

In one of the most well-studied organisms in the life sciences, Escherichia coli, we still do not fully understand what causes populations to die. This is largely due to the technological difficulties of studying bacterial cell death. This study provides an avenue to studying how and why E. coli populations, and perhaps other microbes, transition from stationary phase to death phase by exploring how ethanol and other alcohols delay the onset of death. Here, we demonstrate that alcohols are acting as signaling molecules to achieve the delay in death phase. This study not only offers a better understanding of a fundamental process but perhaps also provides a gateway to studying the dynamics between ethanol and microbes in the human gastrointestinal tract.
KEYWORDS: Escherichia coli, alcohol, death phase, delayed bacterial death
ABSTRACT
When Escherichia coli K-12 is inoculated into rich medium in batch culture, cells experience five phases. While the lag and logarithmic phases are mechanistically fairly well defined, the stationary phase, death phase, and long-term stationary phase are less well understood. Here, we characterize a mechanism of delaying death, a phenomenon we call the “alcohol effect,” where the addition of small amounts of certain alcohols prolongs stationary phase for at least 10 days longer than in untreated conditions. We show that the stationary phase is extended when ethanol is added above a minimum threshold concentration. Once ethanol levels fall below a threshold concentration, cells enter the death phase. We also show that the effect is conferred by the addition of straight-chain alcohols 1-propanol, 1-butanol, 1-pentanol, and, to a lesser degree, 1-hexanol. However, methanol, isopropanol, 1-heptanol, and 1-octanol do not delay entry into death phase. Though modulated by RpoS, the alcohol effect does not require RpoS activity or the activities of the AdhE or AdhP alcohol dehydrogenases. Further, we show that ethanol is capable of extending the life span of stationary-phase cultures for non-K-12 E. coli strains and that this effect is caused in part by genes of the glycolate degradation pathway. These data suggest a model where ethanol and other shorter 1-alcohols can serve as signaling molecules, perhaps by modulating patterns of gene expression that normally regulate the transition from stationary phase to death phase.
IMPORTANCE In one of the most well-studied organisms in the life sciences, Escherichia coli, we still do not fully understand what causes populations to die. This is largely due to the technological difficulties of studying bacterial cell death. This study provides an avenue to studying how and why E. coli populations, and perhaps other microbes, transition from stationary phase to death phase by exploring how ethanol and other alcohols delay the onset of death. Here, we demonstrate that alcohols are acting as signaling molecules to achieve the delay in death phase. This study not only offers a better understanding of a fundamental process but perhaps also provides a gateway to studying the dynamics between ethanol and microbes in the human gastrointestinal tract.
INTRODUCTION
The mechanism(s) modulating bacterial death during batch culture is poorly understood (1–2), and debate continues regarding whether the transition from stationary phase to death phase in Escherichia coli is a stochastic or a “programmed” process akin to a form of bacterial apoptosis (1–6). In typical batch culture in rich media, E. coli K-12 strains will begin to die after 1 to 2 days in stationary phase, resulting in the loss of viability of 99% of cells, with the surviving ∼1% of the population transitioning into long-term stationary phase (1, 7, 8). Not surprisingly, there is an inherent difficulty in studying bacterial cell death mechanisms since most experimental approaches focus on the surviving subpopulations. While previous studies have utilized computer simulation models and synthetic biological techniques to study population-level bacterial death (9–10), an alternative method is to characterize modes of delaying the transition to death phase (11–13). Farrell and Finkel characterized one such method of temporary death delay through buffering of the growth medium (13). These authors showed that by keeping a constant pH of 7.0 in Luria-Bertani (LB) complex growth medium, the stationary phase could be extended for multiple days. This is the case in both wild-type and rpoS-attenuated mutant strains with severely diminished functionality. Both this work and that of Farrell and Finkel describe two of the main modes of delaying death.
The “alcohol effect,” or the “alcohol-induced delay of death,” was first observed in the mid-1990s by members of Roberto Kolter’s laboratory (11, 12) when they sought to determine whether there was a protein synthesis-dependent mechanism for entry into death phase, using chloramphenicol as a translation inhibitor (14, 15). This experiment meant to elucidate a role for active protein synthesis being required for the transition into death phase (1–4, 7, 8). Although it was initially observed that the addition of sublethal amounts of the antibiotic could cause a delay in the initiation of death phase, control experiments revealed that chloramphenicol was not the causal agent. Instead, it was the ethanol solvent that led to the maintenance of the stationary phase (12). Traditionally, studies have focused on adding nearly lethal concentrations of alcohol to better understand ethanol tolerance for biofuel production studies (16, 17). Some of the common physiological effects resulting from addition of high doses of alcohol include increased membrane permeability and slowed outgrowth of E. coli populations compared to the wild type (17–20). The phenomenon, where sublethal alcohol additions actually confer a potentially positive benefit, was subsequently dubbed the “alcohol effect.”
Vulić and Kolter (11) previously demonstrated that the onset of death phase can be delayed when ethanol, 1-propanol, or 1-butanol is added after 24 h of batch culture incubation. These authors also showed that an rpoS-null mutant strain “lost” the effect and that strains without the AdhE alcohol dehydrogenase still exhibited the delay. These data suggested that the effect does not require the catabolism of ethanol as a carbon source (21) but instead that some regulated change is required. Here, we more extensively characterize the physiological, genetic, and transcriptomic factors associated with alcohol-mediated delay of death phase, including a further exploration of the requirement of a functional rpoS gene.
RESULTS
Ethanol addition prolongs stationary phase in a dose-dependent manner.
To define the minimum concentration of ethanol that can produce the delayed death effect, 1 to 6 μl of 95% ethanol was added to 5 ml batch cultures of E. coli on day 1 of incubation (Fig. 1A), which corresponds to stationary-phase populations. We found 5 μl (∼17.6 mM) to be the minimum amount necessary to cause a 1-day delay in death (Fig. 1A) since the addition of 1 to 4 μl (∼3.5 to 14.1 mM) had no effect.
FIG 1.
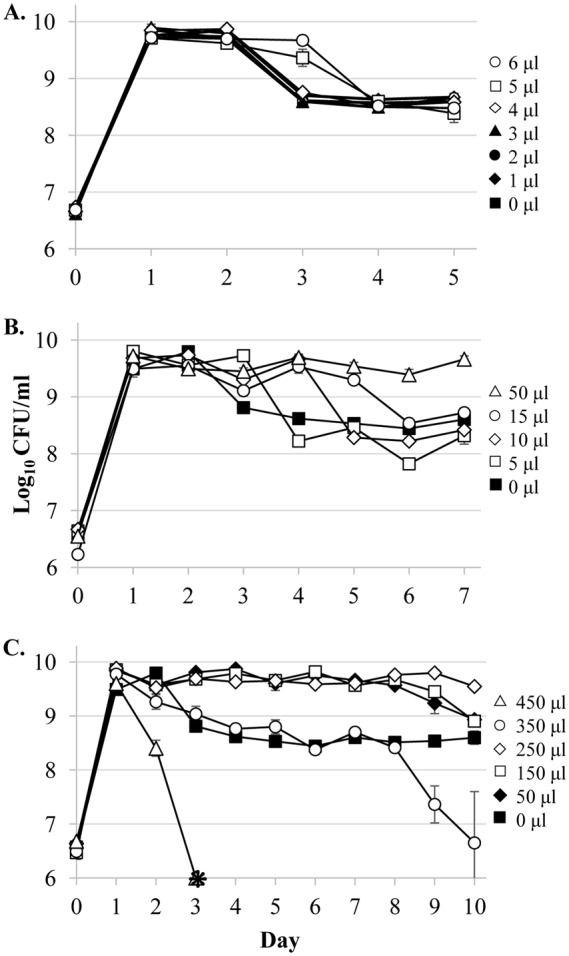
The wild-type strain treated with different concentrations of ethanol on day 1 shows a dose-dependent alcohol effect. To determine the minimum and maximum concentrations necessary to manifest a delay of death effect, various concentrations of 95% ethanol were added to 5-ml LB cultures. (A) Wild-type cultures were checked for the minimum concentration that generates an effect. Symbols: untreated (closed squares), 1 μl (closed diamonds, ∼3.5 mM), 2 μl (closed circles, ∼7.0 mM), 3 μl (closed triangles, ∼10.6 mM), 4 μl (open diamonds, ∼14.1 mM), 5 μl (open squares, ∼17.6 mM), 6 μl (open circles, ∼21.1 mM). (B) E. coli exhibits a dose-dependent effect. Cultures were either treated with 0 μl (closed squares), 5 μl (open squares), 10 μl (open diamonds, ∼35.2 mM), 15 μl (open circles, ∼52.8 mM), or 50 μl (open triangles, ∼176 mM). (C) Different concentrations were added to cultures to identify the maximum concentration able to generate an effect. Cultures were treated with 0 μl (closed squares), 50 μl (closed diamonds), 150 μl (open squares, ∼528 mM), 250 μl (open diamonds, ∼880 mM), 350 μl (open circles, ∼1.2 M), or 450 μl (open triangles, ∼1.6 M). Error bars represent the standard errors of replicates (n = 3). Asterisks indicate viable cell counts below the limit of detection (<1,000 CFU/ml).
Incrementally increasing the amount of added ethanol further extends the length of stationary phase (Fig. 1B). Adding 10 μl (∼35.2 mM) delays death by 2 days, while a 15-μl (∼52.8 mM) addition delays death by 3 days. When 50 μl (∼176 mM) of ethanol is added on day 1, cells remain in stationary phase until at least day 7.
This effect is not strain specific. In addition to the W3110 lineage strain ZK126 (22), we tested the following: another W3110 strain, the Keio Collection parental strain BW25113 from the BD792 background (23–24); PFM2 from the MG1655 lineage (25); and 14 strains from the ECOR collection of E. coli natural isolate “reference” strains (26) (see Fig. S1 in the supplemental material). An additional six E. coli strains isolated from human and canine fecal samples (obtained from the laboratory of I. Ehrenreich) were also tested (data not shown). Every strain tested exhibited the effect except for two strains from the ECOR collection (ECOR-37 and ECOR-40); one showed no effect (Fig. S1G), and one showed extreme sensitivity to the presence of ethanol (Fig. S1I).
To determine the maximum concentration for which the ethanol effect is observed, doses of 50 to 450 μl (∼1.6 M) were added on day 1 of incubation (Fig. 1C). Dosages of 50 and 150 μl (∼528 mM) delay death for 7 days, while a dose of 250 μl (∼880 mM) prolongs stationary phase until after day 10 in batch culture. Cultures receiving 350 μl (∼1.2 M) or 450 μl of ethanol do not exhibit the delay of death phenotype, with cells receiving the highest dose showing a significant loss of viability starting on day 2. This shift from beneficial to harmful concentrations of ethanol likely reflects the balance between the alcohol potentially serving as a beneficial signaling molecule, rather than a toxic denaturant (27).
Further, this effect appears to be phase-specific phenomenon. When alcohol is added to cultures on day 0 (lag phase), day 1 (stationary phase), or day 2 (stationary phase), E. coli populations exhibit the delayed death phenotype. However, if alcohol is added after the transition into death phase (day 3 or later) there is no phenotypic difference compared to untreated cultures (data not shown).
Since increasing concentrations of ethanol cause dose-dependent delays in death, we next sought to verify that the cells present in the medium were responsible for the depletion of ethanol. To test this, we compared the extracellular concentrations of ethanol in the presence or absence of active cultures over time (Fig. 2). A portion (5 μl) of ethanol was added on day 1, and the ethanol concentration was determined over 4 days. By day 3, ethanol is almost entirely depleted from the culture medium in the presence of cells (Fig. 2), correlating with the transition of the population into death phase. In the control culture lacking E. coli cells, the decrease of ethanol in the culture medium is much slower, with concentrations of ∼10 mM still present after 4 days of incubation.
FIG 2.
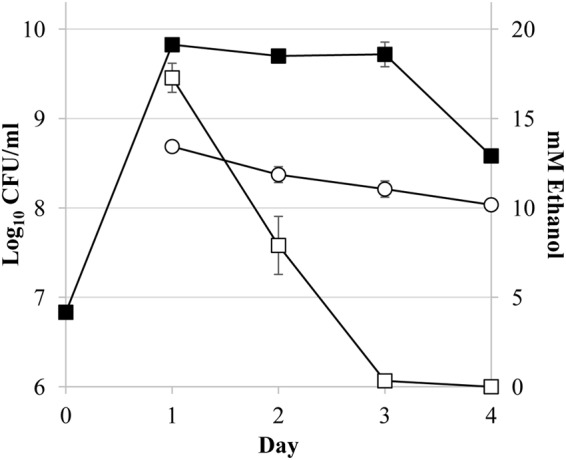
Depletion of ethanol over time in active cultures. Viable cell counts of wild-type cells over 4 days in LB medium are shown (closed squares). On day 1, 5 μl of 95% ethanol (∼17.6 mM) was added to 5-ml cultures, or no cell controls were prepared. The ethanol concentration measurements (mM) are indicated for ZK126 cultures (open squares) and no-cell controls (open circles). Ethanol is depleted from cultures immediately prior to the onset of death phase and at a higher rate than with the no-cell control. Error bars the represent standard errors of replicates (n = 3).
Maintenance of a minimum ethanol concentration is required to prolong stationary phase.
Given that greater doses of ethanol lead to a longer delay of entry into death phase, we next determined whether small daily doses of ethanol, ensuring that ethanol is always present in the medium, would similarly increase the length of stationary phase. Starting on day 1, 5 μl of ethanol was added daily to wild-type cultures (Fig. 3), which prolonged stationary phase for more than 9 days. In contrast, a single addition of 5 μl of ethanol resulted in only a single day of delayed death (Fig. 1A and 3). Conversely, cells treated with a one-time dose of 50 μl of ethanol, corresponding to more than the total amount added over the 9-day time course, die sooner than those treated with smaller daily amounts (Fig. 1C). Further, cells treated with later administrations of ethanol, such as on day 2, do show an “alcohol effect” as long as populations are still in stationary phase when the alcohol is added (data not shown). Populations are not affected by a dose of ethanol after the onset of death phase.
FIG 3.
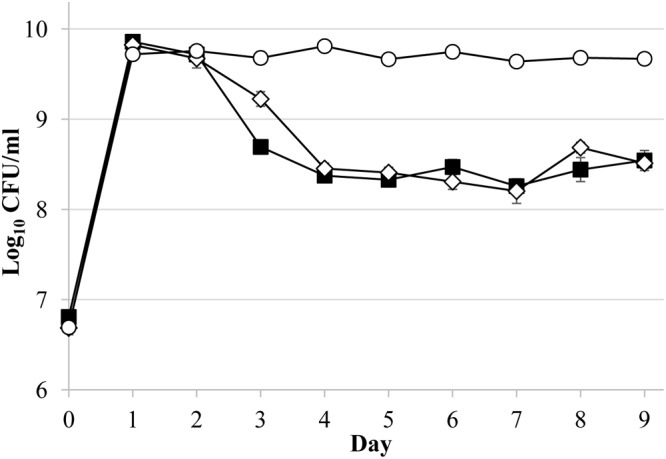
Daily ethanol addition results in a prolonged stationary-phase effect. Ethanol (5 μl) was added daily (open circles) to ZK126 cultures in LB medium, and viable cell counts (CFU/ml) were measured. Untreated cultures are indicated by closed squares, and cultures treated with a single dose on day 1 are represented as open diamonds. Error bars represent the standard errors of replicates (n = 3).
Death is delayed in the presence of other short, straight-chain alcohols between two and six carbons in length.
Cultures treated with equimolar concentrations of either 1-propanol, 1-butanol, or 1-pentanol are able to delay death phase of E. coli for 1 to 2 days longer than with the addition of ethanol (Fig. 4; Table 1). While the addition of an equimolar amount of 1-hexanol causes loss of viability, the addition of half the dose prolongs the stationary phase, although not to the same extent as other short-chain alcohols. The addition of 1-heptanol and 1-octanol proved lethal at the concentrations tested. Surprisingly, neither the addition of equimolar amounts of methanol nor the addition of equimolar amounts of 2-propanol induces the effect (Fig. 4; Table 1). Similarly, the addition of other diols and amines that have structural similarity to ethanol, such as ethanolamine, 3-amino-1-propanol, and ethylene glycol, does not cause the effect (Table 1).
FIG 4.
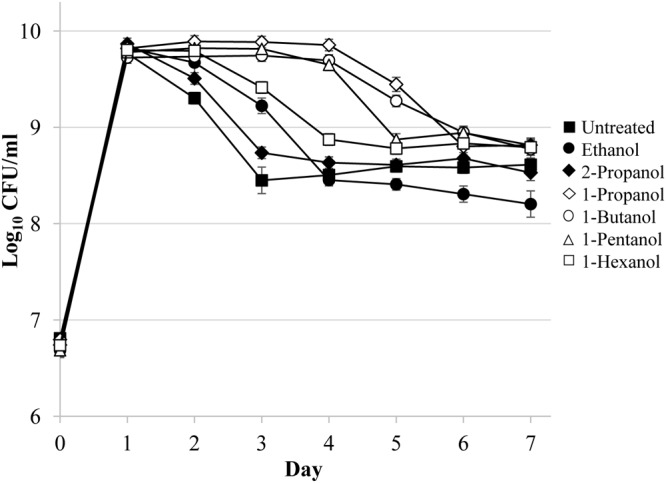
Other n-alcohols can produce the alcohol effect. Viable counts of cultures determined with either 17.6 mM 1-propanol (open diamonds), 1-butanol (open circles), or 1-pentanol (open triangles) or 5 μl of (8.0 mM) 1-hexanol (open squares) added to 1-day-old cultures are shown. 2-Propanol (closed diamonds) had no effect compared to untreated cultures (closed squares). Error bars represent the standard errors of replicates (n = 3).
TABLE 1.
Chemicals tested for delayed death effect
| Compound | Chemical formula | Concn (mM) | Phenotypea |
|---|---|---|---|
| Methanol | CH3OH | 17.6–24.7 | – |
| Ethanol | CH3CH2OH | 17.6 | + |
| 1-Propanol | CH3(CH2)2OH | 17.6 | + |
| 2-Propanol | CH3CHOHCH3 | 13.1–17.6 | – |
| 1-Butanol | CH3(CH2)3OH | 17.6 | + |
| 1-Pentanol | CH3(CH2)4OH | 17.6 | + |
| 1-Hexanol | CH3(CH2)5OH | 8.0–17.6b | +/– |
| 1-Heptanol | CH3(CH2)6OH | 4.8–17.6c | – |
| 1-Octanol | CH3(CH2)6CH2OH | 3.8–17.6d | – |
| Ethanolamine | H2NCH2CH2OH | 6.6–17.6c | – |
| 3-Amino-1-propanol | H2NCH2CH2CH2OH | 5.2–17.6c | – |
| 4-Amino-1-butanol | H2N(CH2)4OH | 4.3–17.6c | – |
| Ethylene glycol | HOCH2CH2OH | 7.2–17.6 | – |
| 1,3-Propanediol | HOCH2CH2CH2OH | 5.5–17.6 | – |
+, has delayed death; –, no delay; +/–, has delayed death at lower but not equimolar (∼17.6 mM) concentrations.
Lethal at equimolar (∼17.6 mM) concentrations and also has an effect at lower concentrations.
Lethal at equimolar concentrations and has no effect at lower concentrations.
Lethal at all concentrations tested.
The “alcohol effect” is modulated by RpoS activity.
Previously, Vulić and Kolter (11) reported that the ethanol effect is a stationary phase-specific phenomenon and depends on the activity of RpoS, which either directly or indirectly regulates the expression of approximately 23% of the genome (28). They showed that a rpoS-null mutant strain shows no effect when treated with ethanol. We sought to verify and expand upon these findings by testing for the effect using a more frequently sampled time course with the isogenic rpoS-null mutant strain ZK1000 (29) where a kanamycin resistance gene cassette has replaced the rpoS gene, completely disrupting its function.
The rpoS-null strain was treated with ethanol, and viable cell counts were determined several times a day over 3 days of incubation. The rpoS-null mutant still shows a prolonged stationary-phase effect, although it is shorter than for wild-type strains (Fig. 5), suggesting that while not essential, RpoS modulates the effect. This was likely previously unreported (11) because the effect’s induction occurs at an earlier time for the rpoS mutant, and the timing for this event would have been missed due to less frequent sampling compared to this study. Further, an rpoS mutant with reduced activity exhibits a robust phenotype similar to that exhibited by the wild-type strain (data not shown).
FIG 5.
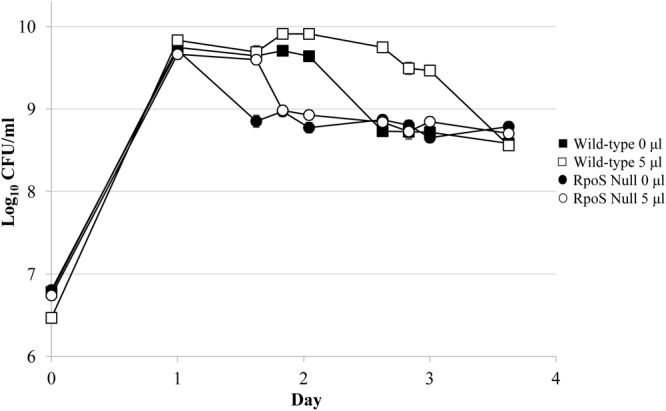
An rpoS-null mutant strain exhibits the alcohol effect. The rpoS-null strain ZK1000 shows a small effect when 5 μl of ethanol (open circles) was added on day 1 of growth compared to untreated cultures (closed circles). Wild-type cultures treated (5 μl of ethanol; open squares) or not treated (closed squares) served as controls. Error bars represent the standard errors of replicates (n = 3).
Alcohol dehydrogenase activity is not essential to prolong the stationary phase.
One possible mechanism of the “alcohol effect” is the metabolism of ethanol as a carbon source (21). To address this, survival patterns were determined for mutants lacking both of E. coli’s alcohol dehydrogenase genes, adhE and adhP (30–31). Cultures of mutant cells were treated with various amounts of either ethanol or 1-butanol, an alcohol that shows an effect in wild-type cells but is not metabolized (32–34), and viable cell counts were determined over 4 days (Fig. 6).
FIG 6.

An alcohol dehydrogenase double-mutant strain exhibits the alcohol effect. To determine whether ethanol is being metabolized as a carbon source, an alcohol dehydrogenase adhE adhP double-mutant strain (open symbols) or wild-type cells (closed symbols) were tested. Double-mutant cultures were treated with either no ethanol (open squares) or 5-μl (17.6 mM; open diamonds) or 50-μl (open circles) ethanol additions on day 1 to LB cultures, and viable cell counts (CFU/ml) were determined. In addition, 17.6 mM 1-butanol (open triangles) was added on day 1. Error bars represent the standard errors of replicates (n = 2).
Without treatment (Fig. 6), the alcohol dehydrogenase double mutant behaves like the wild-type strain, experiencing 2 days in stationary phase. While the addition of 5 μl ethanol on day 1 causes only a slight effect, the addition of 50 μl of ethanol causes a prolonged extension of stationary phase (Fig. 6). Cultures treated with equimolar amounts of 1-butanol also show an effect, further indicating that alcohol catabolism is not required to cause a delay in the onset of death phase.
Enzymes involved in the glycolate degradation pathway may help modulate the alcohol effect.
Given that the essential causal gene(s) involved in the alcohol effect remains unknown after testing several hypotheses, we next chose to analyze the transcriptome of the E. coli populations in the presence or absence of alcohol treatment by transcriptome sequencing (RNA-seq). Wild-type cultures were either left untreated or treated with 10 μl (∼35.2 mM) ethanol after 24 h. After 1 h of additional incubation, mRNA was prepared from culture samples and submitted for sequencing. Transcriptome data were normalized, and pairwise comparisons were made between duplicate treated and untreated populations (35).
An analysis of the most highly induced genes showed that three of the four genes identified are involved in the glycolate/glyoxylate degradation pathway: glxR, glcD, and gcl (21) (Table 2; Tables S1 and S2). To determine whether any of these genes modulate the alcohol effect, single-gene knockout mutations were constructed by bacteriophage P1 transduction. When treated with 5 μl of ethanol, all three single mutants have longer delayed death phenotypes compared to wild-type cells (Fig. 7B; Fig. S2), though to various degrees. Both of the untreated glxR and glcD mutant strains only remain in stationary phase for 1 day before entering death phase (Fig. 7A; Fig. S2A and B); the untreated gcl mutant strain survives 2 days of stationary phase before dying, similar to the wild type (Fig. 7A; Fig. S2C). Interestingly, the gcl-null mutant also shows a severely reduced butanol effect compared to the wild type, while the glxR- and glcD-null populations are unaffected with respect to the addition of butanol (Fig. 7C).
TABLE 2.
Most significantly upregulated genes 1 h after ethanol addition
| Gene | Synonyms | Description | Fold change | P |
|---|---|---|---|---|
| glxR | glxB1, ybbQ | Tartronate semialdehyde reductase 2 | 36.25 | 7.10e–57 |
| hyi | ybbG, gip | Hydroxypyruvate isomerase | 19.83 | 1.28e–36 |
| gcl | Glyoxylate carboligase | 12.05 | 2.88e–75 | |
| glcD | yghM, gox | Glycolate dehydrogenase, putative FAD-linked subunit | 11.46 | 2.14e–94 |
FIG 7.
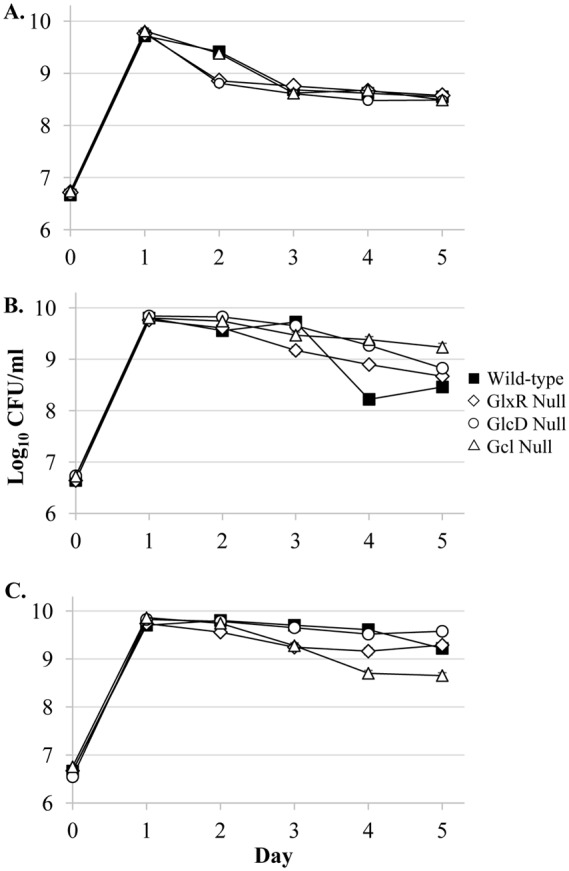
Single-gene null mutations of the glycolate degradation pathway genes glxR, glcD, and gcl cause altered delayed-death phenotypes. RNA-sequencing analysis (Table 2; Tables S1 and S2) showed significant upregulation of three genes involved in the glycolate/glyoxylate degradation pathway: glxR, glcD, and glxR. To test whether these genes play a role in the causing the “alcohol effect,” single-mutant knockouts were made of each (19). Wild-type populations (closed squares), GlxR-null populations (open diamonds), GlcD-null populations (open circles), and Gcl-null populations (open triangles) were examined. (A) Untreated cultures; (B) cultures treated with 5 μl (∼17.6 mM) ethanol; (C) cultures treated with ∼17.6 mM 1-butanol. Error bars represent the standard errors of replicates (n = 3).
DISCUSSION
We show that the addition of ethanol, and several other straight-chain alcohols, causes a delay in the onset of death phase, leading to a prolonged stationary phase during batch culture incubation in a rich medium. Increasing concentrations of ethanol delay death in a dose-dependent manner until toxic levels are reached (Fig. 1B and C). This dose dependency is likely caused by the presence of ethanol above a minimum threshold (Fig. 1A). We also show that ethanol is depleted at a higher rate in the presence of cells (Fig. 2) and that once ethanol is no longer detected in the culture medium, populations enter death phase. We posit that the observed faster depletion of ethanol is due to the cells themselves acting as a sink for the alcohols which can readily pass through the cell membrane (36); apparently, this occurs at a rate faster than simple evaporation. We initially proposed several models to explain the “alcohol effect”: (i) alcohol is being metabolized as a carbon source (21), (ii) the presence of ethanol or other alcohols in cultures triggers a stress response, or (iii) alcohols are serving as signaling molecules.
E. coli possesses a natural ethanol degradation pathway (21), so while it is plausible that ethanol could be utilized as a carbon source whose metabolism might lead to a delay of entry into death phase, data from several experiments argue against this model. We show that the alcohol dehydrogenase double-mutant strain displays the alcohol-induced delayed death phenotype (Fig. 6). Also, treatment with 1-butanol (Fig. 6), which cannot be catabolized as a nutrient by the E. coli strains used in this study (Table 3) (32–34), still induces the effect. Together, these data suggest that the metabolism of ethanol, or any alcohol, as a nutrient is not responsible for the delayed entry into death phase.
TABLE 3.
E. coli strains used in this study
| Strain | Relevant genotype/origin | Nomenclature | Source or reference(s) |
|---|---|---|---|
| ZK126 | W3110 ΔlacU169 tna-2 | Wild type | 22 |
| PFM2 | MG1655 ΔpyrE748 rph+ IS186 | Wild type | 25 |
| BW25113 | BD792 rrnB3 ΔlacZ4787 hsdR514 Δ(araBAD)567 Δ(rhaBAD)568 rph-1 | Wild type | 23, 24 |
| ZK1000 | ZK126 rpoS::Kan | RpoS null | 29 |
| SF2602 | ZK126 adhE adhP::Kan | Double mutant | This study |
| SF2603 | ZK126 gcl::Kan | Gcl null | This study |
| SF2604 | ZK126 glcD::Kan | GlcD null | This study |
| SF2605 | ZK126 glxR::Kan | GlxR null | This study |
| ECOR-04 | Human; Iowa | Natural isolate | 26 |
| ECOR-13 | Human; Sweden | Natural isolate | 26 |
| ECOR-14 | Human; Sweden | Natural isolate | 26 |
| ECOR-15 | Human; Sweden | Natural isolate | 26 |
| ECOR-28 | Human; Iowa | Natural isolate | 26 |
| ECOR-29 | Kangaroo rat; Nevada | Natural isolate | 26 |
| ECOR-37 | Marmoset; Washington (zoo) | Natural isolate | 26 |
| ECOR-38 | Human; Iowa | Natural isolate | 26 |
| ECOR-40 | Human; Sweden | Natural isolate | 26 |
| ECOR-51 | Human infant; Massachusetts | Natural isolate | 26 |
| ECOR-62 | Human; Sweden | Natural isolate | 26 |
| ECOR-63 | Human; Sweden | Natural isolate | 26 |
| ECOR-68 | Giraffe; Washington (zoo) | Natural isolate | 26 |
| ECOR-71 | Human; Sweden | Natural isolate | 26 |
We next speculated that the presence of alcohol in the culture medium may trigger a protective stress response through one or more alternative sigma factors. The “alcohol effect” is a stationary phase-specific phenomenon (11), and RpoS is a global regulator of E. coli’s stress response (28, 37–41). In the absence of added alcohol, the rpoS-null mutant strain behaves differently than the wild-type ZK126 strain. The rpoS mutant strain has a shorter stationary phase of only 1 day in untreated medium, compared to 2 days for the wild type (Fig. 5), and enters death phase before day 2. However, the rpoS-null strain still exhibits the effect (Fig. 5), suggesting that while RpoS may modulate stationary-phase activities, it is not essential to prolong the stationary phase in the presense of added alcohol.
Previous reports have shown that ethanol stresses the E. coli cell envelope by increasing its permeability, affecting the growth rate (17–20). A key regulator involved in the response to envelope stress, including that caused by ethanol, is the alternative sigma factor RpoE (42–43). While other work has described a role for RpoE in responding to the increased membrane stress caused by ethanol, the vast majority of the studies describing RpoE’s role in alcohol stress involve doses of ethanol that are significantly higher (∼2.5 to 5%) than those used in this study (∼0.1 to 0.2%) (16, 17, 43). Further, no significant changes in the gene expression of members of the RpoE regulon directly regulated by the sigma factor were detected in the RNA-seq experiment.
In addition to 1-butanol, other noncatabolizable straight-chain alcohols also cause the delayed-death-phase effect (Fig. 4). Straight-chain n-alcohols containing between two and six carbons lead to the alcohol effect. Equimolar concentrations of straight-chain alcohols containing between three and five carbons not only induce the effect but also result in the cells staying an additional day in stationary phase. The increased amphiphilicity of these longer alcohols could result in greater membrane permeability (18), allowing more alcohol to enter the cells, causing a prolonged effect. 1-Hexanol, while lethal at higher concentrations, causes an effect when added at a half dose. The intermediate dose response of 1-hexanol likely represents a balance between the positive life extension alcohol effect versus the alcohol’s toxicity. Further, the 1-carbon alcohol, methanol, and the branched alcohol, 2-propanol, do not induce the effect, while 1-heptanol either causes no effect or proves lethal to cells. We conclude that to cause the death delay phenotype, alcohols require straight-chain structures between two and six carbons in length. The fact that 2-propanol does not induce the effect, whereas 1-propanol does, further supports a model where the alcohol may be directly interacting with some protein as a signaling molecule and the presence of the hydroxyl group at the 2-position interferes with the interaction.
Other groups have previously performed transcriptomic analyses of ethanol treatment to E. coli (16–17), but those studies were done while performing directed evolution to yield strains with increased ethanol tolerance. Therefore, significantly higher concentrations of ethanol were added to cultures compared to this study. Here, using lower concentrations, we identify three ethanol-induced genes involved in the glycolate/glyoxylate degradation pathway (21) that alter the delayed death phenotype when knocked out: glxR (tartronate semialdehyde reductase), glcD (glycolate dehydrogenase), and gcl (glyoxylate carboligase) (Table 2; Fig. 7; Tables S1 and S2). These three genes serve as enzymes in the first three steps of the glycolate degradation pathway (21). This same pathway appears to be essential for E. coli to grow on either glycolate or glyoxylate as a sole carbon source and also feeds into gluconeogenesis. These genes were not noted in the previous ethanol transcriptome studies as responding to the addition of excess ethanol (16–17). However, a recent study showed that genes involved in the glycolate degradation pathway, including all three of the genes identified here, are upregulated in the presence of butanol (44). Here, all three null strains show various degrees of increased delayed death compared to the wild type, whereas only the gcl-null populations show a reduced, but still present, alcohol effect with equimolar amounts of butanol (Fig. 7C). Given that butanol cannot be catabolized by our parental E. coli strain (only by nonengineered E. coli strains [32–34]), these data support a model where Gcl plays an important role in modulating the alcohol effect. Both glxR and glcD mutants, like the rpoS-null strain, also exhibit shortened stationary-phase lengths in the absence of added ethanol (Fig. 7A), suggesting that both of these genes contribute to the fitness of untreated stationary-phase populations.
A null mutant strain of the second most highly upregulated gene in the transcriptomic analysis, hyi (Table 2), showed no phenotypic difference compared to wild-type cultures (data not shown). This is probably due to the fact that, while coexpressed with the glxR and gcl genes in the same operon, hyi does not act in the glycolate/glyoxylate degradation pathway (21).
The “alcohol effect” is not a strain-specific phenomenon. It occurs in the vast majority of laboratory and natural isolate E. coli strains that we have tested (Fig. S1), as well as in other genera associated with humans, including strains of Pseudomonas, Streptococcus, and Klebsiella (data not shown); interestingly, non-human-associated Vibrio and Shewanella strains show no effect (data not shown). Ethanol is present in many different natural environments, including the human gastrointestinal tract (45–46), and multiple different natural isolates of E. coli strains have ethanol oxidation pathways (47). Recent studies looking at the change in microbial diversity due to alcohol consumption in mouse and human models show dramatic shifts in microbial diversity profiles (48–50). However, little information is availabile distinguishing how microbes in the gut are affected by alcohols present from bacterial fermentation versus from excess human host consumption.
In our experimental system, it appears likely that ethanol and other straight-chain alcohols containing between two and six carbons may be serving primarily as signaling molecules, ultimately delaying the onset of death phase in cell populations. Given the structual similarity between alcohols and glycolate, it is possible that the alcohol is allosterically mimicking glycolate to bind to the GlcC regulator (51), causing a deprepression of the glycolate degradation pathway. This could, in turn, result in an increase in gluconeogenesis, providing additional carbon and energy sources available through increased scavenging of detrital nutrients during stationary phase and thus delaying death.
Despite being one of the best-studied organisms, we still do not understand what triggers populations of E. coli to die. This study has the potential to shed light on the mechanisms by which signaling molecules can impact community dynamics. A better grasp of the mechanisms underlying the “alcohol effect” may also improve our understanding of the interplay between ethanol and bacteria, whether that is in the laboratory or in natural environments like the poorly understood host-microbe dynamics of the human gastrointestinal tract.
MATERIALS AND METHODS
Bacterial strains, culture media, and growth conditions.
E. coli strains used in the study are listed in Table 3, with most experiments performed using the E. coli K-12 strain ZK126, derived from the W3110 lineage (22). Other strains discussed included Pseudomonas aeruginosa PA-14, Shewanella oneidensis MR-1, Vibrio harveyi B392, Streptococcus spp., and Klebsiella spp. (laboratory isolates from human fecal samples). Unless stated otherwise, cultures were inoculated from frozen 20% glycerol stocks into 5 ml of LB (Lennox) medium (Difco) in 18- by 150-mm borosilicate test tubes (Thermo Fisher) and incubated at 37°C with aeration using TC-7 rolling drums (New Brunswick Scientific, Edison, NJ). Cells from overnight cultures were then inoculated at 1:1,000 (vol/vol) into 5 ml of LB culture. After 24 h of incubation, alcohols (Koptec; Sigma) were added to different specified concentrations. For experiments requiring many replicate cultures, one large volume of LB medium was inoculated from the overnight culture and then aliquoted into test tubes. Other strains tested include the rpoS-null mutant strain ZK1000 of the ZK126 lineage (29), strain PFM2 (25) of the MG1655 lineage, strain BW25113 (the parent of the Keio Collection of gene knockouts [23, 24]), and the E. coli Reference Collection (ECOR) strains (26) listed in Table 3. The isogenic adhE adhP::Kan (SF2602) double-mutant strain and the gcl::Kan (SF2603), glcD::Kan (SF2604), and glxR::Kan (SF2605) single-mutant strains were constructed by a combination of P1 transduction and FLP-FRT recombination to remove the kanamycin gene cassette interrupting the genes of interest (adhE), as described previously (24).
Monitoring cell growth, cell survival, and culture pH.
Viable cell counts were determined by serial dilution at indicated time points and plating on LB agar (52); the limit of detection for this method of titering is ≥1,000 CFU/ml (52). Where appropriate, the pH was measured using 6.0 to 10.0 range pH paper with ∼0.3-pH unit increments (EMD Chemicals, La Jolla, CA).
Ethanol colorimetric concentration assay.
Ethanol concentration in cultures was measured using a colorimetric assay (BioVision, Inc., Milpitas, CA) according to the manufacturer’s instructions, using a standard curve generated with known concentrations of ethanol ranging from 2 to 20 mM. Briefly, in a sterile biological hood, samples of culture medium were obtained and resuspended in ethanol assay buffer. Then, 50-μl samples were transferred to a 96-well plate with a lid (Corning). Next, 46 μl of enzyme assay buffer, 2 μl of enzyme mix, and 2 μl of enzyme probe were added, and the plates were incubated at room temperature for 1 h with no light exposure. After incubation, the optical density at 570 nm was determined for each sample and compared to the standard curve. As appropriate, several different sample volumes were obtained to ensure that the ethanol concentrations were within the linear range of the assay.
RNA-sequencing preparation, sequencing, and analysis.
E. coli K-12 strain ZK126 was inoculated into 5 ml of LB medium from frozen a 20% glycerol stock and grown at 37°C as described above. After 24 h of incubation, 10 μl (∼35.2 mM) of 95% ethanol (Koptec; Sigma) was added to duplicate cultures, with an untreated pair of cultures serving as the negative control. Treated and untreated cultures were incubated for an additional hour. The mRNA was then purified from 0.5 ml of each bacterial culture using an RNeasy minikit (Qiagen). Samples were sent to BGI Americas Corporation (Cambridge, MA) for library preparation and sequencing using an Illumina HiSeq 4000 platform. The 100-bp paired-end reads were aligned to the E. coli K-12 W3110 genome. Normalized counts (in transcripts per million), accounting for total number of reads and gene size, were calculated using SAMtools, Bowtie2, TopHat2, and HTSeq (53–55). EdgeR (Bioconductor) software was used to analyze differential expression between treatments (35).
Supplementary Material
ACKNOWLEDGMENTS
We thank Jonathan Lee, Nicole Ratib, Namita Shroff, Christopher Corzett, Karin Kram, Katie Orban, and Melisa Osborne for helpful suggestions and comments on the manuscript. We are especially grateful to Lacey Westphal and Jonathan Lee for advice on the RNA-sequencing analysis, and we thank Ian Ehrenreich for providing strains.
This study was supported, in part, by U.S. Army Research Office grants W911NF1410318 and W911NF1210321.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02113-18.
REFERENCES
- 1.Finkel SE. 2006. Long-term survival during stationary phase: evolution and the GASP phenotype. Nat Rev Microbiol 4:113–120. doi: 10.1038/nrmicro1340. [DOI] [PubMed] [Google Scholar]
- 2.Navarro Llorens JM, Tormo A, Martínez-García E. 2010. Stationary phase in Gram-negative bacteria. FEMS Microbiol Rev 34:476–495. doi: 10.1111/j.1574-6976.2010.00213.x. [DOI] [PubMed] [Google Scholar]
- 3.Bayles KW. 2014. Bacterial programmed cell death: making sense of a paradox. Nat Rev Microbiol 12:63–69. doi: 10.1038/nrmicro3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aizenman E, Engelberg-Kulka H, Glaser G. 1996. An Escherichia coli chromosomal “addiction module” regulated by 3′,5′-bispyrophosphate: a model for programmed bacterial cell death. Proc Natl Acad Sci U S A 93:6059–6063. doi: 10.1073/pnas.93.12.6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandra B, Ramisetty M, Raj S, Ghosh D. 2016. Escherichia coli MazEF toxin-antitoxin system does not mediate programmed cell death. J Basic Microbiol 56:1398–1402. doi: 10.1002/jobm.201600247. [DOI] [PubMed] [Google Scholar]
- 6.Allocati N, Masulli M, Di Ilio C, De Laurenzi V. 2015. Die for the community: an overview of programmed cell death in bacteria. Cell Death Dis 6:e1609. doi: 10.1038/cddis.2014.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kram KE, Finkel SE. 2014. Culture volume and vessel affect long-term survival, mutation frequency, and oxidative stress of Escherichia coli. Appl Environ Microbiol 80:1732–1738. doi: 10.1128/AEM.03150-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kram KE, Finkel SE. 2015. Rich medium composition affects Escherichia coli survival, glycation, and mutation frequency during long-term batch culture. Appl Environ Microbiol 81:4442–4450. doi: 10.1128/AEM.00722-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Avrani S, Bolotin E, Katz S, Hershberg R. 2017. Rapid genetic adaptation during the first four months of survival under resource exhaustion. Mol Biol Evol 34:1758–1769. doi: 10.1093/molbev/msx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanouchi Y, Pai A, Buchler NE, You L. 2012. Programmed stress-induced altruistic death in engineered bacteria. Mol Syst Biol 8:626. doi: 10.1038/msb.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vulić M, Kolter R. 2002. Alcohol-induced delay of viability loss in stationary phase cultures of Escherichia coli. J Bacteriol 184:2898–2905. doi: 10.1128/JB.184.11.2898-2905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazar S. 1996. Survival of Escherichia coli in stationary phase. PhD thesis Harvard University, Cambridge, MA. [Google Scholar]
- 13.Farrell MJ, Finkel SE. 2003. The growth advantage in stationary-phase phenotype conferred by rpoS mutations is dependent on the pH and nutrient environment. J Bacteriol 185:7044–7052. doi: 10.1128/JB.185.24.7044-7052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rendi R, Ochoa S. 1962. Effect of chloramphenicol in cell-free preparations on protein synthesis of Escherichia coli. J Biol Chem 237:3711–3713. [PubMed] [Google Scholar]
- 15.Kohanski MA, Dwyer DJ, Collins JJ. 2010. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol 8:423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haft RJF, Keating DH, Schwaegler T, Schwalbach MS, Vinokur J, Tremaine M, Peters JM, Kotlajich MV, Pohlmann EL, Ong IM, Grass JA, Kiley PJ, Landick R. 2014. Correcting direct effects of ethanol on translation and transcription machinery confers ethanol tolerance in bacteria. Proc Natl Acad Sci 111:E2576–E2585. doi: 10.1073/pnas.1401853111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horinouchi T, Tamaoka K, Furusawa C, Ono N, Suzuki S, Hirasawa T, Yomo T, Shimizu H. 2010. Transcriptome analysis of parallel-evolved Escherichia coli strains under ethanol stress. BMC Genomics 11:579. doi: 10.1186/1471-2164-11-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ly HV, Longo ML. 2004. The influence of short-chain alcohols on interfacial tension, mechanical properties, area/molecule, and permeability of fluid lipid bilayers. Biophys J 87:1013–1033. doi: 10.1529/biophysj.103.034280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ingram LO. 1976. Adaption of membrane lipids to alcohols. J Bacteriol 125:670–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ingram LO, Vreeland NS. 1980. Differential effects of ethanol and hexanol on the Escherichia coli cell envelope. J Bacteriol 144:481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark D, Cronan J. 7 October 2005, posting date Two-carbon compounds and fatty acids as carbon sources. EcoSal Plus 2005. doi: 10.1128/ecosalplus.3.4.4. [DOI] [PubMed] [Google Scholar]
- 22.Zambrano MM, Siegele DA, Almirón M, Tormo A, Kolter R. 1993. Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science 259:1757–1760. doi: 10.1126/science.7681219. [DOI] [PubMed] [Google Scholar]
- 23.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H, Popodi E, Tang H, Foster PL. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci 109:e2774–e2783. doi: 10.1073/pnas.1210309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ochman H, Selander RK. 1984. Standard reference strains of Escherichia coli from natural populations. J Bacteriol 157:690–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yomano LP, York SW, Ingram LO. 1998. Isolation and characterization of ethanol- tolerant mutants of Escherichia coli KO11 for fuel ethanol production. J Ind Microbiol Biotechnol 20:132–138. doi: 10.1038/sj.jim.2900496. [DOI] [PubMed] [Google Scholar]
- 28.Wong GT, Bonocora RP, Schep AN, Beeler SM, Lee Fong AJ, Shull LM, Batachari LE, Dillon M, Evans C, Becker CJ, Bush EC, Hardin J, Wade JT, Stoebel DM. 2017. Genome-wide transcriptional response to varying RpoS levels in Escherichia coli K-12. J Bacteriol 199:e00755-16. doi: 10.1128/JB.00755-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohannon DE, Connell N, Keener J, Tormo A, Espinosa-Urgel M, Zambrano MM, Kolter R. 1991. Stationary-phase-inducible “gearbox” promoters: differential effects of katF mutations and role of σ70. J Bacteriol 173:4482–4492. doi: 10.1128/jb.173.14.4482-4492.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudolph FB, Purich DL, Fromm HJ. 1968. Coenzyme a-linked aldehyde dehydrogenase from Escherichia coli. J Biol Chem 243:5539–5545. [PubMed] [Google Scholar]
- 31.Shafqat J, Höög J, Hjelmqvist L, Oppermann UCT, Ibáñez C, Jörnvall H. 1999. An ethanol-inducible MDR ethanol dehydrogenase/acetaldehyde reductase in Escherichia coli: structural and enzymatic relationships to the eukaryotic protein forms. Eur J Biochem 263:305–311. doi: 10.1046/j.1432-1327.1999.00323.x. [DOI] [PubMed] [Google Scholar]
- 32.Clark DP, Rod ML. 1987. Regulatory mutations that allow the growth of Escherichia coli on butanol as carbon source. J Mol Evol 25:151–158. doi: 10.1007/BF02101757. [DOI] [PubMed] [Google Scholar]
- 33.Tseng H, Prather KLJ. 2012. Controlled biosynthesis of odd-chain fuels and chemicals via engineered modular metabolic pathways. Proc Natl Acad Sci 109:17925–17930. doi: 10.1073/pnas.1209002109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang K, Sawaya MR, Eisenberg DS, Liao JC. 2008. Expanding metabolism for biosynthesis of nonnatural alcohols. Proc Natl Acad Sci U S A 105:20653–20658. doi: 10.1073/pnas.0807157106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. 2013. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8:1765–1786. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 36.Cooper GM. 2000. The cell: a molecular approach, 2nd ed Sinauer Associates, Sunderland, MA. [Google Scholar]
- 37.Hengge R. 2009. Proteolysis of σS (RpoS) and the general stress response in Escherichia coli. Res Microbiol 160:667–676. doi: 10.1016/j.resmic.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 38.Battesti A, Majdalani N, Gottesman S. 2011. The RpoS-mediated general stress response in Escherichia coli. Annu Rev Microbiol 65:189–213. doi: 10.1146/annurev-micro-090110-102946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hengge-Aronis R. 2002. Signal transduction and regulatory mechanisms involved in control of the σS (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev 66:373–395. doi: 10.1128/MMBR.66.3.373-395.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lange R, Hengge-Aronis R. 1991. Growth phase-regulated expression of bolA and morphology of stationary-phase Escherichia coli cells are controlled by the novel sigma factor σS. J Bacteriol 173:4474–4481. doi: 10.1128/jb.173.14.4474-4481.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Membrillo-Hernández J, Lin EC. 1999. Regulation of expression of the adhE gene, encoding ethanol oxidoreductase in Escherichia coli: transcription from a downstream promoter and regulation by fnr and RpoS. J Bacteriol 181:7571–7579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayden JD, Ades SE. 2008. The extracytoplasmic stress factor, σE, is required to maintain cell envelope integrity in Escherichia coli. PLoS One 3:e1573. doi: 10.1371/journal.pone.0001573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao H, Wei D, Yang Y, Shang Y, Li G, Zhou Y, Ma Q, Xu Y. 2017. Systems-level understanding of ethanol-induced stresses and adaptation in Escherichia coli. Sci Rep 7:44150. doi: 10.1038/srep44150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Si HM, Zhang F, Wu AN, Han RZ, Xu GC, Ni Y. 2016. DNA microarray of global transcription factor mutant reveals membrane-related proteins involved in n-butanol tolerance in Escherichia coli. Biotechnol Biofuels 9:114. doi: 10.1186/s13068-016-0527-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halsted CH, Robles EA, Mezey E. 1973. Distribution of ethanol in the human gastrointestinal tract. Am J Clin Nutr 26:831–834. doi: 10.1093/ajcn/26.8.831. [DOI] [PubMed] [Google Scholar]
- 46.Cederbaum AI. 2012. Alcohol metabolism. Clin Liver Dis 16:667–685. doi: 10.1016/j.cld.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salaspuro V, Nyfors S, Heine R, Siitonen A, Salaspuro M, Jousimies-Somer H. 1999. Ethanol oxidation and acetaldehyde production in vitro by human intestinal strains of Escherichia coli under aerobic, microaerobic, and anaerobic conditions. Scand J Gastroenterol 34:967–973. [DOI] [PubMed] [Google Scholar]
- 48.Dubinkina VB, Tyakht AV, Odintsova VY, Yarygin KS, Kovarsky BA, Pavlenko AV, Ischenko DS, Popenko AS, Alexeev DG, Taraskina AY, Nasyrova RF, Krupitsky EM, Shalikiani NV, Bakulin IG, Shcherbakov PL, Skorodumova LO, Larin AK, Kostryukova ES, Abdulkhakov RA, Abdulkhakov SR, Malanin SY, Ismagilova RK, Grigoryeva TV, Ilina EN, Govorun VM. 2017. Links of gut microbiota composition with alcohol dependence syndrome and alcohol liver disease. Microbiome 5:141. doi: 10.1186/s40168-017-0359-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kosnicki KL, Penprase JC, Cintora P, Torres PJ, Harris GL, Brasser SM, Kelley ST. 2018. Effects of moderate, voluntary ethanol consumption on the rat and human gut microbiome. Addict Biol doi: 10.1111/adb.12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartmann P, Seebauer CT, Schnabl B. 2015. Alcohol liver disease: the gut microbiome and liver cross talk. Alcohol Clin Exp Res 39:763–775. doi: 10.1111/acer.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pellicer MT, Fernandez C, Badía J, Aguilar J, Lin EC, Baldom L. 1999. Cross-induction of glc and ace operons of Escherichia coli attribute to pathway intersection. J Biol Chem 274:1745–1752. doi: 10.1074/jbc.274.3.1745. [DOI] [PubMed] [Google Scholar]
- 52.Kraigsley AM, Finkel SE. 2009. Adaptive evolution in single species bacterial biofilms. FEMS Microbiol Lett 293:135–140. doi: 10.1111/j.1574-6968.2009.01526.x. [DOI] [PubMed] [Google Scholar]
- 53.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trapnell C, Pachter L, Salzberg S. 2009. TopHat: discovering splice junctions with RNA-seq. Bioinformatics 25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anders S, Pyl PT, Huber W. 2015. HTSeq: a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.