Cellular and Histological Response
Compared with control rats, animals in the LPS-instilled groups had higher total neutrophil counts in BALF; nebulized heparin limited the increase in neutrophils ( Fig. 2a ). There were no major differences in total macrophage counts among groups ( Fig. 2b ). Compared with the LPS/saline group, only heparin pre-treatment led to a decrease in the total number of cells in BALF (Sal/Sal: 16.8 ± 7.7, LPS/Sal: 45.7 ± 5.7, LPS/Hep: 40.8 ± 6.8, Hep/LPS/Hep: 31.1 ± 3.3; p < 0.05 vs. LPS/Sal group). The total concentrations of BALF proteins ( Fig. 2d ) and the wet-to-dry ratio ( Fig. 2c ) were significantly greater in LPS-instilled animals than in control animals, suggesting increased permeability of the alveolocapillary barrier. Nebulized heparin markedly reduced total BALF proteins and the wet-to-dry ratio. Histological analysis of lung tissue detected evidence of lung injury (haemorrhage, interstitial oedema, peribronchial and intra-alveolar infiltration, and alveolar pneumocyte hyperplasia) in LPS-instilled rats; lung injury was considerably reduced only in the Hep/LPS/Hep rats ( Fig. 3 ).
Fig. 2.
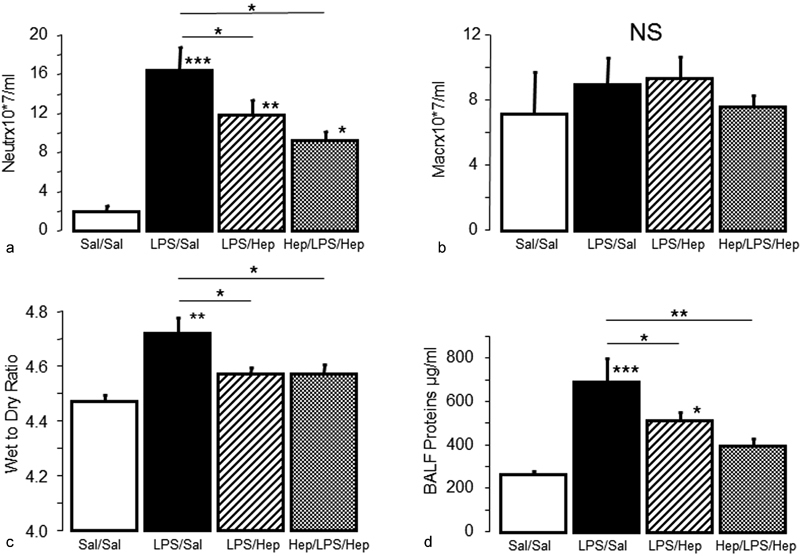
Bronchoalveolar lavage analysis and wet/dry weight ratio . Absolute ( a ) neutrophil (PMN) and ( b ) alveolar macrophage (AM) cell counts in the bronchoalveolar lavage fluid of rats 24 hours after the induction of the injury. ( c ) Wet/dry weight ratio and ( d ) protein concentration in the bronchoalveolar lavage fluid. Data are presented as mean ± SEM. ANOVA followed by the post hoc Fisher's PLSD test were used. * p < 0.05; ** p < 0.001; *** p < 0.0001.
Fig. 3.
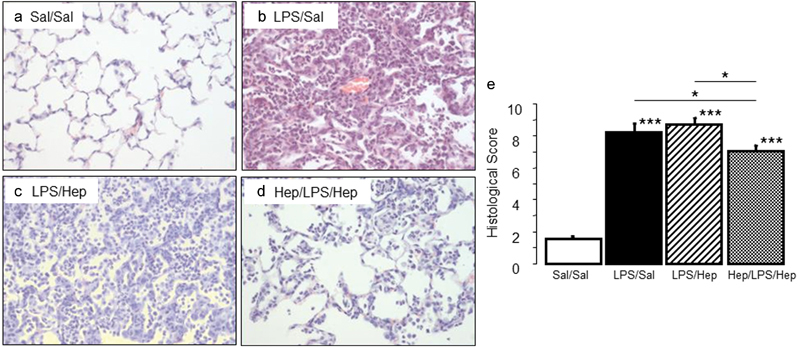
Lung tissue analysis . ( a–d ) Representative images of haematoxylin and eosin staining lung tissue sections and ( e ) histological score in animals 24 hours after induction of the injury. Original magnification ×200. Data are presented as mean ± SEM. ANOVA followed by the post hoc Fisher's PLSD test were used. * p < 0.05; ** p < 0.001; *** p < 0.0001.
Coagulation Effects
LPS instillation increased lung-tissue levels of TF, TATc, FDP and PAI-1 compared with control animals ( Fig. 4a–d respectively). Nebulized heparin reduced lung TF, but TATc and FDP levels decreased only in the Hep/LPS/Hep group. PAI-1 levels were not altered after heparin nebulization ( Fig. 4d ). Heparin had no effects on systemic coagulation, as no changes were observed in the aPTT (data not shown).
Fig. 4.
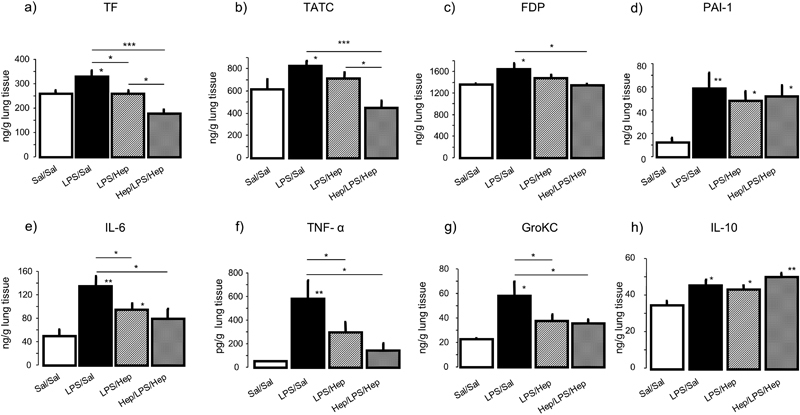
Coagulant effectors and cytokines . ( a ) Tissue factor (TF), ( b ) thrombin–anti-thrombin complexes (TATc), ( c ) fibrin degradation products (FDPs), ( c ) plasminogen activator inhibitor type-1 activity (PAI-1), ( e ) IL-6, ( f ) TNF-α, ( g ) GRO-κC and ( h ) IL-10 concentrations were measured in lung homogenate of animals 24 hours after induction of the injury. Data are presented as mean ± SEM. ANOVA followed by the post hoc Fisher's PLSD test were used. * p < 0.05; ** p < 0.001; *** p < 0.0001.
Inflammatory Response
In lung homogenates, concentrations of the proinflammatory cytokines IL-6, TNF-α and GRO-κC diminished in both groups of animals nebulized with heparin compared with the LPS group ( Fig. 4e–g respectively). Levels of IL-10 were higher in all the rat groups compared with control ( Fig. 4h ).
Macrophage pathway
Alveolar macrophages were isolated from BALF (98% purity; Supplementary Material 1 ). Fig. 5 reports the gene expression of M1 proinflammatory cytokines (TNF-α [ Fig. 5a ] and iNOS [ Fig. 5b ]) and M2 regulatory and reparative markers (IL-10 [ Fig. 5c ] and arginase-1 [ Fig. 5d ]) from alveolar macrophages. Nebulized heparin–deactivated alveolar macrophages stimulated with LPS; TNF-α, iNOS and arginase-1 expression significantly decreased in both LPS/Hep and Hep/LPS/Hep rats, but IL-10 expression significantly decreased only in the Hep/LPS/Hep group.
Fig. 5.
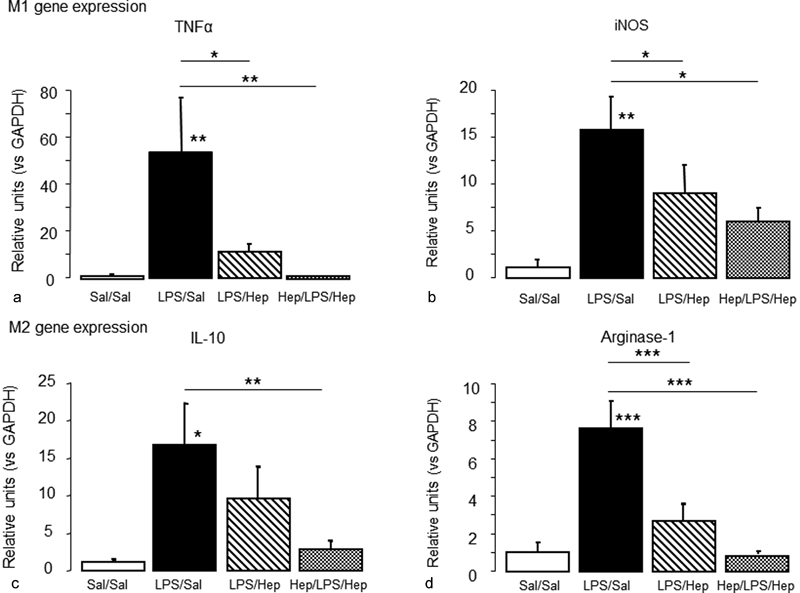
Activation of alveolar macrophages. Gene expression of proinflammatory (M1): ( a ) TNF-α, ( b ) iNOS and alternative (M2), ( c ) IL-10, ( d ) arginase-1 mediators in alveolar macrophages isolated from BALF of animals 24 hours after induction of the injury. Data are presented as mean ± SEM. ANOVA followed by the post hoc Fisher's PLSD test were used. * p < 0.05; ** p < 0.001; *** p < 0.0001.
Fig. 6 reports the expression of genes involved in proinflammatory and coagulation pathways analysed in alveolar macrophages. LPS had no effect on TGF-β expression ( Fig. 6a ) but increased the expression of Smad 3 ( Fig. 6b ) and Smad 2 ( Fig. 6c ). Nebulized heparin diminished the increase in Smad 3 expression in both groups, but diminished the increase in Smad 2 expression only in the Hep/LPS/Hep group. LPS increased the expression of IRAK1 ( Fig. 6d ), p-selectin ( Fig. 6e ) and CCL-2 ( Fig. 6f ) in alveolar macrophages; CCL-2 and p-selectin expression were lower in the Hep/LPS/Hep group. LPS increased expression in alveolar macrophages of TF ( Fig. 6g ), PAI-1 ( Fig. 6h ) and plasminogen ( Fig. 6i ), but only pre-treatment with nebulized heparin mitigated the increase in plasminogen expression.
Fig. 6.
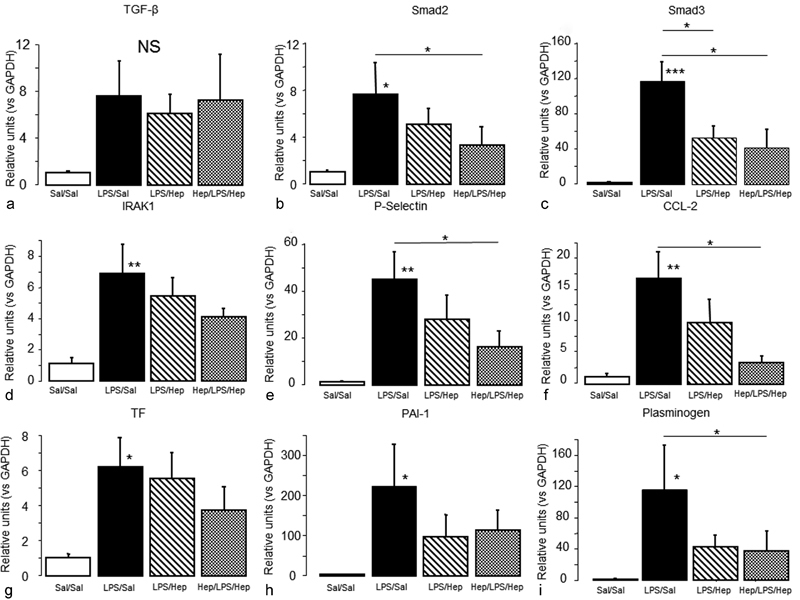
Inflammatory and coagulation pathways of alveolar macrophages . Gene expression of TGF-β pathway: ( a ) TGF-β, ( b ) Smad 2 and ( c ) Smad 3, NF-κ≡ pathway, ( d ) IRAK-1, ( e ) p-selectin and ( f ) CCL-2 and coagulation pathway, ( g ) TF, ( h ) PAI-1 and ( i ) plasminogen in alveolar macrophages isolated from BALF of animals 24 hours after induction of the injury. Data are presented as mean ± SEM. ANOVA followed by the post hoc Fisher's PLSD test were used. * p < 0.05; ** p < 0.001; *** p < 0.0001.
Discussion
The pathogenesis of ARDS involves both proinflammatory and procoagulant mediators, and the breakdown of the epithelial and endothelial barrier results in pulmonary oedema and infiltration of neutrophils in the alveolar space. In this rat model of LPS-induced ALI, administrating nebulized heparin diminished recruitment of neutrophils into the lung and attenuated pulmonary coagulopathy and inflammation without producing systemic bleeding. Part of this positive effect of heparin might be ascribed to the alveolar macrophages, in which the expression of markers of TGF-β effectors, NF-κ≡ and coagulation pathways was decreased after heparin nebulization.
Administering anti-coagulants directly to the lungs allows higher dosages and increases local efficacy without producing systemic bleeding. 19 We tested the effects of two nebulized heparin treatment regimens. Rats in the LPS/Hep group received two doses after ALI induction, while rats in the Hep/LPS/Hep group received an additional dose before LPS. Although the prophylactic pre-treatment plus treatment (Hep/LPS/Hep) model can help elucidate mechanisms, the treatment (LPS/Hep) model better reflects the clinical situation. Heparin reduced the number of neutrophils and their recruitment in both groups. Furthermore, heparin diminished lung permeability, reducing the concentration of BALF proteins and oedema in both groups. However, only in the pre-treatment group, heparin decreased the number of total BALF cells and lung injury.
In the first stages of ALI/ARDS, proinflammatory mediators inhibit natural anti-coagulant factors and induce procoagulant activity. 18 In our ALI model, heparin nebulization decreased pulmonary coagulation and inflammation. Previous studies showed that nebulized heparin could reduce pulmonary coagulopathy in an animal model of endotoxemia 20 or pneumonia, 30 although heparin did not produce any changes on inflammation. This result could be attributed to the different timing and dosage of heparin. In Hofstra et al's experimental model, 20 heparin was given every 6 hours, while in our model the time interval between each heparin nebulization was 4 hours (heparin biological lifetime is ∼1.5 hours), although we administrated fewer doses (three to the pre-treatment group and two to the treatment group).
Alveolar macrophages play important roles in both the development and resolution of ALI/ARDS. 5 6 7 In our ALI model, the expression of both proinflammatory (M1) and anti-inflammatory (M2) markers in alveolar macrophages increased after LPS administration, with a predominant activation of the proinflammatory M1 phenotype. Heparin attenuated the response of alveolar macrophages during ALI, reducing M1 (TNF-α, iNOS) and M2 (IL-10, arginase-1) markers to basal levels.
It has been shown that heparin anti-inflammatory effect could be produced by the inhibition of NF-κ≡ nuclear translocation into the lung, 18 26 reducing the expression of TNF-α and IL-6. 25 28 31 This is consistent with our results. Nebulized heparin decreased proinflammatory cytokines in lung tissue and the expression of NF-κ≡ effectors in alveolar macrophages. Moreover, some studies described that heparin ameliorated lung injury induced by LPS in rats via the inhibition of nitric oxide synthase expression and the TGF-β/Smad pathway. 25 In our model, heparin was also able to reduce the expression of TGF-β effectors in alveolar macrophages.
The expression of TF by inflammatory cells such as macrophages acts as one of the primary initiators of thrombosis. 32 Also, the release of TNF-α and IL-1β results in an increased TF expression. During ARDS, it is known that alveolar macrophages increase their PAI-1 activity, inhibiting fibrin degradation and promoting clots formation. 10 In our LPS model, higher levels of TF, TATc, PAI-1 and plasminogen were found, mimicking clinical ARDS. 10 33 Nebulized heparin decreased TF and TATc in lung tissue. It is known that TF expression on the monocytes surface induces their interaction with platelets and endothelial cells through the union of p-selectin. 34 Nebulized heparin significantly reduced p-selectin in alveolar macrophages. Furthermore, heparin was able to reduce plasminogen in alveolar macrophages, indicating that heparin may increase fibrinolysis through these cells. Accordingly, we ascertained that alveolar macrophages promote the deposition of thrombus and formation of clots, confirming that they are one of the main actors in the link between inflammation and coagulation.
This study has some limitations. First, LPS administration is a common ALI model that mimics human ARDS only in part, because this model cannot reflect the heterogeneous aetiology and management of ARDS. Second, the dose of heparin was chosen from previous studies based on the efficacy of the nose exposure system, the evaporative water loss during nebulization and the biological lifetime, and we cannot know whether lower doses would have similar effects. Third, our Hep/LPS/Hep group received an additional dose of heparin compared with the LPS/Hep group; since the early administration of heparin before LPS instillation might have reduced the development of the injury, it is not possible to know whether the differences between these two groups were due to the timing of administration or to the total dose administered; moreover, 24 hours may not have been long enough to detect some important histological changes in the LPS/Hep group.
Our experimental model focused on the acute phase of lung injury. A prolonged treatment of nebulized heparin and its effect in a late phase of ARDS need additional studies to determine its long-term effects.
Conclusion
In our rat model of ALI, nebulized heparin was effective in attenuating pulmonary coagulopathy and inflammation. Some of the effects of heparin seem to be related with alveolar macrophages. Preclinical data need to be transferred to clinical studies to confirm the potential benefit of nebulized heparin in ARDS patients. A better understanding of the mechanisms involved in the pathogenesis of ARDS might open new fields for the treatment of this disease.
What Is Known on This Topic
Acute respiratory distress syndrome (ARDS) is associated with pulmonary activation of coagulation and inflammation. Previous studies suggest that anti-coagulants may help restore the coagulation cascade and treat ARDS.
It has been shown that nebulized heparin was able to improve pulmonary coagulopathy in animal models of acute lung injury (ALI) and in ARDS patients without producing systemic bleeding.
In vitro findings recently indicated that heparin may exert its effects through alveolar macrophages.
What This Paper Adds
Our data confirmed that heparin nebulized directly into the lungs reduced pulmonary coagulopathy in a rat model of LPS-induced ALI without producing systemic bleeding. We also demonstrated that nebulized heparin significantly ameliorated lung injury, decreasing inflammation, permeability and neutrophils infiltration.
We ascertained that heparin may act on alveolar macrophages limiting the inflammatory response through the reduction of TGF-β effectors, NF-κ≡ and coagulation pathways.
In our rat model of ALI, nebulized heparin was effective in attenuating pulmonary coagulopathy and inflammation. Some of the effects of heparin seem to be related with alveolar macrophages. Preclinical data need to be transferred to clinical studies to confirm the potential benefit of nebulized heparin in ARDS patients.
Grant Support
This work was supported by CIBERES, Institut d'Investigació i Innovació Parc Taulí and Ministerio de Economía y Competitividad-Instituto de Salud Carlos III (PI12/02548).
Conflict of Interest The authors have declared that no conflict of interest exists.
The work was performed in Institut d'Investigació i Innovació Parc Taulí (I3PT), Sabadell, Spain.
Author Contribution
L. C., M. C.-R., R. G.-P. and A. A. designed the experiments; L. C., M. C.-R., R. G.-P., M. N. G. and J. T. performed the experiments; L. C., M. C.-R. and R. G.-P. analysed the data and, together with A. A., interpreted the data; M. C.-R., L. C., R. G.-P. and A. A. wrote the manuscript and L. B. and A. A. helped to revise the manuscript. All the authors approved the last version of the manuscript.
These authors contributed equally
Supplementary Material
References
- 1.Bellani G, Laffey J G, Pham T et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(08):788–800. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 2.Rubenfeld G D, Caldwell E, Peabody E et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353(16):1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 3.Ranieri V M, Rubenfeld G D, Thompson B T et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 4.Johnson E R, Matthay M A. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv. 2010;23(04):243–252. doi: 10.1089/jamp.2009.0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnston L K, Rims C R, Gill S E, McGuire J K, Manicone A M. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am J Respir Cell Mol Biol. 2012;47(04):417–426. doi: 10.1165/rcmb.2012-0090OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aggarwal N R, King L S, D'Alessio F R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol. 2014;306(08):L709–L725. doi: 10.1152/ajplung.00341.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol. 2011;2:65. doi: 10.3389/fimmu.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bastarache J A, Wang L, Geiser T et al. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax. 2007;62(07):608–616. doi: 10.1136/thx.2006.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzales J N, Kim K M, Zemskova M A et al. Low anticoagulant heparin blocks thrombin-induced endothelial permeability in a PAR-dependent manner. Vascul Pharmacol. 2014;62(02):63–71. doi: 10.1016/j.vph.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ware L B, Bastarache J A, Wang L. Coagulation and fibrinolysis in human acute lung injury--new therapeutic targets? Keio J Med. 2005;54(03):142–149. doi: 10.2302/kjm.54.142. [DOI] [PubMed] [Google Scholar]
- 11.Günther A, Mosavi P, Heinemann Set al. Alveolar fibrin formation caused by enhanced procoagulant and depressed fibrinolytic capacities in severe pneumonia. Comparison with the acute respiratory distress syndrome Am J Respir Crit Care Med 2000161(2, Pt 1):454–462. [DOI] [PubMed] [Google Scholar]
- 12.Angus D C. Drotrecogin alfa (activated)...a sad final fizzle to a roller-coaster party. Crit Care. 2012;16(01):107. doi: 10.1186/cc11152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warren B L, Eid A, Singer P et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286(15):1869–1878. doi: 10.1001/jama.286.15.1869. [DOI] [PubMed] [Google Scholar]
- 14.Abraham E, Reinhart K, Opal S et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290(02):238–247. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 15.Eisele B, Lamy M, Thijs L G et al. Antithrombin III in patients with severe sepsis. A randomized, placebo-controlled, double-blind multicenter trial plus a meta-analysis on all randomized, placebo-controlled, double-blind trials with antithrombin III in severe sepsis. Intensive Care Med. 1998;24(07):663–672. doi: 10.1007/s001340050642. [DOI] [PubMed] [Google Scholar]
- 16.Laterre P F, Opal S M, Abraham E et al. A clinical evaluation committee assessment of recombinant human tissue factor pathway inhibitor (tifacogin) in patients with severe community-acquired pneumonia. Crit Care. 2009;13(02):R36. doi: 10.1186/cc7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Sun J F, Cui X et al. The effect of heparin administration in animal models of sepsis: a prospective study in Escherichia coli-challenged mice and a systematic review and metaregression analysis of published studies. Crit Care Med. 2011;39(05):1104–1112. doi: 10.1097/CCM.0b013e31820eb718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacLaren R, Stringer K A. Emerging role of anticoagulants and fibrinolytics in the treatment of acute respiratory distress syndrome. Pharmacotherapy. 2007;27(06):860–873. doi: 10.1592/phco.27.6.860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuinman P R, Dixon B, Levi M, Juffermans N P, Schultz M J. Nebulized anticoagulants for acute lung injury - a systematic review of preclinical and clinical investigations. Crit Care. 2012;16(02):R70. doi: 10.1186/cc11325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofstra J J, Vlaar A P, Cornet A D et al. Nebulized anticoagulants limit pulmonary coagulopathy, but not inflammation, in a model of experimental lung injury. J Aerosol Med Pulm Drug Deliv. 2010;23(02):105–111. doi: 10.1089/jamp.2009.0779. [DOI] [PubMed] [Google Scholar]
- 21.Rehberg S, Yamamoto Y, Sousse L E et al. Advantages and pitfalls of combining intravenous antithrombin with nebulized heparin and tissue plasminogen activator in acute respiratory distress syndrome. J Trauma Acute Care Surg. 2014;76(01):126–133. doi: 10.1097/TA.0b013e3182ab0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dixon B, Santamaria J D, Campbell D J. A phase 1 trial of nebulised heparin in acute lung injury. Crit Care. 2008;12(03):R64. doi: 10.1186/cc6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dixon B, Schultz M J, Hofstra J J, Campbell D J, Santamaria J D. Nebulized heparin reduces levels of pulmonary coagulation activation in acute lung injury. Crit Care. 2010;14(05):445. doi: 10.1186/cc9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elsharnouby N M, Eid H E, Abou Elezz N F, Aboelatta Y A. Heparin/N-acetylcysteine: an adjuvant in the management of burn inhalation injury: a study of different doses. J Crit Care. 2014;29(01):1820–1.82E6. doi: 10.1016/j.jcrc.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 25.Mu E, Ding R, An X, Li X, Chen S, Ma X. Heparin attenuates lipopolysaccharide-induced acute lung injury by inhibiting nitric oxide synthase and TGF-β/Smad signaling pathway. Thromb Res. 2012;129(04):479–485. doi: 10.1016/j.thromres.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Anastase-Ravion S, Carreno M P, Blondin C et al. Heparin-like polymers modulate proinflammatory cytokine production by lipopolysaccharide-stimulated human monocytes. J Biomed Mater Res. 2002;60(03):375–383. doi: 10.1002/jbm.10112. [DOI] [PubMed] [Google Scholar]
- 27.Hochart H, Jenkins P V, Smith O P, White B. Low-molecular weight and unfractionated heparins induce a downregulation of inflammation: decreased levels of proinflammatory cytokines and nuclear factor-kappaB in LPS-stimulated human monocytes. Br J Haematol. 2006;133(01):62–67. doi: 10.1111/j.1365-2141.2006.05959.x. [DOI] [PubMed] [Google Scholar]
- 28.Camprubí-Rimblas M, Guillamat-Prats R, Lebouvier T et al. Role of heparin in pulmonary cell populations in an in-vitro model of acute lung injury. Respir Res. 2017;18(01):89. doi: 10.1186/s12931-017-0572-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puig F, Herrero R, Guillamat-Prats R et al. A new experimental model of acid- and endotoxin-induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol. 2016;311(02):L229–L237. doi: 10.1152/ajplung.00390.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofstra J J, Cornet A D, de Rooy B F et al. Nebulized antithrombin limits bacterial outgrowth and lung injury in Streptococcus pneumoniae pneumonia in rats. Crit Care. 2009;13(05):R145. doi: 10.1186/cc8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang M, He J, Mei B, Ma X, Huo Z. Therapeutic effects and anti-inflammatory mechanisms of heparin on acute lung injury in rabbits. Acad Emerg Med. 2008;15(07):656–663. doi: 10.1111/j.1553-2712.2008.00146.x. [DOI] [PubMed] [Google Scholar]
- 32.Dosquet C, Weill D, Wautier J L. Cytokines and thrombosis. J Cardiovasc Pharmacol. 1995;25 02:S13–S19. doi: 10.1097/00005344-199500252-00004. [DOI] [PubMed] [Google Scholar]
- 33.Levi M, Schultz M. The inflammation-coagulation axis as an important intermediate pathway in acute lung injury. Crit Care. 2008;12(02):144. doi: 10.1186/cc6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindmark E, Tenno T, Siegbahn A. Role of platelet P-selectin and CD40 ligand in the induction of monocytic tissue factor expression. Arterioscler Thromb Vasc Biol. 2000;20(10):2322–2328. doi: 10.1161/01.atv.20.10.2322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.