

Abstract
Although it has been proposed that NOD-like receptor protein 3 (NLRP3) inflammasome activation may have an important contribution to the onset of bladder pain syndrome/interstitial cystitis (BPS/IC), as of today there is still insufficient evidence to accept or to reject this hypothesis. However, taking into consideration that inflammasomes have been already shown as important mediators of cyclophosphamide-induced bladder inflammation and that some studies have also revealed human bladder epithelium expresses high levels of NLRP3, such a hypothesis seems to be reasonable. The purpose of this review is to discuss a scenario that NLRP3 inflammasome is a crucial player in the development of this disease. Identification of a novel mediator of bladder inflammation and pain could lead to emerging new therapeutic strategy and the first causative therapy.
Keywords: BPS/IC, inflammasome, NLRP3, OAB, sterile inflammation
Bladder pain syndrome/interstitial cystitis
Bladder pain syndrome/interstitial cystitis (BPS/IC) is characterized by persistent or recurrent pain or discomfort in the region of the urinary bladder that increases with bladder filling and coexists with other lower urinary tract symptoms such as urinary urgency, frequency, or nocturia.1,2 The BPS/IC can be diagnosed, if the symptoms persist for more than 6 weeks/6 months in the absence of other possible causes.1,3
The disease does not constitute a direct threat to life, but it contributes significantly to a vast decline in the quality of life in almost all its domains.4–6 Although the description of the disease has been present in the literature for many years, its pathogenesis, natural course, and pathological mechanisms are not yet fully understood.
The necessity to recognize pathogenetic mechanisms involved in BPS/IC determines the possibility of developing effective treatment. What seems to be particularly important in the face of epidemiological data, which estimates that in Western countries the prevalence of BPS/IC may reach even 1,700–6,530 cases per 100,000 among females7–9 and 1,300–4,200 cases per 100,000 in males.5,6,9
In studies focused on bladder wall morphology in the course of BPS/IC, various pathological changes were described in all histological layers.10 The most frequently reported are: reduction in urothelium thickness,10–12 disturbances in the glycosaminoglycan (GAG) layer,10,13 abnormal smooth muscle structure,10,14 microvasculature and nerve density upregulation,15–18 mast cell infiltration, and fibrosis.14,17–19 Despite numerous studies to date, the nature of BPS/IC is not yet fully understood. It seems that the phenomena occurring in the course of the disease form a self-perpetuating vicious circle,20 whose main assumptions are presented in Figure 1. The emanation of these events is the inflammation constituting a key element of the events cascade at BPS/IC. The stimulation observed in this disease survives the condition of inflammation into a chronic form, obtaining independence from the triggering factors.20,21
Figure 1.
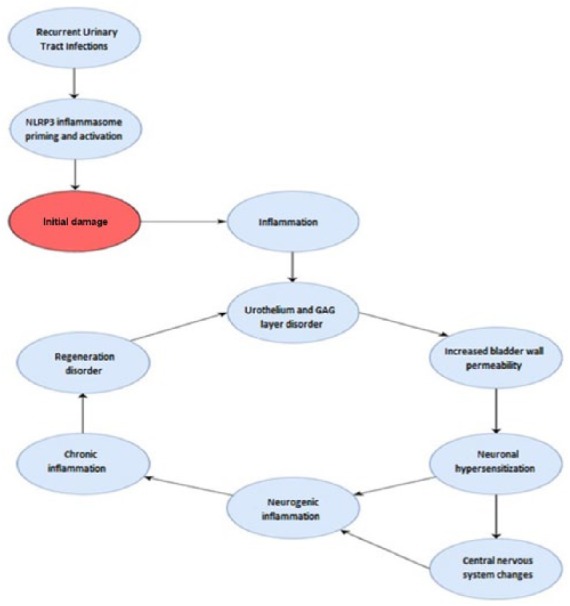
The self-perpetuating vicious circle that leads to continuously deepening pathology, resulting in full-blown BPS/IC.
Urothelium and GAG disorders
The bladder epithelium is coated with extracellular matrix layer, composed mainly of GAG, collagen, elastin, fibronectin, and laminin. In the bladder, the most common representatives of the GAG family are hyaluronic acid, heparin as well as sulphates: keratan, dermatan, heparan, and chondroitin.10,13,22,23 These molecules are polarized and form a highly hydrated protective layer.10 Damage to the GAG and urothelium barrier increases the permeability of the bladder mucosa to the potassium ions, allowing them to penetrate deep into the wall. This causes changes in electrochemical balance and activation of nerve endings, triggering a painful stimulus. The induced disorder induces local inflammation, stimulates chemotaxis, and activates mast cells. The stimulated neurons may also secrete proinflammatory substances in antidromic route that support neurogenic inflammation.10,20
On the other hand, inflammation can be induced by the active secretion of urothelial cells. Experimental studies have shown that mechanical or chemical stimuli acting on the bladder epithelium, cause the secretion of numerous neurotransmitters such as adenosine triphosphate, acetylcholine, nitric oxide, or prostaglandins.10,24,25 This can contribute to the development of inflammation.
Neurogenic and neuroendocrine factors
Neuroendocrine pathways, including the sympathetic system and the hypothalamic–pituitary–adrenal axis, are able to modulate inflammation. In the course of antidromic neurosecretion, they can induce spontaneous neurogenic inflammation.26–29 In the course of BPS/IC, disturbances in the functions of these systems have been observed. This causes dysfunction of transmission pathways between the bladder and the nervous system, in particular sensory neurons.10,20
Moreover, in patients suffering from BPS/IC, the activity of brainstem and hypothalamus was increased, stimulating the secretion of corticoliberin [or corticotropin releasing hormone (CRH)] and adrenocorticotropic hormone (ACTH).10 In addition to central activity, CRH demonstrates a peripheral proinflammatory effect, promoting mast cell activation in various tissues, including the bladder wall.30 Subsequent anomalies in ACTH secretion are associated with increased catecholamine production, as evidenced by the increase in catecholamine metabolites in the urine in this population.31–33
Pathologies in bladder wall are accompanied by changes in central nervous system. Continuous stimulation modifies gene expression and secretion of neurotransmitters, causing the phenomenon of paving stimuli. This strongly facilitates the development and maintenance of neurogenic inflammation.20,34
Inflammation
Inflammation is a key element in pathology of BPS/IC. Numerous studies have shown an increase in the levels of proinflammatory cytokines in patients with BPS/IC.17,20,35,36 Morphological features of inflammation were also visible in biopsies of the bladder wall. In addition to mast cell infiltration, the influx of other immune cells, mainly eosinophils and macrophages, was also reported.15,18 The main chemotactic factors in BPS/IC are proteins from the C-C subfamily: CCL2, CCL4 and CCL11, similar to other causes of chronic inflammation of the bladder.26,37
The systemic emanation of the inflammatory process in patients with BPS/IC is an increase in the concentration of acute phase proteins in the serum. The increased levels of nerve growth factor (NGF), tumor necrosis factor (TNFα), and interleukin (IL)-1β, IL-6, and IL-8 have been described.10,17,20
The pathogenesis of BPS/IC
The pathogenesis of BPS/IC is most likely multifaceted; the exact cause, however, remains unknown. A nonmicrobial inflammation is widely recognized as a key pathological mechanism that mediates or promotes the development of bladder pathology associated with urinary symptoms and BPS/IC symptoms .38–42 Despite that, it cannot be ruled out that the very initial steps of BPS/IC are provoked by pathogens.
The most convincing theory seems to be hypothesis proposing that BPS/IC is initiated by a defective bladder lining.43–48 Epithelial dysfunction, arising from an alteration in the composition of the GAG layer lining the bladder, might be initially caused by recurring urinary tract infection (rUTI). It must be underlined here that medical records of BPS/IC patients very often mention such infections in the past. Although they are not a direct cause of BPS/IC symptoms (any infection must be ruled out first to diagnose BPS/IC), they lead to urothelium damage and may allow metabolites to access underlying cells where they would act as a danger-associate molecular patterns (DAMPs).
The most common cause of the infection is Gram-negative urinary pathogen Escherichia coli which is also the source of lipopolysaccharide (LPS; 75–95% of rUTI cases are caused by E. coli).49 According to the two-signal model which will be presented in the next chapter, LPS is providing a ‘signal I’.50 Then, specific agents acting as a ‘signal II’, for example, DAMPs, lead to the inflammasome activation, and subsequently to processing of pro-IL-1β and finally to releasing its bioactive form IL-1β. As was suggested by,51 IL-1β represents an inducible danger signal. The releasing of IL-1β could trigger pyroptosis and sloughing of urothelial cells.52,53 Thus, the outcome of frequently rUTIs will be initial defect of barrier lining the urothelium and concomitant increase in permeability and sensitivity to diverse agents (please refer to Figure 1).
Overall, epithelial dysfunction results in migration of the urinary solutes (e.g. potassium ions, allergens, chemicals, or drugs) across the abnormally permeable urothelium. Hence, even innocuous urinary solutes such as dietary metabolites may become noxious stimuli; urinary potassium absorption or specific interactions between different components with the urothelium cause the inflammation, mast cell invasion, and activation of the submucosal nerve filaments.44–46,54 In accordance with that, results of survey studies indicated that a high occurrence of dietary factor sensitivity made bladder symptoms worse in the population of patients with BPS/IC.55–58 For instance, a study conducted by researchers from the University of South Florida revealed that nearly all (96%) of the 598 respondents indicated that certain foodstuffs made symptoms worse.59
Although the effects of foods and beverages on BPS/IC symptoms have been acknowledged for many years, the exact mechanism has never been elucidated.40,41,60,61 The previously cited studies provide a good explanation that ingesting certain foodstuffs can lead to worsening of BPS/IC symptoms. Owing to the initial epithelial dysfunction, some unidentified dietary metabolites may constitute an endogenous danger signal that activates an innate immune pattern recognition receptor (PRR) contributing to BPS/IC symptoms. If the hypothesis is proven to be correct, such metabolites should be classified as DAMPs. DAMPs are a very heterogenous group composed of compounds as different as uric acid, homocysteine, cholesterol crystals, and many other host derived factors. In the case of BPS/IC patients, potential DAMPs are so diversified that they constitute a problem difficult to eliminate. As a result, it seems more reasonable to inhibit one common mediator. NOD-like receptor protein 3 (NLRP3) inflammasome appears to be a good molecular target, regardless of which metabolite causes symptoms.
Inflammasomes
The inflammasomes are supramolecular, signaling complexes that sense signals through the recognition of patterns found on various molecules (e.g. molecules that penetrated into the bladder wall). Then inflammasomes promote inflammation by maturation and release of proinflammatory cytokines, IL-1β and IL-18.53
Inflammasome can be formed by nucleotide-binding oligomerization domain and leucine-rich repeat-containing receptors (NLRs) and absent in melanoma 2- like receptors (AIM-2). Depending on inflammasome type, it may consist of apoptosis associated speck-like protein containing a caspase recruitment domain (ASC), NACHT, LRR and PYD domains-containing protein 3 (NALP3), caspase 1, and in some cases caspase 5. The exact composition of an inflammasome depends on the activator that initiates inflammasome formation. Of all inflammasomes types, by far the most studied is NLRP3 (inflammasomes are named after the PRRs that organize them).
Regardless of inflammasome type, expression levels are usually low in most cell types at the steady state and the process of inflammasomes formation is tightly regulated. A ‘two-signal’ model has been proposed to explain the regulation of an active inflammasome in a cell.
Signal I
Before a functional NLRP3 inflammasome can be formed, NLRP3 expression must achieve a sufficient level. Furthermore, a suitable level of pro-IL-1β must be also accumulated. This is a process known also as priming and it is required to promote the transcriptional expression of key components of the inflammasome. The priming step is an effect of activation of non-LPR receptors, for example, toll-like receptor 4 (TLR4). TLRs are cell surface molecules that sample molecular patterns in the endosomal and extracellular spaces. After a contact with an activating stimulus (i.e. signal I) such as LPS, they induce nuclear factor (NF)-κB pathway, which stimulates the transcription of pro-IL-1β and NLRP3 protein as well as other inflammasome components.62,63
Signal II
The second step, inflammasome activation, is initiated either by the same or by additional stimuli. In response to pathogen- and/or damage-associated molecular patterns (PAMPs and/or DAMPs, respectively), cytoplasmic nucleotide oligomerization domain-like receptors (NOD-like receptors or NLRs, they are part of PRR) become activated. This step induces the functional activity of the NLRP3 inflammasome. In the absence of signal II, pro-IL-1b is upregulated but usually fails to be processed or released.
NLR recognizes molecular patterns in various molecules released from damaged cells (DAMP) or patterns present in antigens produced by pathogens (PAMP).64
(1) The PAMP group is composed of exogenous molecules carried by pathogens and represent well-known immunogenic components of bacteria, for example, flagellin or LPS .65–67 Depending on inflammasome type, different microbial signals can trigger the inflammasome formation. PAMPs can induce vigorous production of proinflammatory cytokines, contributing to intracellular inflammatory cascades. This is why LPS is considered a pivotal trigger of several inflammatory diseases.68–70 Furthermore, an E. coli infection is accompanied by cell and tissue injuries triggering the production and secretion of DAMPs.
(2) The DAMP group is composed of endogenic molecules released from injured or necrotic cells. The most common endogenic substances derived from the dying or damaged cells are listed in Table 1.
Table 1.
A list of common inflammasome activators.
| The most common endogenic substances derived from the dying or injured host cells (DAMPs): |
| Mitochondria derived effector molecules such as mitochondrial reactive oxygen species (mROS), mitochondrial oxidized DNA 71–73 |
| Intracellular molecules such as ATP, DNA as well as proteins derived from extracellular matrix. 74 |
| Uric acid, the product of purine catabolism75 |
| Homocysteine, amino acid derived from protein catabolism involving methionine.76 |
| Glycosoaminoglycans,74,77,78 described in the text. |
| Cholesterol crystals or oxidative products of cholesterol, which may serve as a DAMPs.64,74,79 |
| The most common PAMPs: |
| Flagellin 80 |
| LPS 65 |
| Others: |
| Environmental irritants such as asbestos, alum, aluminum salts, and silica.71,74,79 |
| NLRP3 may be activated by trauma and excessive exercises that produce some chemokines. 81 |
Upon activation, NLRs assemble in the cytosol to form a multimeric structure, known as an inflammasome.62 During that process their recruit the adaptor protein (ASC), which in turn recruits pro-caspase-1. The next step is pro-caspase-1 self-cleavage to its active analog. Assembled inflammasome controls the activation of caspase-1, resulting in the processing of inactive forms of proinflammatory cytokines, for example, pro-IL-1β and pro-Il-18 into their mature, bioactive, secreted forms (IL-1β and IL-18, respectively). In addition, pyroptotic cell death may be induced.52,53
Inflammasomes play a crucial role in the innate immunity and have been referred to as the ‘central processing unit’ of the inflammatory response.64,82 They activate inflammatory caspases to produce interleukins. The mature interleukins, IL-1β and IL-18, trigger classic components of the inflammation and coordinate the initiation of an inflammatory response.83 Active forms attract mast cells and launch releasing of histamine by mast cells which results in swelling and inflammatory infiltration (worth of notion is the fact that both elevated levels of IL-1β and IL-18 and strong mastocystosis are the well-known features of BPS/IC).84,85 In general, the activation of inflammasomes results in alarm which is raised to alert adjacent cells.
Much attention has been given to the NLRP3 inflammasome, as it has recently been reported as a central pathogenic mechanism of several chronic degenerative diseases. NLRP3 inflammasome composed of three proteins: a sensor (nucleotide-binding oligomerization domain-like receptor pyrin domain-containing-3 ][NLRP3]), an adaptor protein ASC (apoptosis associated speck like protein containing a CARD) and catalytic protein (cysteine protease procaspase-1). It is expressed in the cytosol of monocytes, dendritic cells, neutrophils, lymphocytes and epithelial cells. In epithelium localization of NLRP3 inflammasome is restricted to stratified, non-keratinizing epithelia, including bladder urothelium and ureter .86,87 Its activation is considered as ubiquitous in different organs and cells. Upon detecting inflammatory signals, the inflammasome sensor molecule induces a rapid polymerization of the adaptor ASC into large helical filaments (‘specks’), which represents the hallmark of inflammasome activation. By recognizing a wide variety of pathogenic microorganisms, for example, E. coli as well as host-derived sterile stressors associated with tissue injury, NLRP3 is strongly engaged in innate immune system.74,88,89
Inflammasome-based etiology of BPS/IC
Taking into consideration that defects of urothelial GAG component allows penetration of such a wide variety of compounds as environmental particles, various allergens, chemicals, drugs, and toxins (e.g. those from tobacco) into the bladder wall it may be assumed that some of them would act as a DAMP and trigger the activation of specific NLR present in the cytoplasm of uroepithelial cells, finally resulting in formation of inflammasomes.87,90
Exogenous stimuli (such as endogenously generated molecules including DAMPs), do not only activate inflammasome, but also stimulates production of reactive oxygen species (ROS) leading to further tissue injuries.76 Damaged tissues in turn release a new set of DAMPs pouring further oil on the fire. Despite it is still the body’s natural defense, the described phenomena combine and create a self-perpetuating vicious circle that leads to continuously deepening pathology, resulting in full-blown BPS/IC (it is also worth noting the fact that in such approach urine plays a role in human health not only as a waste excretion but also as a vehicle for bioactive agents contributing in development of the disease).91
In summary, in the case of BPS/IC, we have to consider the following scenario: (1) LPS derived from E. coli primes inflammasome formation; (2) urinary solutes or some molecules (e.g. derived from damaged cells) that penetrated the bladder wall due to defective bladder lining activate inflammasome;92 (3) now this multiprotein complex is capable of producing bioactive interleukins, which in turn intensify inflammatory response; (4) tissues that have been already injured are in further contact with increasingly numerous danger factors; (5) as a result more and more cells become injured and release the next sets of DAMP. The consequence is a process, stretched over time, of converting microbial inflammation (at the very beginning), into completely uncontrolled nonmicrobial inflammation accompanied by overproduction of interleukins and infiltration of mast cells. At the macroscopic level the described pathology accounts for symptoms of BPS/IC that is, pain, urinary frequency, and urgency.93
Conclusion
NLRP3 is evolving as a new common pathogenic mechanism for different diseases. Recent studies revealed that it contributes to the development of, for example, acute myocardial infarction, atherosclerosis, various chronic metabolic disease such as diabetes mellitus, gout, and obesity.76 Today, this inflammasome has been attributed to more than 20 autoinflammatory diseases .94,95
Diseases described here can be treated by inhibition of NLRP3 activation as well as by antagonizing the action of IL-1β. To attenuate the activation of the inflammasome within urinary bladder a few therapeutic options may currently be considered.96 For instance, two antidiabetic drugs, glyburide and SGLT2 inhibitors (called gliflozins), have been shown to suppress inflammasome activation. The former was actually the first compound identified as an inhibitor of the NLRP3 inflammasome. Although its NLRP3 inhibiting properties have been widely acknowledged for many years, its exact mechanism of actions remains elusive.97 The latter, gliflozins, are one of the newest antidiabetic drugs on the market. The basic mechanism of their action is spatially related to the area of upper urinary tract; precisely it involves inhibiting sodium–glucose transport proteins (SGLT-2) in the kidney. Taking into consideration that the ability of gliflozins to inhibit NLRP3 inflammasome has been already proven (as of today within heart muscle)98, it would be particularly interesting to verify whether this compound would also be able to attenuate inflammation within the lower urinary tract.
BPS/IC has been referred to as ‘the great enigma of medicine’. Treatment attempts to improve or remove the problem do not produce any permanent cure. Quite the contrary, the results are disappointing as there is a significant unmet medical need for new and better treatment options for patients who have BPS/IC. Huge amounts of work remain to be done before an efficient therapy can be offered to BPS/IC patients. However, thanks to recent scientific findings the first step, identification of the potential molecular target, has already been made.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Karol Borys Tudrej, Medical University of Warsaw, Banacha 1, Warszawa, Mazowieckie, 02-097, Poland.
Tomasz Piecha, Medical University of Warsaw, Warszawa, Mazowieckie, Poland.
Małgorzata Kozłowska-Wojciechowska, Medical University of Warsaw, Warszawa, Mazowieckie, Poland.
References
- 1. Hanno PM, Erickson D, Moldwin R, et al. Diagnosis and treatment of interstitial cystitis/bladder pain syndrome: AUA guideline amendment. J Urol 2015; 193: 1545–1553. [DOI] [PubMed] [Google Scholar]
- 2. van de, Merwe JP, Nordling J, Bouchelouche P, et al. Diagnostic criteria, classification, and nomenclature for painful bladder syndrome/interstitial cystitis: an ESSIC proposal. Eur Urol 2008; 53: 60–67. [DOI] [PubMed] [Google Scholar]
- 3. Engeler D, Baranowski AP, Borovicka J, et al. EAU guidelines on chronic pelvic pain. European Association of Urology; 2017. [DOI] [PubMed] [Google Scholar]
- 4. Clemens JQ, Link CL, Eggers PW, et al. Prevalence of painful bladder symptoms and effect on quality of life in black, Hispanic and white men and women. J Urol 2007; 177: 1390–1394. [DOI] [PubMed] [Google Scholar]
- 5. Suskind AM, Berry SH, Ewing BA, et al. The prevalence and overlap of interstitial cystitis/bladder pain syndrome and chronic prostatitis/chronic pelvic pain syndrome in men: results of the RAND Interstitial Cystitis Epidemiology male study. J Urol 2013; 189: 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suskind AM, Berry SH, Suttorp MJ, et al. Health-related quality of life in patients with interstitial cystitis/bladder pain syndrome and frequently associated comorbidities. Qual Life Res 2013; 22: 1537–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ibrahim IA, Diokno AC, Killinger KA, et al. Prevalence of self-reported interstitial cystitis (IC) and interstitial-cystitis-like symptoms among adult women in the community. Int Urol Nephrol 2007; 39: 489–495. [DOI] [PubMed] [Google Scholar]
- 8. Berry SH, Bogart LM, Pham C, et al. Development, validation and testing of an epidemiological case definition of interstitial cystitis/painful bladder syndrome. J Urol 2010; 183: 1848–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berry SH, Elliott MN, Suttorp M, et al. Prevalence of symptoms of bladder pain syndrome/interstitial cystitis among adult females in the United States. J Urol 2011; 186: 540–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Patnaik SS, Lagana AS, Vitale SG, et al. Etiology, pathophysiology and biomarkers of interstitial cystitis/painful bladder syndrome. Arch Gynecol Obstet 2017; 295: 1341–1359. [DOI] [PubMed] [Google Scholar]
- 11. Graham E, Chai TC. Dysfunction of bladder urothelium and bladder urothelial cells in interstitial cystitis. Curr Urol Rep 2006; 7: 440–446. [DOI] [PubMed] [Google Scholar]
- 12. Lynes WL, Flynn SD, Shortliffe LD, et al. The histology of interstitial cystitis. Am J Surg Pathol 1990; 14: 969–976. [DOI] [PubMed] [Google Scholar]
- 13. Lucon M, Martins JR, Leite KR, et al. Evaluation of the metabolism of glycosaminoglycans in patients with interstitial cystis. Int Braz J Urol 2014; 40: 72–79. [DOI] [PubMed] [Google Scholar]
- 14. Horn T, Holm NR, Hald T. Interstitial cystitis. Ultrastructural observations on detrusor smooth muscle cells. APMIS 1998; 106: 909–916. [PubMed] [Google Scholar]
- 15. Rosamilia A, Igawa Y, Higashi S. Pathology of interstitial cystitis. Int J Urol 2003; 10(Suppl.): S11–S15. [DOI] [PubMed] [Google Scholar]
- 16. Nazif O, Teichman JM, Gebhart GF. Neural upregulation in interstitial cystitis. Urology 2007; 69(4 Suppl.): 24–33. [DOI] [PubMed] [Google Scholar]
- 17. Kim HJ. Update on the pathology and diagnosis of interstitial cystitis/bladder pain syndrome: a review. Int Neurol J 2016; 20: 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim A, Han JY, Ryu CM, et al. Histopathological characteristics of interstitial cystitis/bladder pain syndrome without Hunner lesion. Histopathology 2017; 71: 415–424. [DOI] [PubMed] [Google Scholar]
- 19. Richter B, Roslind A, Hesse U, et al. YKL-40 and mast cells are associated with detrusor fibrosis in patients diagnosed with bladder pain syndrome/interstitial cystitis according to the 2008 criteria of the European Society for the Study of Interstitial Cystitis. Histopathology 2010; 57: 371. [DOI] [PubMed] [Google Scholar]
- 20. Grover S, Srivastava A, Lee R, et al. Role of inflammation in bladder function and interstitial cystitis. Ther Adv Urol 2011; 3: 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sant GR, Kempuraj D, Marchand JE, et al. The mast cell in interstitial cystitis: role in pathophysiology and pathogenesis. Urology 2007; 69(4 Suppl.): 34–40. [DOI] [PubMed] [Google Scholar]
- 22. Hurst RE, Meerveld BG, Wisniewski AB, et al. Increased bladder permeability in interstitial cystitis/painful bladder syndrome. Transl Androl Urol 2015; 4: 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hurst RE, Zebrowski R. Identification of proteoglycans present at high density on bovine and human bladder luminal surface. J Urol 1994; 152: 1641–1645. [DOI] [PubMed] [Google Scholar]
- 24. Hanna-Mitchell AT, Beckel JM, Barbadora S, et al. Non-neuronal acetylcholine and urinary bladder urothelium. Life Sci 2007; 80: 2298–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fernandes VS, Hernandez M. The role of nitric oxide and hydrogen sulfide in urinary tract function. Basic Clin Pharmacol Toxicol 2016; 119(Suppl. 3): 34–41. [DOI] [PubMed] [Google Scholar]
- 26. Jhang JF, Kuo HC. Pathomechanism of interstitial cystitis/bladder pain syndrome and mapping the heterogeneity of disease. Int Neurourol J 2016; 20(Suppl. 2): S95–S104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol 2002; 20: 125–163. [DOI] [PubMed] [Google Scholar]
- 28. Littlejohn G. Neurogenic neuroinflammation in fibromyalgia and complex regional pain syndrome. Nat Rev Rheumatol 2015; 11: 639–648. [DOI] [PubMed] [Google Scholar]
- 29. Cooper MS, Clark VP. Neuroinflammation, neuroautoimmunity, and the co-morbidities of complex regional pain syndrome. J Neuroimmune Pharmacol 2013; 8: 452–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Theoharides TC, Stewart JM. Genitourinary mast cells and survival. Transl Androl Urol 2015; 4: 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chelimsky G, Heller E, Buffington CA, et al. Co-morbidities of interstitial cystitis. Front Neurosci 2012; 6: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chelimsky G, McCabe NP, Janata J, et al. Autonomic testing of women with interstitial cystitis/bladder pain syndrome. Clin Auton Res 2014; 24: 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Charrua A, Pinto R, Taylor A, et al. Can the adrenergic system be implicated in the pathophysiology of bladder pain syndrome/interstitial cystitis? A clinical and experimental study. Neurourol Urodyn 2015; 34: 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lai HH, Gardner V, Ness TJ, et al. Segmental hyperalgesia to mechanical stimulus in interstitial cystitis/bladder pain syndrome: evidence of central sensitization. J Urol 2014; 191: 1294–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang YH, Peng CH, Liu HT, et al. Increased pro-inflammatory cytokines, C-reactive protein and nerve growth factor expressions in serum of patients with interstitial cystitis/bladder pain syndrome. PLoS One 2013; 8: e76779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu BL, Yang F, Zhan HL, et al. Increased severity of inflammation correlates with elevated expression of TRPV1 nerve fibers and nerve growth factor on interstitial cystitis/bladder pain syndrome. Urol Int 2014; 92: 202–208. [DOI] [PubMed] [Google Scholar]
- 37. Grigorescu B, Powers K, Lazarou G. Update on urinary tract markers in interstitial cystitis/bladder pain syndrome. Female Pelvic Med Reconstr Surg 2016; 22: 16–23. [DOI] [PubMed] [Google Scholar]
- 38. Gillespie L. Metabolic appraisal of the effects of dietary modification on hypersensitive bladder symptoms. Br J Urol 1993; 72: 293–297. [DOI] [PubMed] [Google Scholar]
- 39. Gillespie L. You don’t have to live with cystitis! New York: Rawson Associates, 1996. [Google Scholar]
- 40. Parsons CL, Greenberger M, Gabal L, et al. The role of urinary potassium in the pathogenesis and diagnosis of interstitial cystitis. J Urol 1998; 159: 1862–1866. [DOI] [PubMed] [Google Scholar]
- 41. Parsons CL, Zupkas P, Parsons JK. Intravesical potassium sensitivity in patients with interstitial cystitis and urethral syndrome. Urology 2001; 57: 428–432. [DOI] [PubMed] [Google Scholar]
- 42. Osborne J. Wine, beer and spirits – do they trigger IC flares? A patient survey reveals surprising results. IC Optimist 2010; 7: 15–17. [Google Scholar]
- 43. Butrick CW, Howard FM, Sand PK. Diagnosis and treatment of interstitial cystitis/painful bladder syndrome: a review. J Womens Health (Larchmt) 2010; 19: 1185–1193. [DOI] [PubMed] [Google Scholar]
- 44. Hurst RE. Structure, function, and pathology of proteoglycans and glycosaminoglycans in the urinary tract. World J Urol 1994; 12: 3. [DOI] [PubMed] [Google Scholar]
- 45. Hurst RE, Roy JB, Min KW, et al. A deficit of chondroitin sulfate proteoglycans on the bladder uroepithelium in interstitial cystitis. Urology 1996; 48: 817. [DOI] [PubMed] [Google Scholar]
- 46. Gülpınar Ö, Esen B, Kayış A, et al. Clinical comparison of intravesical hyaluronic acid and chondroitin sulfate therapies in the treatment of bladder pain syndrome/interstitial cystitis. Neurourol Urodyn 2018; 37: 257–262. [DOI] [PubMed] [Google Scholar]
- 47. Janssen DA. The distribution and function of chondroitin sulfate and other sulfated glycosaminoglycans in the human bladder and their contribution to the protective bladder barrier. J Urol 2013; 189: 336–342. [DOI] [PubMed] [Google Scholar]
- 48. Theoharides TC, Sant GR. A pilot open label study of cystoprotek in interstitial cystitis. Int J Immunopathol Pharmacol 2005; 18: 183–188. [DOI] [PubMed] [Google Scholar]
- 49. Beutler B. Tlr4: central component of the sole mammalian LPS sensor. Curr Opin Immunol 2000; 12: 20–26. [DOI] [PubMed] [Google Scholar]
- 50. Cullen SP, Kearney CJ, Clancy DM, et al. Diverse activators of the NLRP3 inflammasome promote IL-1β secretion by triggering necrosis. Cell Rep 2015; 11: 1535–1548. [DOI] [PubMed] [Google Scholar]
- 51. Cullen SP, Kearney CJ, Clancy DM, et al. Diverse activators of the NLRP3 inflammasome promote IL-1β secretion by triggering necrosis. Cell Rep 2015; 11: 1535–1548. [DOI] [PubMed] [Google Scholar]
- 52. Bauer C, Duewell P, Mayer C, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010; 59: 1192–1199. [DOI] [PubMed] [Google Scholar]
- 53. Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res 2015; 8: 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marinkovic SP, Moldwin R, Gillen LM, et al. The management of interstitial cystitis or painful bladder syndrome in women. BMJ 2009; 339: 337–342. [DOI] [PubMed] [Google Scholar]
- 55. Bologna RA, Gomelsky A, Lukban JC, et al. The efficacy of calcium glycerophosphate in the prevention of food-related flares in interstitial cystitis. Urology 2001; 57: 119–120. [DOI] [PubMed] [Google Scholar]
- 56. Gordon B, Shorter B, Sarcona A, et al. Nutritional considerations for patients with interstitial cystitis/bladder pain syndrome. J Acad Nutr Diet 2015; 115: 1372–1379. [DOI] [PubMed] [Google Scholar]
- 57. Koziol JA, Clark DC, Gittes RF, et al. The natural history of interstitial cystitis: a survey of 374 patients . J Urol 1993; 149: 465–469. [DOI] [PubMed] [Google Scholar]
- 58. Shorter B, Lesser M, Moldwin R, et al. Effect of comestibles on symptoms of interstitial cystitis. J Urol 2007; 178: 145–152. [DOI] [PubMed] [Google Scholar]
- 59. Bassaly R, Downes K, Hart S. Dietary consumption triggers in interstitial cystitis/bladder pain syndrome patients. Female Pelvic Med Reconstr Surg 2011; 17: 36–39. [DOI] [PubMed] [Google Scholar]
- 60. Nguan C, Franciosi LG, Butterfield N, et al. A prospective, double-blind, randomized cross-over study evaluating changes in urinary pH for relieving the symptoms of interstitial cystitis. BJU Int 2005; 95: 91–94. [DOI] [PubMed] [Google Scholar]
- 61. Osborne J. Wine, beer and spirits – do they trigger IC flares? A patient survey reveals surprising results. IC Optimist 2010; 7: 15–17. [Google Scholar]
- 62. Bauernfeind FG, Horvath G, Stutz A, et al. NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183: 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. O’Neill LA. Signal transduction pathways activated by the IL-1 receptor/toll-like receptor superfamily. Curr Top Microbiol Immunol 2002; 270: 47–61. [PubMed] [Google Scholar]
- 64. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of pro IL-1beta. Mol Cell 2002; 10: 417–426. [DOI] [PubMed] [Google Scholar]
- 65. Lamkanfi M, Dixit VM. Modulation of inflammasome pathways by bacterial and viral pathogens. J Immunol 2011; 187: 597–602. [DOI] [PubMed] [Google Scholar]
- 66. Mao K, Chen S, Chen M. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res 2013; 23: 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hughes FM, Turner DP, Purves JT. The potential repertoire of the innate immune system in the bladder: expression of pattern recognition receptors in the rat bladder and a rat urothelial cell line (MYP3 cells). Int Urol Nephrol 2015; 47: 1953–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hoshino K, Takeuchi O, Kawai T, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 1999; 162: 3749–3752. [PubMed] [Google Scholar]
- 69. Brown GT, Narayanan P, Li W, et al. Lipopolysaccharide stimulates platelets through an IL-1β autocrine loop. J Immunol 2013; 191: 5196–5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Trent MS, Stead MC, Tran AX, et al. Diversity of endotoxin and its impact on pathogenesis. J Endotoxin Res 2006;12: 205–223. [DOI] [PubMed] [Google Scholar]
- 71. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol 2013; 13: 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shimada K, Crother TR, Karlin J, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012; 36: 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature 2010; 469: 221–225. [DOI] [PubMed] [Google Scholar]
- 74. Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440: 228–232. [DOI] [PubMed] [Google Scholar]
- 75. Braga TT, Forni MF, Correa-Costa M, et al. Soluble uric acid activates the NLRP3 inflammasome. Sci Rep 2017; 7: 39884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Conley SM, Abais JM, Boini KM, et al. Inflammasome activation in chronic glomerular diseases. Curr Drug Targets 2017; 18: 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ouyang X, Ghani A, Mehal WZ. Inflammasome biology in fibrogenesis. Biochim Biophys Acta 2013; 1832: 979–988. [DOI] [PubMed] [Google Scholar]
- 78. Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140: 821–832. [DOI] [PubMed] [Google Scholar]
- 79. Hornung V, Bauernfeind F, Halle A, Samstad EO, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008; 9: 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhao Y, Yang J, Shi J, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011; 477: 596–600. [DOI] [PubMed] [Google Scholar]
- 81. Li H, Miao W, Ma J, et al. Acute exercise-induced mitochondrial stress triggers an inflammatory response in the myocardium via NLRP3 inflammasome activation with mitophagy. Oxid Med Cell Longev 2016; 2016: 1–11; Article ID: 1987149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Di Virgilio F. The therapeutic potential of modifying inflammasomes and NOD-like receptors. Pharmacol Rev 2013; 65: 872–905. [DOI] [PubMed] [Google Scholar]
- 83. Li P, Allen H, Banerjee S, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 1995; 80: 401–411. [DOI] [PubMed] [Google Scholar]
- 84. Johansson SL, Fall M. Clinical features and spectrum of light microscopic changes in interstitial cystitis. J Urol 1990; 143: 1118–1124. [DOI] [PubMed] [Google Scholar]
- 85. Purves JT, Hughes FM., Jr. Inflammasomes in the urinary tract: a disease-based review. Am J Physiol Renal Physiol 2016; 311: F653–F662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Purves JT, Hughes FM., Jr Inflammasomes in the urinary tract: a disease-based review. Am J Physiol Renal Physiol 2016; 311: F653–F662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kummer JA, Broekhuizen R, Everett H, et al. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem 2007; 55: 443–452. [DOI] [PubMed] [Google Scholar]
- 88. Rathinam VAK, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol 2012; 13: 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Martinon F, Petrilli V, Mayor A, et al. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440: 237–241. [DOI] [PubMed] [Google Scholar]
- 90. Hughes FM, Vivar NP, Kennis JG, et al. Inflammasomes are important mediators of cyclophosphamide-induced bladder inflammation. Am J Physiol Renal Physiol 2014; 306: F299–F308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Urinology Think Tank Writing Group. Urine: waste product or biologically active tissue? Neurourol Urodyn 2018; 37: 1162–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hughes FM, Kennis JG, Youssef MN, et al. The NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome mediates inflammation and voiding dysfunction in a lipopolysaccharide-induced rat model of cystitis. J Clin Cell Immunol 2016; 7: 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Han XM, Wu XH, Li B, et al. The effects of intravesical therapy with hyaluronic acid for painful bladder syndrome: preliminary Chinese experience and systematic review. Taiwan J Obstet Gynecol 2015; 54: 240. [DOI] [PubMed] [Google Scholar]
- 94. Jones HD, Crother TR, Gonzalez-Villalobos RA, et al. The NLRP3 inflammasome is required for the development of hypoxemia in LPS/mechanical ventilation acute lung injury. Am J Respir Cell Mol Biol 2014; 50: 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Próchnicki T, Mangan MS, Latz E. Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Research 2016; 5 (F1000 Faculty Rev): 1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Guo H, Callaway JB, Ting JP-Y, et al. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015; 21: 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lamkanfi M, Mueller JL, Vitari AC, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 2009; 187: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ye Y, Bajaj M, Yang HC, Perez-Polo JR, et al. SGLT-2 Inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. Further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Cardiovasc Drugs Ther 2017; 31: 119–132. [DOI] [PubMed] [Google Scholar]