

Abstract
Alternative oxidase (AOX) is a non‐mammalian enzyme that can bypass blockade of the complex III‐IV segment of the respiratory chain (RC). We crossed a Ciona intestinalis AOX transgene into RC complex III (cIII)‐deficient Bcs1l p.S78G knock‐in mice, displaying multiple visceral manifestations and premature death. The homozygotes expressing AOX were viable, and their median survival was extended from 210 to 590 days due to permanent prevention of lethal cardiomyopathy. AOX also prevented renal tubular atrophy and cerebral astrogliosis, but not liver disease, growth restriction, or lipodystrophy, suggesting distinct tissue‐specific pathogenetic mechanisms. Assessment of reactive oxygen species (ROS) production and damage suggested that ROS were not instrumental in the rescue. Cardiac mitochondrial ultrastructure, mitochondrial respiration, and pathological transcriptome and metabolome alterations were essentially normalized by AOX, showing that the restored electron flow upstream of cIII was sufficient to prevent cardiac energetic crisis and detrimental decompensation. These findings demonstrate the value of AOX, both as a mechanistic tool and a potential therapeutic strategy, for cIII deficiencies.
Keywords: BCS1L, complex III, GRACILE syndrome, mitochondrial disorder, respiratory chain
Subject Categories: Cardiovascular System; Genetics, Gene Therapy & Genetic Disease
Introduction
Mitochondrial disorders are the most common class of inherited errors of metabolism. However, effective treatments are lacking, and their clinical management remains largely supportive (Pfeffer et al, 2013). In patients with RC cIII (ubiquinol:cytochrome c oxidoreductase) deficiency, mutations in several genes encoding either cIII subunits or assembly factors have been identified. These compromise cIII enzymatic activity and result in a wide variety of clinical manifestations (Fernandez‐Vizarra & Zeviani, 2015). BCS1L mutations are the most common cause of cIII deficiency, with various neonatal and adult phenotypes described worldwide (Fernandez‐Vizarra & Zeviani, 2015), the most severe and prevalent of them being GRACILE syndrome (fetal growth restriction, aminoaciduria, cholestasis, liver iron overload, lactic acidosis, and early death during infancy) (Fellman et al, 1998; Visapää et al, 2002). BCS1L is a mitochondrial inner membrane translocase required for Rieske iron–sulfur protein (RISP, UQCRFS1) topogenesis and incorporation into cIII (Nobrega et al, 1992; Cruciat et al, 1999). Homozygous Bcs1l c.A232G (Bcs1l p.S78G) knock‐in mice bearing the GRACILE syndrome‐analogous mutation recapitulate many of the clinical manifestations, including growth failure, progressive hepatopathy, kidney tubulopathy, and, in a C57BL/6JCrlBomTac background, short survival of 35 days (Levéen et al, 2011; Kotarsky et al, 2012; Rajendran et al, 2016; Purhonen et al, 2017). In the slightly different C57BL/6JCrl substrain, the homozygotes develop the same early manifestations but do not succumb to the early metabolic crisis. This extends their survival to over 150 days (Purhonen et al, 2017) and brings additional later‐onset phenotypes, such as cerebral astrogliosis (Tegelberg et al, 2017).
Under physiological conditions, quinols that transport electrons in the mitochondrial inner membrane are efficiently oxidized by cIII, with electron transfer via cytochrome c and cytochrome c oxidase (complex IV, cIV) to oxygen (Brand, 2010; El‐Khoury et al, 2014). However, plants and some lower organisms, but not mammals, express alternative oxidases (AOXs) that transfer electrons directly from quinols to oxygen without proton translocation. Their main role is to maintain electron flow when the cIII‐cIV segment of the RC is impaired, limiting production of ROS and supporting redox and metabolic homeostasis (McDonald & Vanlerberghe, 2004; El‐Khoury et al, 2014). Ciona intestinalis AOX has been cloned and expressed in human cultured cells (Hakkaart et al, 2006), fruit flies and mice (El‐Khoury et al, 2013; Szibor et al, 2017). In these models, AOX is inert under non‐stressed conditions, most likely because it accepts electrons only when the quinone pool is highly reduced (Hoefnagel & Wiskich, 1998; Castro‐Guerrero et al, 2004), such as under inhibition or overload of cIII or cIV (Dassa et al, 2009). Accordingly, upon inhibition of cIII or cIV by mutations or chemical inhibitors, ectopic AOX can maintain respiration and prevent cell death (Dassa et al, 2009; Fernandez‐Ayala et al, 2009). We set out to test whether AOX expression could prevent the detrimental effects of cIII deficiency in a mammalian model, by restoring electron flow upstream of cIII. To this end, we crossed mice carrying a broadly expressed AOX transgene (Szibor et al, 2017) with the Bcs1l c.A232G mice and assessed disease progression, organ manifestations, and metabolism in the homozygotes with and without AOX expression.
Results
Broadly expressed AOX triples the life span of cIII‐deficient Bcs1l p.S78G mice
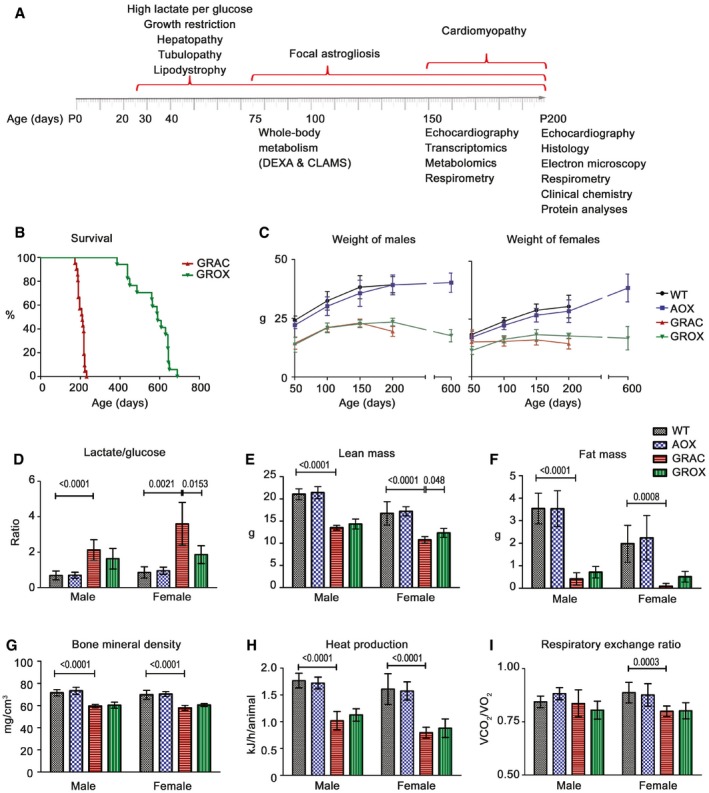
To assess the effect of cIII bypass on the survival and tissue manifestations in cIII‐deficient mice, we bred cohorts of wild‐type and Bcs1l mutant mice with or without a Ciona intestinalis AOX transgene. Hereafter, we will refer to the Bcs1l p.S78G homozygotes as GRAC (as an abbreviation of GRACILE syndrome) mice. The Bcs1l p.S78G homozygotes carrying AOX transgene will be referred to as GROX mice (GRAC + AOX). Figure 1A shows a timeline of the appearance of the previously reported and novel phenotypes in GRAC mice, as well as the assessments included in this study. The GRAC mice reached the criteria of euthanasia between postnatal day 180 (P180) and P220, with median survival to P210 (Fig 1B). In contrast, the GROX mice showed no signs of terminal deterioration or spontaneous deaths at P200 and survived to a median age of 590 days (Fig 1B). To assess whether the extended survival was due to an overall improvement in energy metabolism, we measured growth, whole‐body metabolism, and body composition in young adult mice. The GRAC mice were growth restricted (Fig 1C and E) and had increased lactate‐to‐glucose ratio (Fig 1D), low fat mass (Fig 1F), bone density (Fig 1G), and heat production (Fig 1H) and, in females, low respiratory exchange ratio (Fig 1I). Unexpectedly, AOX had no or only small effect on these parameters (Fig 1C–I), suggesting that the AOX‐mediated extension of survival depended on a tissue or cell‐type specific pathology rather than whole‐body energy metabolism.
Figure 1. AOX expression prolongs the survival of cIII‐deficient Bcs1l p.S78G mice without affecting growth or whole‐body metabolism.
-
ASchematic presentation of the multiorgan manifestations, described in this study or previously, in homozygous Bcs1l c.A232G (GRAC) mice, and the time points of the investigations performed in this study.
-
BSurvival curves of homozygous Bcs1l mutant mice without (GRAC) and with (GROX) alternative oxidase (AOX) expression (n = 18–21/group). The median survival of GRAC mice was 210 days and of GROX mice was 589 days with no gender difference.
-
CWeight of mice from P50 to P200 (n > 10/group) and at P600 (n = 4–6/group).
-
DBlood lactate‐to‐glucose ratio at P200 (n = 8/group).
-
E–GDual‐energy X‐ray absorptiometry (DEXA) analysis (n = 10/group) of (E) lean mass, (F) fat mass, and (G) bone mineral density at P98.
-
H, IIndirect calorimetric measurement (n = 10/group) of (H) heat production and (I) respiratory exchange ratio at P77.
AOX permanently prevents lethal cardiomyopathy and alleviates renal and cerebral manifestations
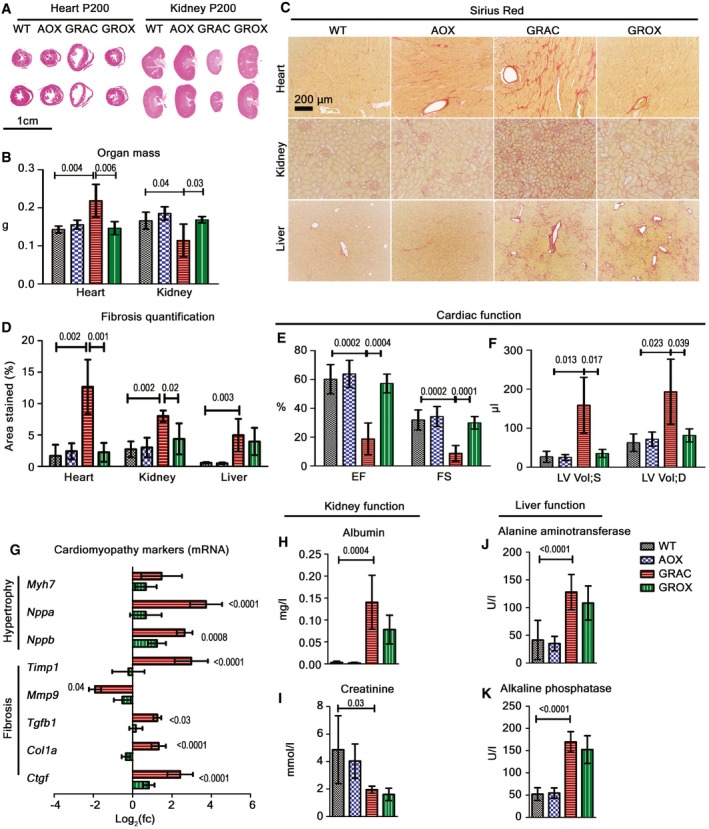
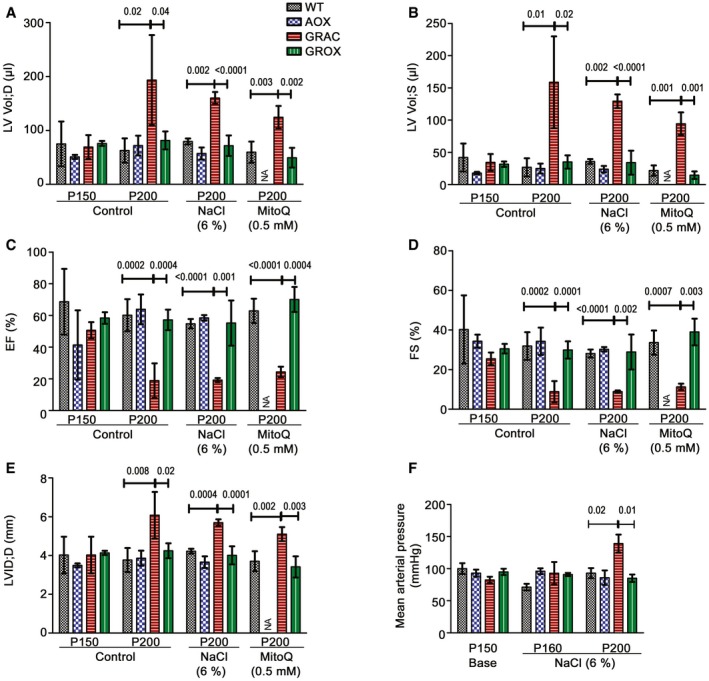
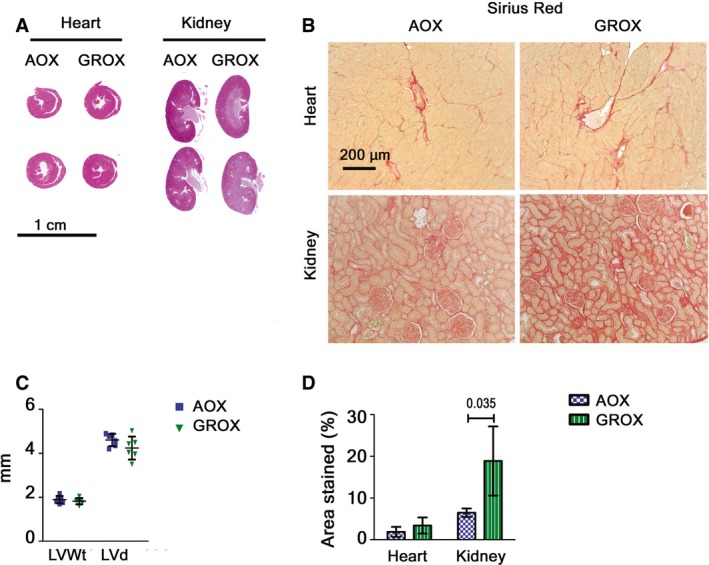
Histopathological analysis of autopsy samples from end stage (P200) mice showed two novel phenotypes not previously reported in studies of younger GRAC mice: cortical kidney atrophy and cardiomegaly with dilated ventricles (Fig 2A and B). Fibrosis was prominent in liver, kidney, and heart (Fig 2C and D). Suspecting cardiomyopathy as the cause of death, we assessed cardiac functions at several time points. Echocardiography showed minimal functional changes at P150 (Fig EV1A–F), but severe dilated cardiomyopathy, focal fibrosis, decreased ejection fraction, and fractional shortening at P200 (Fig 2A and F), indicating end‐stage cardiomyopathy. Strikingly, the GROX littermates had normal heart size (Fig 2A and B), no fibrosis (Fig 2C and D), and overtly normal cardiac function (Fig 2E and F), explaining their extended survival. mRNA expression of key markers for cardiac hypertrophy and fibrosis was significantly altered already in the presymptomatic (P150) GRAC hearts, and these changes were largely prevented in the GROX mice (Fig 2G). A kidney stress test with salt‐enriched (6% w/w) chow starting at P150 had no effect on cardiac function at P200, or on survival, and blood pressure was only increased at the end stage in the salt‐fed mice when compared to P150 baseline (Fig EV1A–F), consistent with a primary cardiomyopathy. Remarkably, the GROX mice had normal‐sized and non‐fibrotic heart throughout their life span, to over P600 (Fig EV2A–D).
Figure 2. AOX prevents lethal cardiomyopathy and progression of renal tubulopathy to kidney atrophy.
-
AHematoxylin–eosin‐stained cross sections of heart and kidney at P200.
-
BWeight of heart and kidney at P200 (n = 4/group).
-
C, DSirius Red staining (C) for fibrosis in liver, heart, and kidney at P200, and (D) quantification of fibrosis in myocardium, kidney cortex, and liver (n = 5–7).
-
E, FEchocardiography data (n = 4–6/group) showing (E) ejection fraction (EF) and fractional shortening (FS). (F) Systolic (LV Vol;S) and diastolic (LV Vol;D) left ventricle volume in mice.
-
GExpression of cardiac hypertrophy and fibrosis‐associated genes in the presymptomatic (P150) heart (n = 6/group).
-
H, I24‐h excretion of (H) albumin and (I) creatinine in urine at P200 (n = 4/group).
-
J, KLiver enzymes (J) alanine aminotransferase (ALT) and (K) alkaline phosphatase (ALP) in plasma at P200 (n = 8/group).
Figure EV1. Salt and antioxidant feeding have no effect on cardiac functions at P200.
-
A–EEchocardiography data from mice fed standard or 6% NaCl‐supplemented chow or drinking water with 0.5 mM mitoQ (n = 4–6/group). Left ventricle volume at (A) diastolic (LV Vol;D) and (B) systolic (LV Vol;S) states, (C) ejection fraction (EF), (D) fractional shortening (FS), and (E) left ventricle internal dimensions (LVID; d‐D).
-
FMean arterial blood pressure in the salt‐fed GRAC mice at baseline (P150), at P160, and at end stage (P200) (n = 4–6/group).
Figure EV2. AOX provides permanent protection from cardiomyopathy and kidney atrophy.
-
ACross‐sections of heart and kidney from old (P600) AOX and GROX mice.
-
BHeart and kidney fibrosis.
-
CLeft ventricle wall thickness (LVWt) and diameter (LVd) measured from heart cross‐sections (n = 6/group).
-
DQuantification of Sirius Red staining (n = 6–7/group, Mann–Whitney U‐test).
Kidneys of GRAC mice showed proximal tubulopathy with fibrosis (Fig 2C and D) and tubular degeneration (decreased tubular mass) at P200 (Appendix Fig S1). In GROX mice, the kidney mass and apparent tubular volume were preserved (Fig 2A and B). On the basis of the proliferation marker Ki67 and apoptosis marker cleaved caspase‐3, this was likely due to decreased apoptosis rather than increased regeneration (Appendix Fig S1I and J). However, AOX had only minor effect on other histological lesions (Appendix Fig S1A–G) or functional parameters: albuminuria, hematuria, and urinary creatinine (Fig 2H and I, Appendix Fig S1H). At P600, the kidneys were severely fibrotic but still of normal size, indicating long‐term protection from tubular atrophy (Fig EV2A, B and D). Surprisingly, AOX had no effect on the liver fibrosis (Fig 2C and D) or the elevated liver enzymes (Fig 2J and K) at end stage. The cause of death of the GROX mice remains unknown, but eventual deterioration due to the progressing kidney and liver disease is an obvious possible explanation.
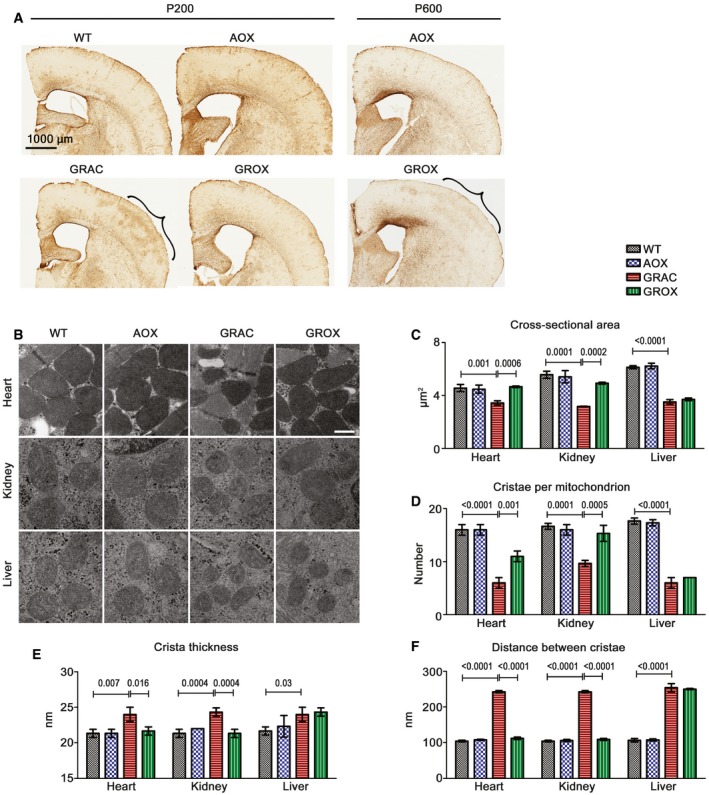
GRACILE syndrome patients have no encephalopathy, but the brains of GRAC mice show peculiar focal astrogliosis in the primary barrel field of the somatosensory cortex (S1BF) (Tegelberg et al, 2017). Staining for glial fibrillary acidic protein (GFAP) showed that the astrogliosis was almost fully prevented by AOX at P200 (Fig 3A).
Figure 3. AOX ameliorates cerebral astrogliosis and maintains mitochondrial ultrastructure in rescued tissues.
-
AGFAP staining for cerebral astrocytes. The barrel field of the primary somatosensory cortex (S1BF) is highlighted with brackets.
-
BMitochondrial ultrastructure in cardiomyocytes, kidney tubular cells, and hepatocytes at P200 as visualized by electron microscopy. Scale bar 1 μm.
-
C–F(C) Average cross‐sectional area of mitochondrion, (D) number of cristae per mitochondrion, (E) crista thickness, and (F) average distance between cristae in mitochondria (n = 3 mice/group).
AOX preserves mitochondrial ultrastructure in rescued tissues
Previous studies have shown disrupted hepatic mitochondrial ultrastructure in younger Bcs1l mutant mice (Levéen et al, 2011; Purhonen et al, 2017). In the P150 GRAC mice, electron microscopy showed abnormal mitochondrial ultrastructure in hepatocytes as well as in kidney tubular cells, and, importantly, in cardiomyocytes already at onset of the cardiomyopathy (Fig 3B). The mitochondria were smaller and contained fewer cristae, which were thicker than in WT mitochondria. AOX fully or partially prevented these changes in cardiomyocytes and kidney tubular cells, but not in hepatocytes, (Fig 3C–F), correlating faithfully with the rescue of tissue pathology.
AOX relieves cardiac metabolic stress and prevents decompensation
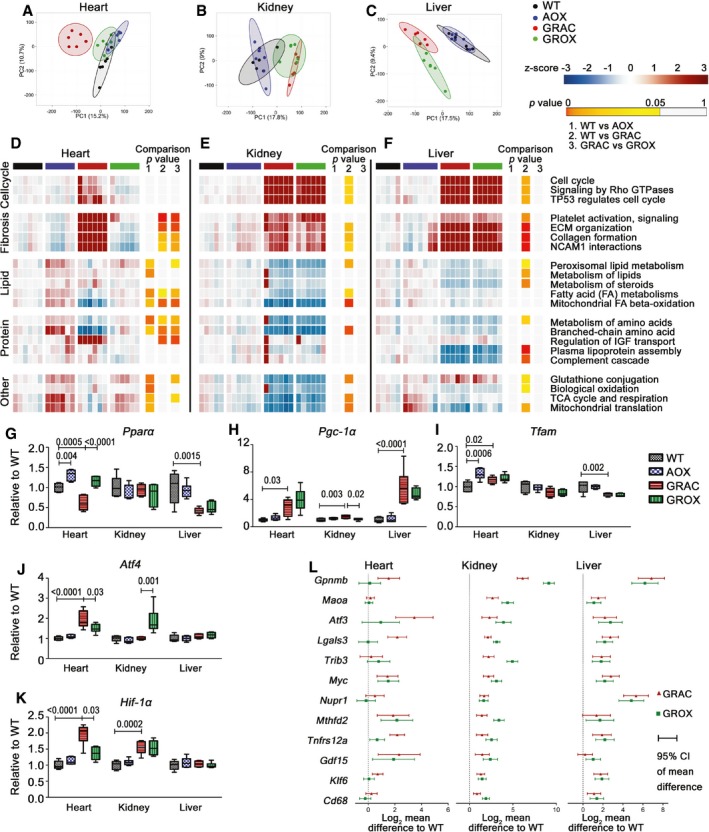
Seeking a mechanism for the remarkably tissue‐specific rescue effects of AOX, we performed transcriptomics and metabolomics at P150, at the onset of the cardiomyopathy. The three affected tissues showed marked global transcriptional changes, including in the GRAC heart despite it being functionally normal at this stage (Fig 4A–C). In correlation with the histological findings, AOX normalized the expression of only few genes in the liver, but of about 25% of dysregulated genes in the kidney and about 50% in the heart (Appendix Fig S2A–C). The most robustly upregulated gene sets were related to extracellular matrix organization, a signature of tissue remodeling and fibrosis (Fig 4D–F, Appendix Table S1), and cell cycle in liver and kidney, but not in the heart (Fig 4D–F) reflecting differential regenerative capacity of these tissues. As expected, energy metabolism‐related gene expression was altered in all three tissues (Fig 4D–F, Appendix Fig S2D). Alternative oxidase almost fully prevented these changes in heart, but not in kidney or liver (Fig 4D–F). Notably, AOX had a significant effect on gene expression in the Bcs1l wild‐type heart, including upregulation of genes related to mitochondrial function (Fig 4D, G, I and Appendix Fig S2D). Expression of the major cardiac metabolic regulator HIF‐1α and the metabolic stress‐inducible transcriptional regulators ATF3 and ATF4 (Kalfon et al, 2017; Quiros et al, 2017) was elevated in GRAC hearts and normalized by AOX (Fig 4J and K), indicating relieved metabolic stress. Upregulation of PGC‐1α, the master regulator of mitochondrial biogenesis, and the mitochondrial transcription factor TFAM, in the GRAC heart suggested an attempt for compensatory mitochondrial biogenesis (Fig 4G–I). However, Western blot analysis using the abundance of RC complex subunits as a proxy showed no significant changes in mitochondrial mass between the groups (Fig EV3I) in any tissue. We previously identified an upregulated set of genes, which we designated cIII stress signature, in P45 Bcs1l mutant livers (Purhonen et al, 2017). This gene set was also highly upregulated in all three GRAC tissues. Despite rescue of kidney tubular mass, AOX amplified the cIII stress signature specifically in this tissue, but not in the heart or liver (Fig 4L).
Figure 4. AOX mitigates cardiac, but not hepatic or renal, metabolic stress‐related gene expression changes.
-
A–CPrincipal component analysis of transcriptome data from (A) heart, (B) kidney, and (C) liver.
-
D–FHeat map visualization of pathway enrichment analysis (Reactome database). Full pathway analysis is provided in Appendix Table S1. Benjamin–Hochberg FDR‐corrected P‐values are color labeled as indicated for the three comparisons.
-
G–KGene expression of major transcriptional regulators of energy metabolism (PPAR‐α, HIF‐1α), mitochondrial biogenesis (PGC‐1α, TFAM), and stress responses (ATF4) in heart at P150 (n = 6/group).
-
LExpression of cIII stress signature genes in heart, kidney, and liver (n = 6/group). Error bars represent 95% confidence interval of mean difference.
Figure EV3. AOX does not affect mitochondrial mass in the affected tissues.
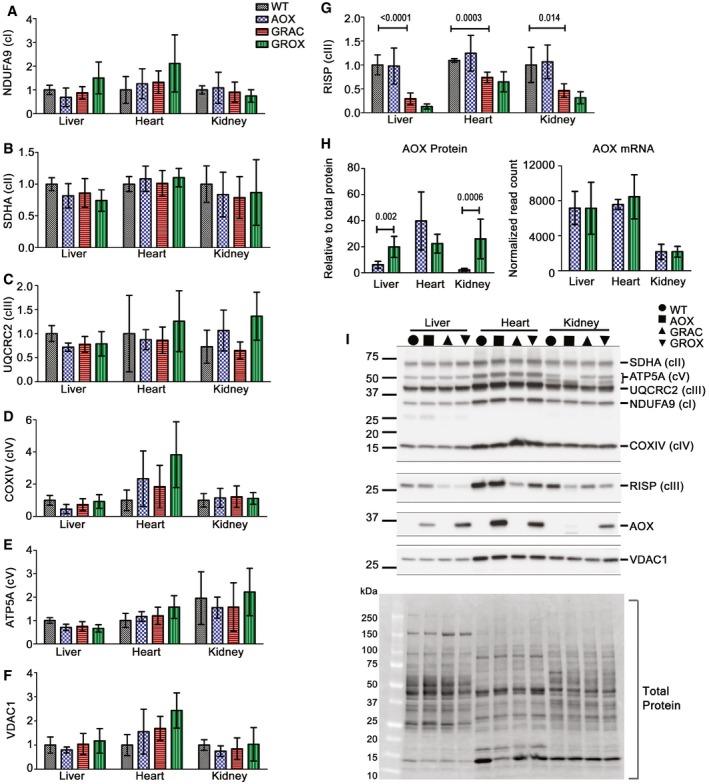
-
A–GQuantification (n = 5–6/group) of mitochondrial respiratory chain subunits by Western blotting with antibodies against (A) NDUFA9 (cI), (B) SDHA (cII), (C) UQCRC2 subunit (cIII), (D) COXIV (cIV), (E) ATP5A (cV), (F) VDAC1, and (G) RISP (cIII).
-
HProtein (n = 6/group) and mRNA expression of AOX (n = 6/group).
-
IA representative Western blot with one sample per group and total protein visualization.
Alternative oxidase is under the strong synthetic CAG promoter and expressed in all tissues in the Rosa26 AOX mice (Szibor et al, 2017). In our mice, AOX mRNA expression was similar in heart and liver and somewhat lower in kidney (Fig EV3H). In total tissue lysates from the AOX mice, the amount of AOX protein was considerably higher in heart than in liver or kidney (Fig EV3H). However, this difference was mainly due to the higher mitochondrial mass in heart, as shown by the mitochondrial loading control VDAC1 and also by most respiratory chain subunits (Fig EV3I). Interestingly, the amount of AOX protein was affected by the Bcs1l mutation (AOX vs. GROX mice) so that the amount in the three GROX tissues was almost identical (Fig EV3H), which essentially rules out that the differences in rescue would be due to different levels of AOX expression.
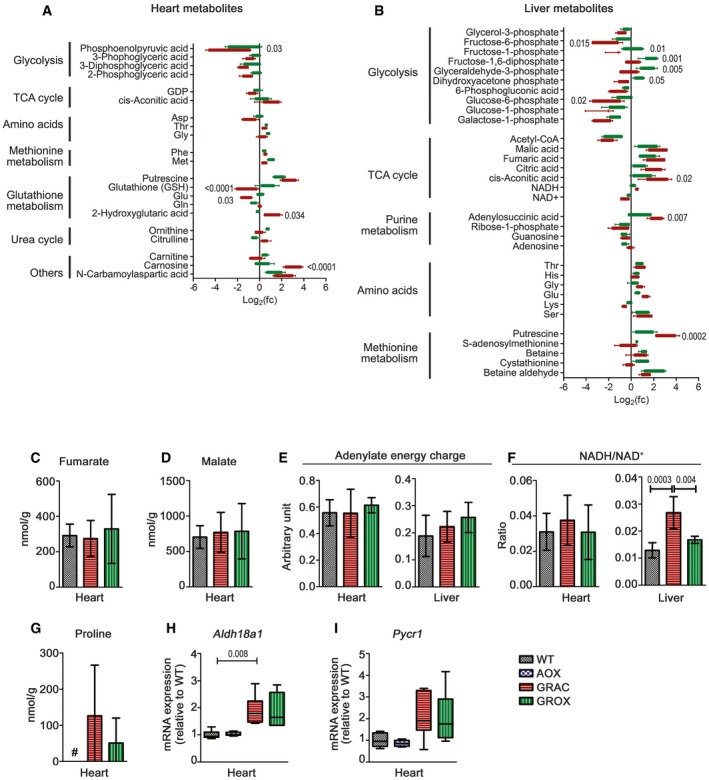
The metabolomics revealed only modest cardiac metabolite changes at the onset (P150) of the cardiomyopathy (Fig 5A, Appendix Table S2). Nevertheless, several three‐carbon glycolytic intermediates were depleted, and these tended to be normalized by AOX (Fig 5A). In line with this, the gene expression of PPAR‐1α, the major driver of fatty acid utilization, was downregulated in GRAC but normal in GROX heart (Fig 4G). The TCA cycle metabolites malate and fumarate, adenylate energy charge, and NADH/NAD+ ratio, which all could be affected by both the cIII blockade and AOX, were not changed in the GRAC or GROX heart tissue (Fig 5C–F). Interestingly, the amino acid proline, which has been long known to accumulate in conditions with lactic academia (Kowaloff et al, 1977), was below detection limit in WT heart but increased to approximately 200 nmol/g in GRAC heart. The proline accumulation was partially prevented by AOX (Fig 5G). Concurrently, glutamate, the biosynthetic precursor of proline, was decreased to about 50% in GRAC mice and normalized to WT level in GROX heart tissue (Fig 5A). The mRNA levels of glutamate γ‐semialdehyde synthetase (P5CS, ALDH18A1) and pyrroline‐5‐carboxylate reductase 1 (P5CR, PYCR1), both driving proline synthesis from glutamate, were upregulated 1.8‐fold and 1.7‐fold, respectively, in the GRAC heart (Fig 5H and I).
Figure 5. AOX has little effect on cardiac energy metabolites but attenuates proline accumulation at onset of disease.
-
ASignificantly altered metabolites (FDR < 0.2; P < 0.05, n = 5/group) in presymptomatic (P150) heart tissue of GRAC (red) and GROX (green) mice.
-
BSignificantly altered metabolites (FDR < 0.2; P < 0.05, n = 5/group) in liver tissue of GRAC (red) and GROX (green) mice.
-
C–FConcentrations of the TCA cycle intermediates (C) fumarate and (D) malate, (E) adenylate energy charge, and (F) NADH/NAD+ ratio (n = 5/group).
-
G–I(G) Proline concentration in heart tissue at P200 (# below detection limit) and mRNA expression of proline synthesis‐related genes (n = 6/group) (H) Aldh18a1 and (I) Pycr1 at P150.
Targeted metabolomics of liver tissue (Fig 5B, Appendix Table S2) showed significant metabolite changes characteristic of failing energy metabolism and glycogen depletion, as previously shown in juvenile mice (Kotarsky et al, 2012). These included decreased hexose phosphates, several other glycolytic intermediates, acetyl‐CoA and NAD+, and elevated TCA cycle intermediates and amino acids, the latter suggesting protein degradation for fuel. NADH/NAD+ ratio was increased (Fig 5F), in line with our previously published data showing decreased hepatic NAD+ (Purhonen et al, 2018). Proline level was not changed in liver (Appendix Table S2). Alternative oxidase had only a minor effect on the hepatic metabolite levels (Fig 5B, E, and F).
AOX restores cardiac mitochondrial respiration
To investigate whether the AOX rescue effect was linked to improved RC assembly and function, we first performed blue native gel electrophoresis (BNGE) analyses. These confirmed the loss of RISP from free cIII dimer (cIII2) and from the cI‐cIII2 supercomplex (SC1) in GRAC tissues (Fig 6A). Surprisingly, the amount of this fully assembled cIII2 was significantly increased in the GROX heart when compared to GRAC (Fig 6B and C). In the GROX liver and kidney, the change was opposite, with lower amount of RISP in cIII2 (Fig 6B and C).
Figure 6. AOX modifies RISP assembly and rescues mitochondrial respiration in tissue‐specific fashion.
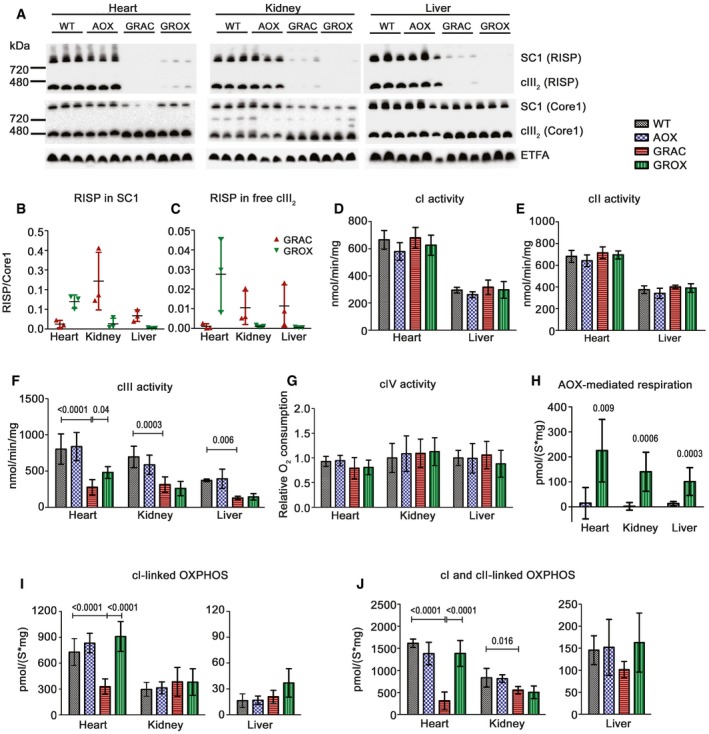
-
ABlue native PAGE analysis of RISP and CORE1 assembled into free cIII2 and supercomplexes (SCs).
-
B, CRISP per CORE1 ratio in (B) SC1 and (C) free cIII2 (n = 3/group).
-
D–FSpectrophotometric assay of (D) cI, (E) cII, and (F) cIII activity in isolated mitochondria at P200 (n = 4–6/group).
-
GcIV activity as measured using an oxygraph (n = 7/group).
-
Hn‐Propyl gallate‐sensitive AOX‐mediated cI‐ and cII‐linked state 3 respiration in AOX and GROX mice (n = 7/group, Mann‐Whitney U‐test).
-
I, JcI‐linked (I), and cI‐ and cII‐linked (J) state 3 respiration in isolated heart, kidney, and liver mitochondria (n = 8/group). We used malate, pyruvate, and glutamate to generate NADH for the cI. Subsequently, we added the cII substrate, succinate, to obtain cI‐ and cII‐linked respiration in the presence of saturating ADP.
Spectrophotometrically measured cIII activity in isolated mitochondria was decreased to < 50% of WT values in all three tissues at P150, a threshold for the appearance of hepatic pathology in juvenile mice (Levéen et al, 2011). Despite that AOX should theoretically not affect cIII activity, it was partially restored in GROX heart mitochondria (Fig 6F), in line with the improved RISP assembly. The activities of cI, cII, and cIV were unchanged in heart and liver (Fig 6D, E and G).
Respirometry confirmed AOX‐mediated respiration in all three GROX tissues at P150, as measured using the AOX inhibitor n‐propyl gallate (nPG). Respiration did not respond to nPG in AOX mice (with wild‐type Bcs1l and normal cIII activity), which indicates that AOX was catalytically active only in the mutants (Fig 6H). The cIII dysfunction did not limit state 3 respiration of liver and kidney mitochondria (Fig 6I and J) when driven by cI‐linked substrates (malate, pyruvate, and glutamate to generate NADH), but clearly did so in the heart (Fig 6I). Further stimulation of respiration through cII by addition of succinate revealed cIII deficiency in all three tissues in GRAC mice, with the most severe effect in the heart (Fig 6J). Remarkably, cI‐ and cII‐linked state 3 respiration (malate, pyruvate, glutamate, and succinate at ADP saturation) were rescued to wild‐type level in GROX heart mitochondria (Fig 6I and J). A similar effect was found in state 3 respiration of heart and liver at P200 (Fig EV4A and B).
Figure EV4. Respiration and ROS damage markers in heart, kidney, and liver tissue at P200.
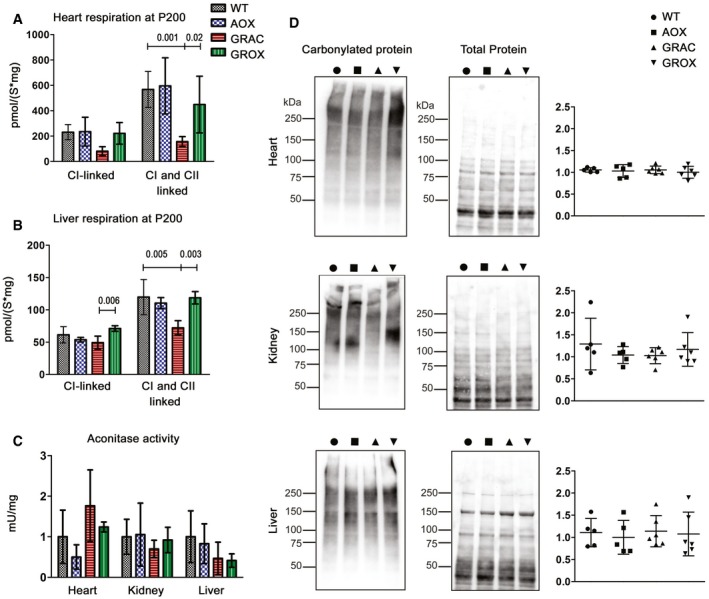
-
A, BCI‐linked, and cI‐ and cII‐linked respiration in (A) heart and (B) liver homogenates at P200 (n = 4–6/group).
-
CAconitase activity, a marker of ROS damage, in mitochondrial fractions from frozen tissues (n = 4/group).
-
DAnalysis of carbonylated proteins in total tissue lysates using on‐filter labeling with 2,4‐dinitrophenylhydrazine and detection with anti‐dinitrophenol antibody. Representative Western blots and quantification are shown.
AOX does not affect ROS damage or defense but normalizes cardiac NO‐related gene expression
Limiting excessive ROS production from the RC has been posited as a major mechanism of action of AOX (El‐Khoury et al, 2014). To assess ROS production in the affected GRAC tissues, we measured hydrogen peroxide (H2O2) emission from isolated mitochondria at P150. The Amplex Red‐peroxidase assay showed that total H2O2 emission was decreased in GROX heart and kidney as compared to GRAC (Fig 7A). However, analysis of gene expression related to ROS defense and damage showed that glutathione synthesis and conjugation were upregulated only in the GRAC liver (Fig 7B) and were not affected by AOX. GRAC hearts showed no upregulation of ROS defense at all (Fig 7B) despite the most severe cIII dysfunction. Total tissue glutathione was not changed in GRAC heart or liver but was increased in both GROX tissues (Fig 7C). Mitochondrial aconitase activity, a sensitive marker of ROS damage to iron–sulfur clusters (Yan et al, 1997), and protein carbonylation (Fig EV4C and D) were not significantly different between the genotypes. Urinary isoprostanes, a marker of systemic oxidative stress (Fig 7D), was elevated in both GRAC and GROX. Immunohistochemical staining for the lipid peroxidation product 4‐hydroxynonenal was increased in all three GRAC tissues, but the increase was not prevented by AOX (Fig 7E and F). Finally, we fed the GRAC mice the mitochondria‐targeted ROS scavenger mitoQ, which has shown beneficial effects in mouse models of cardiac ischemia–reperfusion injury (Adlam et al, 2005). MitoQ feeding started at P150 had no effect on the survival of the mice by P200 or on their cardiac function (Fig EV1A–F).
Figure 7. AOX normalizes ROS production by isolated mitochondria but has little effect on ROS markers in tissues.
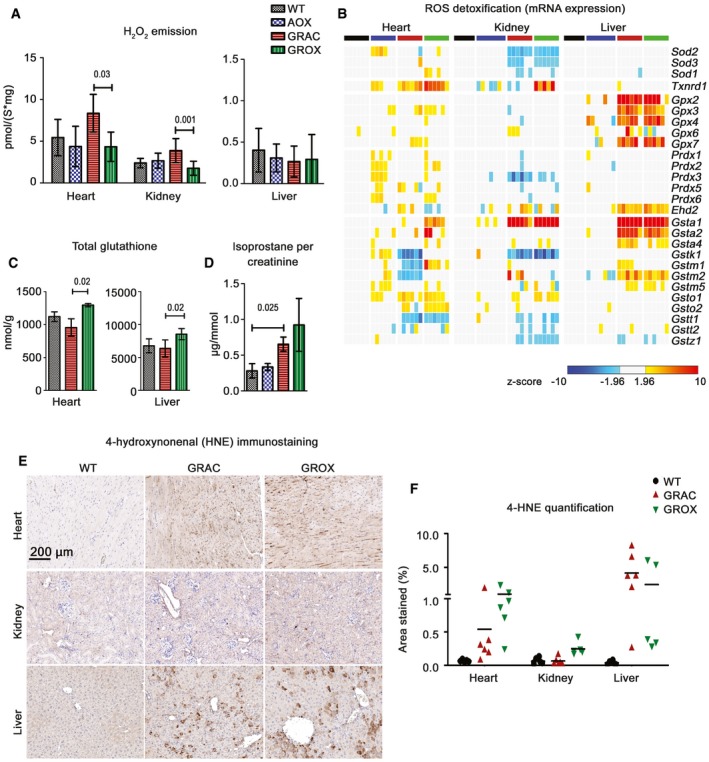
-
AAmplex Red‐based detection of H2O2 emission, a surrogate for respiratory chain‐derived ROS, during cI‐ and cII‐linked state 3 respiration (n = 6/group).
-
BGene expression related to ROS scavenging.
-
CTotal glutathione in heart and liver tissue (n = 5/group).
-
DUrinary isoprostanes per creatinine as a measure of oxidative stress (n = 4/group).
-
E, FRepresentative images (E) of tissue sections immunostained with an antibody against 4‐hydroxynonenal (4‐HNE), a lipid peroxidation marker. (F) Quantification of the 4‐HNE immunostaining.
Since ROS damage was clearly not instrumental in the cardiac rescue by AOX, we looked at nitric oxide (NO) metabolism that is particularly important in the cardiovascular system and known to interact chemically with ROS and the respiratory chain (Carnicer et al, 2013). We observed a significant increase in NO signaling‐related gene expression specifically in GRAC hearts, and these changes were attenuated by AOX (Fig 8A). Decreased mRNA expression of central targets of NO signaling, ryanodine receptor (RyR2), sarco(endo)plasmic reticulum Ca2+‐ATPase Serca2 (Atp2a2), and phospholamban (Pln) in GRAC heart suggested decreased cardiac contraction‐related Ca2+ channel function (Fig 8B–D). AOX upregulated these Ca2+ channel genes in both Bcs1l mutant and wild‐type mice. Expression of endothelial NO synthase (Nos3) was increased in GRAC heart (Fig 8E), and, most strikingly, expression of neuronal NO synthase (nNos, Nos1) was increased over 10‐fold in the GRAC heart with a significant attenuation in GROX mice. Western blot analysis confirmed similar upregulation of NOS1 protein (Fig 8F and G). Increased gene expression of dimethylarginine dimethylaminohydrolase 1 (Ddah1), which hydrolyzes the endogenous NOS inhibitors dimethylarginine and monomethylarginine (Fig 8H), and increased product/substrate (citrulline/arginine) ratio (Fig 8I) in GRAC heart suggested increased NO production. However, neither protein nitrotyrosine nor total nitrites were increased in the GRAC hearts (Fig 8J and K).
Figure 8. AOX normalizes cardiac nitric oxide‐related gene expression.
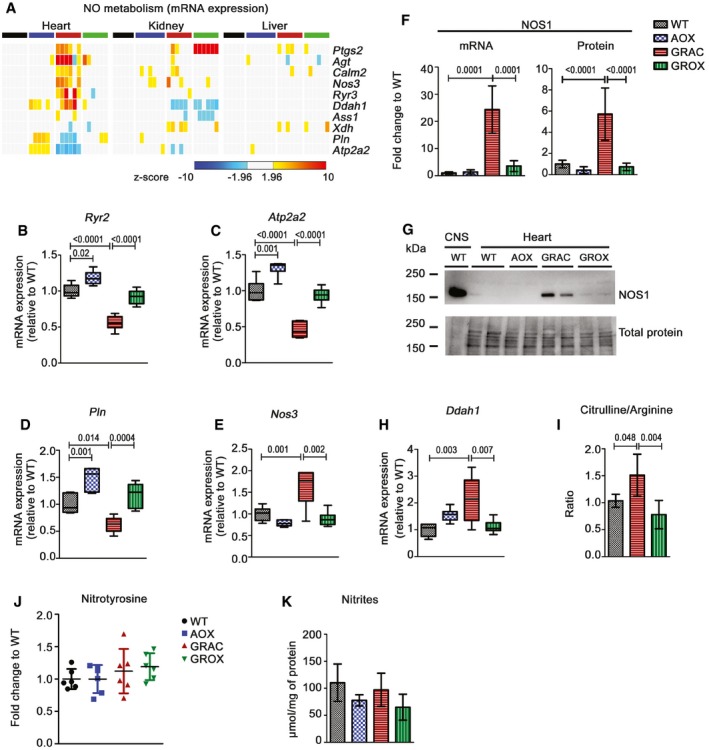
-
AHeat map visualization of the expressions of selected nitric oxide (NO)‐related genes.
-
B–EGene expression of Ca2+ channel genes (B) ryanodine receptor, RYR2, (C) sarco(endo)plasmic reticulum Ca2+‐ATPase (SERCA2 or ATP2A2), (D) phospholamban (PLN), and (E) endothelial nitric oxide synthase NOS3 (n = 6/group).
-
F, GExpression of neuronal nitric oxide synthase (NOS1) mRNA (F) and (G) protein with a representative Western blot. WT brain was used as positive control in the first lane (G).
-
HCardiac gene expression of dimethylarginine dimethylaminohydrolase 1 (DDAH1) (n = 6/group).
-
ICardiac citrulline‐to‐arginine ratio (n = 5/group).
-
JCardiac protein nitrotyrosine quantified from Western blots (n = 6/group).
-
KNitrite concentration in heart tissue (n = 4/group).
Discussion
Here, we show that AOX, a non‐mammalian enzyme that can bypass cIII blockade by shunting electrons directly from the quinone pool to oxygen, is able to permanently prevent lethal cardiomyopathy and alleviate multiple other pathologies in cIII‐deficient GRAC mice. The GRAC mice, initially bred in C57BL/6JBomTac background (Levéen et al, 2011), have turned out particularly useful in mechanistic and interventional studies (Rajendran et al, 2016; Purhonen et al, 2017, 2018) due to their postnatal symptom‐free period and short survival. In the current study, we found that, in the C57BL/6JCrl background, the homozygotes survive to median P210 and develop lethal cardiomyopathy after P150, a manifestations not seen in the GRACILE syndrome patients with early neonatal lethality, but a common manifestation in other mitochondrial disorders. AOX provided a full functional rescue of the cardiomyopathy, restored cI‐ and cII‐linked respiration to wild‐type levels, and abrogated the signature of metabolic stress responses. Heart muscle is highly dependent on mitochondrial respiration and operates at constant ATP and phosphocreatine concentrations (Ventura‐Clapier et al, 2011; Guzun et al, 2015). Our results clearly indicate that the pathogenesis mechanism leading to cardiomyopathy in GRAC mice is insufficiency of electron flow, resulting in disturbed redox and metabolic homeostasis. Cardiac hypertrophy and a metabolic switch from fatty acid to glucose utilization are hallmarks of an adaptive response to chronic stress (Hansson et al, 2004; Ventura‐Clapier et al, 2011; Tuomainen & Tavi, 2017), reflected in the GRAC mice by depletion of glycolytic intermediates, downregulation of PPAR‐α, and the upregulation of ATF4 (Quiros et al, 2017), PGC‐1α, and TFAM. These changes were fully or partially prevented in GROX mice, further implying that the underlying metabolic stress was relieved. The surprising partial rescue of cIII assembly and activity by AOX in cardiac mitochondria was similar to what we observed in liver mitochondria upon attenuation of hepatopathy by ketogenic diet (Purhonen et al, 2017) and could be secondary to the general improvement in mitochondrial structure and function.
GRACILE syndrome patients and GRAC mice display proximal kidney tubulopathy (Fellman et al, 1998; Levéen et al, 2011), which we found to progress to kidney atrophy in the older mice. Tubular epithelial cells have a high energy demand, are rich in mitochondria, and thus susceptible to damage from RC dysfunction (Emma et al, 2012). The typical atrophic changes in the GRAC kidney were efficiently prevented, and tubular mass was preserved by AOX during the entire life span of the GROX mice. However, the compromised cI‐ and cII‐linked respiration and pathological gene expression changes were not improved in samples prepared from the whole kidney, which suggest that the cIII bypass mainly affected a limited cell population, most likely the tubular epithelial cells.
Surprisingly, AOX provided no long‐term histological or functional benefit in the liver. Correspondingly, it did not correct the early‐onset systemic manifestation growth restriction and lipodystrophy, which are likely dependent on the liver. Even though ex vivo oxygen consumption at saturating substrate concentration showed that AOX was able to support respiration in liver mitochondria, this conferred no functional improvement in liver. One explanation would be that the metabolic threshold for AOX activation was not passed in the liver in vivo. Indeed, loss of cIII activity and phosphorylating respiration were not as prominent in the liver as in the heart. The apparent further loss of RISP from cIII in the presence of AOX could simply reflect relaxation of the need to keep cIII assembled and running when AOX replaces its ubiquinol oxidase activity. It is also possible that disturbances in the numerous anabolic and catabolic functions of hepatic mitochondria contribute to the pathogenesis in this tissue, but are not affected by the cIII bypass. Finally, AOX transgene expression was somewhat different in different tissues, as shown in this study and previously (Szibor et al, 2017), but this did not correlate with the rescue effect in the tissues we studied. Thus, the disparity in AOX rescue effect was likely due to differences in the severity of the respiration defect in different tissues.
Respiratory chain blockade may lead to increased ROS production via electron leak to oxygen (Murphy, 2009; Brand, 2010). Although loss of RISP inactivates the quinol oxidation site of cIII and thus should decrease rather than increase ROS, cIII dysfunction may lead to ROS production at cI and cII (Korde et al, 2011; Sena et al, 2013). Indeed, we observed increased ROS production in GRAC heart and kidney mitochondria, and this was prevented by AOX. Increased ROS may cause oxidative damage to proteins and other molecules, which, in turn, should invoke a protective detoxification response. However, expression of glutathione peroxidases and glutathione‐S‐transferases was induced only in the GRAC liver. In the heart and kidney, apart from locally increased lipid peroxidation, there were no consistent signs of ROS damage or detoxification response. The lack of transcriptional response to oxidative damage was particularly striking in the GRAC heart, ruling this out as a significant early pathogenetic mechanism, although it may still play a role in end‐stage disease. Dogan et al (2018) recently reported that AOX expression in a Cox15 knock‐out model of cIV deficiency exacerbated myopathy and decreased survival. Harmful perturbation of redox signaling via AMPK/PGC‐1α was suggested as a mechanism. Interestingly, AOX did not worsen any of the multiple visceral and systemic phenotypes we thoroughly investigated in the cIII‐deficient GRAC mice. Neither did we observe worsening in any behavioral or motor activity parameters (our behavioral scoring and additional unpublished phenotyping data from the German Mouse Clinic) that could have suggested development of skeletal myopathy, not otherwise present in GRAC mice, upon AOX expression. Moreover, feeding GRAC mice the mitochondria‐targeted antioxidant mitoQ had no effect on the progression of cardiomyopathy. Compound heterozygous or homozygous missense mutations in COX15 have been identified in patients presenting with cardiomyopathy and/or encephalopathy, but not skeletal myopathy (Alfadhel et al, 2011). Whether the findings of Dogan et al (2018), showing adverse effect of AOX expression in the mice with Cre‐mediated skeletal muscle‐specific Cox15 knock‐out, apply to other models of cIV deficiency and/or mitochondrial myopathy remains to be investigated. Nevertheless, given that we found obvious differences in the response to AOX‐mediated cIII bypass in different tissues in our model, it is possible that the responses would be disparate in other models, depending on the exact nature of the mutation and respiration defect.
We found that NOS1, an enzyme generating NO, a major modulator of cardiac function and calcium fluxes (Pechanova et al, 2015), was highly upregulated specifically in the GRAC heart, and this was to large extent prevented in GROX hearts. NOS1 and NOS3 upregulation was accompanied by changes in NO‐ and Ca2+ channel‐related gene expression, both alleviated by AOX. NO itself is relatively inert but reacts rapidly with superoxide, the primary ROS emitted by the RC, forming highly reactive and cytotoxic peroxynitrite (Pechanova et al, 2015). Peroxynitrite can impair cardiac function via multiple mechanisms, including inhibition of cI and cIV by S‐nitrosation and nitrosylation, respectively (Burwell & Brookes, 2008; Chouchani et al, 2013; Pechanova et al, 2015), which in the case of GRAC mice could exacerbate the cardiomyopathy via a vicious circle‐type mechanism. However, cIV activity was similar in all groups and lack of increased protein tyrosine nitration and total nitrates suggests NOS uncoupling or loss of function despite highly increased expression, rather than increased NO production in the GRAC heart. These findings imply that impaired NO metabolism should be further investigated as a pathological process in mitochondrial cardiomyopathy.
In summary, our findings represent the first proof‐of‐concept that RC bypass can alleviate pathological manifestations in a genetic mouse model of a human mitochondrial disorder. While restoring (e.g., viral delivery) or correcting (e.g., CRISPR/Cas9 genome editing) the mutated gene in the affected tissue is theoretically the most efficient treatment strategy in monogenic diseases, and has shown promise in animal models (Logan et al, 2014; Ohmori et al, 2017), this requires gene‐specific tools and is only under development for mtDNA mutations. Therefore, as an enzyme that can potentially alleviate cIII or cIV blockade due to a wide variety of mutations, AOX is worth further preclinical investigations, e.g., using viral delivery to the affected tissues in mouse models. Such experiments should shed light on the possible immunological adverse effects associated with the introduction of a non‐mammalian protein into juvenile or adult tissues. Alternatively, there is suggestive evidence that beneficial cIII bypass‐like effects can be achieved pharmacologically, for example with vitamin K2 (Vos et al, 2012) or methylene blue (Atamna et al, 2008). Our results urge the search for novel pharmacological bypass strategies potentially translatable to patients. Finally, the AOX transgenic mice provide an excellent tool to dissect the mechanisms underlying the wide variety of manifestations in mitochondrial disorders, the understanding of which is a prerequisite for the development of novel therapies.
Materials and Methods
Animal breeding and husbandry
Heterozygous knock‐in (Bcs1l c.A232G) mice in the C57BL/6JCrl (RRID:IMSR_JAX:000664) background and AOX transgenic mice (Szibor et al, 2017) were maintained in the animal facilities of University of Helsinki, Finland. All mice were housed in individually ventilated cages with 12‐h light/12‐h dark cycle at a temperature of 22–23°C, and water and food (2018 Teklad global 18% protein rodent diet, Envigo) available ad libitum. The animal studies were approved by the animal ethics committee of the State Provincial Office of Southern Finland (ESAVI/6142/04.10.07/2014 and ESAVI/6365/04.10.07/2017) and were performed according to FELASA (Federation of Laboratory Animal Science Associations) guidelines.
Bcs1l c.A232G homozygotes survive to over P150 in the C57BL/6JCrl background (Purhonen et al, 2017). The two mouse strains were crossed to generate double heterozygous mice, which were backcrossed to the C57BL/6JCrl background for several generations. Wild‐type or Bcs1l heterozygous animals, littermates whenever possible, were used as wild‐type (WT) controls. Mice carrying a single copy of the AOX transgene (AOX mice), littermates whenever possible, were used as a second control group. Both genders were used in the experiments and were not separated unless separate analysis for males and females is indicated in Results. Genotyping for the Bcs1l mutation and AOX transgene was performed as described (Levéen et al, 2011; Szibor et al, 2017).
Mouse health was monitored by manual behavioral scoring and weighing according to the ethical permit and as described elsewhere (Rajendran et al, 2016; Purhonen et al, 2017). The time points for assessments were chosen to verify early and late‐onset manifestations: growth and survival data from weaning on, whole‐body metabolism and DEXA data between 10 and 14 weeks of age (at German Mouse Clinic), presymptomatic cardiac data at 5 months (P150), end‐stage disease for GRAC mice at 6–7 months (P200), and survival and tissue histology of surviving GROX mice at up to 22 months (P680). The total number of experimental mice used in this study was 301.
Assessment of body composition and whole‐body metabolism
At the German Mouse Clinic (GMC), the mice were maintained from 8 weeks of age until sacrificing at week 21 according to the GMC housing conditions (www.mouseclinic.de) and German laws. All tests were approved by the responsible authority of the Regierung von Oberbayern. Twenty mice per genotype (WT, AOX, GRAC, and GROX), 10 mice per gender, were phenotyped between ages P56 and P112. The determination of energy expenditure by indirect calorimetry was performed at P77. Dual‐energy X‐ray absorptiometry (DEXA) was used to measure bone density, fat mass, and lean mass at P98.
Salt provocation and antioxidant administration
Powdered chow (2018 Teklad global 18% protein, Envigo) was supplemented to contain 6% NaCl (w/w), moistened, and manually compressed into pellets. The pellets were air dried for 24 h and stored frozen. Control diet was prepared similarly but without the addition of NaCl. The NaCl‐supplemented chow was administered ad libitum from P150 onwards. For antioxidant feeding, drinking water was supplemented with 0.50 mM mitoquinol mesylate (mitoQ, a kind gift from MitoQ Ltd, New Zealand) starting from P150.
Echocardiography
The cardiac dimensions and ejection volume of mice were measured by using Vevo 2100 imaging system (Fujifilm). B‐mode and M‐mode were used to measure left ventricle dimensions, and the average values were used for analysis.
Clinical chemistry and urine analysis
Alanine aminotransferase and alkaline phosphatase were measured from plasma, and albumin and creatinine from urine samples using Siemens ADVIA1650 analyzer. For urine collection, the mice were kept in metabolic cages for 24 h under 12‐h/12‐h light/dark cycle. Other urine analyses were performed by pipetting 15 μl of urine per each test area on human urine strips (Combur10 Test UX, Roche), followed by reading of the values in Urisys 1100 urine analyzer (Roche).
Sample collection
For end point sacrificing and sample collection, the mice were not fasted for longer than 2–3 h due to the hypoglycemia associated with the mutant phenotype. All samples were collected at the same time of the day during the light period of the mice. The mice were anesthetized with pentobarbital, blood was collected into Li‐heparin tubes by cardiac puncture, and plasma was separated. Organs were immediately dissected, and snap‐frozen in liquid nitrogen or immersed in 10% buffered formalin solution. Approximately 1 mm3 pieces of tissue were fixed in 1.5% glutaraldehyde and 1.5% paraformaldehyde in 0.1 M Sörensen buffer pH 7.2 for electron microscopy. Brains (n = 6 per genotype at P200; n = 8 for AOX and n = 10 for GROX at P600) were immersion‐fixed in 4% paraformaldehyde in 0.1 M sodium phosphate buffer pH 7.4 for 48 h followed by cryoprotection in 30% sucrose/0.05% sodium azide in 50 mM Tris‐buffered saline (TBS). Frozen coronal sections (40 μm) were cut through cerebrum, while cerebella were cut sagittally and stored in cryoprotectant solution as described (Tegelberg et al, 2017). Blood glucose and lactate levels were measured using commercial quick sticks and meters (Freestyle lite and Lactate Pro LT‐1710). In living animals, the ventral tail artery was punctured under light isoflurane anesthesia to measure glucose and lactate.
Histology and electron microscopy
Tissue sections were stained with standard methods to assess general histology (hematoxylin–eosin) and fibrosis (Sirius Red). The area stained by Sirius Red was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA) from 5 to 10 random fields. Tissues were prepared for transmission electron microscopy using standard methods and examined in a Philips/FEI CM 100 BioTWIN transmission electron microscope (Hillsboro, OR) at 60 kV. Images were captured in side‐mounted Olympus Veleta camera (Center Valley, PA) with a resolution of 2,048 × 2,048 pixels (2k × 2k).
Immunohistochemistry
One‐in‐twelve series of free‐floating cryosections were stained as previously described (Bible et al, 2004). Primary antibody against glial fibrillary acidic protein (GFAP: Z0334, DAKO, Agilent Technologies) diluted 1:4,000 in 10% normal serum in TBS‐T was incubated overnight at 4°C. Standard paraffin sections were immunostained with primary antibodies against Ki67, cleaved caspase‐3, and 4‐HNE (see Appendix Table 3), using Vectastain Elite peroxidase reagents (Vector Laboratories) and standard protocols.
Western blot analyses
Frozen tissues were homogenized in RIPA buffer (50 mM Tris, pH 8, 150 mM NaCl2, 1% Triton X‐100, 0.5% sodium deoxycholate, 0.1% SDS) with protease inhibitors (Complete mini, Roche). Protein concentration was measured using Bradford reagent (Bio‐Rad). Approximately 10 μg of protein per lane was run on Mini‐PROTEAN StainFree© precast gradient gels (Bio‐Rad). StainFree© dye was activated by UV light for one minute and proteins transferred to PVDF membrane using Trans‐Blot Turbo Transfer System (Bio‐Rad). The filters were then imaged for total protein, blocked with 5% milk powder in TBST, and stained with primary antibodies (Appendix Table S3). Appropriate HRP‐conjugated secondary antibodies and enhanced chemiluminescence were used for the detection. Band intensities, as recorded with ChemiDoc MP digital imager (Bio‐Rad), were normalized to total protein.
Isolation of mitochondria
Liver and kidney samples were minced with scissors and then homogenized with a teflon‐glass potter homogenizer containing homogenization buffer (225 mM mannitol, 75 mM sucrose, 10 mM Tris–HCl, 1 mM EGTA, 1 mg/ml albumin, and pH set to 7.4 at +4°C). For respirometry, a crude mitochondrial fraction was obtained by a two‐step differential centrifugation (10 min at 800 g and 10 min at 7,800 g). For other assays, differential centrifugation was continued, including a 19% Percoll solution (GE Healthcare), and the mitochondria were stored as pellets at −80°C. Heart mitochondria were isolated similarly, but the tissue was minced in trypsin–EDTA solution (Thermo Scientific). Trypsin was inactivated with fetal bovine serum and removed before homogenization. All steps were carried at 0–4°C.
Blue native gel electrophoresis
Digitonin‐solubilized mitochondria were prepared and subjected to BNGE as described (Davoudi et al, 2014).
Respirometry
Oxygen consumption by isolated mitochondria was measured polarographically in Mir05 buffer (110 mM sucrose, 60 mM lactobionic acid, 20 mM taurine, 20 mM HEPES, 10 mM KH2PO4, 3 mM MgCl2, 0.5 mM EGTA, and 1 g/l fatty acid free bovine serum albumin, pH 7.1) in a 2‐ml chamber (OROBOROS Instruments). Sample, substrates, inhibitors, and uncouplers were injected in following order: (i) 1 mM malate, 5 mM pyruvate and 5 mM glutamate; (ii) sample; (iii) 1.25 mM ADP; (iv) 10 μM cytochrome c; (v) 10 mM succinate; (vi) 100 μM propyl gallate; (vii) 1 μg/ml oligomycin A; (viii) carbonyl cyanide 4‐(trifluoromethoxy) phenylhydrazone (FCCP) titration to maximum respiration; (ix) 0.5 μM rotenone; and (x) 1 μg/ml antimycin A. Complex IV activity was measured using 2 mM ascorbate and 0.5 mM N,N,N′,N′‐tetramethyl‐p‐phenylenediamine (TMPD) as substrates. Chemical and oxygen‐dependent TMPD auto‐oxidation was determined after addition of 10 mM sodium azide.
Mitochondrial H2O2 emission
The oxygraph was equipped with a fluorometer and set for Amplex Red/horseradish peroxidase (HRP) assay to measure mitochondrial H2O2 emission simultaneously with respirometry (Makrecka‐Kuka et al, 2015). To validate the assay, HRP‐independent and catalase‐insensitive conversion of Amplex Red to the fluorescent resorufin was initially tested. Liver and kidney, but not heart, mitochondria possessed high rate of HRP‐independent artificial Amplex Red oxidation due to presence of mitochondrial carboxylesterases (Miwa et al, 2016). This signal was inhibited by adding 100 μM phenylmethylsulfonyl fluoride. Final assay conditions in Mir05 buffer were 10 μM Amplex Ultra Red (Invitrogen), 1 U/ml HRP, 6 U/ml superoxide dismutase, 100 μM PMSF (for liver and kidney), and substrates for cI‐ and cII‐linked respiration as described in the previous chapter. The assay was calibrated with a 0.4 nmol bolus of H2O2 in every respiratory state for which results are reported. As a background for heart and kidney mitochondria, the rate of fluorescence was recorded when mitochondria were kept without any substrates. For liver mitochondria, background was taken as a catalase‐insensitive (700 U/ml) fluorescence flux.
Enzyme activity and lipid peroxidation assays
The activity of cIII in isolated mitochondria was measured using a spectrophotometric assay as described previously (Davoudi et al, 2014). Aconitase activity (MAK051, Sigma) and urinary isoprostanes (EA85, Oxford Biomedical Research) were measured using the indicated commercial kits. Carbonylated proteins were labeled on‐filter with 2,4‐dinitrophenylhydrazine (phosphoric acid solution, 42215, Sigma‐Aldrich) and detected with anti‐dinitrophenol antibody (Oxidized Protein Western Blot Kit, ab178020, Abcam) as described (Colombo et al, 2016). Nitrites in heart tissue were measured using Griess reagent (03553, Sigma‐Aldrich). Isoprostanes were quantified from urine using a commercial ELISA kit (ab175819, Abcam).
Transcriptomics
For RNA sequencing, rRNA was removed and the sequencing libraries were prepared using Illumina TruSeq Stranded Total RNA Library Prep Kit with Ribo‐Zero Human/Mouse/Rat. Libraries were sequenced on Illumina NextSeq 500 instrument using 75 bp kit. Adapter sequences and low‐quality reads were removed from the data using cutadapt (Martin, 2011), and data were further screened for remaining rRNA reads using SortMeRNA (Kopylova et al, 2012). The data were mapped to M. musculus genome GRCm38.p4 using STAR (Dobin et al, 2013). The original genome sequence and annotation were augmented with sequence and annotation for AOX. Count data were processed in R using GenomicFeatures and GenomicAlignments (Lawrence et al, 2013), and the differential expression analysis was carried out using DESeq2 (Love et al, 2014). PCA plots were generated using ClustVis online tool (www.biit.cs.ut.ee/clustvis).
Pathway enrichment analysis
Enrichment analyses were performed using filtered gene sets (|FC| > 1.5, P < 0.05) against Reactome database using www.mousemine.org. To visualize pathway changes, heat maps were generated by Gitool.2.3.1 with WT mean‐centered Z‐score values and gene mapping for selected pathways as described (Gundem & Lopez‐Bigas, 2014; Perez‐llamas & Lopez‐Bigas, 2011). Pathway heat maps were generated based on Z‐score, showing upregulation (brown color) and downregulation (blue color) of each pathway. P‐values for group comparison (labeled 1, 2, and 3) were calculated using one‐way ANOVA followed by selected comparisons and the Benjamin–Hochberg FDR correction.
Metabolomics
Targeted quantitative metabolomics covering 116 metabolites was performed using capillary electrophoresis–mass spectrometry (CE‐TOMFS and CE‐QqQMS) at Human Metabolome Technologies Inc., Japan (http://humanmetabolome.com/en/). Details of the protocol are available upon request. We omitted the AOX group in the metabolomics since ectopic AOX was shown to be inert in healthy wild‐type mice (Szibor et al, 2017).
Statistics (excluding transcriptomics)
For normally distributed data, one‐way ANOVA followed by Tukey's test or by selected comparisons using t‐tests with Welch's correction was used. Normality of data was assessed by the d'Agostino–Pearson omnibus normality test, and equality of variances by Bartlett's test. Data not compatible with parametric tests were assessed by Kruskal–Wallis and Mann–Whitney U‐tests. Survival curves were analyzed using log‐rank Mantel–Cox test. GraphPad Prism 7 software (GraphPad Software Inc., La Jolla, CA, USA) was used for the statistical analyses. Group sizes (n) and the statistical tests used are described in figure legends, and exact P‐values are shown in the graphs.
Data availability
Additional raw data and detailed protocols are available from the authors upon request. Transcriptomics data have been deposited to ArrayExpress database at EMBL‐EBI under the accession ID E‐MTAB‐7416 (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-7416).
Author contributions
HTJ, MS, VF, and JK invented the concept to combine the mouse models; HTJ and MS provided the mouse strain with broad AOX expression; VF and JK group combined the strains; JRa, JP, ST, EM, VF, and JK conceived and designed the experiments; HF, VG‐D, and MHA conceived and supervised phenotyping experiments at the GMC; JRa performed the animal experiments (echocardiography, metabolic sample collection, sickness scoring, tissue sampling); JRa performed the laboratory analyses (histology, protein analyses, BNGE, respirometry, cIII activity, metabolomics and transcriptome pathway enrichment analysis, ROS markers); JP performed respirometry, histology, immunohistochemistry quantitation, and Amplex Red assay analyses; ST performed brain histology and immunohistochemistry analyses; O‐PS and PA performed transcriptomics analysis; JK performed immunohistochemistry and ROS marker analyses; MM performed electron microscopy analyses; JRo and HF performed indirect calorimetry and DEXA analysis; JRo and HF analyzed the data from GMC; JRa, JP, ST, VF, and JK analyzed the data from Helsinki; EM, HTJ, and MS participated in interpretation of the results; JRa, JP, VF and JK wrote the manuscript draft; all authors revised the manuscript and have contributed substantially to the work reported.
Conflict of interest
MS is a shareholder of a commercial enterprise that is dedicated to developing therapeutics based on AOX. The authors declare that they have no conflict of interest.
The paper explained.
Problem
Mitochondrial diseases are genetic disorders of energy metabolism that can affect any one or several organs of the body, including skeletal muscle, heart, brain, and visceral organs. Nowadays, early diagnosis of mitochondrial disease is often possible using modern molecular genetics, but treatment options remain scarce. Mutations in the BCS1L gene are the most common cause of mitochondrial diseases affecting the respiratory chain complex III (cIII). GRACILE syndrome (Growth Restriction, Aminoaciduria, Cholestasis, liver Iron overload, Lactic acidosis, and Early death), with neonatal lethality, is the most severe of them. Due to the strikingly similar disease in all GRACILE syndrome patients, Prof. Fellman's group previously introduced the underlying missense mutation (Bcs1l c.A232G, Bcs1l p.S78G) into mice. This mouse model faithfully recapitulates most of the symptoms of the patients. In search for novel strategies to alleviate cIII deficiency‐related pathology, we crossed the Bcs1l c.A232G mice with transgenic mice expressing alternative oxidase (AOX), a mitochondrial inner membrane enzyme that can bypass respiratory electron flow blockade when the quinol oxidation capacity of cIII is compromised.
Results
Mice homozygous for the Bcs1l c.A232G mutation survived approximately 200 days and died of dilating cardiomyopathy, a novel late‐onset phenotype in the C57BL/6JCrl background. In contrast, the homozygotes carrying the AOX transgene lived approximately 600 days and never developed the cardiomyopathy. AOX also ameliorated the severe kidney disease and focal astrogliosis of the brain in the homozygotes. Surprisingly, AOX did not correct the liver disease, poor growth, and loss of white fat, suggesting different disease mechanism in different tissues. AOX corrected the abnormal ultrastructure of mitochondria and mitochondrial respiration in those tissues, in which the tissue pathology was alleviated. Heart and kidney mitochondria from the homozygotes showed elevated reactive oxygen species (ROS) production, but analyses of ROS damage suggested that the beneficial effects of AOX were not ROS‐related. Instead, we found that AOX normalized cardiac gene expression related to nitric oxide metabolism and signaling, a major modulator of cardiovascular functions. We conclude that AOX efficiently prevented tissue pathology in the Bcs1l mutant mouse model of cIII deficiency by restoring respiration, preferably in the most highly oxidative tissues such as heart.
Impact
Our study in the patient mutation knock‐in model of cIII deficiency is the first proof‐of‐concept that bypass of the cIII‐cIV segment of the respiratory electron transfer can alleviate pathological manifestations in a physiologically relevant genetic mouse model of a human mitochondrial disorder. These findings highlight the potential of AOX as a tool to unravel disease mechanisms, and also urge further studies of AOX in preclinical models, e.g., using viral delivery to the affected tissues in mouse models, as well as studies to search for novel pharmacological bypass strategies potentially translatable to patients.
For more information
(i) GRACILE syndrome: https://www.omim.org/entry/603358.
(ii) BCS1L gene: https://www.omim.org/entry/603647.
(iii)United Mitochondrial Disease Foundation (UMDF): https://www.umdf.org/.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 6
Source Data for Figure 8
Acknowledgements
We thank Prof. Hannu Sariola for expert assistance with tissue histopathology; Dr. Eric Dufour for advice on respirometry; Praveen Dhandapani for the AOX genotyping protocol; Elisa Altay, Päivi Leinikka, and Nada Bechara‐Hirvonen for expert technical assistance; the staff of the DNA Sequencing and Genomics Laboratory, University of Helsinki, for running the RNAseq; the staff of the BioEM Lab (at C‐Cina), Biozentrum, University of Basel, the Core Facility for Integrated Microscopy, Panum Institute, University of Copenhagen, and Ola Gustafsson of Microscopy Facility at the Department of Biology, Lund University, for providing the electron microscopy facilities and assistance; and finally Dr. Sanna Marjavaara for helpful initial discussions. This study was supported by grants from Academy of Finland (grant 259296 to VF, 256615 and 272376 to HTJ), Swedish Research Council (grant 521‐2011‐3877 to VF), European Research Council (Advanced Grant 232738 to HTJ), Finska Läkaresällskapet (to VF), Foundation for Pediatric Research in Finland (to VF), Folkhälsan Research Center (to VF, JK), and German Federal Ministry of Education and Research (Infrafrontier grant 01KX1012 to MHdA).
EMBO Mol Med (2019) 11:e9456
See also: A Saada (January 2019)
References
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA (2005) Targeting an antioxidant to mitochondria decreases cardiac ischemia‐reperfusion injury. FASEB J 19: 1088–1095 [DOI] [PubMed] [Google Scholar]
- Alfadhel M, Lillquist YP, Waters PJ, Sinclair G, Struys E, McFadden D, Hendson G, Hyams L, Shoffner J, Vallance HD (2011) Infantile cardioencephalopathy due to a COX15 gene defect: report and review. Am J Med Genet A 155a: 840–844 [DOI] [PubMed] [Google Scholar]
- Atamna H, Nguyen A, Schultz C, Boyle K, Newberry J, Kato H, Ames BN (2008) Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J 22: 703–712 [DOI] [PubMed] [Google Scholar]
- Bible E, Gupta P, Hofmann SL, Cooper JD (2004) Regional and cellular neuropathology in the palmitoyl protein thioesterase‐1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis 16: 346–359 [DOI] [PubMed] [Google Scholar]
- Brand MD (2010) The sites and topology of mitochondrial superoxide production. Exp Gerontol 45: 466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burwell LS, Brookes PS (2008) Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia‐reperfusion injury. Antioxid Redox Signal 10: 579–599 [DOI] [PubMed] [Google Scholar]
- Carnicer R, Crabtree MJ, Sivakumaran V, Casadei B, Kass DA (2013) Nitric oxide synthases in heart failure. Antioxid Redox Signal 18: 1078–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro‐Guerrero NA, Krab K, Moreno‐Sanchez R (2004) The alternative respiratory pathway of Euglena mitochondria. J Bioenerg Biomembr 36: 459–469 [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding SJ, James AM, Cocheme HM, Reinhold J, Lilley KS et al (2013) Cardioprotection by S‐nitrosation of a cysteine switch on mitochondrial complex I. Nat Med 19: 753–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo G, Clerici M, Garavaglia ME, Giustarini D, Rossi R, Milzani A, Dalle‐Donne I (2016) A step‐by‐step protocol for assaying protein carbonylation in biological samples. J Chromatogr B 1019: 178–190 [DOI] [PubMed] [Google Scholar]
- Cruciat CM, Hell K, Folsch H, Neupert W, Stuart RA (1999) Bcs1p, an AAA‐family member, is a chaperone for the assembly of the cytochrome bc(1) complex. EMBO J 18: 5226–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassa EP, Dufour E, Goncalves S, Paupe V, Hakkaart GA, Jacobs HT, Rustin P (2009) Expression of the alternative oxidase complements cytochrome c oxidase deficiency in human cells. EMBO Mol Med 1: 30–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoudi M, Kotarsky H, Hansson E, Fellman V (2014) Complex I function and supercomplex formation are preserved in liver mitochondria despite progressive complex III deficiency. PLoS One 9: e86767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan SA, Cerutti R, Beninca C, Brea‐Calvo G, Jacobs HT, Zeviani M, Szibor M, Viscomi C (2018) Perturbed redox signaling exacerbates a mitochondrial myopathy. Cell Metab 28: 764–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Khoury R, Dufour E, Rak M, Ramanantsoa N, Grandchamp N, Csaba Z, Duvillie B, Benit P, Gallego J, Gressens P et al (2013) Alternative oxidase expression in the mouse enables bypassing cytochrome c oxidase blockade and limits mitochondrial ROS overproduction. PLoS Genet 9: e1003182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Khoury R, Kemppainen KK, Dufour E, Szibor M, Jacobs HT, Rustin P (2014) Engineering the alternative oxidase gene to better understand and counteract mitochondrial defects: state of the art and perspectives. Brit J Pharmacol 171: 2243–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emma F, Bertini E, Salviati L, Montini G (2012) Renal involvement in mitochondrial cytopathies. Pediatr Nephrol 27: 539–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellman V, Rapola J, Pihko H, Varilo T, Raivio KO (1998) Iron‐overload disease in infants involving fetal growth retardation, lactic acidosis, liver haemosiderosis, and aminoaciduria. Lancet 351: 490–493 [DOI] [PubMed] [Google Scholar]
- Fernandez‐Ayala DJ, Sanz A, Vartiainen S, Kemppainen KK, Babusiak M, Mustalahti E, Costa R, Tuomela T, Zeviani M, Chung J et al (2009) Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab 9: 449–460 [DOI] [PubMed] [Google Scholar]
- Fernandez‐Vizarra E, Zeviani M (2015) Nuclear gene mutations as the cause of mitochondrial complex III deficiency. Front Genet 6: 134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundem G, Lopez‐Bigas N (2014) Sample‐level enrichment analysis unravels shared stress phenotypes among multiple cancer types. Genome Med 4: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzun R, Kaambre T, Bagur R, Grichine A, Usson Y, Varikmaa M, Anmann T, Tepp K, Timohhina N, Shevchuk I et al (2015) Modular organization of cardiac energy metabolism: energy conversion, transfer and feedback regulation. Acta Physiol 213: 84–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkaart GA, Dassa EP, Jacobs HT, Rustin P (2006) Allotopic expression of a mitochondrial alternative oxidase confers cyanide resistance to human cell respiration. EMBO Rep 7: 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson A, Hance N, Dufour E, Rantanen A, Hultenby K, Clayton DA, Wibom R, Larsson NG (2004) A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain‐deficient mouse hearts. Proc Natl Acad Sci USA 101: 3136–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefnagel MHN, Wiskich JT (1998) Activation of the plant alternative oxidase by high reduction levels of the Q‐Pool and pyruvate. Arch Biochem Biophys 355: 262–270 [DOI] [PubMed] [Google Scholar]
- Kalfon R, Koren L, Aviram S, Schwartz O, Hai T, Aronheim A (2017) ATF3 expression in cardiomyocytes preserves homeostasis in the heart and controls peripheral glucose tolerance. Cardiovasc Res 113: 134–146 [DOI] [PubMed] [Google Scholar]
- Kopylova E, Noe L, Touzet H (2012) SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28: 3211–3217 [DOI] [PubMed] [Google Scholar]
- Korde AS, Yadav VR, Zheng YM, Wang YX (2011) Primary role of mitochondrial Rieske iron‐sulfur protein in hypoxic ROS production in pulmonary artery myocytes. Free Radic Biol Med 50: 945–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotarsky H, Keller M, Davoudi M, Levéen P, Karikoski R, Enot DP, Fellman V (2012) Metabolite profiles reveal energy failure and impaired beta‐oxidation in liver of mice with complex III deficiency due to a Bcs1l mutation. PLoS One 7: e41156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaloff EM, Phang JM, Granger AS, Downing SJ (1977) Regulation of proline oxidase activity by lactate. Proc Natl Acad Sci USA 74: 5368–5371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ (2013) Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levéen P, Kotarsky H, Morgelin M, Karikoski R, Elmer E, Fellman V (2011) The GRACILE mutation introduced into Bcs1l causes postnatal complex III deficiency: a viable mouse model for mitochondrial hepatopathy. Hepatology 53: 437–447 [DOI] [PubMed] [Google Scholar]
- Logan GJ, de Alencastro G, Alexander IE, Yeoh GC (2014) Exploiting the unique regenerative capacity of the liver to underpin cell and gene therapy strategies for genetic and acquired liver disease. Int J Biochem Cell Biol 56: 141–152 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makrecka‐Kuka M, Krumschnabel G, Gnaiger E (2015) High‐Resolution respirometry for simultaneous measurement of oxygen and hydrogen peroxide fluxes in permeabilized cells, tissue homogenate and isolated mitochondria. Biomolecules 5: 1319–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J 17: 10–12 [Google Scholar]
- McDonald A, Vanlerberghe G (2004) Branched mitochondrial electron transport in the Animalia: presence of alternative oxidase in several animal phyla. IUBMB Life 56: 333–341 [DOI] [PubMed] [Google Scholar]
- Miwa S, Treumann A, Bell A, Vistoli G, Nelson G, Hay S, von Zglinicki T (2016) Carboxylesterase converts Amplex red to resorufin: implications for mitochondrial H2O2 release assays. Free Radic Biol Med 90: 173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobrega FG, Nobrega MP, Tzagoloff A (1992) Bcs1, a novel gene required for the expression of functional Rieske iron sulfur protein in Saccharomyces cerevisiae . EMBO J 11: 3821–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori T, Nagao Y, Mizukami H, Sakata A, Muramatsu SI, Ozawa K, Tominaga SI, Hanazono Y, Nishimura S, Nureki O et al (2017) CRISPR/Cas9‐mediated genome editing via postnatal administration of AAV vector cures haemophilia B mice. Sci Rep 7: 4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechanova O, Varga ZV, Cebova M, Giricz Z, Pacher P, Ferdinandy P (2015) Cardiac NO signalling in the metabolic syndrome. Brit J Pharmacol 172: 1415–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐llamas C, Lopez‐Bigas N (2011) Gitools: analysis and visualisation of genomic data using interactive heat‐maps. PLoS One 6: e19541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Horvath R, Klopstock T, Mootha VK, Suomalainen A, Koene S, Hirano M, Zeviani M, Bindoff LA, Yu‐Wai‐Man P et al (2013) New treatments for mitochondrial disease‐no time to drop our standards. Nat Rev Neurol 9: 474–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purhonen J, Rajendran J, Mörgelin M, Uusi‐Rauva K, Katayama S, Krjutskov K, Einarsdottir E, Velagapudi V, Kere J, Jauhiainen M et al (2017) Ketogenic diet attenuates hepatopathy in mouse model of respiratory chain complex III deficiency caused by a Bcs1l mutation. Sci Rep 7: 957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purhonen J, Rajendran J, Tegelberg S, Smolander OP, Pirinen E, Kallijärvi J, Fellman V (2018) NAD+ repletion produces no therapeutic effect in mice with respiratory chain complex III deficiency and chronic energy deprivation. FASEB J 32: 5913–5926 [DOI] [PubMed] [Google Scholar]
- Quiros PM, Prado MA, Zamboni N, D'Amico D, Williams RW, Finley D, Gygi SP, Auwerx J (2017) Multi‐omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216: 2027–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran J, Tomašić N, Kotarsky H, Hansson E, Velagapudi V, Kallijärvi J, Fellman V (2016) Effect of high‐carbohydrate diet on plasma metabolome in mice with mitochondrial respiratory chain complex III deficiency. Int J Mol Sci 17: E1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H et al (2013) Mitochondria are required for antigen‐specific T cell activation through reactive oxygen species signaling. Immunity 38: 225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szibor M, Dhandapani PK, Dufour E, Holmstrom KM, Zhuang Y, Salwig I, Wittig I, Heidler J, Gizatullina Z, Gainutdinov T et al (2017) Broad AOX expression in a genetically tractable mouse model does not disturb normal physiology. Dis Model Mech 10: 163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegelberg S, Tomašić N, Kallijärvi J, Purhonen J, Elmer E, Lindberg E, Nord DG, Soller M, Lesko N, Wedell A et al (2017) Respiratory chain complex III deficiency due to mutated BCS1L: a novel phenotype with encephalomyopathy, partially phenocopied in a Bcs1l mutant mouse model. Orphanet J Rare Dis 12: 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuomainen T, Tavi P (2017) The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp Cell Res 360: 12–18 [DOI] [PubMed] [Google Scholar]
- Ventura‐Clapier R, Garnier A, Veksler V, Joubert F (2011) Bioenergetics of the failing heart. Biochim Biophys Acta Mol Cell Res 1813: 1360–1372 [DOI] [PubMed] [Google Scholar]
- Visapää I, Fellman V, Vesa J, Dasvarma A, Hutton JL, Kumar V, Payne GS, Makarow M, Van Coster R, Taylor RW et al (2002) GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L . Am J Hum Genet 71: 863–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M, Esposito G, Edirisinghe JN, Vilain S, Haddad DM, Slabbaert JR, Van Meensel S, Schaap O, De Strooper B, Meganathan R et al (2012) Vitamin K2 Is a mitochondrial electron carrier that rescues Pink1 deficiency. Science 336: 1306–1310 [DOI] [PubMed] [Google Scholar]
- Yan LJ, Levine RL, Sohal RS (1997) Oxidative damage during aging targets mitochondrial aconitase. Proc Natl Acad Sci USA 94: 11168–11172 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 6
Source Data for Figure 8
Data Availability Statement
Additional raw data and detailed protocols are available from the authors upon request. Transcriptomics data have been deposited to ArrayExpress database at EMBL‐EBI under the accession ID E‐MTAB‐7416 (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-7416).