

Summary
Dendritic cells (DCs) are professional antigen‐presenting cells that play a key role in directing T‐cell responses. Regulatory T (Treg) cells possess an immunosuppressive ability to inhibit effector T‐cell responses, and Notch ligand Jagged1 (Jag1) is implicated in Treg cell differentiation. In this study, we evaluated whether bone marrow‐derived DCs genetically engineered to express Jag1 (Jag1‐DCs) would affect the maturation and function of DCs in vitro and further investigated the immunoregulatory ability of Jag1‐DCs to manipulate T helper type 2 (Th2) ‐mediated allergic asthma in mice. We produced Jag1‐DCs by adenoviral transduction. Overexpression of Jag1 by ovalbumin (OVA) ‐stimulated Jag1‐DCs exhibited increased expression of programmed cell death ligand 1 (PD‐L1) and OX40L molecules. Subsequently, co‐culture of these OVA‐pulsed Jag1‐DCs with allogeneic or syngeneic CD4+ T cells promoted the generation of Foxp3+ Treg cells, and blocking PD‐L1 using specific antibodies partially reduced Treg cell expansion. Furthermore, adoptive transfer of OVA‐pulsed Jag1‐DCs to mice with OVA‐induced asthma reduced allergen‐specific immunoglobulin E production, airway hyperresponsiveness, airway inflammation, and secretion of Th2‐type cytokines (interleukin‐4, interleukin‐5, and interleukin‐13). Notably, an increased number of Foxp3+ Treg cells associated with enhanced levels of transforming growth factor‐β production was observed in Jag1‐DC‐treated mice. These data indicate that transgenic expression of Jag1 by DCs promotes induction of Foxp3+ Treg cells, which ameliorated Th2‐mediated allergic asthma in mice. Our study supports an attractive strategy to artificially generate immunoregulatory DCs and provides a novel approach for manipulating Th2 cell‐driven deleterious immune diseases.
Keywords: asthma, dendritic cell, Jagged1, regulatory T cell, T helper type 2
Introduction
Asthma is chronic airway inflammation characterized by different endotypes, and a wide variety of environmental stimulators can cause this disease.1 The most frequent endotype of asthma is allergic (or atopic) asthma.2 Allergic asthma, a type of T helper type 2 (Th2) ‐associated asthma, is induced by Th2 responses and allergen‐specific immunoglobulin E (IgE).3 The production of cytokines, including interleukin‐4 (IL‐4), IL‐5, and IL‐13, by Th2 cells produces importantly critical effects on asthmatic responses. Interleukin‐4 causes B cells to produce a high level of circulating IgE, and this IgE can sensitize mast cells and basophils after allergen exposure. Additionally, IL‐5 promotes eosinophil maturation and activation and results in the release of cytotoxic proteins from eosinophils that destroy the airway.4 In particular, IL‐13 enhances the development of airway hyperresponsiveness (AHR) by increasing epithelial permeability and mucus production.5 Current therapeutic methods for asthma include an inhaled corticosteroid to suppress lung inflammation and β 2 agonists as bronchodilators.6 These standard therapies are able to relieve asthma syndromes in patients. However, asthma in some patients remains poorly controlled. Almost 10% of individuals with asthma need high doses of inhaled corticosteroid, and 1% require long‐term daily treatment with oral steroids for asthma management. In addition, in most of this steroid‐dependent population, asthma is severe and steroid‐resistant.7 Hence, the need for targeted therapies for severe asthma and novel treatments with sustained effects highlights that new therapeutic strategies for treating asthma are urgently required.
In asthma, regulatory T (Treg) cells play a critical role in controlling immune homeostasis in the airway. Many studies reported that Treg cells possess an immunosuppressive ability to dampen Th2‐type inflammation and induce specific immune tolerance.8, 9, 10 Hence, inhibition of Th2 cell responses through induction of Treg cells is a good strategy to treat Th2‐mediated allergic asthma. Treg cells are a heterogeneous population, and forkhead box protein 3 (Foxp3) ‐expressing Treg cells can be either thymus‐derived (t)Treg cells or peripherally derived (p)Treg cells. The tTreg subset is characterized by the constitutively high expression of CD25 and of Foxp3, whereas pTreg cells divide into Foxp3+ Treg cells and Foxp3− IL‐10+ type 1 regulatory cells.11 These Treg cells perform their suppressive function through cell contact mechanisms and/or release of IL‐10 and transforming growth factor‐β (TGF‐β).12 Recently, some interventions focused on dendritic cells (DCs), because DCs play roles in determining T‐cell differentiation in the face of allergen exposure.13, 14 In particular, DCs may promote Foxp3+ Treg cell differentiation by expressing such immunoregulatory molecules as programmed cell death ligand 1 (PD‐L1), OX40L, and inducible co‐stimulatory ligand (ICOSL) by DCs.15, 16, 17 As immunomodulatory molecules are major contributors to the decision of T cells to either activate or suppress a specific immune response, an attractive approach to altering DC functions is to genetically modify them to overexpress such immunomodulatory molecules.
Recent findings have revealed the important roles of the Notch signaling pathway in T‐cell differentiation.18, 19 This signaling is mediated by Notch ligand–receptor interactions between neighboring cells. In mammals, Notch receptors contain four members (Notch1–4), and Notch ligands include members of the Delta‐like (Dll) family (Dll1, Dll3, and Dll4) and Jagged (Jag) family (Jag1 and Jag2).20 Although both Notch receptors and their ligands are expressed by DCs, there is little information about the effects of Jag1 signaling on DC maturation and activation. One study showed that culturing DCs with Jag1‐expressing cells up‐regulated expressions of maturation markers and the production of IL‐12 by DCs.21 On the other hand, low expressions of Notch1 and Notch2 were detected in naive CD4+ T cells, and expressions of all receptors were induced in T cells after activation.22 Hence, during DC and T‐cell interactions, T‐cell differentiation can be controlled by Notch signaling. To accomplish that, the Jag group promotes Th2 or Treg responses, whereas the Dll group directs T‐cell polarization toward Th1.19, 23, 24 Several reports showed that Jag1 ligands possess immunosuppressive properties. A recent study demonstrated that adoptive transfer of isolated OX40L+ Jag1+ bone marrow‐derived DCs (BMDCs) led to Treg cell expansion and suppression of experimental autoimmune thyroiditis in recipient mice.25 Another report revealed that stimulation of CD4+ T cells with Jag1 facilitated antigen‐specific Treg cell production and function.26 The Jag1/Notch pathway influences the generation of CD4+ Foxp3+ Treg cells by triggering recruitment of the Notch‐specific transcription factor, CSL/RBP‐Jk, to the Foxp3 promoter and by inducing the Notch target gene, Hey1.27 Moreover, one study identified that knockdown of Jag1 expression in mesenchymal stromal cells reduced the CD4+ CD25+ Foxp3+ Treg cell population in an murine model of asthma.28 Collectively, those results highlight the potential application of gene therapy to effectively induce Treg cells by providing strong Jag1 stimulation.
In this study, we aimed to alter the functional behavior of BMDCs through Notch ligand Jag1 gene modification in vitro and subsequently observe what the effects of such modulation were on the phenotype and function of DCs. A Jag1‐expressing adenovirus (Ad‐Jag1) was constructed and transduced into BMDCs (Jag1‐DCs). The activation and function of Jag1‐DCs were subsequently evaluated. Furthermore, we evaluated the effects of Jag1‐DCs on the potential to suppress established Th2‐mediated allergic asthma in a mouse model. Herein, we provide a new treatment method that facilitates the clinical use of genetically modified DCs to ameliorate Th2‐mediated allergic diseases.
Materials and methods
Mice
Female BALB/c (H‐2d) and C57BL/6 (H‐2b) mice were obtained from the National Laboratory Animal Center and Laboratory Animal Center of National Taiwan University (Taipei, Taiwan). OT‐II mice expressing a transgenic T‐cell receptor that is specific for the peptide sequence 323–339 of ovalbumin (OVA) were kindly provided by Prof. Shih‐Jen Liu (National Health Research Institutes, Miaoli, Taiwan). Mice were maintained in the Animal Center of Taipei Medical University (TMU). All animals used for the experiments were 5–9 weeks of age. Animal care and handing protocols were approved by the animal use committee of the College of Medicine, TMU (approval no.: LAC‐2013‐0207).
Preparation of an adenovirus containing the Jag1 construct
The JAG1 gene was cloned from the pYX‐Asc‐Jag1 plasmid (a kind gift from Dr. Michael J. Bevan, University of Washington, Seattle, WA), subcloned into XbaI–NotI sites of the pShuttle2‐CMV vector, and then inserted into the pAdeno‐X vector after digestion with PI‐Sce I and I‐Ceu I (TaKaRa Bio, Mountain View, CA) to obtain Ad‐Jag1. As a control, Ad‐mock was made from the pAdeno‐X vector, which did not carry the JAG1 transgene. Both vectors were linearized with PacI and transfected into the human transformed embryonic kidney AD293 cell line to generate the Ad‐Jag1 virus. The adenovirus was propagated and amplified from infected AD293 cells. High‐titer adenoviral stocks were purified by successive banding by cesium chloride density‐gradient centrifugation. The purified viruses were further dialyzed and then stored at −80° until the experiment. Viral titers were measured by an end‐point dilution assay.
Preparation of BMDCs
Mouse BMDCS were prepared from tibial and femoral bone marrow of 5‐ to 9‐week‐old BALB/c mice. Bone marrow cells were cultured in RPMI‐1640 combined with 5% fetal bovine serum complete medium with granulocyte–macrophage colony‐stimulating factor (GM‐CSF) (500 U/ml; PeproTech, Rocky Hill, NJ) and IL‐4 (500 U/ml; Thermo Fisher Scientific, Rockford, IL) on day 0. Fresh medium containing GM‐CSF and IL‐4 was used to replace the old medium on day 2. Non‐adherent cells were collected, and the old medium was replaced with medium containing GM‐CSF and IL‐4 on day 4. On day 6 of culture, BMDCs (106 cells/well) were infected with the Ad‐Jag1 or Ad‐mock using different multiplicities of infection (MOIs) of 10, 50, 100, 200, 500, or 1000 in 5% fetal bovine serum–RPMI‐1640. At 4 hr after transduction, cells were moved to fresh medium containing GM‐CSF and IL‐4 for another 16–18 hr of culture. These immature DCs were then stimulated with 50 μg per well OVA protein (Sigma‐Aldrich, St. Louis, MO) at 37° for 24 hr to induce DC maturation and activation.
Reverse transcription polymerase chain reaction
To detect Jag1 messenger (m)RNA expression, virus‐transduced DCs (106 cells) were harvested, and total RNA was extracted according to the TRIzol method (Sigma‐Aldrich). Complementary (c)DNA was generated from 1 μg of total RNA using a high‐capacity cDNA RT kit (Thermo Fisher Scientific) in the presence of 1 μl of total cDNA and specific primers for murine Jag1 (forward, 5′‐CGGCTCTAGAATGCGGTCCCCACGGACGC‐3′ and reverse, 5′‐CCCAGCGGCCGCTATACGATGTATTCCATCCGGTTC‐3′). A Taq DNA polymerase 2× Master Mix RED kit (Ampliqon, Odense M, Denmark) was used to amplify Jag1 genes. The level of β‐actin mRNA was used as a control.
Flow cytometric analysis
We used flow cytometry to detect the transduction efficiency of the adenovirus and Jag1 protein expression by DCs. In addition, surface molecule expressions on OVA‐pulsed and virus‐transduced DCs were detected. Cells were harvested and stained with fluorescein isothiocyanate (FITC) ‐conjugated anti‐CD11c or phycoerythrin (PE)‐conjugated anti‐I‐A/I‐E (MHC class II), anti‐CD80, anti‐CD86, ICOSL, PD‐L1, OX40L (Thermo Fisher Scientific), and anti‐Jag1 (BioLegend, San Diego, CA) antibodies. For the CD4+ CD25+ Foxp3+ Treg cell analysis, T cells were stained with the following antibodies from Thermo Fisher Scientific: PE‐Cy5‐conjugated anti‐CD4, PE‐conjugated anti‐CD25, and FITC‐conjugated anti‐Foxp3 antibodies. Intracellular staining of Foxp3 was performed according to the manufacturer's protocol (BioLegend). Staining with isotype control antibodies was performed in all experiments. Following incubation with the antibodies, cells were washed and analyzed using FACSCalibur (Becton Dickinson, Mountain View, CA). Data were processed using the cellquest pro program (Becton Dickinson).
Determination of cytokine levels
Supernatants of treated DCs were collected, and cytokine levels of IL‐1β, IL‐6, and tumor necrosis factor‐α (TNF‐α) (Thermo Fisher Scientific) were determined by enzyme‐linked immunosorbent assay (ELISA). In asthma models, quantities of eotaxin, keratinocyte‐derived chemokine, TGF‐β, IL‐4, IL‐5, IL‐10, IL‐13, and interferon‐γ (IFN‐γ) from bronchoalveolar lavage fluid (BALF) were determined using ELISA kits (R&D Systems, Minneapolis, MN).
Induction of Treg cells in vitro
Splenocytes from C57BL/6 or OT‐II transgenic mice were isolated, resuspended as signal cells in MACS buffer, and stained with anti‐CD4 magnetic beads at 107 cells/20 μl (Miltenyi Biotec, Auburn, CA), which were incubated at 4° for 15 min. Cells were then washed and resuspended in MACS buffer to positively select CD4+ T cells. To identify whether the PD‐L1/PD‐1 pathway is involved in inducing Treg cells, anti‐PD‐L1 antibodies (BioLegend) were used to block the interaction between DCs and T cells. For the mixed lymphocyte reaction (MLR), 106 cells/well of purified CD4+ T cells from C57BL/6 mice were cultured with allogeneic DCs that were infected with Ad‐mock (at an MOI of 500) or Ad‐Jag1 (at an MOI of 500) and incubated with or without 10 μg/ml anti‐PD‐L1 antibodies, and then 50 μg/ml of the OVA protein was added at a DC : T‐cell ratio of 1 : 2 in the presence of IL‐2 (20 units/ml). For the antigen‐specific T‐cell response, treated DCs from BALB/c mice were co‐cultured with CD4+ T cells from OT‐II transgenic mice, and the DC : T‐cell ratio was the same as that used in the MLR. After 5 days of culture, numbers of CD25+ Foxp3+ Treg cells were analyzed by flow cytometry.
Sensitization, airway challenge, and adoptive transfer experiments
All groups of BALB/c mice (n = 5 per group) at 6 weeks of age were sensitized by an intraperitoneal injection of 50 μg OVA plus 4 mg alum (Thermo Fisher Scientific) on day 1. Mice were boosted with 30 μg OVA and the same dosage of alum on days 14, 28, and 44. On days 51 and 52, mice were intranasally challenged with 100 μg OVA and then further forced to inhale 5% OVA in 0·9% NaCl for 30 min daily for three consecutive days (days 53, 54, and 55). The AHR was measured 1 day after the final challenge, and mice were killed for further assays. Four groups of mice were respectively injected intravenously with 5 × 105 OVA‐stimulated mock DCs (at an MOI of 500), OVA‐stimulated Jag1‐DCs (at an MOI of 50 or 500), and OVA‐stimulated Ad‐uninfected DCs (control DCs) on day 45. OVA‐sensitized and OVA‐challenged mice without DC transfer served as an asthmatic positive control (PC) group.
OVA‐specific serum antibody assay
Serum samples were collected from mice on day 56. OVA‐specific IgE serum antibody titers were measured using an ELISA kit (Becton Dickinson Biosciences, San Jose, CA). The OVA‐specific IgE standard was derived by pooling sera from OVA‐sensitized mice. The concentration of standard serum was arbitrarily assigned as 1 ELISA unit (EU). Results are expressed as a ratio of the value to the standard.
Measurement of the AHR
After final exposure to the aerosol, the AHR of the mice was measured as an increase in the pulmonary resistance after challenge with aerosolized methacholine (MCh; Sigma‐Aldrich). Tracheotomized mice were anesthetized with 100 μl pentobarbital (10 mg/ml) and 50 μl zoletil (5 mg/ml). Ventilation was achieved at a rate of 150 breaths/min (model 683; Harvard Apparatus, South Natick, MA) and a tidal volume of 0·3 ml with a positive end‐expiratory pressure of 2–4 cmH2O using a ventilator. Increasing concentrations (1–32 mg/ml) of the MCh aerosol were administered by nebulization. After each MCh challenge, data were continuously collected for 3 min, and maximum values of the lung resistance (R L) (labview, National Instruments, Dallas, TX) were taken to represent changes in these functional parameters.
Determination of airway inflammation and lung tissue histology
After assessing the AHR, the lungs were immediately lavaged three times via the tracheal cannula with 1 ml of Hanks' balanced salt solution (HBSS). The recovered BALF was collected for the chemokine and cytokine assays. The total cell number in the BALF was counted using a hemocytometer and cytospin onto microscope slides. Cells were stained with Liu's staining solution (Chi I Pao, Taipei, Taiwan), and different cell types were classified as macrophages, eosinophils, neutrophils, and lymphocytes, based on standard morphological criteria. Numbers of the four types of cells were obtained by counting at least 200 cells in randomly selected fields of the slide. The lungs were immediately removed and cut into two parts. One half of a lung was fixed in 10% neutral‐buffered formalin after the lavage and embedded in paraffin wax. Five‐micrometer sections were prepared and stained with hematoxylin and eosin (H&E) and periodic acid‐Schiff (PAS). Stained slices were scanned with a digital camera and analyzed with imagej software (National Institutes of Health, Maryland, USA) to quantify the degree of inflammation and mucus production. The other halves of the lung and spleen were prepared for a CD4+ CD25+ Foxp3+ Treg cell analysis by flow cytometry.
Detection of Foxp3+ Treg cells in the lungs and spleen
Half of a lung from each group of mice was extracted and enzymatically digested with 4 mg/ml collagenase A (Sigma‐Aldrich) for 90 min at 37°. Then, cell suspensions and digested tissues were collected and passed through a 0·07‐mm nylon cell strainer. After further centrifugation, cells were washed, and the cell number was determined. Spleens from each group of mice were collected and ground into single‐cell suspensions. After centrifugation, ACK lysis buffer was used to remove red blood cells, which were then washed with HBSS. To determine the number of CD4+ CD25+ Foxp3+ Treg cells, these cells were stained with the Ture‐Nuclear™ transcription factor buffer set (BioLegend) and measured by flow cytometry.
Data analysis
Results are expressed as the mean ± standard error of the mean (SEM). Statistical analyses were performed using a one‐way analysis of variance (anova) followed by Dunnett's post‐hoc test. P values of < 0·05 were considered statistically significant.
Results
Ad‐Jag1 infection enhanced Jag1 gene and protein expressions by DCs
We prepared a recombinant adenovirus carrying the mouse JAG1 gene (Ad‐Jag1), and BMDCs from BALB/c mice were further infected with Ad‐Jag1 or Ad‐mock viral particles at various MOIs (of 10, 50, 200, 500, or 1000). After 24 hr of incubation, infected and uninfected DCs were pulsed with the OVA protein to induce DC maturation and activation. JAG1 mRNA expression was analyzed with RT‐PCR. As shown in Fig. 1(a), JAG1 mRNA exhibited low constitutive expression in uninfected DCs and Ad‐mock‐infected DCs (mock DCs), whereas levels of JAG1 mRNA were obviously enhanced in Ad‐Jag1‐infected DCs (Jag1‐DCs). In addition, surface expression of the JAG1 protein on DCs was identified by flow cytometry. Data showed that Ad‐Jag1 infection resulted in an increased percentage of Jag1+ DCs in a dose‐dependent manner (Fig. 1b). In contrast, low Jag1 expression was detected in immature DCs, Ad‐uninfected DCs (control DCs), and mock DCs.
Figure 1.
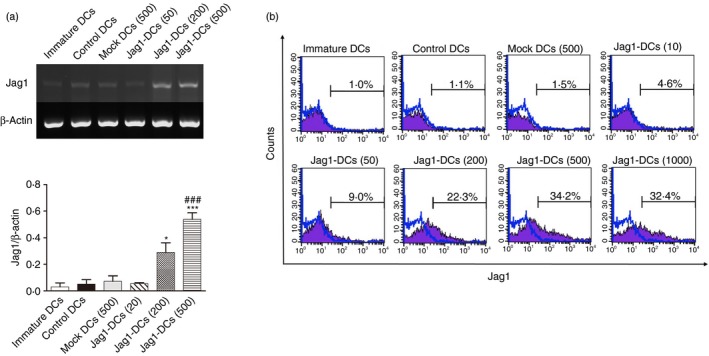
Levels of Jag1 (a) mRNA and (b) protein expressions following transduction of bone marrow‐derived dendritic cells (DCs) with the adenovirus vector, Ad‐Jag1. DCs were generated from bone marrow of BALB/c mice and cultured for 6 days. These DCs were infected with various multiplicities of infection (MOIs; of 10, 50, 200, 500, and 1000) of Ad‐Jag1 or Ad‐mock (500 MOI) for 24 hr, and then stimulated with ovalbumin (OVA; 50 μg/ml) for another 24 hr. Cells were collected, and Jag1 expression was detected by RT‐PCR and FACS analyses. Ratios of Jag1 to β‐actin were calculated, and results are expressed as the mean ± SEM of three independent experiments. Representative flow cytometric profiles of expression of Jag1 molecules on DCs are shown. *P < 0·05, ***P < 0·001 versus control DCs. ### P < 0·001 versus mock DCs.
Ad‐Jag1 infection promoted expressions of immunomodulatory molecules by DCs
It is well known that fully matured DCs display long‐lasting peptide–MHC class II complexes, co‐stimulatory molecules (CD40, CD80, and CD86) and intercellular adhesion molecules (CD54). These mature DCs also secrete high levels of pro‐inflammatory cytokines, such as TNF‐α, IL‐1β, and IL‐6.29 We wanted to ascertain whether overexpression of Jag1 modulated the phenotype or cytokine production by OVA‐stimulated DCs. Compared with control DCs and mock DCs, DCs infected with the adenovirus at various MOIs displayed no different effects on TNF‐α, IL‐1β, or IL‐6 production (Fig. 2a). Moreover, we used flow cytometry to investigate the phenotype profiles of Ad‐Jag1‐infected DCs. Results revealed that CD80 and CD86 molecules were up‐regulated in DCs after Ad‐Jag1 infection, whereas MHC II expression did not differ in these Ad‐infected DCs (Fig. 2b). We also analyzed expression levels of PD‐L1, OX40L, and ICOSL by Ad‐infected DCs. Notably, Jag1‐DCs expressed markedly increased levels of PD‐L1 and OX40L compared with control DCs and mock DCs. Collectively, Ad‐Jag1 infection did not reduce DC maturation, but it induced high expression levels of tolerance‐associated molecules; hence, we speculated that Jag1‐DCs may have the potential to induce Treg differentiation.
Figure 2.

Adenovirus (Ad)‐Jag1 infection changed the phenotype and cytokine pattern of dendritic cells (DCs). (a) Levels of pro‐inflammatory cytokines [interleukin‐1β (IL‐1β), IL‐6, and tumor necrosis factor‐α (TNF‐α)] from Ad‐Jag1‐infected DCs following ovalbumin (OVA) stimulation. On day 6 of culture, DCs were infected with Ad‐Jag1 (at MOIs of 50, 200, and 500) or Ad‐mock (at an MOI of 500) for 24 hr. After incubation, DCs were treated with OVA (50 μg/ml) for another 24 hr. Supernatants were harvested and analyzed using ELISA kits. (b) Expression levels of CD80, CD86, MHC class II, PD‐L1, OX40L, and ICOSL on Ad‐Jag1‐infected DCs were detected by flow cytometry. Representative flow cytometric profiles of expressions of these surface molecules on gated live CD11c+ cells are shown. The mean fluorescence intensity (MFI) was calculated, and results are expressed as the mean ± SEM of three independent experiments. *P < 0·05, **P < 0·01, ***P < 0·001 versus control DCs. # P < 0·05, ## P < 0·01, ### P < 0·001 versus mock DCs.
Jag1‐DCs induced Foxp3+ Treg cell differentiation in vitro
Dendritic cells are the most important professional antigen‐presenting cells and play a crucial role in the direction of T‐cell polarization. Therefore, we used an allogeneic MLR to clarify whether Jag1‐DCs possessed the ability to induce Treg cell differentiation. Furthermore, because the PD‐L1/PD‐1 pathway is involved in the induction of Treg cells by DCs, anti‐PD‐L1‐blocking antibodies were used to identify this possible mechanism. Allogeneic CD4+ T cells were purified and co‐cultured with different groups of DCs. As shown in Fig. 3(a), infection of DCs with Ad‐Jag1 at an MOI of 500 obviously promoted Foxp3+ Treg cell differentiation compared with control DCs and mock DCs. Furthermore, syngeneic OVA‐specific OT‐II CD4+ T cells were co‐cultured with differently treated DCs, and T‐cell polarization was measured. Consistent with the above results, the OT‐II CD4+ T‐cell population driven by OVA‐pulsed Jag1‐DCs induced a higher number of Treg cells than those driven by control DCs and mock DCs (Fig. 3b). Notably, anti‐PD‐L1 antibody treatment caused a partial reduction in the population of Foxp3+ Treg cells in both allogeneic and syngeneic systems. Taken together, these results demonstrated that overexpression of Jag1 by DCs enhanced their immunomodulatory ability to drive Foxp3+ Treg cell differentiation and implied that a direct interaction of PD‐L1 with PD‐1 receptors on T cells was required for Treg cell expansion by Jag1‐DCs.
Figure 3.
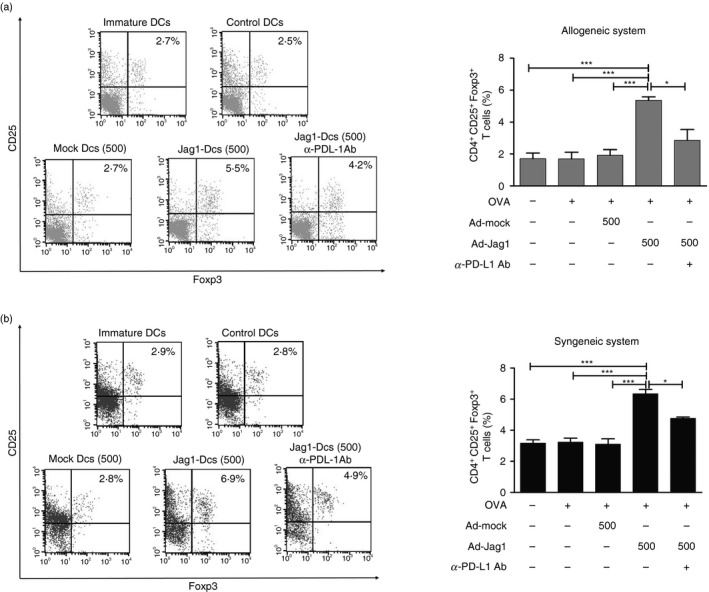
Jag1‐overexpressing dendritic cells (DCs) enhanced Foxp3+ regulatory T‐cell expansion in vitro. (a) In the mixed lymphocyte reaction, Jag1‐DCs from BALB/c mice were co‐cultured with naive CD4+ T cells from C57BL/6 mice. (b) For the ovalbumin (OVA) ‐specific CD4+ T‐cell response, differently treated DCs from BALB/6 mice were co‐cultured with syngeneic OT‐II CD4+ T cells. Anti‐PD‐L1 antibodies were used to block the PD‐L1/PD‐1 signaling pathway between DCs and T cells. After 5 days of culture, CD4+ T cells were collected and further analyzed by flow cytometry. Representative dot plots show expressions of CD25 and Foxp3 on gated CD4+ T cells. The percentage was calculated, and results are expressed as the mean ± SEM of three independent experiments. *P < 0·05, ***P < 0·001 versus Jag1‐DCs.
Jag1‐DC treatment reduced OVA‐specific IgE production and the severity of AHR in mice with OVA‐induced asthma
Based on the above in vitro results, we proposed that induction of Treg cells by Jag1‐DCs may be beneficial as a regulator in situations where an immune response needs to be suppressed. Hence, we further examined therapeutic effects of Jag1‐DCs in an OVA‐induced asthmatic animal model. All groups of mice were exposed to OVA sensitization and challenge. The treatment protocol is shown in Fig. 4(a). OVA‐stimulated control DCs, mock DCs, Jag1‐DCs (at 50 MOI), and Jag1‐DCs (at 500 MOI) were, respectively, transferred into four groups of OVA‐sensitized mice. Positive control mice (the PC group) received phosphate‐buffered saline injection instead of DCs. Serum samples were collected on day 56, and levels of OVA‐specific IgE were assayed by ELISA. In the PC, control DC, and mock DC groups, high levels of IgE production were observed. Conversely, administration of Jag1‐DCs at an MOI of 500 [Jag1‐DCs (500)] efficiently inhibited OVA‐specific IgE production (Fig. 4b). Some reports suggested that IL‐4, a Th2‐type cytokine, promotes IgE production.4 Our data indicated that Jag1‐DC treatment reduced Th2 responses in mice with allergic asthma. Further, we investigated whether administration of Jag1‐DCs could attenuate the development of AHR. After exposure to increasing concentrations of MCh, the degree of AHR was markedly increased in all groups of mice except Jag1‐DC‐treated mice (Fig. 4c). Results indicated that treatment with Jag1‐DCs caused a reduction in the OVA‐induced AHR.
Figure 4.
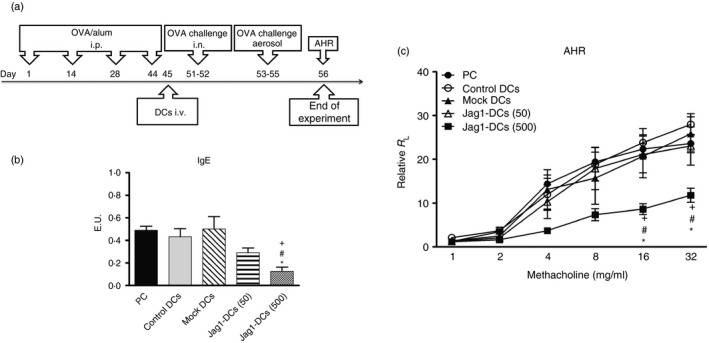
Treatment of Jag1‐DCs reduced anti‐ovalbumin (OVA) IgE production and the severity of airway hyperresponsiveness in mice with OVA‐induced asthma. (a) Procedures of animal sensitization and challenge are briefly summarized. Mice were randomized into five groups. On days 1, 14, 28, and 44, all groups of mice were sensitized by an intraperitoneal injection of the OVA allergen (100 μg/mice). On day 45, 5 × 105 OVA‐stimulated control dendritic cells (DCs), mock DCs [at a multiplicity of infection (MOI) of 500], and Jag1‐DCs (at MOIs of 50 and 500) were respectively transferred into four groups of mice. Positive control (PC) mice were injected with phosphate‐buffered saline (PBS) instead of cells. These mice were further challenged with OVA for five consecutive days (days 51–55), and the airway hyperresponsiveness (AHR) was measured 1 day after the last challenge (on day 56). Serum was collected on day 56. (b) Jag1‐DCs inhibited OVA‐specific IgE production and (c) the development of AHR in mice with OVA‐induced asthma. Results are expressed as the mean ± SEM of five mice in each group. *P < 0·05, ***P < 0·001 versus the PC group. # P < 0·05, ## P < 0·01 versus the control DC group. + P < 0·05, ++ P < 0·01 versus the mock DC group.
Jag1‐DCs suppressed allergen‐induced lung inflammation and the production of Th2 cytokines
To assess the anti‐allergic effects of Jag1‐DCs on allergic asthma, the accumulation of inflammatory cells in the BALF and lungs was observed. As shown in Fig. 5(a), PC mice exhibited increased numbers of total cells, eosinophils, and neutrophils in BALF. In addition, H&E‐ and PAS‐stained lung sections from the PC group showed clear infiltration of inflammatory cells around the bronchioles and mucus production by goblet cells (Fig. 5b). Similarly, administration of control DCs, mock DCs, or Jag1‐DCs (50) had no effect of alleviating the degree of airway inflammation. Unlike in PC mice, Jag1‐DC (500) treatment significantly diminished inflammatory cell recruitment and mucus secretion. Furthermore, mediators in the lungs were collected and assayed. BALF from PC mice was found to have markedly increased secretion of Th2 cytokines (IL‐4, IL‐5, and IL‐13) (Fig. 5c). Additionally, eotaxin and keratinocyte‐derived chemokine are chemokines for recruiting eosinophils and neutrophils, and these mediators had also increased in PC mice. Similar patterns were shown in control DC‐, mock DC‐, and Jag1‐DCs (50)‐treated mice. In contrast, Jag1‐DCs (500) treatment resulted in significant down‐regulation of the production of these mediators. Both IL‐10 and TGF‐β were proposed to have important anti‐inflammatory effects in the immune system and can be produced by Treg cells.11 In addition, IFN‐γ is a Th1‐type cytokine.30 Results showed that mice treated with a high dose of Jag1‐DCs (500) expressed a significantly enhanced level of TGF‐β in the lungs compared with those expressed in other groups. However, Jag1‐DC treatment had no effects on IL‐10 or IFN‐γ production in this organ. As expected, similar cytokine patterns were shown in culture supernatants of OVA‐restimulated splenocytes in these mice (see Supplementary material, Fig. S1). Together, these results indicated that Jag1‐DCs were able to inhibit Th2‐mediated responses and might induce TGF‐β‐producing Treg cells but produced no induction of IFN‐γ‐producing Th1 or IL‐10‐producing Treg cells during the development of OVA‐induced allergic asthma.
Figure 5.
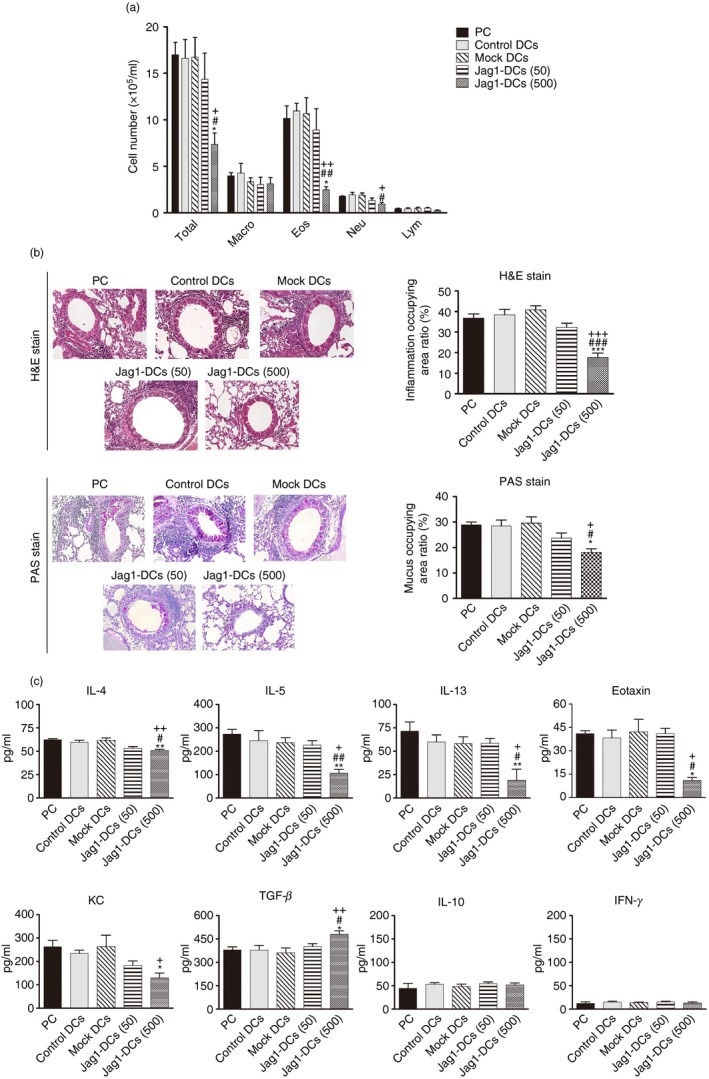
Effect of Jag1‐dendritic cells (DCs) on airway inflammation and production of mediators in bronchoalveolar lavage fluid (BALF). (a) Each group of mice was killed after measuring pulmonary function parameters. Cells from the BALF were stained with Liu's dye. Numbers of total cells, eosinophils, neutrophils, macrophages, and lymphocytes were counted. (b) Lung sections were stained with H&E and PAS to measure inflammatory cells and mucus production around the airway. Inflammatory changes and mucus production are respectively presented as percentages of the inflamed area and the PAS‐positive area. (c) BALF was collected and analyzed for interleukin‐4 (IL‐4), IL‐5, IL‐10, IL‐13, interferon‐γ (IFN‐γ), transforming growth factor‐β (TGF‐β), eotaxin, and keratinocyte‐derived chemokine (KC) contents by ELISA kits. Results are expressed as the mean ± SEM of five mice in each group. *P < 0·05, **P < 0·01, ***P < 0·001 versus the positive control (PC) group. # P < 0·05, ## P < 0·01, ### P < 0·001 versus the control DC group. + P < 0·05, ++ P < 0·01, +++ P < 0·001 versus the mock DC group.
Jag1‐DCs increased the generation of Treg cells in vivo
Based on the in vitro findings that Jag1‐DCs could promote Foxp3+ Treg cell development, we further identified the population of Foxp3+ Treg cells in Jag1‐DC‐treated mice. As shown in Fig. 6(a,b), there were low percentages of Foxp3+ Treg cells in the PC, control DC, and mock DC groups. In contrast, the Jag1‐DC (500)‐treated groups presented significantly increased frequencies of Foxp3+ Treg cells in both the lungs and spleen. Moreover, we used flow cytometry analysis to observe TGF‐β expression by Jag1‐DC‐induced Treg cells in vivo. The treatment process in the animal model is shown in the Supplementary material (Fig. S2a). As shown in the Supplementary material (Fig. S2b), the high‐dose Jag1‐DC‐treated mice expressed significantly increased percentages of TGF‐β‐producing Foxp3+ Treg cells compared with the control DC group. This evidence revealed that Jag1‐DC (500) treatment promotes the frequencies of TGF‐β‐producing Foxp3+ Treg cell differentiation in vivo. Overall, these data indicated that Jag1‐DCs might relieve the allergic syndrome through induction of TGF‐β‐producing Foxp3+ Treg cells to suppress effector Th2 responses.
Figure 6.
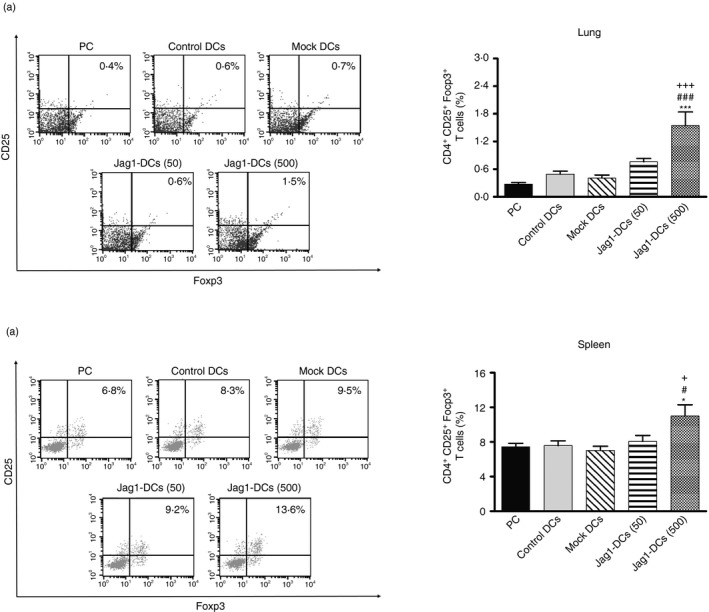
Jag1‐dendritic cell (DC) treatment enhanced the generation of Foxp3+ regulatory T (Treg) cells. The percentage of Foxp3+ Treg cells in (a) the lungs and (b) spleen. Lung and spleen cells from each group were analyzed by flow cytometry. Representative dot plots show expressions of CD25 and Foxp3 by gated CD4+ T cells. The percentage was calculated, and results are expressed as the mean ± SEM of five mice in each group. *P < 0·05, ***P < 0·001 versus the positive control (PC) group. # P < 0·05, ### P < 0·001 versus the control DC group. + P < 0·05, +++ P < 0·001 versus the mock DC group.
Discussion
Human adenoviruses have been developed as replication‐deficient gene delivery vectors and are used to treat cancers and allergic diseases.31, 32, 33 These vectors allow the transmission of genes to host nuclei but do not insert their genes into the host chromosome. Therefore, there is a low probability of disturbing vital cellular genes or processes.34 Today, replication‐competent adenoviral vectors have shown promising results in clinical trials.35, 36 On the other hand, as major contributors to the decision between immunity and tolerance, DCs are the focus of various immune interventions.37, 38, 39, 40 An attractive conceptual approach to enhancing the immunosuppressive capacity of DCs is their genetic modification so that they express immunomodulatory molecules, which induce Treg cells to inhibit effector T‐cell responses. Our previous study revealed that DCs transduced with the IL‐10 and IL‐12 genes by adenoviruses possessed the ability to promote IL‐10+ IFN‐γ + CD4+ Treg cell development and suppress asthmatic airway inflammation.41 Our current study is the first to demonstrate that overexpression of Notch ligand Jag1 by DCs enhanced their immunoregulatory capacity and exerted anti‐allergic effects on Th2‐mediated allergic asthma in mice by inducing Foxp3+ Treg cells. This study provides evidence showing that Jag1‐DCs may act as immunomodulators in allergic asthma.
In this study, we constructed Jag1‐DCs, and results clearly showed that the Jag1 protein was overexpressed by Jag1‐DCs, and these OVA‐stimulated Jag1‐DCs exhibited up‐regulated CD80 and CD86 expressions. However, using CD80/CD86 double‐knockout mice, Gopisetty et al.25 reported that either CD80 or CD86 on BMDCs was not required for the expansion of Treg cells. Hence, according to this evidence, we suspected that CD80 and CD86 might not affect Jag1‐DCs by causing expanded Treg cells. Pro‐inflammatory cytokines have a crucial role in lymphocyte activation and inducing an inflammatory response.42 Some reports documented that recombinant adenovirus‐infected DCs displayed enhanced levels of pro‐inflammatory cytokine production,43, 44 whereas our data showed no enhancement of these cytokines. We suggest that Ad‐Jag1 infection might not enhance DC‐mediated inflammatory responses.
In the results of the MLR and antigen‐specific T‐cell response assay, we confirmed that Jag1‐DCs significantly promoted the development of Foxp3+ Treg cells. One study revealed that soluble Jag1 released from Sertoli cells triggered Treg cell differentiation by activating the Foxp3 promoter.27 Other studies reported that transgenic expression of Jag1 by Epstein–Barr virus‐positive lymphoblastoid cell lines induced antigen‐specific Treg cells and thereby reduced the alloreactivity of T cells.26, 45 Moreover, Lin et al.46 transduced the Jag1 gene into the DC2.4 cell line by an adenovirus, and they demonstrated that blockade of the CD40 pathway enhanced the immune tolerance by Jag1‐overexpressing DC2.4 cells during heart transplantation. Collectively, these results indicate that Jag1 signaling is critical for inducing the generation of Treg cells. In our study, it was interesting to note that Jag1‐DCs expressed enhanced levels of PD‐L1 and OX40L surface molecules. We further demonstrated the important role of PD‐1 signaling in the induction of Treg cell expansion by engaging the PD‐1 receptor on T cells by PD‐L1 on Jag1‐DCs. Foxp3+ Treg cells expressed high levels of PD‐1, and a role for the PD‐L1/PD‐1 pathway in the development of Treg cells was supported by many studies. One study demonstrated that DCs expressing a great number of PD‐L1 molecules induced more Foxp3+ CD25+ CD4+ Treg cells upon co‐culture with CD4+ T cells.47 Conversely, DCs from PD‐L1‐deficient mice failed to expand such Treg cells in vitro.17 Importantly, by inhibiting the Notch signaling pathway in a lipopolysaccharide‐tolerant THP1 cell model, Pan et al. found that the Notch signaling pathway was involved in up‐regulating PD‐L1 expression in monocytes.48 On the other hand, the role of OX40L in the induction of Treg cells is controversial. Although a study by Gopisetty et al. demonstrated that OX40L‐ and Jagged1‐induced co‐signaling by BMDCs was required for Treg cell expansion,25 most reports supported up‐regulation of OX40L on DCs being implicated in the induction of Th2 responses and allergic asthma.49, 50 In addition, using a human model of allergic asthma, OX40L blockade did not alter the circulating Treg frequency.51 Hence, further work is needed to clarify the question of whether the expression of OX40L by Jag1‐DCs plays a role in regulating Treg cell induction.
In our animal model of OVA‐induced asthma, we demonstrated that Jag1‐DC treatment efficiently reduced Th2 cytokine production in the BALF, the severity of AHR, and eosinophil infiltration in the airway. In addition, enhanced numbers of CD4+ CD25+ Foxp3+ Treg cells associated with increased levels of TGF‐β production were observed in the lungs and spleen of Jag1‐DC‐treated mice. However, levels of IL‐10 secretion showed no differences among these groups of mice. One of the Treg populations, IL‐10‐producing Treg cells, also called type 1 regulatory cells, suppress immune responses by producing high levels of IL‐10.10 Hence, according to this result, we predicted that OVA‐stimulated Jag1‐DCs could not promote the induction of IL‐10‐producing Treg cells. In addition, although transfer of OVA‐pulsed Jag1‐DCs induced a low frequency of Foxp3+ Treg cells in vivo, these Treg cells specific for the OVA allergen might be more effective in suppressing effector Th2 responses in allergic asthma than were polyclonally activated Treg cells. The Treg cell response was proposed to protect against asthma by blocking T‐cell proliferation, reducing DC activation, and inhibiting Th2 cell responses.11 However, although Jag1‐DCs were shown to influence the Th2 response, and hence pulmonary inflammation, we are unsure whether suppression of the Th2 allergic response is a direct consequence of an enhanced Treg cell response. Further work is needed to additionally demonstrate that depletion of Treg cells with anti‐CD25 antibodies abrogates the protective effect of Jag1‐DCs to provide important proof that the protection is mediated by Treg cells.
In conclusion, our data demonstrated that the increased expression of PD‐L1 by Jag1‐DCs is required for the enhanced generation of Foxp3+ Treg cells in vitro. Furthermore, we proved that treatment of mice with Jag1‐DCs can ameliorate allergic asthma, and the suppressive mechanism may be associated with induction of the Foxp3+ Treg cell population to inhibit the Th2 response (Fig. 7). These results indicate that Ad‐Jag1‐infected DCs may be a novel candidate for use as an immunomodulator to prevent or treat allergic asthma.
Figure 7.
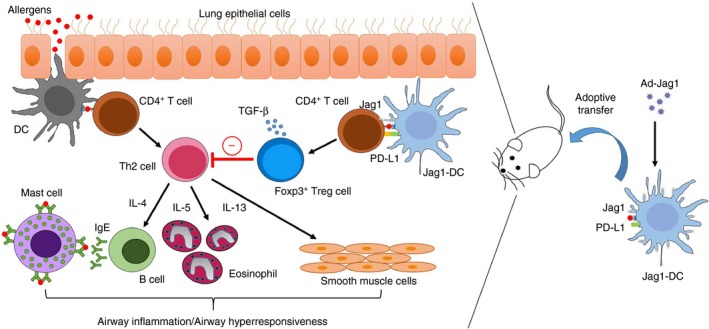
Jag1‐dendritic cells (DCs) attenuate T helper type 2 (Th2) ‐mediated allergic airway inflammation. The Jag1 gene‐modified DCs express up‐regulated Jag1 and PD‐L1 molecules. These modified DCs promote the CD4+ CD25+ Foxp3+ Treg cell expansion and ameliorate Th2‐mediated allergic airway inflammation in mice.
Disclosures
The authors declare that they have no financial conflicts of interest.
Supporting information
Figure S1. Effect of Jag1‐DCs on cytokine production of OVA‐restimulated splenocytes in allergic asthmatic mice.
Figure S2. Adoptive transfer of Jag1‐DCs increased frequencies of CD4+ Foxp3+ TGF‐β + Treg cells in a mouse model.
Acknowledgement
This study was supported by research grants (MOST103‐2320‐B‐038‐032‐MY3 and MOST106‐2320‐B‐038‐020) from the Ministry of Science and Technology, Taiwan.
References
- 1. Froidure A, Mouthuy J, Durham SR, Chanez P, Sibille Y, Pilette C. Asthma phenotypes and IgE responses. Eur Respir J 2016; 47:304–19. [DOI] [PubMed] [Google Scholar]
- 2. Comberiati P, Di Cicco ME, D'Elios S, Peroni DG. How much asthma is atopic in children? Front Pediatr 2017; 5:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fahy JV. Type 2 inflammation in asthma – present in most, absent in many. Nat Rev Immunol 2015; 15:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol 2015; 16:45–56. [DOI] [PubMed] [Google Scholar]
- 5. Ingram JL, Kraft M. IL‐13 in asthma and allergic disease: asthma phenotypes and targeted therapies. J Allergy Clin Immunol 2012; 130:829–42. [DOI] [PubMed] [Google Scholar]
- 6. Fanta CH. Asthma. N Engl J Med 2009; 360:1002–14. [DOI] [PubMed] [Google Scholar]
- 7. Mukherjee M, Svenningsen S, Nair P. Glucocortiosteroid subsensitivity and asthma severity. Curr Opin Pulm Med 2017; 23:78–88. [DOI] [PubMed] [Google Scholar]
- 8. Noval Rivas M, Chatila TA. Regulatory T cells in allergic diseases. J Allergy Clin Immunol 2016; 138:639–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stelmaszczyk‐Emmel A. Regulatory T cells in children with allergy and asthma: it is time to act. Respir Physiol Neurobiol 2015; 209:59–63. [DOI] [PubMed] [Google Scholar]
- 10. Palomares O, Martín‐Fontecha M, Lauener R, Traidl‐Hoffmann C, Cavkaytar O, Akdis M et al Regulatory T cells and immune regulation of allergic diseases: roles of IL‐10 and TGF‐β . Genes Immun 2014; 15:511–20. [DOI] [PubMed] [Google Scholar]
- 11. Palomares O, Akdis M, Martin‐Fontecha M, Akdis CA. Mechanisms of immune regulation in allergic diseases: the role of regulatory T and B cells. Immunol Rev 2017; 278:219–36. [DOI] [PubMed] [Google Scholar]
- 12. Yu H, Paiva R, Flavell RA. Harnessing the power of regulatory T‐cells to control autoimmune diabetes: overview and perspective. Immunology 2018; 153:161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol 2017; 8:121–33. [DOI] [PubMed] [Google Scholar]
- 14. Hu J, Wan Y. Tolerogenic dendritic cells and their potential applications. Immunology 2011; 132:307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zheng J, Chan PL, Liu Y, Qin G, Xiang Z, Lam KT et al ICOS regulates the generation and function of human CD4+ Treg in a CTLA‐4 dependent manner. PLoS ONE 2013; 8:e82203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Willoughby J, Griffiths J, Tews I, Cragg MS. OX40: structure and function – what questions remain? Mol Immunol 2017; 83:13–22. [DOI] [PubMed] [Google Scholar]
- 17. Liu H, Bakthavatsalam R, Meng Z, Li Z, Li W, Perkins JD et al PD‐L1 signal on liver dendritic cells is critical for Foxp3+CD4+CD25+ Treg and liver tolerance induction in mice. Transplant Proc 2013; 45:1853–5. [DOI] [PubMed] [Google Scholar]
- 18. Fazio C, Ricciardiello L. Inflammation and Notch signaling: a crosstalk with opposite effects on tumorigenesis. Cell Death Dis 2016; 7:e2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amsen D, Helbig C, Backer RA. Notch in T cell differentiation: all things considered. Trends Immunol 2015; 36:802–14. [DOI] [PubMed] [Google Scholar]
- 20. Zhou ZD, Kumari U, Xiao ZC, Tan EK. Notch as a molecular switch in neural stem cells. IUBMB Life 2010; 62:618–23. [DOI] [PubMed] [Google Scholar]
- 21. Weijzen S, Velders MP, Elmishad AG, Bacon PE, Panella JR, Nickoloff BJ et al The Notch ligand Jagged‐1 is able to induce maturation of monocyte‐derived human dendritic cells. J Immunol 2002; 169:4273–8. [DOI] [PubMed] [Google Scholar]
- 22. Koyanagi A, Sekine C, Yagita H. Expression of Notch receptors and ligands on immature and mature T cells. Biochem Biophys Res Commun 2012; 418:799–805. [DOI] [PubMed] [Google Scholar]
- 23. Mochizuki K, He S, Zhang Y. Notch and inflammatory T‐cell response: new developments and challenges. Immunotherapy 2011; 3:1353–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dongre A, Surampudi L, Lawlor RG, Fauq AH, Miele L, Golde TE et al Non‐canonical notch signaling drives activation and differentiation of peripheral CD4+ T cells. Front Immunol 2014; 5:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gopisetty A, Bhattacharya P, Haddad C, Bruno JC Jr, Vasu C, Miele L et al OX40L/Jagged1 cosignaling by GM‐CSF‐induced bone marrow‐derived dendritic cells is required for the expansion of functional regulatory T cells. J Immunol 2013; 190:5516–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yvon ES, Vigouroux S, Rousseau RF, Biagi E, Amrolia P, Dotti G et al Overexpression of the Notch ligand, Jagged‐1, induces alloantigen‐specific human regulatory T cells. Blood 2003; 102:3815–21. [DOI] [PubMed] [Google Scholar]
- 27. Campese AF, Grazioli P, de Cesaris P, Riccioli A, Bellavia D, Pelullo M et al Mouse Sertoli cells sustain de novo generation of regulatory T cells by triggering the notch pathway through soluble JAGGED1. Biol Reprod 2014; 90:53. [DOI] [PubMed] [Google Scholar]
- 28. Cahill EF, Tobin LM, Carty F, Mahon BP, English K. Jagged‐1 is required for the expansion of CD4+ CD25+ FoxP3+ regulatory T cells and tolerogenic dendritic cells by murine mesenchymal stromal cells. Stem Cell Res Ther 2015; 6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kapsenberg ML. Dendritic‐cell control of pathogen‐driven T‐cell polarization. Nat Rev Immunol 2003; 3:984–93. [DOI] [PubMed] [Google Scholar]
- 30. Tumes DJ, Papadopoulos M, Endo Y, Onodera A, Hirahara K, Nakayama T. Epigenetic regulation of T‐helper cell differentiation, memory, and plasticity in allergic asthma. Immunol Rev 2017; 278:8–19. [DOI] [PubMed] [Google Scholar]
- 31. Kanerva A, Raki M, Hemminki A. Gene therapy of gynaecological diseases. Expert Opin Biol Ther 2007; 7:1347–61. [DOI] [PubMed] [Google Scholar]
- 32. Haviv YS, Blackwell JL, Kanerva A, Nagi P, Krasnykh V, Dmitriev I et al Adenoviral gene therapy for renal cancer requires retargeting to alternative cellular receptors. Cancer Res 2002; 62:4273–81. [PubMed] [Google Scholar]
- 33. Fu CL, Chuang YH, Chau LY, Chiang BL. Effects of adenovirus‐expressing IL‐10 in alleviating airway inflammation in asthma. J Gene Med 2006; 8:1393–9. [DOI] [PubMed] [Google Scholar]
- 34. Lee CS, Bishop ES, Zhang R, Yu X, Farina EM, Yan S et al Adenovirus‐mediated gene delivery: potential applications for gene and cell‐based therapies in the new era of personalized medicine. Genes Dis 2017; 4:43–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim KH, Dmitriev IP, Saddekni S, Kashentseva EA, Harris RD, Aurigemma R et al A phase I clinical trial of Ad5/3‐Δ24, a novel serotype‐chimeric, infectivity‐enhanced, conditionally‐replicative adenovirus (CRAd), in patients with recurrent ovarian cancer. Gynecol Oncol 2013; 130:518–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu FC, Hou LH, Li JX, Wu SP, Liu P, Zhang GR et al Safety and immunogenicity of a novel recombinant adenovirus type‐5 vector‐based Ebola vaccine in healthy adults in China: preliminary report of a randomised, double‐blind, placebo‐controlled, phase 1 trial. Lancet 2015; 385:2272–9. [DOI] [PubMed] [Google Scholar]
- 37. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12:265–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gross CC, Wiendl H. Dendritic cell vaccination in autoimmune disease. Curr Opin Rheumatol 2013; 25:268–74. [DOI] [PubMed] [Google Scholar]
- 39. Sabado RL, Bhardwaj N. Directing dendritic cell immunotherapy towards successful cancer treatment. Immunotherapy 2010; 2:37–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garcia‐Gonzalez P, Ubilla‐Olguin G, Catalan D, Schinnerling K, Aguillon JC. Tolerogenic dendritic cells for reprogramming of lymphocyte responses in autoimmune diseases. Autoimmun Rev 2016; 15:1071–80. [DOI] [PubMed] [Google Scholar]
- 41. Hsu CY, Leu SJ, Chiang BL, Liu HE, Su HC, Lee YL. Cytokine gene‐modulated dendritic cells protect against allergic airway inflammation by inducing IL‐10+IFN‐γ +CD4+ T cells. Gene Ther 2010; 17:1011–21. [DOI] [PubMed] [Google Scholar]
- 42. Kochupurakkal NM, Kruger AJ, Tripathi S, Zhu B, Adams LT, Rainbow DB et al Blockade of the programmed death‐1 (PD1) pathway undermines potent genetic protection from type 1 diabetes. PLoS ONE 2014; 9:e89561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Korst RJ, Mahtabifard A, Yamada R, Crystal RG. Effect of adenovirus gene transfer vectors on the immunologic functions of mouse dendritic cells. Mol Ther 2002; 5:307–15. [DOI] [PubMed] [Google Scholar]
- 44. Hirschowitz EA, Weaver JD, Hidalgo GE, Doherty DE. Murine dendritic cells infected with adenovirus vectors show signs of activation. Gene Ther 2000; 7:1112–20. [DOI] [PubMed] [Google Scholar]
- 45. Vigouroux S, Yvon E, Wagner HJ, Biagi E, Dotti G, Sili U et al Induction of antigen‐specific regulatory T cells following overexpression of a Notch ligand by human B lymphocytes. J Virol 2003; 77:10872–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lin Y, Chen W, Li J, Yan G, Li C, Jin N et al Overexpression of Jagged‐1 combined with blockade of CD40 pathway prolongs allograft survival. Immunol Cell Biol 2015; 93:213–7. [DOI] [PubMed] [Google Scholar]
- 47. Wong TH, Chen HA, Gau RJ, Yen JH, Suen JL. Heme oxygenase‐1‐expressing dendritic cells promote Foxp3+ regulatory T cell differentiation and induce less severe airway inflammation in murine models. PLoS ONE 2016; 11:e0168919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pan T, Liu Z, Yin J, Zhou T, Liu J, Qu H. Notch signaling pathway was involved in regulating programmed cell death 1 expression during sepsis‐induced immunosuppression. Mediators Inflamm 2015; 2015:539841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burrows KE, Dumont C, Thompson CL, Catley MC, Dixon KL, Marshall D. OX40 blockade inhibits house dust mite driven allergic lung inflammation in mice and in vitro allergic responses in humans. Eur J Immunol 2015; 45:1116–28. [DOI] [PubMed] [Google Scholar]
- 50. Froidure A, Shen C, Pilette C. Dendritic cells revisited in human allergic rhinitis and asthma. Allergy 2016; 71:137–48. [DOI] [PubMed] [Google Scholar]
- 51. Baatjes AJ, Smith SG, Dua B, Watson R, Gauvreau GM, O'Byrne PM. Treatment with anti‐OX40L or anti‐TSLP does not alter the frequency of T regulatory cells in allergic asthmatics. Allergy 2015; 70:1505–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of Jag1‐DCs on cytokine production of OVA‐restimulated splenocytes in allergic asthmatic mice.
Figure S2. Adoptive transfer of Jag1‐DCs increased frequencies of CD4+ Foxp3+ TGF‐β + Treg cells in a mouse model.