

Abstract
Serratia marcescens is a Gram-negative bacterial species that can be found in a wide range of environments like soil, water and plant surfaces, while it is also known as an opportunistic human pathogen in hospitals and as a plant growth promoting bacteria (PGPR) in crops. We have used a pangenome-based approach, based on publicly available genomes, to apply whole genome multilocus sequence type schemes to assess whether there is an association between source and genotype, aiming at differentiating between isolates from nosocomial sources and the environment, and between strains reported as PGPR from other environmental strains. Most genomes from a nosocomial setting and environmental origin could be assigned to the proposed nosocomial or environmental MLSTs, which is indicative of an association between source and genotype. The fact that a few genomes from a nosocomial source showed an environmental MLST suggests that a minority of nosocomial strains have recently derived from the environment. PGPR strains were assigned to different environmental types and clades but only one clade comprised strains accumulating a low number of known virulence and antibiotic resistance determinants and was exclusively from environmental sources. This clade is envisaged as a group of promissory MLSTs for selecting prospective PGPR strains.
Introduction
Serratia marcescens is a Gram-negative rod-shaped bacterium of the Enterobacteriaceae family that has been isolated from different environmental and nosocomial sources. Its importance as an opportunistic human pathogen has been acknowledged in the last decades, when S. marcescens has been signaled as responsible for a range of symptoms in hospitalized patients including septicemia1, meningitis, infections of the urinary tract2–4, eyes5, bloodstream6 and organs of the respiratory apparatus7. Putative and confirmed virulence determinants have been recognized including the production of hemolysins (ShlA), proteases, siderophores and lipopolysaccharides8. Virulence factors of indirect action but whose presence increase virulence are related to bacteria motility and antibiotic resistance. However, some of these virulence factors, most notably those of secondary importance, may also confer competence for any strain to strive in different hosts like insects and plants surfaces, and also soil and water. A particular case that has drawn great interest is the presence of genetic determinants of intrinsic and acquired resistance to antibiotics, linked to the origin and spread of multidrug resistant strains (MDR) between the environment and hospitals, and the impact of this secondary factor as an enhancer of the virulence of the nosocomial strains9–11.
Being an ubiquitous microorganism, environmental S. marcescens has been isolated from water and soil, plants, insects, foods and machinery. Plant roots and their adjacent soil -the rhizosphere- can host S. marcescens and other species that positively interact with plants, enhancing nutrition, stress tolerance and health and therefore have been considered plant growth promoting rhizobacteria (PGPR)12–14. The rhizosphere of wild and cultivated plants is also known to host opportunistic human pathogens like Burkholderia cepacia, Pseudomonas aeruginosa and Stenotrophomonas maltophilia, although little is known about the virulence of these environmental strains relative to the clinical strains15,16. For S. marcescens, its double life-strategy as a human pathogen and as a PGPR has arisen doubts about its use in agriculture. In particular, there is a need to understand whether the nosocomial and environmental populations of S. marcescens can be clearly defined and whether strains promoting plant growth can be genotyped with high precision to undoubtly track them down in the field after their deployment in agricultural settings.
Whole genome sequencing (WGS) has emerged as an ultimate typing tool that fits any bacterial species, study type, and laboratory17. Whole genome multilocus sequence typing (wgMLST) has been proposed as a very useful and practical method based on WGS to distinguish strains within epidemic settings18 and between epidemic and unrelated specimens17. To apply MLST based on WGS, an allele database for the population of a bacterial organism must first be set.
In this work we have used the PGAdb-builder19, a recently available web-based tool, to create a pangenome allele database for this species and to apply wgMLST schemes to genomes of nosocomial, environmental and PGPR strains of S. marcescens available at GenBank to address questions regarding the link between origin, PGPR status and genotype. In addition, we explored the annotated genes in the genomes of selected S. marcescens to weigh the presence of some known virulence, antibiotic resistance and PGP determinants. The ultimate goal is to use the growing resource of genomic information to draw preliminary conclusions on the diversity and genetic make-up of S. marcescens populations and their suitability to be used as PGPR.
Results
Serratia marcescens pangenome
The average nucleotide identity (ANI) of whole genome sequences of 49 selected isolates, representing diverse environments and countries and including ATCC 13880 type strain, confirmed that 45 strains could be assigned to S. marcescens as they showed ANI values > 95.0% with S. marcescens type strain ATCC 13880 (Table 1). Within this group, most of the strains of nosocomial origin, all three strains from insects and 2 strains known as PGPR had ANI values between 95.0 and 96.0, while most environmental, 2 nosocomial and 2 PGPR strains had ANI values > 96.0 (Table 1). Among the four strains with ANI values below 95.0, that were not considered as S. marcescens and were no further analyzed, MSU97 -reported as a PGPR strain- had the lowest ANI value (93.77).
Table 1.
Strains used in this study, source, country, GenBank genome accession, ANIm % and assembly parameters.
| Strain Codea | Strain Name | Source of isolation | Country | Sequence Number | ANIm | Contigs N50-L50 |
|---|---|---|---|---|---|---|
| 1 | 2880STDY5683032 | Nosocomial pathogen/blood | UK | GCF_001538705.1 | 95.64 | 251,419-7 |
| 2 | 2880STDY5682907 | Nosocomial pathogen/blood | UK | GCF_001538645.1 | 95.67 | 278,529-7 |
| 3 | 2880STDY5683006 | Nosocomial pathogen/blood | UK | GCF_001537785.1 | 95.67 | 340,665-6 |
| 4 | SM1890 | Nosocomial/urinary tract infection | Germany | GCA_900108825.1 | 95.34 | 94,867-24 |
| 5 | LCT-SM213 | Derived from CGMCC 1.1857 | China | GCF_000264275.1 | 98.90 | 299,433-5 |
| Na | W2.3 | Sick fish | Malaysia | GCF_000292365.1 | 94.44 | 216,941-8 |
| 6 | WW4 | Paper machine | Taiwan | GCF_000336425.1 | 96.96 | complete |
| 7 | VGH 107 | Snake-bite wound | Taiwan | GCF_000342205.1 | 97.03 | 220,690--10 |
| 8 | MC620 | Stool of patient with bacteremia | USA | GCF_000418815.1 | 95.33 | 86,897-21 |
| 9 | MC6001 | Blood sample of individual with bacteremia | USA | GCF_000418835.1 | 95.32 | 85,735-22 |
| 10 | EGD-HP20 | Tannery waste water | India | GCF_000465615.2 | 97.00 | 189,845-10 |
| 11 | Db-11 | Derived from Db10/insect pathogen (Drosophila) | Sweden | GCF_000513215.1 | 96.07 | complete |
| 12 | BIDMC 50 | Bronchoalveolar lava | USA | GCF_000521905.1 | 95.45 | 685,952- 3 |
| Na | H1q | Carnivorous plant phytotelma | Malaysia | GCF_000633335.1 | 94.44 | 345,525-4 |
| Na | PH1a | Carnivorous plant phytotelma | Malaysia | GCF_000633555.1 | 94.44 | 374,527- 4 |
| 13 | ATCC 14041 | Unknown | USA | GCF_000695485.1 | 98.91 | 113,020- 15 |
| 14 | ATCC 13880 T | Pond water | Unknown | GCF_000735445.1 | — | 238,240- 7 |
| 15 | FDAARGOS_65 | Endotracheal aspirate | USA | GCF_000783915.2 | 95.64 | complete |
| 16 | SM39 | Nosocomial pathogen/septicemia | Japan | GCF_000828775.1 | 95.65 | complete |
| 17 | 90–166 | Field-grown plant | USA | GCF_001007555.1 | 95.32 | 632,250- 4 |
| 18 | CAV1492 | Nosocomial pathogen/respiratory | USA | GCF_001022215.1 | 95.32 | complete |
| 19 | RSC-14 | Solanum nigrum plant | South Korea | GCF_001280365.1 | 95.47 | complete |
| 20 | SmUNAM836 | Bronchial sample/ obstructive pneumonia | Mexico | GCF_001294565.1 | 95.64 | complete |
| 21 | B3R3 | Zea mays plant | China | GCF_001417865.2 | 97.05 | Complete |
| 22 | ano2 | Anopheles stephensi gut contents | China | GCF_001853495.1 | 95.40 | 232,610- 4 |
| 23 | Sicaria (Ss1) | Apis mellifera hemolymph | USA | GCF_001889685.1 | 95.42 | 105742- 18 |
| Na | MSU97 | Plant surface | Venezuela | GCF_001902635.1 | 93.77 | 197,047- 10 |
| 24 | 189 | Feces of neonate/asymptomatic | Russia | GCF_001908015.1 | 95.64 | 100,707- 16 |
| 25 | AS1 | Anopheles stephensi | USA | GCF_001932655.1 | 95.40 | 3,366,121- 1 |
| 26 | BJL200 | Soil | Unknown | GCF_001940505.1 | 96.91 | 41,521- 45 |
| 27 | 19 F | Atelopus zeteki skin | USA | GCF_001975745.1 | 96.92 | 618,117- 3 |
| 28 | 1274 | Agave sisalana plant | Brazil | GCF_002007925.1 | 95.40 | 1,503,251- 2 |
| 29 | CAPREX_SY13 | Ghanaian yam (plant) | England | GCF_002029205.1 | 98.94 | 498,095- 4 |
| 30 | MEW06 | Lake water | China | GCF_002094145.1 | 97.07 | 335,101- 6 |
| 31 | D-1 | Clinical/blood simple | USA | GCF_002104105.1 | 95.66 | 565,574- 3 |
| 32 | Z6 | Contaminated soil | China | GCF_002108655.1 | 97.07 | 652,363-4 |
| 33 | S217 | Petroleum contaminated soil | India | GCF_002205475.1 | 96.99 | 119,928- 14 |
| 34 | UMH2 | Bacteremia | USA | GCF_002220515.1 | 95.48 | complete |
| 35 | UMH8 | Bacteremia | USA | GCF_002220535.1 | 99.44 | complete |
| 36 | UMH1 | Bacteremia | USA | GCF_002220615.1 | 96.07 | complete |
| 37 | at10508 | Chronic obstructive pulmonary disease/lavage | Austria | GCF_002250685.1 | 95.33 | 120,371- 14 |
| 38 | EGD-HP20_1 | Tannery waste | India | GCF_002265665.1 | 96.90 | 25,890-65 |
| 39 | KHCo-24B | Gossypium hirsutum | India | GCF_002592035.1 | 97.06 | 306,793-7 |
| 40 | 12TM | Pharyngeal secretions | Romania | GCF_002810285.1 | 95.66 | complete |
| 41 | AR_0027 | Clinical/patient associated | Unknown | GCF_002947235.1 | 95.67 | complete |
| 42 | AR_0091 | Clinical/patient associated | Unknown | GCF_002996885.1 | 95.62 | complete |
| 43 | SMB2099 | Clinical | Germany | GCF_900029885.1 | 95.65 | complete |
| 44 | SM1978 | Rectal swab at hospital admission | Germany | GCF_900108835.1 | 95.34 | 41,390-41 |
| 45 | UENF-22GI | Cattle vermicompost | Brazil | SAMN03892645 | 98.92 | * |
aOrigin of the strains is indicated as nosocomial (bold), environmental (black), unknown (italic), PGPR reported status (bold/italic).
Na: strain code not assigned due to ANI < 95.
*UENF-22GI genome was downloaded from https://www.ncbi.nlm.nih.gov/Traces/wgs/?display = contigs&page = 1 and N50 – L50 parameters were not available.
The pangenome of S. marcescens built from 45 genome assemblies comprised a total of 19469 genes. The core genome -made of loci present in >95% of genomes- contained 3155 genes (16.2%), the dispensable genome contained 7847 genes (40.3%), while unique or exclusive genes -present in a single genome- were 8467 (43.5%). The automatically annotated genes were 4989, including core, dispensable and unique genes. The database identification number of this pangenome of S. marcescens is 1718436483 and can be accessed directly at http://wgmlstdb.imst.nsysu.edu.tw/disProfileDB.php?folder=1718436483.
Classification of nosocomial, environmental and PGPR strains based on WGS MLST
Two dendrograms with bootstrap values were generated based on allelic sequences of a maximum of 19469 genes by the Build_wgMLSTtree module, which uses PHYLIP program UPGMA clustering algorithm. The first dendrogram was based on genes that are included in the core genome (Fig. 1a). A central node divided the dendrogram in two sectors: one that gathered mostly strains of environmental origin (clade 1) and another that gathered mostly strains of nosocomial origin (clade 2). These two sectors were considered to represent environmental and nosocomial cgMLST respectively, and the subclades and strains in each sector were defined as environmental-type and nosocomial-type, independently from their origin.
Figure 1.
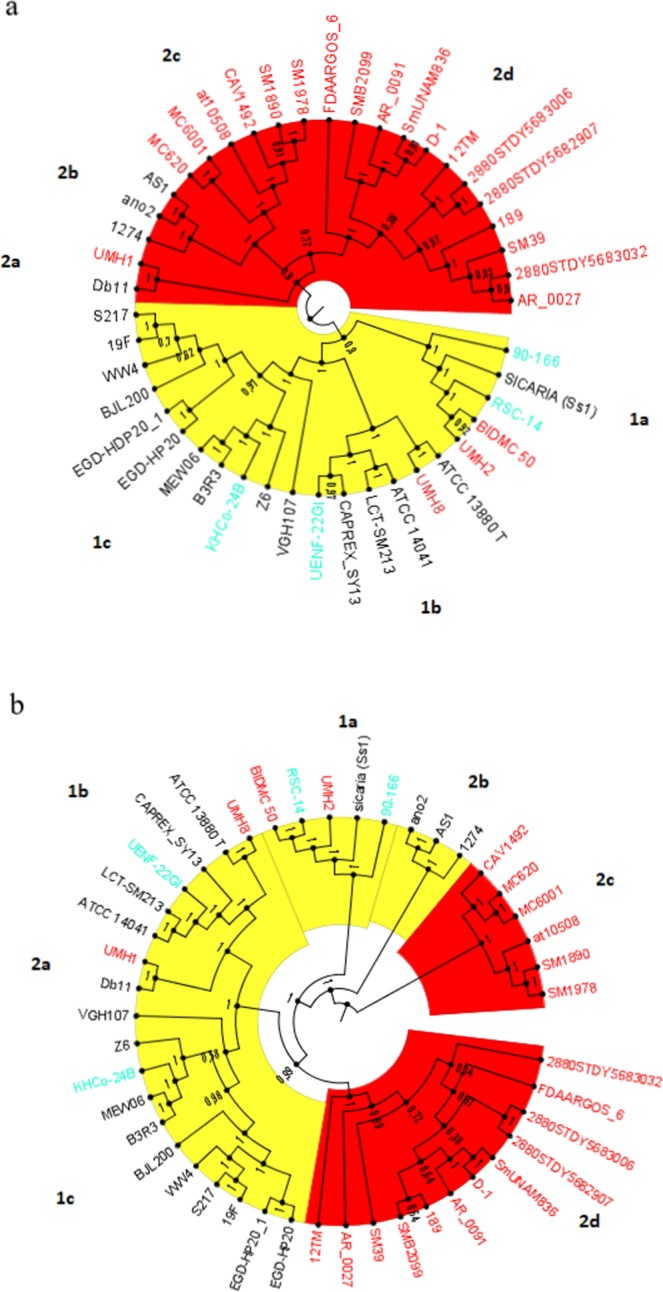
Dendrograms based on genetic distance between allelic sequences of 1a core genes and 1b whole genome genes of 45 Serratia marcescens strains representing nosocomial, environmental and PGPR strains. Taxa were colored according to the origin of the strain: clinical (red), environmental (black), PGPR (green) while clades were numbered and colored according to the assigned genomic type in the cgMLSTs: clinical (red), environmental/PGPR (yellow).
Regarding the origin, nosocomial subclades 2c and 2d defined by cgMLST were integrated exclusively by strains from clinical origin. The sister nosocomial subclade 2b was integrated by three strains from environmental origin, including two strains from mosquitoes, and subclade 2a by one strain of nosocomial origin and one of environmental origin. Environmental clade 1 defined by cgMLST was formed mostly but not exclusively by strains of environmental origin, along with UMH8, UMH2 and BIDMC 50 of nosocomial origin. Also, environmental clade 1 could be separated in three sub-subclades (a, b, c). One of these subclades (1c) was formed exclusively by strains of environmental origin, while 1a and 1b contained 3 isolates of nosocomial origin. S. marcescens genomes with known PGPR activity were assigned to the environmental subclades 1a, 1b, 1c.
When the dendrogram was based on alleles of all genes (wgMLST), some re-ordering could be observed (Fig. 1b). Although in general subclades maintained their integrity, the most notorious rearrangement was the separation of nosocomial subclade 2b and 2c and environmental subclade 1a, which were detached from their original clades and formed independent clades. Besides, subclade 2a from the nosocomial clade, was relocated within environmental subclade 1b, adding one more strain of nosocomial origin (UMH1) to an environmental clade. The assignment of PGPR genomes to their subclusters was maintained for all strains.
Analysis of genomes based on presence/absence of virulence genes
An ordination of strains based on presence/absence of known genes related to virulence (Supplementary Table S1) was carried out by means of correspondence analysis (Fig. 2). This allowed for the clear separation of strains of clinical origin (red dots) in three groups, in the same fashion as these strains were grouped in the wgMLST tree: two main groups of strains of nosocomial origin separated along the horizontal axis whereas a group of 4 clinical isolates with an environmental wgMLST further separated along the vertical axis, forming a group with strains from environmental origin and those with reported PGPR status. Genomes showed different numbers of accumulated virulence, resistance and PGPR related genes (Supplementary Tables S1–S3; Fig. 2). The strains that ranked highest in accumulated number of virulence-related genes were in subclades 2c and 2d in Fig. 1a, those that ranked highest in antibiotic resistance genes were from clade 2c, while the strains accumulating more PGPR-related genes belonged with clade 1b and 1c. Genes coding for Beta-lactamases were present only in strains of nosocomial origin, mostly from clade 2c (Supplementary Table S2).
Figure 2.

Correspondence analysis based on the presence/absence of 21 known virulence-related genes of Serratia marcescens strains of clinical (red) and environmental (black) origin, including strains reported as PGPR (green). Strains that accumulated higher numbers of known virulence, antibiotic resistance and PGP-related genes are indicated by skulls, underline and a leaf respectively.
Discussion
Serratia marcescens behaves as an opportunistic human pathogen in hospitals, where it affects immunocompromised patients2–7,10. At the same time, this species has been isolated from environmental sources like soils, water, insects and plants surfaces including the rhizosphere, where it can exert positive plant growth promotion effects12–14. The wide niche and functional diversity of this species raises questions about the genetic background that defines and separates nosocomial strains from environmental strains, and PGPR strains from other environmental strains. This is relevant to the discussions regarding the existence and distinction of nosocomial and non-nosocomial populations and the presence of genes related with virulence and antibiotic resistance in strains of this species that could be selected and used as PGPR.
In this work, we have attempted to assess differences between strains of S. marcescens of clinical and environmental origin based on the analysis of whole genome sequences of a panel of 45 strains - representing clinical and environmental populations from different countries- that were available at the GenBank assembly database.
This analysis implied a progressive approach in which strains were first grouped according to core genome MLST and whole genome MLST schemes. While the former was based on the allele variants of genes that are present in more that 95% of the tested strains, the latter was based on allele variants present also in the genes of the flexible genome or even exclusive genes, partly related to strain specific features like virulence and antibiotic resistance20.
A second step of the analysis was based on the presence/absence of some genes that are known to be involved in these traits. This added a bias towards clinical genomes regarding the presence of virulence/resistance determinants and towards PGPR genomes regarding the presence of PGP determinants. Therefore, although the correspondence analysis -based on the presence of virulence genes- was done with genomes of all origins, further comparisons and considerations were done within genomes of nosocomial and environmental origin by separate.
ANI value of 95.0 was used as a cut off threshold to circumscribe the species, which led to the exclusion of four strains that could hardly be considered part of S. marcescens species, including MSU97, a proposed PGPR strain of S. marcescens21. In a comparative genomic study of clinical and environmental S. marcescens22, this strain showed 1265 strain specific genes, a digital DNA to DNA hybridization (dDDH) value under 68%, and an isolated position in a phylogenetic tree based on 10 concatenated single copy core genes. In keeping with recent guidelines for bacterial species definition23, all this information supported our decision to not include MSU97 as part of the pangenome of S. marcescens.
The proposed pangenome of S. marcescens based on 45 strains from highly diverse nosocomial and environmental sources comprised 19469 genes, higher than 13614 genes reported from 205 clinical isolates in UK and Ireland10 and 16456 genes reported from a panel of 35 strains of S. marcescens from environmental and nosocomial origin, including MSU9722. Interestingly, only 16% of the genes were part of the core genome and 84% of the genes were either part of the flexible genome or were present in single strains, in nearly equal parts. This high number of exclusive and accessory genes can reflect the adaptability of S. marcescens to different niches and functions. Similar results regarding the high genomic variability of this species have been reported10,20,22.
The cgMLST scheme allowed for a segregation of most strains according to their origin, except in the case of 3 strains of nosocomial origin and 3 of environmental origin that were grouped with their alternative cgMLST. The position of the 3 strains that originated from hospitals within the environmental clades in the cgMLST and 4 strains in the wgMLST tree suggests that these nosocomial strains are derived from the non-clinical environment and therefore indicates that environmental strains of S. marcescens might develop as opportunistic pathogens in nosocomial infections.
Still, it is noticeable that the majority of strains from hospitals from different countries showed nosocomial MLSTs, which is indicative of the existence of a predominantly clinical population of S. marcescens that can be distinguished from the environmental counterparts. Within this population, the existence of two clades -2c and 2d- that are maintained in both MLST schemes implies that both subpopulations differ not only in the core genes but also in their particular accessory genes. This was also noted by Moradigaravard et al.10 in 2016, who found that nosocomial S. marcescens core genome revealed a highly structured population comprising distinct clades, with each clade showing a unique combination of accessory genes. In our study, although the analysis of virulence and resistance determinants was not exhaustive, nosocomial strains in clade 2c accumulated more antibiotic resistance genes and Beta-lactamase coding genes in particular. In this regard, multivariate analysis of strains based on presence/absence of virulence genes produced groups similar to those in the cgMLST and wgMLST: clade 2c of nosocomial strains, containing 6 virulent strains, was clearly separated from nosocomial strains that belonged to clade 2d and from the 4 strains of nosocomial origin but environmental wgMLST, that grouped again with the environmental strains. This last verification confirmed that although these 4 strains were obtained from diseased patients, not only their wgMLSTs but their virulence determinant, associated them with environmental strains. The significance of this finding is two folds: it confirms that at least some clinical strains of S. marcescens obtained from hospitalized patients are derived from the environment and therefore that some environmental strains might act as carriers of novel genes for the nosocomial population of S. marcescens, including unknown virulence and resistance determinants that were not considered in this study. Although it is known that the environmental resistome is distinct from the human -associated resistome, the environment can act as a reservoir of resistance determinants that can be present in human and clinical associated resistomes24 and opportunistic pathogens may represent a major conduit through which antibiotic resistance genes move between natural and nosocomial environments25.
On the other hand, the assignment of 3 strains from environmental sources to the nosocomial clade in the cgMLST tree suggests that the environment can host strains that are genetically more similar to nosocomial strains than to other environmental strains. However, when dispensable genes were added to produce the wgMLST tree, these three strains relocated from the nosocomial clade and formed an independent clade. This could reflect that these strains do not possess the set of dispensable genes that characterize the other strains from nosocomial origin and MLST. Two of these three strains (ano2 and AS1) were isolated from Anopheles stephensi – the malaria vector- and are known to be commensal strains capable of exerting control not on the vector but on Plasmodium, the causal agent of malaria26,27. Strain ano2 is known to have many predicted virulence factors including hemolysins, chitinases, flagella formation and lipooligosacharide27, but lacks genes encoding prodigiosins, which can be lethal to mosquitos. These highly specific symbionts might then conserve some of the pathogenicity related genes, needed to cope with the mosquito blood meals and to outcompete other species in the mosquito gut microbiota, as part of their core genome, but lack other genes that might damage the host, which would be no longer present in the dispensable genome. Such specific combination of attributes might explain why these strains separated from the nosocomial clade when all genes were considered.
According to Bruto et al.28, the PGPR status is dependent on the accumulation of a variable assortment of genes. It is noteworthy that the strains that accumulated more PGP-related genes belonged to subclade 1c and 1b in the MLST trees. Subclade 1c contains only environmental strains and one reported PGPR strain. Taken together, this could be an indication that clade 1c is a prospective PGPR clade, with sequence types less probable to be found in nosocomial environments. However, the inclusion of more genomes from clinical origin is necessary to challenge this hypothesis. The fact that no other PGPR have been found within this clade might be a consequence of these strains not yet being tested for their PGP activity. Therefore, clade 1c, with its particular accumulation and assortment of PGP genes and the low number of known virulence and resistance determinants is thought to represent a promissory safe MLST for finding more strains with PGP traits. Yet, because unknown virulence and resistance genes may be present but unnoticed in any environmental strain24, it is foreseen that more detailed bioinformatic studies based on WGS should be implemented on promissory strains from this subclade, to weigh this possibility29.
In conclusion, the proposed pangenome of S. marcescens and both cgMLST and wgMLST schemes can be used to type strains of this species and assign them to environmental or nosocomial MLSTs and clades, with wgMLST scheme producing a more strict segregation, probably reflecting specific functions that reside in genes of the dispensable genome.
The fact that some strains from nosocomial origin had environmental MLSTs highlights the potential role of opportunistic pathogens -like S. marcescens- in the gene flow between the environment and hospitals.
One subclade named 1c was found to comprise only environmental strains, including one reported as PGPR. Some strains in this subclade were also the ones accumulating more PGP traits and fewer known virulence and antibiotic resistance determinants and could therefore be signaled as a promissory clade from which obtain more PGPR. Because strains in this clade may possess unknown virulence or antibiotic resistance genes that have not yet been annotated, their presence should be addressed by different approaches to further sustain the use of strains from this subclade as PGPR.
Methods
Genomes
Genomes of S. marcescens assembled as contigs, scaffolds or complete genomes were downloaded from the National Center for Biotechnology Information (NCBI) Assembly database (https://www.ncbi.nlm.nih.gov/assembly). A total of 49 assemblies comprising 23 nosocomial strains, 24 environmental strains and 2 of unknown origin including reference strains ATCC 13880T were downloaded (Table 1). Nosocomial strains were chosen randomly from a range of countries, with some specific strains included for their known virulence or prevalence based on bibliography9,10. Environmental strains were chosen also from a range of countries and sources (lakes, plants, insects, soils) with 5 strains included for their reported PGPR status (Table 1).
Average nucleotide identity was calculated by MUMer algorithm (ANIm) for 48 strains in relation with ATCC 13880 type strain with JSpecies software30 to verify that they could be considered S. marcescens (ANIMm > 95). Only strains with ANIm higher than 95 % were assigned a strain code (Table 1) and used in subsequent analyses.
Building a S. marcescens pangenome
To build a pangenome for S. marcescens, 45 assemblies with ANI > 95.0 were first uploaded to the Build_PGAdb module of the PGAdb-builder server19 freely available at http://wgmlstdb.imst.nsysu.edu.tw. In this module, genes were annotated using Prokka and paralogous genes were excluded from the pan-genome allele dataset by default. Each orthologous cluster consisted of a protein family (a gene) with 95% sequence identity (default parameter). For allele calling, sequences in a locus with one or more mismatched nucleotides between each other were defined as different alleles. A pie chart was produced showing the number of genes in the core genome (genes shared by at least 95% of the genomes), number of genes exclusive or unique genes (present in a single genome) and the number of genes present in more than 1 but less than 95% of the genomes (the flexible genome). Besides, a list of annotated genes and the sequences of each allele of each annotated gene were produced.
Building MLST trees based on whole genome sequences
Two MLST trees based on WGS were built for the same pangenome: one scheme included genes from the core genome (cgMLST) and the other scheme included all genes in the pangenome (wgMLST). For this, the same 45 genomes were uploaded on the module Build_wgMLSTtreeof the PGAdb-builder server. The uploaded genomes were compared with the built allele database (PGAdb) using BLASTN23 with parameters set by default. Because the same set of assemblies was used to create the pangenome and to build the wgMLST trees, all the alleles of any given gene were expected to be present in at least one sequence. After the allele finding process was finished for each scheme, the allelic sequences in each scheme were created. A dendrogram with bootstrap values, which is calculated by the ETE tool kit24, was then constructed from allelic sequences with the PHYLIP program through use of UPGMA clustering algorithm by default. For each scheme, the Build_wgMLSTtree produced files for presence/absence of loci, alleles present in each loci of each strain, a distance matrix and a dendrogram based on the genetic relatedness of the genomes. Dendrograms were further edited with FigTree31.
Multivariate analysis of genomes based on presence/absence of genes
Genes that are known to be linked to virulence, plant growth promotion and antibiotic resistance were selected from bibliography11,22,28 and used to search in the Prokka annotated gene list produced by the PGAdb_builder. An ordering of strains based on the presence/absence matrix of virulence genes was achieved by multivariate analysis with software PAST32. The strains accumulating the highest number of virulence, resistance and plant growth promotion genes were highlighted on the graph.
Supplementary information
Author Contributions
E.A. and N.A. designed the study, E.A. performed the genomic analysis and wrote the manuscript, E.A. and N.A. analyzed the results. Both authors reviewed the manuscript.
Data Availability
The datasets generated and analyzed during the current study are available in the following links: Pangenome: http://wgmlstdb.imst.nsysu.edu.tw/disProfileDB.php?folder=1718436483 cgMLST_tree, presence/absence of genes and allele database: http://wgmlstdb.imst.nsysu.edu.tw/diswgProfiling.php?folder=12569572 wgMLST_tree, presence/absence of genes and allele database: http://wgmlstdb.imst.nsysu.edu.tw/diswgProfiling.php?folder=1699143652.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-37118-0.
References
- 1.Altemeier WA, Culbertson WR, Fullen WD, Mc Donough JJ. Serratia marcescens septicemia. A new threat in surgery. Arch. Surg. 1969;99:232–238. doi: 10.1001/archsurg.1969.01340140104015. [DOI] [PubMed] [Google Scholar]
- 2.Maki DG, Hennekens CG, Phillips CW, Shaw WV, Bennett JV. Nosocomial urinary tract infection with Serratia marcescens: an epidemiologic study. J. Infect. Dis. 1973;128:579–587. doi: 10.1093/infdis/128.5.579. [DOI] [PubMed] [Google Scholar]
- 3.Okuda T, Endo N, Osada Y, Zen-Yoji H. Outbreak of nosocomial urinary tract infections caused by Serratia marcescens. J. Clin. Microbiol. 1984;20:691–695. doi: 10.1128/jcm.20.4.691-695.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su LH, et al. Extended epidemic of nosocomial urinary tract infections caused by Serratia marcescens. J. Clin. Microbiol. 2003;41:4726–4732. doi: 10.1128/JCM.41.10.4726-4732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Templeton WC, Eiferman MRA, Snyder JW, Melo JC, Raff MJ. Serratia keratitis transmitted by contaminated eyedroppers. Am. J. Ophthalmol. 1982;93:723–726. doi: 10.1016/0002-9394(82)90467-6. [DOI] [PubMed] [Google Scholar]
- 6.Korner RJ, Nicol A, Reeves DS, MacGowan AP, Hows J. Ciprofloxacin resistant Serratia marcescens endocarditis as a complication of non-Hodgkin’s lymphoma. J. Infect. 1994;29:73–76. doi: 10.1016/S0163-4453(94)95141-1. [DOI] [PubMed] [Google Scholar]
- 7.Dessi A, et al. Serratia marcescens infections and outbreaks in neonatal intensive care units. J. Chemother. 2009;21:493–499. doi: 10.1179/joc.2009.21.5.493. [DOI] [PubMed] [Google Scholar]
- 8.Kurz CL, et al. Virulence factors of the human opportunistic pathogen Serratia marcescens identified by in vivo screening. EMBO J. 2003;22:1451–1460. doi: 10.1093/emboj/cdg159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gruber TM, et al. Pathogenicity of pan-drug resistant Serratia marcescens harbouring blaNDM-1. J. Antimicrob. Chemother. 2015;70:26–30. doi: 10.1093/jac/dku482. [DOI] [PubMed] [Google Scholar]
- 10.Moradigaravand D, Boinett CJ, Martin V, Peacock SJ, Parkhill J. Recent independent emergence of multiple multidrug-resistant Serratia marcescens clones within the United Kingdom and Ireland. Gen. Res. 2016;26:1101–1109. doi: 10.1101/gr.205245.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandner-Miranda L, Vinuesa P, Cravioto A, Morales-Espinosa R. The genomic basis of intrinsic and acquired antibiotic resistance in the genus Serratia. Front. Microbiol. 2018;9:828. doi: 10.3389/fmicb.2018.00828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gyaneshwar P, et al. Endophytic colonization of rice by a diazotrophic strain of Serratia marcescens. J. Bacteriol. 2001;183:2634–2645. doi: 10.1128/JB.183.8.2634-2645.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen LM, Tisa LS. Friend or foe? A review of the mechanisms that drive Serratia towards diverse lifestyles. Can. J. Microbiol. 2013;59:627–640. doi: 10.1139/cjm-2013-0343. [DOI] [PubMed] [Google Scholar]
- 14.Singh RP, Jha PN. The multifarious PGPR Serratia marcescens CDP-13 augments induced systemic resistance and enhanced salinity tolerance of wheat (Triticum aestivum L.) PLoS One. 2016;11:1–24. doi: 10.1371/journal.pone.0155026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berg G, Eberl L, Hartmann A. The rhizosphere as a reservoir for oppurtunistic human pathogenic bacteria. Environ. Microbiol. 2005;7:672–1685. doi: 10.1111/j.1462-2920.2005.00891.x. [DOI] [PubMed] [Google Scholar]
- 16.Mendes R, Garbeva P, Raaijmakers J. The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013;37:634–663. doi: 10.1111/1574-6976.12028. [DOI] [PubMed] [Google Scholar]
- 17.Kluytmans-van den Bergh MF, et al. Whole genome multilocus sequence typing of extended-spectrum beta-lactamase-producing Enterobacteriaceae. J. Clin. Microbiol. 2016;54:2919–2927. doi: 10.1128/JCM.01648-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu YY, Chen CC, Chiou CS. Construction of a pan-genome allele database of Salmonella enterica serovar enteritidis for molecular subtyping and disease cluster identification. Front. Microbiol. 2016;7:2010. doi: 10.3389/fmicb.2016.02010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu YY, Chen CC, Chiou CS. PGAdb-Builder: A web service tool for creating pan-genome allele database for molecular fine typing. Sci. Rep. 2016;6:1–5. doi: 10.1038/s41598-016-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iguchi A, et al. Genome evolution and plasticity of Serratia marcescens, an important multidrug-resistant nosocomial pathogen. Genome Biol. Evol. 2014;6:2096–2110. doi: 10.1093/gbe/evu160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matilla MA, Udaondo Z, Krell T, Salmonda GPC. Genome sequence of Serratia marcescens MSU97, a plant-associated bacterium that makes multiple antibiotics. Genome Announc. 2017;5:e01752–16. doi: 10.1128/genomeA.01752-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matteoli, F. et al. Genome sequencing and assessment of plant growth-promoting properties of a Serratia marcescens strain isolated from vermicompost. BMC Genomics, 1–28. [DOI] [PMC free article] [PubMed]
- 23.Chun, J. et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. [DOI] [PubMed]
- 24.Gibson MK, Forsberg KJ, Dantas G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 2015;9:207–216. doi: 10.1038/ismej.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forsberg KJ, et al. Bacterial phylogeny structures soil resistomes across habitats. Nature. 2014;509:612–616. doi: 10.1038/nature13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang S, et al. Driving mosquito refractoriness to Plasmodium falciparum with engineered symbiotic bacteria. Science. 2017;357:1399–1402. doi: 10.1126/science.aan5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen S, Blom J, Walker ED. Genomic, physiologic, and symbiotic characterization of Serratia marcescens strains isolated from the mosquito Anopheles stephensi. Front. Microbiol. 2017;8:1483. doi: 10.3389/fmicb.2017.01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruto M, Prigent-Combaret C, Muller D, Moënne-Loccoz Y. Analysis of genes contributing to plant-beneficial functions in plant growth-promoting rhizobacteria and related Proteobacteria. Sci. Rep. 2014;4:1–10. doi: 10.1038/srep06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paterson J, et al. The contribution of genome mining strategies to the understanding of active principles of PGPR strains. FEMS. Microbiol. Ecol. 2017;93:1–12. doi: 10.1093/femsec/fiw249. [DOI] [PubMed] [Google Scholar]
- 30.Richter M, Rosselló-Móra R, Oliver Glöckner F, Peplies J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2015;32:929–931. doi: 10.1093/bioinformatics/btv681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rambaut, A. FigTree, a graphical viewer of phylogenetic trees. Available from, http://tree.bio.ed.ac.uk/software/figtree/ (2007).
- 32.Hammer Ø, Harper DAT, Ryan PD. PAST: Paleontological statistics software package for education and data analysis. Palaeontologia electronica. 2001;4:1–9. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available in the following links: Pangenome: http://wgmlstdb.imst.nsysu.edu.tw/disProfileDB.php?folder=1718436483 cgMLST_tree, presence/absence of genes and allele database: http://wgmlstdb.imst.nsysu.edu.tw/diswgProfiling.php?folder=12569572 wgMLST_tree, presence/absence of genes and allele database: http://wgmlstdb.imst.nsysu.edu.tw/diswgProfiling.php?folder=1699143652.