

Abstract
Patients with cancer are at high risk for both venous and arterial thrombotic complications. A variety of factors account for the greater thrombotic risk, including the underlying malignancy and numerous cancer-directed therapies. The occurrence of an acute thrombotic event in patients with cancer is associated with substantial morbidity and mortality. Acute coronary syndrome (ACS) represents a particularly important cardiovascular complication in cancer patients. With cardio-vascular risk factors becoming more prevalent in an aging cancer population that is surviving longer, questions pertaining to the appropriate management of vascular toxicity are likely to assume even greater value in the coming years. In this article, we review the current understanding of ACS in patients with cancer. The predisposition to thrombosis in a malignant host and the cancer treatments most commonly associated with vascular toxicity are reviewed. Risk prediction and management strategies are discussed, and discrepancies in the clinical evidence are highlighted.
Keywords: Acute coronary syndrome (ACS), anti-cancer therapy, coronary artery disease, radiation therapy, thromboembolism
Introduction
Thromboembolic events occur in about 20% of patients with cancer with wide variations in reported frequencies related to differences in study populations (1-4). These events portend a worse prognosis; cancer patients with venous thromboembolism (VTE) have a 2–3× higher mortality risk than cancer patients without VTE and a 8× higher mortality risk than non-cancer patients with VTE (5,6). Even more, among cancer outpatients, thromboembolism is the second-leading cause of death (7). In fact, in striking contrast to the attention devoted to the venous circulation when it comes to cancer-related thrombosis, the contribution of arterial thromboembolism (ATE) to outpatient mortality ranks even higher than that of VTE (7). This review will provide an overview of arterial events in cancer patients, especially as it pertains to presentations of acute coronary syndrome (ACS) and acute coronary thrombosis.
ACS in the patient with cancer
Acute coronary thrombosis is part of the presentation spectrum of ACS in cancer patients. (8) As outlined in Figure 1, a number of pathological processes can contribute to this presentation. Plaque rupture has historically been considered to be the leading mechanism underlying ACS. However, recent work enabled by optical coherence tomography (OCT) confirms this to be still the case for most presentations of ST segment elevation myocardial infarction (STEMI) but not for non-STEMI (NSTEMI) (9,10). In fact, erosion has been found to be a phenomenon that is much more common than previously thought (11). This is a testimony to the advances of intravascular imaging such as OCT, which has allowed detecting in vivo what was formerly missed using techniques with lower resolution or studying patients post-mortem (12). While erosion is much more prevalent than commonly estimated, specific data on the relative incidence and frequency of this or other mechanisms of ACS in cancer patients are missing at this point. A pertinent roadmap question therefore is: what is the etiology, the relative distribution of types of myocardial infarction (MI) in cancer patients? Vasospasm, supply-demand mismatch in the setting of anemia, for instance, are as important in the differential diagnosis as it coronary thrombosis. The latter can even be due to embolization of thrombotic (and at times even cancer) material into the coronary circulation (13). Thus, even more so in cancer patients the etiology of a presenting ACS needs to be individually defined. This is the first critical step towards appropriate management.
Figure 1.

Illustration of the potential factors contributing to acute coronary syndrome in cancer patients.
It is essential to recall the above mentioned aspects as it pertains to cohort studies which have influenced this field. One of the most influential has been a recent study on 280,000 cancer patients registered in the Surveillance, Epidemiology, and End Results (SEER) database which were individually matched each to a patient from the Medicare database. (14) Accordingly, the results of this analysis apply to patients in Medicare age, and indeed, the average age of the cancer cohort was 77 years. Furthermore, myocardial infarction was identified by ICD-9-CM code 410 in any diagnosis position, therefore encompassing all forms of cardiac infarction, which includes coronary artery plaque rupture, embolism, occlusion, vasospasm, and other forms of thrombosis. As shown in Figure 2, the overall cumulative incidence increased over time in cancer as well as non-cancer patients. The greatest relative increase in ATE risk in cancer patients was seen in the first month after diagnosis (hazard ratio 7.3 for myocardial infarction, 4.5 for ischemic stroke versus non-cancer control). A tapering effect in excess risk among cancer patients was seen thereafter, several cancer types losing their risk after 6 months and only lung cancer maintaining a 2.5-fold increased relative risk (hazard) at 12 months (14). Of further note, there was not only a time-dependent risk but also a stage-dependent risk, i.e. advanced, stage 3 and 4 cancer accounted for the increased risk in ATEs (14). Cancer types most strongly associated with an elevated risk of ATE included lung (HR 9.6), pancreas (HR 6.8), colorectal (HR 6.7) and gastric cancer (HR 6.0), a subset of cancers that also correlate with higher likelihood of venous thrombotic events. These are very peculiar data that prompt discussions on a general thrombotic predisposition linked to the malignancy itself, its treatments, and underlying predisposition (Figure 3).
Figure 2.

Cumulative incidence of arterial thromboembolism (composite of myocardial infarction and ischemic stroke), stratified by cancer patients versus matched controls (left panel) and by cancer stage at the time of cancer diagnosis (right panel). Modified from Navi BB, Reiner AS, Kamel H, et al. Risk of Arterial Thromboembolism in Patients With Cancer. J Am Coll Cardiol 2017;70:926-38, with permission by Elsevier.
Figure 3.
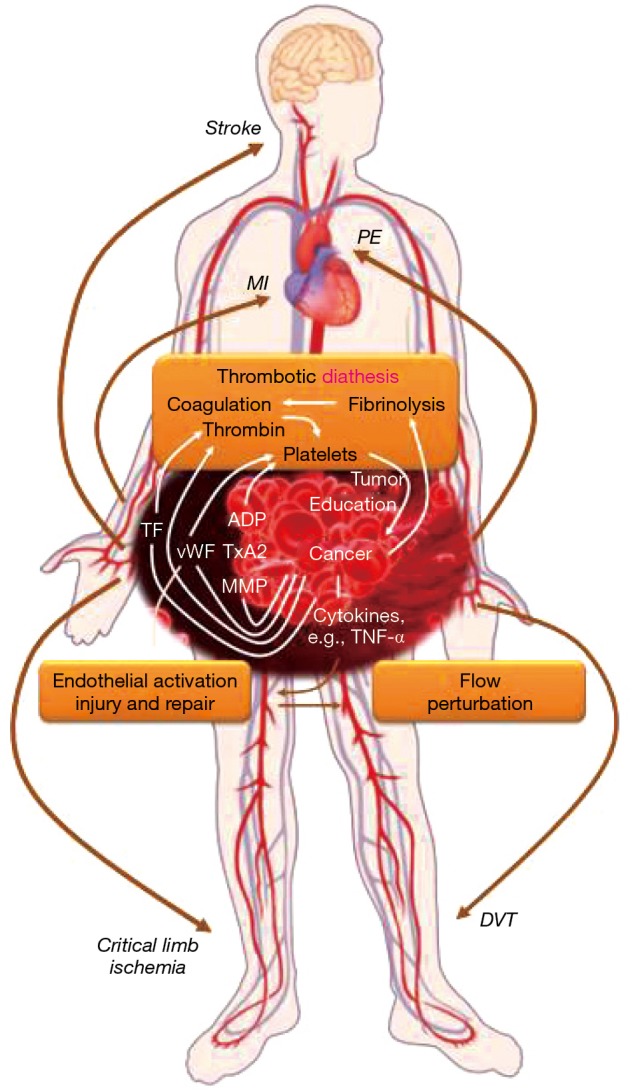
Outlined of the thrombotic triad for arterial and venous thromboembolism in cancer patients.
Predisposition to thrombosis in cancer patients
Virchow’s triad captures the classical concept of vascular thrombosis and the conceptual framework for arterial and venous thrombotic events in cancer patients (Figure 3) (15). The first element of alterations of blood content, i.e., the coagulation and fibrinolytic systems, is very pertinent to cancer patients. Examples include elevated fibrinogen levels and diminished levels of (anti-thrombin) proteins C and S and tissue plasminogen activator (15). Cancer cells can express tissue factor, and the subsequent recruitment of Factor VII/VIIa to the cell surface leads to potent activation of the coagulation cascade, thrombin production, as well as platelet activation (15). Platelet activation is mainly mediated by stimulation of platelet surface receptors (e.g., PAR-1 and PAR-4 receptors, P2Y12 receptor, and thromboxane receptor) in response to various ligands. Importantly, cancer cells can produce these ligands including ADP and thromboxane A2 in addition to tissue factor and thrombin. Over the past years tumor cell-induced platelet aggregation (TCIPA) has been increasingly recognized as well as the fact that a bidirectional (reinforcing) relationship exists between cancer cells and platelets, referred to as the “platelet-cancer loop” (16). Activated platelets support tumor growth and metastases, and last but not least, angiogenesis (16,17). The latter is extremely important for both, the growth and the spread of cancers, and it is inherently linked to vascular endothelial growth factor (VEGF). In this context it is pertinent that tumor-derived VEGF-A triggers von-Willebrand factor (vWF) release from endothelial cells (18). This leads to the activation of platelets, which release several angiogenesis-regulating proteins from their granules with a net pro-thrombotic effect. In cancer patients, several markers of platelet activation are increased including soluble CD40 ligand, soluble P-selectin, platelet factor 4, thrombospondin, beta-thromboglobulin as well as vWF (16,17). Of further note, cancers with a high thrombotic risk such as gastric adenocarcinomas have been shown to express vWF as well (19). This is in addition to the two main natural sources of vWF: platelets and endothelial cells, and as outlined cancer cells can influence these as well. Thus, the first element in the triad of Virchow seems to be primarily influenced by the malignancy itself.
The vascular wall is the second element in triad of Virchow’s, and endothelial cells are integral in this regard. In addition to releasing vWF, thereby promoting platelet activation and aggregation, the loss of expression of thrombomodulin on the endothelial surface reduces the capacity to activate anti-coagulant protein C, thereby adding to the prothrombotic state (15). Endothelial dysfunction also decreases fibrinolytic activity and contributes to abnormal vasoconstriction (20). A number of cancer therapeutics as well as radiation therapy can affect endothelial cells in a profoundly negative manner and might account for most of the modulation of the second element of Virchow’s triad (21).
Flow disturbances constitute the third element in Virchow’s triad. These are thought of in terms of stasis for venous thrombosis. For arterial thrombosis, these might constitute extremes of the shear stress spectrum as well as non-laminar and turbulent flow as seen at flow dividers (22). This third element is likely set by the baseline conditions of the individual patient. In this context it is pertinent to point out that coronary artery disease (CAD) may already be known at the time of cancer diagnosis, as is the case in approximately 20% of patients older than 75 years of age with a new diagnosis of cancer. Alternatively, CAD may be clinically silent and unrecognized, only to emerge in the context of the cancer milieu. It can be unmasked by cancer-directed treatments such as chemotherapies, targeted therapies, and radiation. In addition, it has been suggested that cancer therapy, prior and concurrent, may decrease the compensatory reserve to acute coronary events (23). At present, it remains an open question though: what are the factors that acutely precipitate a coronary event in cancer patients? Why do some but not all patients with the same malignancies, subjected to the same therapies with similar clinical comorbidity profile develop an acute vascular event and why do they do so at certain times?
Vascular toxicity of cancer therapeutics
A number of cancer therapeutics have been associated with vascular toxicity (Table 1) (21). The therapies most commonly used in clinical practice and of relevance in this regard will be outlined in the following.
Table 1. Incidence of vascular toxicities with chemotherapeutics (based on Micromedex® and Lexicomp®).
| Chemotherapeutic | Angina | Acute myocardial infarction | Stroke | Peripheral arterial disease |
|---|---|---|---|---|
| Antimetabolites | ||||
| 5-Flourouracil | +++ | +++ | + | |
| Capecitabine | ++ | +++ | + | |
| Gemcitabine | + | + | ||
| Anti-microtubule agents | ||||
| Paclitaxel | + | + | ||
| Alkylating agents | ||||
| Cisplatin | + | + | + | + |
| Cyclophosphamide | + | + | ||
| Antitumor antibiotics | ||||
| Bleomycin | + | + | + | |
| Vinca alkaloids | ||||
| Vincristine | + | + | ||
| mTOR inhibitors | ||||
| Everolimus | ++ | + | ||
| Temsirolimus | ++++ | |||
| Proteasome inhibitors | ||||
| Bortezomib | + | + | + | |
| Carfilzomib | ++++ | ++++ | ||
| Monoclonal antibodies | ||||
| Bevacizumab | +++ | +++ | +++ | |
| Ramucirumab | ++ | ++ | ||
| Rituximab | + | + | ||
| Vascular endothelial growth factor-receptor fusion molecules | ||||
| Aflibercept | ++ | ++ | ||
| Tyrosine kinase inhibitors | ||||
| Sorafenib | + | ++ | + | |
| Sunitinib | ++++ | + | + | |
| Pazopanib | +++ | ++ | + | |
| Axitinib | + | ++ | + | |
| Regorafenib | + | + | ||
| Cabozantinib | ++ | ++ | ||
| Vandetanib | + | |||
| Lenvatinib | +++ | |||
| Nilotinib | +++ | + | ++ | ++ |
| Ponatinib | ++++ | ++++ | ++++ | +++ |
| Dasatinib | ++ | |||
| Miscellaneous | ||||
| Interferon-alpha 2B | ++++ | ++ | ++ | ++ |
| Thalidomide | + | + | ||
| Lenalidomide | +++ | ++ | ++ |
Symbols reflect frequency: +, <1%; ++, 1–5%; +++, 6–10%; ++++, more than 10%.
Anti-metabolite chemotherapies [i.e., 5-fluorouracil (5-FU), capecitabine] interfere with key signaling pathways that mediate vascular smooth muscle tone and can lead to coronary vasospasm (24,25). Severity and duration of the coronary vasospasm bestow a spectrum of presentations ranging from stable angina, variant angina to unstable angina, acute myocardial infarction and even sudden cardiac death secondary to ventricular fibrillation (26). The extent of abnormal vasoreactivity can involve the microvasculature, as a putative mechanism explaining presentations of Takotsubo’s (apical ballooning/stress cardiomyopathy) (27).
Alkylating agents (i.e., cyclophosphamide, and platinum drugs like cisplatin) are among the most endothelial-toxic drugs. The toxic effect on endothelial cells lining the pericardium and myocardium is the basis for acute hemorrhagic pericarditis and even acute hemorrhagic myocarditis seen with cyclophosphamide when administered at high doses, both of which can be lethal (28,29). Acute myocardial infarctions have been reported as well. Coronary thrombosis has been seen with platinum drugs such as cisplatin, and the extent can be quite extensive, leading to suboptimal outcome of intervention due to thrombotic obstruction of the distal and adjacent vasculature (Figure 4) (30-33). Even distal embolic phenomena have been noted. The underlying mechanism may be the interplay of cytotoxic effects on the endothelium leading to erosion with subsequent activation of platelets and the coagulation cascade. This is further enhanced by cisplatin’s activating effect on platelet phospholipase A2 (34). Of interest, cisplatin levels may remain elevated for many years after therapy, thus posing a potentially protracted risk (35).
Figure 4.

Case of 77-year-old female who developed a non-ST segment elevation myocardial infarction during her third weekly cycle of paclitaxel and carboplatin for adenocarcinoma of unknown primary. Coronary angiogram revealed thrombotic occlusion of the mid left anterior descending artery (LAD) and thrombus in the first diagonal branch (arrow). Intervention restored flow in the LAD, but with TIMI II flow only and thrombotic occlusion of the first diagonal branch. Of interest, no coronary artery disease is otherwise seen and intravascular ultrasound did not show atherosclerotic plaque.
Anti-vascular endothelial growth factor antibodies (i.e., bevacizumab) can exert various effects that can culminate in cardiac ischemic presentations. These include alteration in coronary vasoreactivity secondary to the important role of VEGF in endothelial function including nitric oxide (NO) production, the key endothelium-derived vasorelaxation factor (36). NO is also important for anti-atherosclerotic effects, although the anti-angiogenic effects on the plaque vasculature may counteract any propagating effect on atherosclerotic plaque growth and stability (20,37). Thrombotic effects may be precipitated by interaction with platelets in a manner similar to what is occurring in heparin-induced thrombocytopenia and/or alteration in fibrinolytic capacities by neutralizing the inhibitory effect of VEGF on the expression of PAI-1 in tumor cells (38,39).
Numerous tyrosine kinase inhibitors have been associated with symptoms of myocardial ischemia and with overt myocardial infarction. A number of these inhibit the VEGF signaling pathway, especially sunitinib, sorafenib, and pazopanib, and as shown in elegant experimental studies, internal inhibition of this pathway is much more detrimental for endothelial cell viability than external inhibition (40). The effects extend into the microvasculature and additional inhibition of PDGF-b is detrimental to pericyte health, which destabilizes myocardial microvascular structure (41). Of further interest, multi-targeted TKIs like sorafenib promote apoptotic cell death of c-kit+ stem cells, thereby decreasing repair capacity and increasing mortality after myocardial infarction (42). Cases of coronary vasospasm to the point of myocardial infarction have been reported as well (43,44).
BCR-ABL inhibitors are a second group of multi-targeted TKIs that have been associated with acute coronary events, especially ponatinib and nilotinib, as reviewed extensively before (21,45-51). The risk is higher and emerges earlier with ponatinib (52). Inhibition of the VEGF signaling pathway inhibition might be the common denominator with secondary targets as the differentiating factor (48,53). Both drugs, but ponatinib more potently, suppress endothelial function and induce endothelial apoptosis (54). In experimental models, ponatinib and nilotinib lead to changes consistent with increased plaque vulnerability (55). This might explain some, but not all of the pathophysiology seen with these medications. For one, it is until now not absolutely clear if plaque rupture and thrombus formation bestows the acute events, vasoconstriction, accelerated progression of atherosclerosis with luminal compromise, or reduction in compensatory abilities due to compromised collateral formation and/or microvasculature.
Experimental work on proteasome inhibitors indicated that prolonged, high level inhibition of this central intracellular protein degradation system can cause and aggravate atherosclerosis (56-58). Ischemic heart disease (including presentations of chest tightness, angina, and myocardial infarction) as a side effect of therapy has been reported more so with the second generation proteasome inhibitor carfilzomib than with the first generation agent bortezomib. An integrated analysis of 4 phase II trials with carfilzomib even concluded on 1.5% cardiac mortality rate, and cases of sudden cardiac death occurring within 24 hours from infusion were reported (59). This could be due to acute coronary artery occlusion due to thrombosis, or, as seen with 5-FU due to profound and prolonged vasospasm. In this context it is very pertinent that carfilzomib was found to exert a vasoconstricting effect, aggravating endothelial dysfunction and increasing coronary vascular resistance (60).
With the emergence of immune checkpoint inhibitors, there has been increasing concern regarding the cardiotoxic potential of PD-1 and PD-L1 inhibitors. Given that the PD-1/PD-L1 regulates auto-reactive lymphocytes, it was postulated that blockage of that pathway might lead to deleterious autoimmune diseases. Indeed, animal models and human data indicate that myocarditis may occur with checkpoint inhibition, and this has been subject to various analyses (61-63). Importantly, animal studies have also indicated that especially inhibition of the PD-1/PD-L1 system leads to progression of atherosclerosis with increased plaque inflammation and a predisposition to complications such as plaque rupture (64,65). It has also been shown to contribute to graft vasculopathy in murine hearts and giant cell vasculitis, in keeping with PD-1’s known role in the regulation of immune response (66-69). Isolated cases of myocardial infarction and vasculitis have been reported with immune checkpoint inhibitors (70,71). More comprehensive analyses of the incidence, nature and clinical outcomes of vascular disease in patients treated with PDL-1/CTLA-4 inhibitors is eagerly anticipated.
Radiation therapy has a unique injury potential that can affect all structures of the heart, which has become known as radiation-induced heart disease (72,73). The most sensitive cell population are endothelial cells, which line the pericardium and the endocardium and form the microvascular network in the myocardium. They are also the key element to the health of major arteries. Indeed, coronary artery disease can emerge in not an insubstantial number of breast cancer patients (74-76), as well as Hodgkin lymphoma patients (77). It is also seen after head/neck radiation in the carotid arteries, which allowing for easier serial assessment, which has confirmed the progressive nature of the disease (78,79). Inflammation is an early characteristic that can be sustained, involving the entire wall, and can be followed by vascular fibrosis (80-82). Animal models utilizing atherosclerosis-prone mice confirm that irradiation accelerates atherosclerosis and induces both, an inflammatory and a thrombotic phenotype (83-86). While remaining effective in general atherosclerosis, neither aspirin nor clopidogrel were of sound therapeutic benefit for radiation atherosclerosis in the mouse model (87). The same held true for atorvastatin though it reduced cardiac fibrosis (88). This points to the possibility that the vascular disease process induced by radiation is not exactly the same as the one induced by standard cardiovascular risk factors.
Risk stratification for vascular events in cancer patients
Various tools have been developed to define cancer prognosis and response to therapy, but in a striking contrast there are not many to predict cardiotoxicity risk and even fewer to alert to vascular toxicity. However, cardiovascular toxicities were not subject of major concern until relatively recently. With the improved cancer outcomes and extended life expectancy the morbidity and mortality of non-cancer comorbidities and toxicities bestows a much greater and sometimes profound impact. Historically cardiomyopathy and heart failure have been the main cardiovascular concerns. With the recent advances (as outlined above) vascular toxicities have move more to the forefront of some cancer therapies. For instance, the ischemic risk with VEGF inhibitor therapy is considerably higher than that of cardiomyopathy and heart failure (89). Further, ATEs such as myocardial infarction and stroke can have devastating consequences, let alone the delays and terminations of cancer therapy. Estimating and preparing for cardiac and vascular risk therefore should be an important aspect in the care of cancer patients.
For VEGF inhibitor therapy, limited retrospective data indicate that a history of ATEs and age >65 years increase the risk of ATEs on therapy (90). In patients meeting these high risk criteria, aspirin exerts a preventive effect. In fact, following an ATE in the past these patients meet secondary prevention criteria and abate the debate of primary prevention with aspirin. The same holds true for any other patient undergoing chemotherapy with a history of ATEs, clinical CAD and ASCVD in general though concerns pertaining to anemia and thrombocytopenia need to be taken into account as outlined below.
For patients undergoing cisplatin-based chemotherapy for testicular cancer, 3 or more cardiovascular risk factors define a high risk fingerprint for ATEs (nearly 10-fold higher risk) (91). These patients also have a higher intima-media thickness and higher vWF level at baseline. In these but not in the low risk fingerprint patients significant increases in vWF levels on therapy are seen. Such dynamics may point out greater endothelial vulnerability as well as greater predisposition to platelet activation though confirming data for this conclusion are missing at present. Another important aspect, these usually young and healthy men remain at increased risk for ischemic vascular events over time (92-94). This might relate to the fact that cisplatin level can remain detectable (elevated) for years after therapy as well as the intensification of cardiovascular risk factor exposure (35). Correlation with ASCVD risk scores might be an intuitive step but will likely underestimate the true risk as not factoring in the chemotherapeutic dimension. In comparison with the reference population for which ASCVD risk scores were developed, these patients have an excess risk. A pertinent question is if such risk scores would perform well when the disease process is arterial thrombosis secondary to erosion and possibly related to a general pro-coagulant state. The correlation with VTE risk scores is not defined, i.e., whether there could be a general thrombotic risk score. As outlined above, certain malignancies have a higher risk, likely related to the expression of pro-coagulants and platelet activating factors, which improves with cancer therapy. Thus, the first few months after diagnosis pose the highest risk period when cancer production meets cancer therapy induction of thrombotic risk. If and how to bridge this vulnerable period is not known as often also the highest risk period for bleeding events.
Importantly, ASCVD risk scores such as the Euro Score have been shown to predict events in patients with CML on nilotinib therapy (95). In comparison with a risk factor-matched cohort, patients on nilotinib have a much higher than expected rate of events over time (48). CVD in these patients presents primarily as progressive peripheral arterial disease rather than CAD or CVD. The ability to predict such risk in these patients using general ASCVD scores suggests that the underlying pathophysiology is indeed likely accelerated atherosclerosis rather than another disease process. Whether serial ABIs can detect the predisposition and pace of progression is unknown at present (53). Likewise, whether aggressive ASCVD prevention and treatment will hinder development and/or slow progression is unknown. Models for intervention have been outlined before.
Finally, for therapies that are associated with vasofunctional rather than structural changes, one might argue that there is no need to assess and treat for risk predisposition as easily reversible with intervention. 5-FU and capecitabine are the classical examples. However, depending on the severity and duration of coronary vasospasm and concomitant factors ventricular tachycardia, ventricular fibrillation, sudden cardiac death, (Takotsubo’s) cardiomyopathy, heart failure, hypotension and circulatory shock can develop (96). These are grave complications and one might thus present the counter argument that it might be important to know beforehand and to be prepared though modes of surveillance are not defined. For instance, should these patients be followed with telemetry to detect ST elevation, then to be initiated on vasodilatory therapy? Clearly more work in all of the outlined domains is yet to be done. How can the risk of the individual cancer patient for any given type of vascular toxicity/event be predicted?
Treatment of ACS in cancer patients
When ACS occurs in a patient with cancer, its treatment relies on careful assessment and definition of the type of ACS, its underlying etiology, and the patient’s stage within the continuum of cancer care [before, during (active), or years out (in remission)]. This then enables an individualized approach to the management and application of medical and interventional therapies. The Universal Definition of Myocardial Infarction provides an excellent foundation for the diagnostic framework, and patients can be classified as experiencing unstable angina, NSTEMI, or STEMI with the underlying mechanisms being plaque rupture or plaque erosion with intracoronary thrombus, or supply-demand mismatch due to vasospasm, severe stenosis, anemia, tachycardia, etc. (97). In most cases, coronary imaging remains an essential element, and while coronary computed tomography angiography could be applied in some scenarios, coronary angiography remains the standard. Such an invasive approach, however, is often met with scrutiny in cancer patients. The risk-benefit ratio may be viewed unfavorable, either realistically or unrealistically. It is also important to consider that the context of each case is unique, and angiography cannot be a general rule. Some patients’ cancer prognosis may be too grave, other patients may have decided not to undergo certain procedures or are on hospice care. Also, while the visual confirmation is important (rule out approach), the rule in approach is the usual mandate, and angiography might be considered pointless if not translating into revascularization. The risks and benefits of cardiovascular interventions themselves might be incompletely understood and over- or underestimated. A summary of the available evidence based on studies specifically enrolling cancer patients with ACS is outlined in Table 2 (23,98-104).
Table 2. Studies on cancer patients with acute coronary syndrome.
| Author/Year | ACS type(s) | Cancer population | Comparison group | In-hospital mortality | 1-year mortality | PCI utilization and outcomes | GDMT utilization and outcomes | Additional outcomes/comments |
|---|---|---|---|---|---|---|---|---|
| Kurisu et al. 2012 | NSTEMI and STEMI | 18 pts with active solid tumors (15 of these advanced, 9 with chemotx before AMI) | 59 non-cancer pts | Higher all-cause mortality 22% vs. 5% (P<0.05) and cardiac death 6% vs. 5% (P= NS). 2 in-hospital death due to lung cancer, 1 due to AMI, 1 cerebral hemorrhage | Higher all-cause mortality 46.5% vs. 9% (P<0.001) and cardiac death 5.6% vs. 7.1% (P= NS) | All PCI, 15 BMS, 2 POBA, 1 DES, no difference in angiographic presentation and PCI outcomes | Dismissed less often on beta-blocker (14% vs. 48%, P<0.05) | 4 major bleed (2 GI). Main baseline difference: lower Hb and anemia more common |
| Velders et al. 2013 | STEMI | 208 patients with cancer (diagnosis >3 years in 58%, within 6 mo. in 21%, 23% with h/o chemotx) |
3,215 non-cancer pts | Higher all-cause mortality 9.1% vs. 3.4% (P<0.001) and cardiac mortality 8.7% vs. 3.4% (P<0.001) | Higher all-cause mortality 7-day: 9.7% vs. 3.1% (P<0.001); 1 year: 17.4% vs. 6.5% (P<0.05) and cardiac death 7-day: 9.2% vs. 3.1% (P<0.001); 1 year: 10.7% vs. 5.4% (P<0.05) | Less stenting (92.3% vs. 95.9%, P=0.01), esp. less DES (60.8% vs. 70.8%, P=0.002) | Dismissed less often on beta-blocker (84.2% vs. 90.5%, P=0.005) | Cancer diagnosis w/in 6 months highest all-cause mortality (19%, 23.8%, 50%), and cardiac mortality (19%, 23.8%, 28.9%). Cancer history, esp. cancer diagnosis within 6 months independent predictor of early mortality; felt to be related to anemia, rate 5.2% in non-cancer vs. 11.8% in cancer patients and 65.9% in recent cancer patients) and a higher incidence of cardiogenic shock in recent cancer patients (16.7% vs. 8.7% and 6.1%). Higher use of IABP (6.7% vs. 3.9%, P=0.04) |
| Pothinenie et al. 2017 | STEMI | 48,813 pts: 5,552 breast; 32,246 lung; 12,015 colon cancer pts |
3,745,572 non-cancer pts | 21.6% (all); 36.4% (breast); 28.9% (lung); 12.9% (colon) | N/a | PCI in all pts, with significant reduction in mortality: 7.5% vs. 27.8% (all); 15.4% vs. 41.7% (breast); 10.3% vs. 32.7% (lung); 3.9% vs. 21.8% (colon) |
N/a | – |
| Rohrmann et al. 2018 | NSTEMI; STEMI (50%) | 1,918 cancer pts (specs?) | 33,268 unmatched and 1,918 matched non-cancer pts |
Higher all-cause mortality 10.7% vs. 7.6% (P<0.05) | – | Less like to receive PCI (67.8 vs. 73.4, OR 0.76) | Less likely to receive DAPT (70.3% vs. 74.3%, OR 0.82). Statins (67.5% vs. 70,4%, OR 0.87) | Higher risk of new HF (2.9% vs. 1.8%). Higher risk of CS (6.2% vs. 4.4%). No higher risk of bleeding or AKI. More anemia at baseline |
| BeeMACS group 2018 | NSTEMI; STEMI | 858 cancer pts (specs?) | 13,773 non-cancer pts | No difference | More often re-infarction (8.3% vs. 3.6%, P<0.001), and death and re-infarction (15.2% vs. 5.3%, P<0.001) as well as bleeding (6.5% vs. 3%, P<0.001) at 1 year follow-up. Cancer strongest independent predictor for death and re-infarction [hazard ratio (HR) 2.1, 1.8–2.5, P<0.001] and bleeding (HR 1.5, 1.1–2.1, P=0.015) | Lower rates of DES (35.4% vs. 40.1%, P=0.007) and complete revasc. (55.4% vs. 60.3%, P=0.015), higher rates of POBA (5.4% vs. 3.2%, P=0.002). DES with neutral effect on death (RR 0.7, 0.5–1.1, P=0.07), POBA associated with higher mortality (RR 3.6, 2.1–6.5, P<0.001) | Less likely to receive beta-blockers (74.6% vs. 81.3%, P<0.001) and ACE inhibitors/ARBs (70.6% vs. 75.1%, P=0.004) or statins (90.9% vs. 93.5%, P=0.004). Statin use lowered mortality risk (RR 0.3, 0.2–0.5, P<0.001) | More frequently NSTEMI (32.2% vs. 28.4%, P=0.006) or UA (16% vs. 13.2%, P=0.02) than STEMI, more frequently experience in-hospital bleeding (9.2% vs. 5.4%, P<0.001) and blood transfusion (6.4% vs. 2.4%, P<0.001); most frequent types of bleedings: gastrointestinal (34.1%) and genitourinary (22.5%), the only bleeding type with impact on mortality was intracranial (HR 3.9, 1.7–8.9, P=0.02) |
| Yusuf et al. 2012 | NSTEMI; STEMI | 456 cancer pts (33% hem) | None | – | 26% | – | Aspirin, beta-blocker, statin, and PCI associated with better outcomes; in multivariate analyses only aspirin (OR 0.77) and beta-blocker (OR 0.64) | Previous chemo (OR 1.42) and chest radiation (OR 1.40) independent risk factors for mortality |
| Wang et al. 2016 | STEMI | 261 cancer pts | 1,313 non-cancer pts | In-hospital 7.7% vs. 4.9% (P=0.07). Cardiac 5.8% vs. 4.6% (NS). Non-cardiac 1.9% vs. 0.4% (0.03) |
5-year all-cause (34.2% vs. 16.8%, P<0.001) and non-cardiac mortality (30% vs. 11%, P<0.001) higher in cancer pts. Cardiac mortality (4.2% vs. 5.8%) | – | No significant differences | Lower hemoglobin level, more preprocedural shock and IABP use, less lytic therapy, less GP IIb/IIIa, less DES. Higher risk for HF hospitalization (15% vs. 10%, P<0.05). Difference in long-term death solely due to cancer- related death. Patients diagnosed with cancer within 6 months had the highest risk of in-hospital and long-term mortality |
| Park et al. 2018 | UA; NSTEMI; STEMI | 73 pts with hematological malignancies | – | All-cause l mortality 21.9% (75% non-cardiac etiology) | All-cause mortality 58.9% (70% non-cardiac etiology) | – | Aspirin, beta-blocker, statin, and ACE inh./ARB but not coronary angiography or PCI associated with better survival | Mortality in the first three years was largely driven by non-cardiac death, greater contribution of cardiac death after 6 years |
Several studies evaluated differences and outcomes of cancer patients among those undergoing PCI. A general notion is the lower likelihood of DES implantation in cancer patients. Lower mortality rates were found among those undergoing PCI regardless of cancer type. However, such positive impact seems to be lost once adjusted for other baseline differences or other predictors. Coronary angiography by itself does not seem to change outcomes though there is still a role in ruling out structural epicardial pathology, e.g., in Takotsubo’s, which then directs further care. Furthermore, it is important to differentiate embolus from in situ thrombosis. The former may require thrombectomy only and a comprehensive evaluation for the source of thrombus, including heart chambers, valves (marantic endocarditis), upper/lower deep venous thrombosis with patent foramen ovale. As detailed in our recent analysis in an exemplary hematological malignancy cohort, platelet count and hemoglobin levels were predictive of an invasive approach. Furthermore, patients undergoing angiography tended to receive more recommended medical therapies as well, indicating an all-or-nothing approach.
Among the medial therapies that are proven to be beneficial even after adjusting for confounders are aspirin and beta-blocker (100,104). The benefit of statin and ACE inhibitor/ARB has not been consistently shown. The same holds true for dual antiplatelet therapy (DAPT). Anticoagulation with heparin in the acute setting does not translate into a mortality benefit; but it is current standard of care in ACS (though not without debate) and part of the continuum of care towards an invasive approach and essential in patients with embolic ACS. One has to state also that these data were derived from patients who are not in remission, thus in active therapy mode. For patients who are years out from therapy likely standard guidelines apply as they do for patients without cancer. The most challenging group of cancer patients seems to be those diagnosed just within 6 months prior to ACS presentation. Two studies in STEMI patients confirm that these patients have the highest mortality risk both acutely and thereafter (23,99). These patients seem to be more prone to hemodynamic compromise and have greater needs for hemodynamic support and greater risk of cardiogenic shock. They also are more prone to VT and cardiac arrest. The precise reasons are not clear at present. One study noted a greater burden of left main disease, but whether this relates to an acute boost in atherosclerosis due to the stress of cancer diagnosis and therapy is not known (23). Chemotherapy exposure was not found to significantly increase the acute risk in these patients (23,99).
This being said, hemoglobin levels are lower in these patients, possibly as a reflection of the effects of chemotherapy and presumably with the known prognostic implications of anemia in PCI patients (105-107). The same holds true for thrombocytopenia (108,109). Especially if confronted with both, antiplatelet and invasive therapy might be withheld. However, even in patients with these adversities, aspirin use leads to better outcomes. The current consensus recommendation of SCAI for the care of cancer patients set the lower level of platelet count for aspirin therapy at 10,000 and for DAPT with aspirin and clopidogrel at 30,000 (Figure 5) (73). Ticagrelor and prasugrel should be avoided unless PLT counts are >50,000. There is no cutoff level for diagnostic coronary angiography with a radial or careful 4F femoral approach using a micropuncture kit (+/− ultrasound). In general, all revascularization options are available with platelet counts >50,000 and PCI with bare metal stents (BMS) or drug-eluting stents (DES) with platelet counts >30,000. Furthermore, thrombectomy may be permissible with lower platelet counts though one needs to be prepared and in a position to manage dissections should they occur. Taking complications into consideration, one may want to heed more conservative voices arguing for a universal cutoff of 50,000 for any coronary procedure (110). One important aspect is to factor in the dynamics in blood counts over time. Platelet counts should meet recommended targets not only for the time of the procedure but also for the foreseeable future after the procedure. How safely and effectively transfusions could bridge such periods is unknown.
Figure 5.

Society for Cardiovascular Angiography and Interventions (SCAI) algorithm for acute coronary syndrome management in patients with cancer. Modified from Iliescu CA, Grines CL, Herrmann J, et al. SCAI Expert consensus statement: Evaluation, management, and special considerations of cardio-oncology patients in the cardiac catheterization laboratory. Catheter Cardiovasc Interv 2016;87:E202-23, with permission by Wiley.
These considerations are linked to the duration of DAPT and stent types. Historically, BMS have been used when DAPT could not be extended for more than 4 weeks. However, as shown in the LEADERS FREE trial, new stent designs, namely the polymer-free and carrier-free biolimus A9-coated BioFreedom stent, have allowed for only 1-month DAPT durations even for DES (111,112). These developments have been outlined in the 2017 European recommendations for duration of DAPT, but acknowledging the still limited level of evidence in this area (113). On the on contrary, the 2016 AHA/ACC updates on DAPT take a more conservative (and traditional) approach on BMS and DES (114). As of 2018, the European guidelines on revascularization are further avant-garde in giving a general preference to DES (115). This may address concerns regarding general underutilization of DES in cancer patients (116). However, giving the shifting complexity, in cancer patients a differential rather than a general approach will need to be taken. Accordingly, the likelihood of following general practice guidelines in cancer patients increases with increasing time from cancer diagnosis and treatment. In the early phase, these patients need individualized case, taking into account their procoagulant state, anemia and thrombocytopenia, among other elements.
Along these lines, the relative contributions of ACS care to the overall outcomes do shift with the stage the patient is in within the continuum of cancer care. For instance, in studies that evaluated patients with STEMIs, most of them >6 months, in fact years after cancer therapy, acute, in-hospital mortality was driven by cardiac mortality (23). In distinction, in patients developing ACS with active malignancies, the leading cause of in-hospital death is non-cardiac in etiology (104). Regarding long-term outcomes, in both patient groups, cancer-related mortality takes the lead though cardiac mortality seems to have an additive effect on overall mortality over time, especially once patients survive more than 7 years (23,104). In view of the above, the aggressiveness of cardiovascular therapy needs to be tailored with realistic assessments, expectations and goals. In fact, goal-directed cardiovascular therapy might be the best integrative term for the management approach to these patients.
Conclusions
The vulnerability of cancer patients to cardiovascular disease has long been recognized. Among the factors that contribute to heightened cardiovascular risk are advanced age, other (shared) risk-factors, underlying (pre-existing) disease and anatomic predisposition, the effects of the underlying cancer and its associated treatments. Validated frameworks to assess a cancer patient’s risk of vascular toxicity with any given anti-cancer treatment are very limited. As outlined herein, there is a level of complexity that does not allow for a one-size-fits-all risk prediction model. Cancer patients can develop acute ischemic complications such as ACS for various reasons, which require specific treatment. Vasodilatory therapy is key in those with events precipitated by vasospasm with no role of anticoagulation and antiplatelet therapy. On the other hand, these therapies are key in those with acute coronary thrombosis secondary to coronary emboli or erosion without any underlying atherosclerotic plaque. Percutaneous intervention with stenting is reserved for those with atherosclerotic disease that compromises myocardial perfusion, and patients need to be able to continue DAPT for at least 4 weeks without interruption. While cardiovascular therapy can be of significant benefit, all decisions are to be made within the overall clinical context (goal-directed care). As cancer treatments and survival continue to improve, better documentation and survival continues to lengthen, better documentation of the clinical outcomes of particular subsets of cancer patients and cancer therapies will be required (Table 3). This will include the formation of large registries with attention to detailed analyses of the clinical data. Furthermore, expanding the horizon for cardiovascular toxicity in the same way that genomics and proteomics have heralded an unprecedented era of treatment selection for patients with cancer might lead to major advances. The cardiovascular sequels of cancer-directed therapies can no longer remain disconnected from the management of the underlying cancer itself.
Table 3. Roadmap for augmenting the evidence for the prevention and treatment of cardiovascular disease in patients with cancer.
| Risk prediction |
| Validity of standard cardiovascular risk prediction models should be tested in various cancer types/stages/treatment groups |
| Genomic studies should explore the association between specific SNPs/allele profile and risk for cardiovascular toxicity with anti-cancer drugs |
| Natural history |
| Imaging/biochemical analyses should be performed to determine sub-clinical cardiovascular injury in cancer patients along their disease course |
| Uniform cohorts of cancer patients should be followed longitudinally and independent risk factors for CVD (on/off treatment) delineated |
| Studies should assess the utility of non-pharmacologic interventions (i.e., aerobic activity, diet) in cancer patients with CVD |
| Outcomes |
| Retrospective studies should further evaluate the benefit of various CVD treatments in the cancer population |
| Longer-term data should be collected regarding clinical outcomes of cancer patients with various CVD states on/off treatment |
| Futility of particular CVD-targeted therapies should be determined in patients with high-risk features (very frail, metastatic disease, etc.) |
SNP, single nucleotide polymorphism; CVD, cardiovascular disease.
Acknowledgements
Funding: Research funds provided by National Institute of Health (HL116952).
Footnotes
Conflicts of Interest: Past advisory board involvements: Takeda (ARIAD) Pharmaceuticals, Bristol-Myers-Squibb, Amgen; Current research funds: Amgen.
References
- 1.Blom JW, Doggen CJ, Osanto S, et al. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005;293:715-22. 10.1001/jama.293.6.715 [DOI] [PubMed] [Google Scholar]
- 2.Gomes M, Khorana AA. Risk assessment for thrombosis in cancer. Semin Thromb Hemost 2014;40:319-24. 10.1055/s-0034-1370770 [DOI] [PubMed] [Google Scholar]
- 3.Levitan N, Dowlati A, Remick SC, et al. Rates of initial and recurrent thromboembolic disease among patients with malignancy versus those without malignancy. Risk analysis using Medicare claims data. Medicine (Baltimore) 1999;78:285-91. 10.1097/00005792-199909000-00001 [DOI] [PubMed] [Google Scholar]
- 4.Sallah S, Wan JY, Nguyen NP. Venous thrombosis in patients with solid tumors: determination of frequency and characteristics. Thromb Haemost 2002;87:575-9. 10.1055/s-0037-1613051 [DOI] [PubMed] [Google Scholar]
- 5.Prandoni P, Lensing AW, Cogo A, et al. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med 1996;125:1-7. 10.7326/0003-4819-125-1-199607010-00001 [DOI] [PubMed] [Google Scholar]
- 6.Sørensen HT, Mellemkjaer L, Olsen JH, et al. Prognosis of cancers associated with venous thromboembolism. N Engl J Med 2000;343:1846-50. 10.1056/NEJM200012213432504 [DOI] [PubMed] [Google Scholar]
- 7.Khorana AA, Francis CW, Culakova E, et al. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost 2007;5:632-4. 10.1111/j.1538-7836.2007.02374.x [DOI] [PubMed] [Google Scholar]
- 8.Pathanjali Sharma PV, Babu SC, et al. Arterial thrombosis and embolism in malignancy. J Cardiovasc Surg (Torino) 1985;26:479-83. [PubMed] [Google Scholar]
- 9.Libby P, Pasterkamp G. Requiem for the 'vulnerable plaque'. Eur Heart J 2015;36:2984-7. [DOI] [PubMed] [Google Scholar]
- 10.Pasterkamp G, den Ruijter HM, Libby P. Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat Rev Cardiol 2017;14:21-9. 10.1038/nrcardio.2016.166 [DOI] [PubMed] [Google Scholar]
- 11.Jia H, Abtahian F, Aguirre AD, et al. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. J Am Coll Cardiol 2013;62:1748-58. 10.1016/j.jacc.2013.05.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iliescu CA, Cilingiroglu M, Giza DE, et al. "Bringing on the light" in a complex clinical scenario: Optical coherence tomography-guided discontinuation of antiplatelet therapy in cancer patients with coronary artery disease (PROTECT-OCT registry). Am Heart J 2017;194:83-91. 10.1016/j.ahj.2017.08.015 [DOI] [PubMed] [Google Scholar]
- 13.Takeshita S, Ogata T, Mera H, et al. Multiple Thrombi in the Heart in Trousseau Syndrome Caused by Pancreatic Carcinoma. J Stroke Cerebrovasc Dis 2018;27:e75-7. 10.1016/j.jstrokecerebrovasdis.2017.12.005 [DOI] [PubMed] [Google Scholar]
- 14.Navi BB, Reiner AS, Kamel H, et al. Risk of Arterial Thromboembolism in Patients With Cancer. J Am Coll Cardiol 2017;70:926-38. 10.1016/j.jacc.2017.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tuzovic M, Herrmann J, Iliescu C, et al. Arterial Thrombosis in Patients with Cancer. Curr Treat Options Cardiovasc Med 2018;20:40. 10.1007/s11936-018-0635-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goubran HA, Burnouf T, Radosevic M, et al. The platelet-cancer loop. Eur J Intern Med 2013;24:393-400. 10.1016/j.ejim.2013.01.017 [DOI] [PubMed] [Google Scholar]
- 17.Goubran HA, Stakiw J, Radosevic M, et al. Platelet-cancer interactions. Semin Thromb Hemost 2014;40:296-305. 10.1055/s-0034-1370767 [DOI] [PubMed] [Google Scholar]
- 18.Bauer AT, Suckau J, Frank K, et al. von Willebrand factor fibers promote cancer-associated platelet aggregation in malignant melanoma of mice and humans. Blood 2015;125:3153-63. 10.1182/blood-2014-08-595686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang AJ, Wang M, Wang Y, et al. Cancer cell-derived von Willebrand factor enhanced metastasis of gastric adenocarcinoma. Oncogenesis 2018;7:12. 10.1038/s41389-017-0023-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herrmann J, Lerman A. The endothelium: dysfunction and beyond. J Nucl Cardiol 2001;8:197-206. 10.1067/mnc.2001.114148 [DOI] [PubMed] [Google Scholar]
- 21.Herrmann J, Yang EH, Iliescu CA, et al. Vascular Toxicities of Cancer Therapies: The Old and the New--An Evolving Avenue. Circulation 2016;133:1272-89. 10.1161/CIRCULATIONAHA.115.018347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quillard T, Franck G, Mawson T, et al. Mechanisms of erosion of atherosclerotic plaques. Curr Opin Lipidol 2017;28:434-41. 10.1097/MOL.0000000000000440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang F, Gulati R, Lennon RJ, et al. Cancer History Portends Worse Acute and Long-term Noncardiac (but Not Cardiac) Mortality After Primary Percutaneous Coronary Intervention for Acute ST-Segment Elevation Myocardial Infarction. Mayo Clin Proc 2016;91:1680-92. 10.1016/j.mayocp.2016.06.029 [DOI] [PubMed] [Google Scholar]
- 24.Mosseri M, Fingert HJ, Varticovski L, et al. In vitro evidence that myocardial ischemia resulting from 5-fluorouracil chemotherapy is due to protein kinase C-mediated vasoconstriction of vascular smooth muscle. Cancer Res 1993;53:3028-33. [PubMed] [Google Scholar]
- 25.Südhoff T, Enderle MD, Pahlke M, et al. 5-Fluorouracil induces arterial vasocontractions. Ann Oncol 2004;15:661-4. 10.1093/annonc/mdh150 [DOI] [PubMed] [Google Scholar]
- 26.Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol 2018;10:1758835918780140. 10.1177/1758835918780140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi N, Hata N, Yokoyama S, et al. A case of Takotsubo cardiomyopathy during 5-fluorouracil treatment for rectal adenocarcinoma. J Nippon Med Sch 2009;76:27-33. 10.1272/jnms.76.27 [DOI] [PubMed] [Google Scholar]
- 28.Dhesi S, Chu MP, Blevins G, et al. Cyclophosphamide-Induced Cardiomyopathy: A Case Report, Review, and Recommendations for Management. J Investig Med High Impact Case Rep 2013;1:2324709613480346. 10.1177/2324709613480346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shanholtz C. Acute life-threatening toxicity of cancer treatment. Crit Care Clin 2001;17:483-502. 10.1016/S0749-0704(05)70196-2 [DOI] [PubMed] [Google Scholar]
- 30.Centola M, Lucreziotti S, Cazzaniga S, et al. A rare case of large intracoronary thrombosis in advanced breast cancer patient treated with epirubicin and cisplatin. J Cardiovasc Med (Hagerstown) 2016;17 Suppl 2:e241-3. 10.2459/JCM.0000000000000444 [DOI] [PubMed] [Google Scholar]
- 31.Ito D, Shiraishi J, Nakamura T, et al. Primary percutaneous coronary intervention and intravascular ultrasound imaging for coronary thrombosis after cisplatin-based chemotherapy. Heart Vessels 2012;27:634-8. 10.1007/s00380-011-0222-5 [DOI] [PubMed] [Google Scholar]
- 32.Jafri M, Protheroe A. Cisplatin-associated thrombosis. Anticancer Drugs 2008;19:927-9. 10.1097/CAD.0b013e3283100e9c [DOI] [PubMed] [Google Scholar]
- 33.Karabay KO, Yildiz O, Aytekin V. Multiple coronary thrombi with cisplatin. J Invasive Cardiol 2014;26:E18-20. [PubMed] [Google Scholar]
- 34.Togna GI, Togna AR, Franconi M, et al. Cisplatin triggers platelet activation. Thromb Res 2000;99:503-9. 10.1016/S0049-3848(00)00294-2 [DOI] [PubMed] [Google Scholar]
- 35.Gietema JA, Meinardi MT, Messerschmidt J, et al. Circulating plasma platinum more than 10 years after cisplatin treatment for testicular cancer. Lancet 2000;355:1075-6. 10.1016/S0140-6736(00)02044-4 [DOI] [PubMed] [Google Scholar]
- 36.Touyz RM, Herrmann SMS, Herrmann J. Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J Am Soc Hypertens 2018;12:409-25. 10.1016/j.jash.2018.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herrmann J, Lerman LO, Mukhopadhyay D, et al. Angiogenesis in atherogenesis. Arterioscler Thromb Vasc Biol 2006;26:1948-57. 10.1161/01.ATV.0000233387.90257.9b [DOI] [PubMed] [Google Scholar]
- 38.Meyer T, Robles-Carrillo L, Robson T, et al. Bevacizumab immune complexes activate platelets and induce thrombosis in FCGR2A transgenic mice. J Thromb Haemost 2009;7:171-81. 10.1111/j.1538-7836.2008.03212.x [DOI] [PubMed] [Google Scholar]
- 39.Chen N, Ren M, Li R, et al. Bevacizumab promotes venous thromboembolism through the induction of PAI-1 in a mouse xenograft model of human lung carcinoma. Mol Cancer 2015;14:140. 10.1186/s12943-015-0418-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee S, Chen TT, Barber CL, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell 2007;130:691-703. 10.1016/j.cell.2007.06.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chintalgattu V, Rees ML, Culver JC, et al. Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci Transl Med 2013;5:187ra69. 10.1126/scitranslmed.3005066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duran JM, Makarewich CA, Trappanese D, et al. Sorafenib cardiotoxicity increases mortality after myocardial infarction. Circ Res 2014;114:1700-12. 10.1161/CIRCRESAHA.114.303200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arima Y, Oshima S, Noda K, et al. Sorafenib-induced acute myocardial infarction due to coronary artery spasm. J Cardiol 2009;54:512-5. 10.1016/j.jjcc.2009.03.009 [DOI] [PubMed] [Google Scholar]
- 44.Naib T, Steingart RM, Chen CL. Sorafenib-associated multivessel coronary artery vasospasm. Herz 2011;36:348-51. 10.1007/s00059-011-3444-5 [DOI] [PubMed] [Google Scholar]
- 45.Herrmann J. Tyrosine Kinase Inhibitors and Vascular Toxicity: Impetus for a Classification System? Curr Oncol Rep 2016;18:33. 10.1007/s11912-016-0514-0 [DOI] [PubMed] [Google Scholar]
- 46.Herrmann J, Bell MR, Warren RL, et al. Complicated and Advanced Atherosclerosis in a Young Woman With Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Success and Challenges of BCR/ABL1-Targeted Cancer Therapy. Mayo Clin Proc 2015;90:1167-8. 10.1016/j.mayocp.2015.05.013 [DOI] [PubMed] [Google Scholar]
- 47.Aichberger KJ, Herndlhofer S, Schernthaner GH, et al. Progressive peripheral arterial occlusive disease and other vascular events during nilotinib therapy in CML. Am J Hematol 2011;86:533-9. 10.1002/ajh.22037 [DOI] [PubMed] [Google Scholar]
- 48.Hadzijusufovic E, Albrecht-Schgoer K, Huber K, et al. Nilotinib-induced vasculopathy: identification of vascular endothelial cells as a primary target site. Leukemia 2017;31:2388-97. 10.1038/leu.2017.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valent P, Hadzijusufovic E, Hoermann G, et al. Risk factors and mechanisms contributing to TKI-induced vascular events in patients with CML. Leuk Res 2017;59:47-54. 10.1016/j.leukres.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valent P, Hadzijusufovic E, Schernthaner GH, Wolf D, Rea D, le Coutre P. Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood 2015;125:901-6. 10.1182/blood-2014-09-594432 [DOI] [PubMed] [Google Scholar]
- 51.Douxfils J, Haguet H, Mullier F, et al. Association Between BCR-ABL Tyrosine Kinase Inhibitors for Chronic Myeloid Leukemia and Cardiovascular Events, Major Molecular Response, and Overall Survival: A Systematic Review and Meta-analysis. JAMA Oncol 2016. [Epub ahead of print]. 10.1001/jamaoncol.2015.5932 [DOI] [PubMed] [Google Scholar]
- 52.Nicolini FE, Gagnieu MC, Heiblig M, et al. Cardio-Vascular Events Occurring On Ponatinib In Chronic Phase Chronic Myeloid Leukemia Patients, Preliminary Analysis Of a Multicenter Cohort. Blood 2013;122:4020. [Google Scholar]
- 53.Moslehi JJ, Deininger M. Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J Clin Oncol 2015;33:4210-8. 10.1200/JCO.2015.62.4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gover-Proaktor A, Granot G, Shapira S, et al. Ponatinib reduces viability, migration, and functionality of human endothelial cells. Leuk Lymphoma 2017;58:1455-67. 10.1080/10428194.2016.1239258 [DOI] [PubMed] [Google Scholar]
- 55.Pouwer MG, Pieterman EJ, Verschuren L, et al. The BCR-ABL1 Inhibitors Imatinib and Ponatinib Decrease Plasma Cholesterol and Atherosclerosis, and Nilotinib and Ponatinib Activate Coagulation in a Translational Mouse Model. Front Cardiovasc Med 2018;5:55. 10.3389/fcvm.2018.00055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herrmann J, Lerman LO, Lerman A. On to the road to degradation: atherosclerosis and the proteasome. Cardiovasc Res 2010;85:291-302. 10.1093/cvr/cvp333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herrmann J, Saguner AM, Versari D, et al. Chronic proteasome inhibition contributes to coronary atherosclerosis. Circ Res 2007;101:865-74. 10.1161/CIRCRESAHA.107.152959 [DOI] [PubMed] [Google Scholar]
- 58.Herrmann J, Soares SM, Lerman LO, et al. Potential role of the ubiquitin-proteasome system in atherosclerosis: aspects of a protein quality disease. J Am Coll Cardiol 2008;51:2003-10. 10.1016/j.jacc.2008.02.047 [DOI] [PubMed] [Google Scholar]
- 59.Siegel D, Martin T, Nooka A, et al. Integrated safety profile of single-agent carfilzomib: experience from 526 patients enrolled in 4 phase II clinical studies. Haematologica 2013;98:1753-61. 10.3324/haematol.2013.089334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen-Scarabelli C, Corsetti G, Pasini E, et al. Spasmogenic Effects of the Proteasome Inhibitor Carfilzomib on Coronary Resistance, Vascular Tone and Reactivity. EBioMedicine 2017;21:206-12. 10.1016/j.ebiom.2017.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson DB, Balko JM, Compton ML, et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N Engl J Med 2016;375:1749-55. 10.1056/NEJMoa1609214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahmood SS, Fradley MG, Cohen JV, et al. Myocarditis in Patients Treated With Immune Checkpoint Inhibitors. J Am Coll Cardiol 2018;71:1755-64. 10.1016/j.jacc.2018.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang DY, Salem JE, Cohen JV, et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol 2018;4:1721-8. 10.1001/jamaoncol.2018.3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bu DX, Tarrio M, Maganto-Garcia E, et al. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler Thromb Vasc Biol 2011;31:1100-7. 10.1161/ATVBAHA.111.224709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gotsman I, Grabie N, Dacosta R, et al. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest 2007;117:2974-82. 10.1172/JCI31344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koga N, Suzuki J, Kosuge H, et al. Blockade of the interaction between PD-1 and PD-L1 accelerates graft arterial disease in cardiac allografts. Arterioscler Thromb Vasc Biol 2004;24:2057-62. 10.1161/01.ATV.0000145015.23656.e4 [DOI] [PubMed] [Google Scholar]
- 67.Watanabe R, Zhang H, Berry G, et al. Immune checkpoint dysfunction in large and medium vessel vasculitis. Am J Physiol Heart Circ Physiol 2017;312:H1052-H1059. 10.1152/ajpheart.00024.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weyand CM, Berry GJ, Goronzy JJ. The immunoinhibitory PD-1/PD-L1 pathway in inflammatory blood vessel disease. J Leukoc Biol 2018;103:565-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang H, Watanabe R, Berry GJ, et al. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci U S A 2017;114:E970-9. 10.1073/pnas.1616848114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lyon AR, Yousaf N, Battisti NML, et al. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol 2018;19:e447-58. 10.1016/S1470-2045(18)30457-1 [DOI] [PubMed] [Google Scholar]
- 71.Daxini A, Cronin K, Sreih AG. Vasculitis associated with immune checkpoint inhibitors-a systematic review. Clin Rheumatol 2018. [Epub ahead of print]. 10.1007/s10067-018-4177-0 [DOI] [PubMed] [Google Scholar]
- 72.Lancellotti P, Nkomo VT, Badano LP, et al. Expert consensus for multi-modality imaging evaluation of cardiovascular complications of radiotherapy in adults: a report from the European Association of Cardiovascular Imaging and the American Society of Echocardiography. J Am Soc Echocardiogr 2013;26:1013-32. 10.1016/j.echo.2013.07.005 [DOI] [PubMed] [Google Scholar]
- 73.Iliescu CA, Grines CL, Herrmann J, et al. SCAI Expert consensus statement: Evaluation, management, and special considerations of cardio-oncology patients in the cardiac catheterization laboratory (endorsed by the Cardiological Society of India, and Sociedad Latino Americana de Cardiologia intervencionista). Catheter Cardiovasc Interv 2016;87:E202-23. 10.1002/ccd.26379 [DOI] [PubMed] [Google Scholar]
- 74.Marks LB, Yu X, Prosnitz RG, et al. The incidence and functional consequences of RT-associated cardiac perfusion defects. Int J Radiat Oncol Biol Phys 2005;63:214-23. 10.1016/j.ijrobp.2005.01.029 [DOI] [PubMed] [Google Scholar]
- 75.Prosnitz RG, Hubbs JL, Evans ES, et al. Prospective assessment of radiotherapy-associated cardiac toxicity in breast cancer patients: analysis of data 3 to 6 years after treatment. Cancer 2007;110:1840-50. 10.1002/cncr.22965 [DOI] [PubMed] [Google Scholar]
- 76.Darby SC, Ewertz M, McGale P, et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med 2013;368:987-98. 10.1056/NEJMoa1209825 [DOI] [PubMed] [Google Scholar]
- 77.Heidenreich PA, Schnittger I, Strauss HW, et al. Screening for coronary artery disease after mediastinal irradiation for Hodgkin's disease. J Clin Oncol 2007;25:43-9. 10.1200/JCO.2006.07.0805 [DOI] [PubMed] [Google Scholar]
- 78.Bashar K, Healy D, Clarke-Moloney M, et al. Effects of neck radiation therapy on extra-cranial carotid arteries atherosclerosis disease prevalence: systematic review and a meta-analysis. PLoS One 2014;9:e110389. 10.1371/journal.pone.0110389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wilbers J, Dorresteijn LD, Haast R, et al. Progression of carotid intima media thickness after radiotherapy: a long-term prospective cohort study. Radiother Oncol 2014;113:359-63. 10.1016/j.radonc.2014.10.012 [DOI] [PubMed] [Google Scholar]
- 80.Halle M, Gabrielsen A, Paulsson-Berne G, et al. Sustained inflammation due to nuclear factor-kappa B activation in irradiated human arteries. J Am Coll Cardiol 2010;55:1227-36. 10.1016/j.jacc.2009.10.047 [DOI] [PubMed] [Google Scholar]
- 81.Halle M, Christersdottir T, Back M. Chronic adventitial inflammation, vasa vasorum expansion, and 5-lipoxygenase up-regulation in irradiated arteries from cancer survivors. FASEB J 2016;30:3845-52. 10.1096/fj.201600620R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Modrall JG, Sadjadi J. Early and late presentations of radiation arteritis. Semin Vasc Surg 2003;16:209-14. 10.1016/S0895-7967(03)00026-7 [DOI] [PubMed] [Google Scholar]
- 83.Stewart FA, Heeneman S, Te Poele J, et al. Ionizing radiation accelerates the development of atherosclerotic lesions in ApoE-/- mice and predisposes to an inflammatory plaque phenotype prone to hemorrhage. Am J Pathol 2006;168:649-58. 10.2353/ajpath.2006.050409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gabriels K, Hoving S, Gijbels MJ, et al. Irradiation of existing atherosclerotic lesions increased inflammation by favoring pro-inflammatory macrophages. Radiother Oncol 2014;110:455-60. 10.1016/j.radonc.2014.01.006 [DOI] [PubMed] [Google Scholar]
- 85.Yu T, Parks BW, Yu S, et al. Iron-ion radiation accelerates atherosclerosis in apolipoprotein E-deficient mice. Radiat Res 2011;175:766-73. 10.1667/RR2482.1 [DOI] [PubMed] [Google Scholar]
- 86.Mancuso M, Pasquali E, Braga-Tanaka I, 3rd, et al. Acceleration of atherogenesis in ApoE-/- mice exposed to acute or low-dose-rate ionizing radiation. Oncotarget 2015;6:31263-71. 10.18632/oncotarget.5075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hoving S, Heeneman S, Gijbels MJ, et al. Anti-inflammatory and anti-thrombotic intervention strategies using atorvastatin, clopidogrel and knock-down of CD40L do not modify radiation-induced atherosclerosis in ApoE null mice. Radiother Oncol 2011;101:100-8. 10.1016/j.radonc.2011.09.019 [DOI] [PubMed] [Google Scholar]
- 88.Stewart FA, Seemann I, Hoving S, et al. Understanding radiation-induced cardiovascular damage and strategies for intervention. Clin Oncol (R Coll Radiol) 2013;25:617-24. 10.1016/j.clon.2013.06.012 [DOI] [PubMed] [Google Scholar]
- 89.Abdel-Qadir H, Ethier JL, Lee DS, et al. Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: A systematic review and meta-analysis. Cancer Treat Rev 2017;53:120-7. 10.1016/j.ctrv.2016.12.002 [DOI] [PubMed] [Google Scholar]
- 90.Scappaticci FA, Skillings JR, Holden SN, et al. Arterial thromboembolic events in patients with metastatic carcinoma treated with chemotherapy and bevacizumab. J Natl Cancer Inst 2007;99:1232-9. 10.1093/jnci/djm086 [DOI] [PubMed] [Google Scholar]
- 91.Lubberts S, Boer H, Altena R, et al. Vascular fingerprint and vascular damage markers associated with vascular events in testicular cancer patients during and after chemotherapy. Eur J Cancer 2016;63:180-8. 10.1016/j.ejca.2016.05.022 [DOI] [PubMed] [Google Scholar]
- 92.Haugnes HS, Wethal T, Aass N, et al. Cardiovascular risk factors and morbidity in long-term survivors of testicular cancer: a 20-year follow-up study. J Clin Oncol 2010;28:4649-57. 10.1200/JCO.2010.29.9362 [DOI] [PubMed] [Google Scholar]
- 93.Meinardi MT, Gietema JA, van der Graaf WT, et al. Cardiovascular morbidity in long-term survivors of metastatic testicular cancer. J Clin Oncol 2000;18:1725-32. 10.1200/JCO.2000.18.8.1725 [DOI] [PubMed] [Google Scholar]
- 94.van den Belt-Dusebout AW, Nuver J, de Wit R, et al. Long-term risk of cardiovascular disease in 5-year survivors of testicular cancer. J Clin Oncol 2006;24:467-75. 10.1200/JCO.2005.02.7193 [DOI] [PubMed] [Google Scholar]
- 95.Breccia M, Molica M, Zacheo I, et al. Application of systematic coronary risk evaluation chart to identify chronic myeloid leukemia patients at risk of cardiovascular diseases during nilotinib treatment. Ann Hematol 2015;94:393-7. 10.1007/s00277-014-2231-9 [DOI] [PubMed] [Google Scholar]
- 96.Y-Hassan S , Tornvall P, Törnerud M, et al. Capecitabine caused cardiogenic shock through induction of global Takotsubo syndrome. Cardiovasc Revasc Med 2013;14:57-61. 10.1016/j.carrev.2012.10.001 [DOI] [PubMed] [Google Scholar]
- 97.Thygesen K, Alpert JS, Jaffe AS, et al. Fourth Universal Definition of Myocardial Infarction (2018). J Am Coll Cardiol 2018;72:2231-64. 10.1016/j.jacc.2018.08.1038 [DOI] [PubMed] [Google Scholar]
- 98.Kurisu S, Iwasaki T, Ishibashi K, et al. Comparison of treatment and outcome of acute myocardial infarction between cancer patients and non-cancer patients. Int J Cardiol 2013;167:2335-7. 10.1016/j.ijcard.2012.11.009 [DOI] [PubMed] [Google Scholar]
- 99.Velders MA, Boden H, Hofma SH, et al. Outcome after ST elevation myocardial infarction in patients with cancer treated with primary percutaneous coronary intervention. Am J Cardiol 2013;112:1867-72. 10.1016/j.amjcard.2013.08.019 [DOI] [PubMed] [Google Scholar]
- 100.Yusuf SW, Daraban N, Abbasi N, et al. Treatment and outcomes of acute coronary syndrome in the cancer population. Clin Cardiol 2012;35:443-50. 10.1002/clc.22007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pothineni NV, Shah NN, Rochlani Y, et al. Temporal trends and outcomes of acute myocardial infarction in patients with cancer. Ann Transl Med 2017;5:482. 10.21037/atm.2017.11.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rohrmann S, Witassek F, Erne P, et al. Treatment of patients with myocardial infarction depends on history of cancer. Eur Heart J Acute Cardiovasc Care 2018;7:639-45. 10.1177/2048872617729636 [DOI] [PubMed] [Google Scholar]
- 103.Iannaccone M, D'Ascenzo F, Vadala P, et al. Prevalence and outcome of patients with cancer and acute coronary syndrome undergoing percutaneous coronary intervention: a BleeMACS substudy. Eur Heart J Acute Cardiovasc Care 2018;7:631-8. 10.1177/2048872617706501 [DOI] [PubMed] [Google Scholar]
- 104.Park JY, Guo W, Al-Hijji M, et al. Acute coronary syndromes in patients with active hematologic malignancies - Incidence, management, and outcomes. Int J Cardiol 2019;275:6-12. 10.1016/j.ijcard.2018.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Al-Hijji MA, Gulati R, Lennon RJ, et al. Outcomes of Percutaneous Coronary Interventions in Patients With Anemia Presenting With Acute Coronary Syndrome. Mayo Clin Proc 2018;93:1448-61. 10.1016/j.mayocp.2018.03.030 [DOI] [PubMed] [Google Scholar]
- 106.Nikolsky E, Aymong ED, Halkin A, et al. Impact of anemia in patients with acute myocardial infarction undergoing primary percutaneous coronary intervention: analysis from the Controlled Abciximab and Device Investigation to Lower Late Angioplasty Complications (CADILLAC) Trial. J Am Coll Cardiol 2004;44:547-53. 10.1016/j.jacc.2004.03.080 [DOI] [PubMed] [Google Scholar]
- 107.McKechnie RS, Smith D, Montoye C, et al. Prognostic implication of anemia on in-hospital outcomes after percutaneous coronary intervention. Circulation 2004;110:271-7. 10.1161/01.CIR.0000134964.01697.C7 [DOI] [PubMed] [Google Scholar]
- 108.Hakim DA, Dangas GD, Caixeta A, et al. Impact of baseline thrombocytopenia on the early and late outcomes after ST-elevation myocardial infarction treated with primary angioplasty: analysis from the Harmonizing Outcomes with Revascularization and Stents in Acute Myocardial Infarction (HORIZONS-AMI) trial. Am Heart J 2011;161:391-6. 10.1016/j.ahj.2010.11.001 [DOI] [PubMed] [Google Scholar]
- 109.Yadav M, Genereux P, Giustino G, et al. Effect of Baseline Thrombocytopenia on Ischemic Outcomes in Patients With Acute Coronary Syndromes Who Undergo Percutaneous Coronary Intervention. Can J Cardiol 2016;32:226-33. 10.1016/j.cjca.2015.05.020 [DOI] [PubMed] [Google Scholar]
- 110.McCarthy CP, Steg G, Bhatt DL. The management of antiplatelet therapy in acute coronary syndrome patients with thrombocytopenia: a clinical conundrum. Eur Heart J 2017;38:3488-92. 10.1093/eurheartj/ehx531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tomoda H. Polymer-free Drug-Coated Coronary Stents. N Engl J Med 2016;374:892-3. [DOI] [PubMed] [Google Scholar]
- 112.Carrié D, Menown I, Oldroyd K, et al. Safety and Efficacy of Polymer-Free Biolimus A9-Coated Versus Bare-Metal Stents in Orally Anticoagulated Patients: 2-Year Results of the LEADERS FREE Oral Anticoagulation Substudy. JACC Cardiovasc Interv 2017;10:1633-42. 10.1016/j.jcin.2017.05.033 [DOI] [PubMed] [Google Scholar]
- 113.Valgimigli M, Bueno H, Byrne RA, et al. 2017 ESC focused update on dual antiplatelet therapy in coronary artery disease developed in collaboration with EACTS: The Task Force for dual antiplatelet therapy in coronary artery disease of the European Society of Cardiology (ESC) and of the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J 2018;39:213-60. 10.1093/eurheartj/ehx419 [DOI] [PubMed] [Google Scholar]
- 114.Levine GN, Bates ER, Bittl JA, et al. 2016 ACC/AHA Guideline Focused Update on Duration of Dual Antiplatelet Therapy in Patients With Coronary Artery Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2016;68:1082-115. 10.1016/j.jacc.2016.03.513 [DOI] [PubMed] [Google Scholar]
- 115.Neumann FJ, Sousa-Uva M, Ahlsson A, et al. 2018. ESC/EACTS Guidelines on myocardial revascularization. Eur Heart J 2018 [Epub ahead of print]. [Google Scholar]
- 116.Wang F, Herrmann J. In Reply II-A Differing Opinion on Primary Percutaneous Coronary Intervention in Patients Who Have Had Cancer: Stent Choice in Onco-cardiology Revisited. Mayo Clin Proc 2017;92:1317-8. 10.1016/j.mayocp.2017.05.012 [DOI] [PubMed] [Google Scholar]