

Abstract
Stroke leads to inflammatory and immune response in the brain and immune organs. The gut or gastrointestinal tract is a major immune organ equipped with the largest pool of immune cells representing more than 70% of the entire immune system and the largest population of macrophages in the human body. The bidirectional communication between the brain and the gut is commonly known as brain–gut or gut–brain axis. Stroke often leads to gut dysmotility, gut microbiota dysbiosis, “leaky” gut, gut hemorrhage, and even gut-origin sepsis, which is often associated with poor prognosis. Emerging evidence suggests that gut inflammatory and immune response plays a key role in the pathophysiology of stroke and may become a key therapeutic target for its treatment. Ischemic brain tissue produces damage-associated molecular patterns to initiate innate and adaptive immune response both locally and systemically through the specialized pattern-recognition receptors (e.g., toll-like receptors). After stroke, innate immune cells including neutrophils, microglia or macrophages, mast cells, innate lymphocytes (IL-17 secreting γδ T-cell), and natural killer T-cell respond within hours, followed by the adaptive immune response through activation of T and B lymphocytes. Subpopulations of T-cells can help or worsen ischemic brain injury. Pro-inflammatory Th1, Th17, and γδ T-cells are often associated with increased inflammatory damage, whereas regulatory T-cells are known to suppress postischemic inflammation by increasing the secretion of anti-inflammatory cytokine IL-10. Although known to play a key role, research in the gut inflammatory and immune response after stroke is still in its initial stage. A better understanding of the gut inflammatory and immune response after stroke may be important for the development of effective stroke therapies. The present review will discuss recent advances in the studies of the brain–gut axis after stroke, the key issues to be solved, and the future directions.
Keywords: Brain–gut or gut–brain axis, damage-associated molecular patterns, gut inflammatory and immune response, macrophage, mice, microglia, middle cerebral artery occlusion, regulatory T-cells, stroke, Th1, Th17, and γδ T-cells
Introduction
According to the World Health Organization, cerebrovascular accidents (stroke) are the second leading cause of death and the third leading cause of disability worldwide.[1] In the United States of America, 140,000 Americans died each year with stroke.[2] Stroke is primarily divided into two categories; ischemic stroke (an obstruction within a blood vessel supplying blood to the brain) and hemorrhagic stroke (a rupture in a weakened blood vessel in the brain). Ischemic stroke accounts for about 70%–80% of all strokes. The most ischemic stroke is due to the middle cerebral artery occlusion (MCAO), resulting in the brain tissue damage in the affected territory, which is followed by inflammatory and immune response. In this review article, inflammatory response generally indicates the initial innate immune response after tissue injury, while immune response represents adaptive immune response. This review will mainly discuss the role of the brain–gut axis in inflammatory and immune response after ischemic stroke (or stroke hereafter).
It is generally held that ischemic brain tissue must be reperfused to be rescued after stroke. For that reason, medical professionals have successfully developed two reperfusion or recannulation modalities for the treatment of ischemic stroke: (i) Thrombolysis by intravenous administration of recombinant tissue plasminogen activator and (ii) performance of endovascular thrombectomy to physically remove the blood clot or other occluded materials. However, both thrombolysis and endovascular thrombectomy have significant limitations; as both are carried out within a limited therapeutic window (e.g., within about 4.5 h) after ischemic stroke onset and are often associated with unwanted effects (e.g., hemorrhage). Furthermore, many pathological events, triggered by brain ischemia, continuously develop in the postischemic brain tissue after reperfusion or recanalization, the so-called ischemia-reperfusion injury. Thrombolysis and endovascular thrombectomy modalities, although effective, they can treat only a small proportion of ischemic stroke patients with limited success. Two key issues to be solved currently for the treatment of ischemic stroke are: (i) To prolong the therapeutic window of thrombolysis and endovascular thrombectomy and (ii) to develop therapeutic approaches against tissue ischemia-reperfusion injury.
All clinical trials of stroke therapeutic agents have so far failed to demonstrate the efficacy in human stroke patients, despite showing the protective properties of these agents against brain ischemia-reperfusion injury in animal models of the preclinical settings. The reasons behind the failures are many, but two of them may be significant: (i) current animal stroke models are mostly conducted using young animals, which inadequately reflects stroke in the aging populations and (ii) the major contributor (s) leading to brain ischemia-reperfusion injury have yet to be completely identified. As a result, no effective therapeutic agents are clinically proven to be effective currently in the treatment of brain ischemia-reperfusion injury after stroke.
Bidirectional Communications between Brain and Gut after Stroke
The brain–gut axis or gut–brain axis is often referring to the bidirectional communications between the central nervous system (CNS) and the gastrointestinal (GI) tract (microbiota and immune system) [Figure 1].[3] The bidirectional communications between the gut and the brain after stroke may involve vagus nerve, release of damage associated molecular patterns (DAMPs) and cytokines from the injury site of the brain, release of cytokines from the gut, as well as migration of gut inflammatory or immune cells to the brain injury site [Figure 1].[4]
Figure 1.
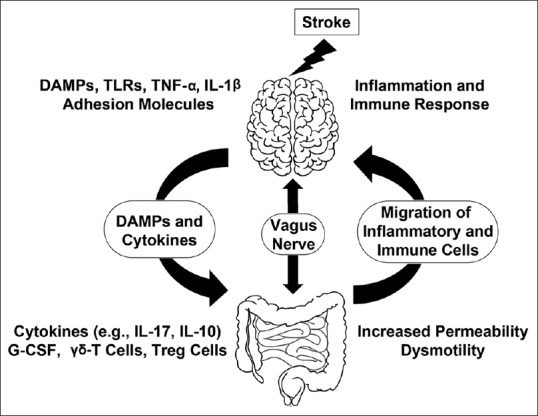
Changes in brain–gut–microbiota axis after stroke. Ischemic brain tissue and activated microglia release DAMPs and cytokines, resulting in the activation of endothelial cells to express adhesion molecules and to recruit inflammatory and immune cells from the circulation to the sites of stroke injury. Meanwhile, release of DAMPs and cytokines as well as activation of the vagus nerve induce gut dysmotility, gut dysbiosis, and increased gut permeability, resulting in translocation of intestinal bacteria and migration of gut inflammatory and immune cells through the circulation into the sites of stroke injury. Treg=Regulatory T-cell, G-CSF=granulocyte colony-stimulating factor, DAMPs=damage-associated molecular patterns
After stroke, up to 50% of patients experience GI complications, including gut dysmotility, gut microbiota dysbiosis, “leaky” gut, gut hemorrhage, and even gut-origin sepsis.[5] Stroke patients associated with GI complications often have poor outcomes, with increased mortality rates and deteriorating neurologic function.[6] Gut dysbiosis or gut-origin sepsis are both reported to occur after stroke in aged animals.[7,8,9,10] The underlying mechanisms of stroke-associated GI complications, as well as the poor stroke outcome, remain understudied.
Human intestine contains trillions of diverse microbes. The loss of microbiota diversity leads to intestinal dysbiosis, resulting in several complex diseases. Gut bacteria are able to produce short-chain fatty acids which play a neuroprotective role. Gut bacteria may also synthesize neurotransmitters (γ-aminobutyric acid, noradrenaline, and dopamine) and modulate activation of the immune system.[11]
The communication between the CNS and the gut takes place through several complex signaling pathways involving vagus nerves to the enteric nervous system (ENS), the neuronal-glial-endothelial interactions, as well as, DAMPs-and cytokines-induced activation of gut inflammatory and immune cells [Figure 1].[12,13,14,15] The afferent fiber of the vagus nerve expresses receptor to sense microbiota metabolite, gut peptides like ghrelin, and leptin to transfer gut information to the CNS.[16,17] In Parkinson's disease (PD), Braak et al. suggested that vagus nerve might be the site of initiation of this disease.[18] Svensson et al. reported that removal of part of the vagus nerve declines the development of PD severity.[19] Winek et al. reported that the poststroke mortality rate was higher in mice pretreated with antibiotics. A higher survival rate was observed in the mice having complex gut microbiota.[20]
Gut microbiota is highly abundant with various types of phyla including Firmicutes, Bacteroidetes, and Actinobacteria which are severely reduced after stroke.[7] Several studies show significant changes in the microbial diversity in the fecal samples of patients with stroke and transient ischemic attack.[21,22] Stanley et al. reported that commensal bacteria residing in the intestinal tract are the major sources of infections in stroke patients after the onset of stroke.[23] Severe stroke leads to gut dysbiosis, as a result of increased Bacteroidetes and decreased species diversity, damage to the intestinal barrier, and decrease in the intestinal movement, resulting in an intestinal inflammatory and immune response and altered immune homeostasis.[7] A range of bacteria and their derivatives in altered gut microbiota regulates intestinal immune homeostasis by communicating with intestinal epithelial cells, mononuclear phagocytes, innate lymphoid cells, dendritic cells, and T and B lymphocytes.[24]
Lymphocytes in the lamina propria and intraepithelial compartments of both the small and large intestine perform regulatory and effector function both locally and systemically.[25] After stroke, activation of gut γδ T-cells may increase ischemic brain injury by migrating to the injury site and by secreting pro-inflammatory cytokines (e.g., IL-17) to attract myeloid cells (neutrophils and monocytes) at the site of injury, whereas activation and migration of CD4+ CD25+ Foxp3+ regulatory T-cells (Tregs) in the injury site may play a protective role against cerebral ischemic injury.[26,27] At the late phase of stroke, dendritic cells in the mesenteric lymph node promote migration of Treg cells to the gut to suppress differentiation of IL-17 producing γδ T-cell, thus reducing migration of γδ T-cell to the brain. In this case, anti-inflammatory cytokine IL-10 is upregulated from Treg cells to reduce the migration of γδ T-cells from the intestine to the meninges of the brain, resulting in downregulation of IL-17 expression, and thus protecting the brain from ischemia-reperfusion brain injury.[28,29] Upon activation, effector CD4+ T-cells may produce neuroprotective IL-4 to restore neuronal tissue homeostasis in the late phase of stroke [Figure 1].[30]
Gut Microbiome and Progression of Inflammatory Events after Stroke
Gut microbiome and immune system have coevolved with both animals and humans for millions of years. The interaction between them is important for keeping animal and human healthy. Experimental stroke in mice has revealed the changes in gut microbiota but the influence of the altered microbiota on inflammation in this disease condition is still incompletely understood.
Commensal microbiota playing important role in maintaining the host immune homeostasis. However, dysbiosis leads to an imbalance of T-cell subpopulations including Th1, Th2, Th17, and Treg to trigger several types of autoimmune and inflammatory disease [Table 1].[31] Th1 cells secrete pro-inflammatory cytokines including IL-2, IL-12, tumor necrosis factor-alpha (TNF-α) and interferon-gamma (IFN-γ) to promote cellular immune response, and thus may be involved in the pathogenesis of stroke.[32] Th2 cells secrete IL-4, IL-5, and IL-13 to promote humoral immune responses against parasites and allergens.[32] A small amount of IL-17 is produced by αβ T-cells (Th17 cells), which is required for antigen-specific priming, whereas the major source of IL-17 is produced by γδ T-cells during acute infection, which does not require prior antigen priming and thus can rapidly induce inflammation.[33,34] In experimental brain ischemia, IL-17 produced by γδ T-cells aggravates the pro-inflammatory response.[26,27] The study of Benakis et al. show that γδ T-cells are abundant in the gut from where they seem to traffic to the leptomeninges structure of the brain after stroke.[29] Treg cells are derived from the same lineage as that of naive CD4 cells, express transcription factor Foxp3, and secrete anti-inflammatory cytokine IL-10 to dampen the excessive immune response.[35] A study of Liesz et al. demonstrates that the absence of Treg cells enhances poststroke activation of resident and invading inflammatory cells including microglia and T-cells in the animal stroke model, suggesting that Treg cells play a key role in dampening postischemic inflammation.[36] Treg cells suppress both the differentiation of Th17 cells and proliferation of γδ T-cells in the gut to maintain the anti-inflammatory environment [Table 1].[37,38,39]
Table 1.
Differentiation of Na651 CD4+ T cells
| Cells | Significance in stroke | Host defense | Differentiation cytokine | Transcription factor | Cytokine produced |
|---|---|---|---|---|---|
| Th1 | Induce inflammation, activation of microglia | Intracellular pathogens | IFN-γ, IL-12 | Tbet | IFN-γ, IL-2 |
| Th2 | Induce inflammation | Large worms (helminths) | IL-2, IL-4 | GATA3 | IL-4, IL-5, IL-13 |
| Th9 | Neuroprotective | Extracellular parasites | IL-9 | Foxo1 | IL-9 |
| Th17 | Activation of MMPs and BBB breakdown | Extracellular pathogens (fungi) | IL-6, IL-23 | RORγt | IL-17A, IL-17F, IL-22 |
| Treg | Suppression of inflammation, neuroprotection | Bacteria and parasites | TGF-β | FoxP3 | TGF-β, IL-10, IL-35 |
| Tfh | Increase early ischemic tissue injury | Defense against extracellular pathogens | IL-6 | Bcl6 | IL-21 |
CD4+ T-cells polarization in various subtypes like Th1, Th2, Th9, Th17, Treg, and Tfh takes place in the presence of specific combination of cytokines to protect host from pathogens and injuries. For the differentiation and production of cytokines every cell type has their own signature transcription factors. Treg: Regulatory T cells, IFN-γ: Interferon-γ, IL: Interleukin, TGF-β: Transforming growth factor-β, BBB: Blood-brain barrier, MMPs: Matrix metalloproteinases
The blood vessels at the injury site after stroke become activated to express cell surface molecules including chemokines, adhesion molecules (e.g., Fibronectin and the α5 β1 and αvβ3 Integrins), intercellular adhesion molecule 1, and vascular cell adhesion molecule 1 to facilitate leukocyte adhesion and infiltration by extravasation across the blood–brain barrier (BBB). Damaged neurons and activated microglia after stroke release cytokines such as IL-4, TNF-α, and IL-1 β.[40,41] After stroke, microglia become active locally, while neutrophils, as well as macrophages, lymphocytes, and dendritic cells infiltrate into the ischemic brain area.[27,42] Activation of microglia is the earliest cellular inflammatory change which peaks at 48 h and remains in the plateau at 96 h after MCAO [Figure 2]. Infiltration of neutrophils to the injury site is also an early event after stroke.[43] Perez-de-Puig et al. demonstrated the migration of neutrophils to the leptomeninges from 6 h onward, to the cortical-basal lamina and cortical Virchow–Robin spaces from 15 h onward, and to the cortical brain parenchyma for 24 h onward after the MCAO.[44] T-cells invade the infarct region from day 1 onward. Their number increases at day 7 and reached a peak at day 14 day in the mouse model of stroke.[45] Xie et al. found that CD4+ and CD8+ T-cells increased in the peri-infarct region for as long as 1-month after experimental ischemic stroke in mice and persisted for years in stroke patients.[46] CD4+ CD25+ Foxp3+ Treg cells accumulate in the ischemic lesion sites for as long as 14–30 days and played an anti-inflammatory role after experimental stroke in mice, which is expected to play a brain protective role by suppression of effector T-cells.[47] In the absence of Treg cell activation, the numbers of resident and infiltrating inflammatory cells such as microglia and T-cells are expanded after experimental stroke in mice [Figure 2 and Table 2].[36]
Figure 2.

Time-dependent migration of inflammatory cells to the site of stroke injury. (a) The numbers of neutrophil, microglia, macrophage, and dendritic and natural killer cells at the site of brain injury at days 1, 3, and 7 in mice after 60-min middle cerebral artery occlusion. (b) The number of CD4+, CD8+, and regulatory T-cells at the site of brain injury at days 7, 14, and 30 in mice after 30-min middle cerebral artery occlusion. Graphs were made with the excellent graphing tool based on the published data from references listed in Table 2
Table 2.
The number of cells at the injury site after stroke
| MCAO duration (min) | Cells | Cells (103) at poststroke days | References | ||||
|---|---|---|---|---|---|---|---|
| 1 | 3 | 7 | 14 | 30 | |||
| 60 | Neutrophils | ~5.1 | ~75 | ~50 | Gelderblom et.al., 2009 | ||
| 60 | Microglia | ~60 | ~55 | ~45 | Gelderblom et.al., 2009 | ||
| 30 | Microglia | ~45 | ~90 | ~110 | Stubbe et.al., 2013 | ||
| 60 | Macrophage | ~20 | ~30 | ~5.1 | Gelderblom et.al., 2009 | ||
| 30 | Macrophage | ~5.1 | ~8.1 | ~8.5 | Stubbe et.al., 2013 | ||
| 60 | CD4+ T-cells | ~0.6 | ~0.75 | ~0.4 | Gelderblom et.al., 2009 | ||
| 60 | CD8+ T-cells | ~1.1 | ~1.5 | ~0.8 | Gelderblom et.al., 2009 | ||
| 30 | CD4+ T-cells | ~0.5 | ~4.5 | ~4.1 | Stubbe et.al., 2013 | ||
| 30 | Treg cells | ~0.1 | ~1.25 | ~1.25 | Stubbe et.al., 2013 | ||
| 90 | CD4+ T-cells | ~4.1 | Crapser et.al., 2016 | ||||
| 90 | CD8+ T-cells | ~2.1 | Crapser et.al., 2016 | ||||
| 60 | γδ T-cells | ~0.5 | Shichita et.al., 2009 | ||||
| 35 | γδ T-cells | ~0.35 | Benakis et.al., 2016 | ||||
The data are derived from the references cited in the table. The estimated number of cells is approximate from mice model of MCAO. MCAO: Middle cerebral artery occlusion, Treg: Regulatory T-cells
Gut Immune Response to Brain Injury
The GI contains a broad range of commensal and pathogenic microorganism. To protect the host from pathogenic microorganisms and dietary antigens, GI encompasses extremely efficient mucosal barrier and a specialized multifaceted immune system, made up of a large population of scattered immune cells and organized lymphoid tissues termed the gut-associated lymphoid tissue.[48] The GI comprised three major entities: the intestinal lumen commensal flora, epithelium, and mucosal immune system. First, the gut system immediately faces trillions of different types of bacteria and is therefore challenged with the daunting task of segregating the underlying tissues from noxious intestinal lumen contents.[49] The release of intestinal lumen bacteria and toxins into the circulation due to increased gut permeability or intestinal injury, the so-called “leaky gut” or, its severe form, “sepsis,” propagates systemic inflammation, and thus is considered the origin of the systemic inflammation or its severe form systemic inflammatory response syndrome (SIRS).[50] SIRS along the gut–brain, gut–lung, and gut–liver axes can result in multiple organ dysfunction syndrome (MODS), followed by mortality.[49,50] For example, stroked brain tissue releases DAMPs that propagates systemic inflammation through activation of the GI immune and inflammatory system, resulting in increased gut permeability, intestinal injury, and even sepsis, followed by systemic inflammation.[51] Second, in order to prevent invasion of the intestinal microorganisms and toxins during transport of nutrients, the gut is equipped with the largest pool of immune cells representing more than 70% of the entire immune system and the largest population of macrophages in the body.[49,50] This explains why severe stroke is often associated with long-term multiple organ complications. Hence, understanding the gut immune response may provide an opportunity for the therapeutic intervention to prevent progressive tissue damage and loss of brain function after stroke.
Damage Associated Molecular Patterns and Sterile Inflammation
Damaged tissue after stroke releases inflammatory molecules and cellular debris, the so-called DAMPs. DAMPs are thought to play a crucial role in initiating an inflammatory and immune response via the specialized pattern-recognition receptors (PRRs) (e.g., Toll-like receptors [TLR]). This type of immune and inflammatory response is also known as sterile inflammation. In comparison, pathogen-associated molecular patterns (PAMPs) from infected microorganism also initiate an inflammatory and immune response through the same PRRs. The term of sterile inflammation is to indicate DAMAPs-elicited, while the term of nonsterile inflammation represents PAMPs-induced inflammatory and immune response, both of which are through the specialized PRR to exert their effects.
Either infection or tissue injury can induce the innate immune system, commonly known as inflammatory response, which is followed by the adaptive immune response, also referring to as immune response. Inflammatory and immune response can be triggered either by PAMPs after microorganism infection or DAMPs after tissue injury. The inflammatory and immune response aims originally to defend the body from microorganism invasion, which is also be used to clear damaged tissues after injury. However, excessive inflammatory or immune response can cause local or remote tissue damage.[52]
Systemic Inflammatory Response Syndrome after Stroke
DAMPs and cytokines produced in brain ischemic tissue are released to the circulation to gain access to immune or lymphoid organs [Figure 1]. This will lead to systemic inflammatory and immune response, or its severe form; the SIRS. For example, high mobility group protein B1 (HMGB-1) is a DAMP molecule. HMGB-1 produced in stroke-damaged tissue can gain access to the circulation through the broken BBB to initiate SIRS after stroke.[53] This may be illustrated by the circulatory cytokine wave or even “cytokine storm” with increased TNF-α, IFN-γ, and IL-6 after stroke.[54] The inflammatory and immune response or SIRS after stroke can be mediated by DAMPs alone in the absence of microorganism infection, or mediated by both DAMPs and PAMPs when stroke is complicated with poststroke leaky gut or gut-origin sepsis.[55] In general, the systemic inflammatory and immune response begin with the innate immune response, which is followed by activation of the adaptive immune cells after stroke. The innate immune response is executed by innate immune cells including neutrophils, microglia and macrophages, mast cells, innate lymphocytes (IL17-secreting γδ T-cells and nature killer T-cells), whereas adaptive immune response is mediated mainly by T and B lymphocytes.
It was originally thought that brain was an immune privileged site due to the presence of highly restrictive BBB and the “absence” of the adaptive immune cells within the brain tissue. However, this concept has been challenged by experimental evidence that the brain has the lymphatic fluid circulation system.[56] This notion is consistent with the fact that both innate (inflammation) and adaptive immune response (activation of T and B lymphocytes) occur in the tissue of CNS disorders including stroke, Alzheimer's disease, PD, Huntington's disease, amyotrophic lateral sclerosis, epilepsy, and traumatic brain injury (TBI).
Recent Advances
In recent years, scientists made several exciting findings to understand how gut microbes listen and regulate the conversation between the brain and the gut in several disease conditions.[3] To understand the complexity of bacteria, the development of metagenomics techniques is highly helpful for sequencing the nucleic acids of the microbes without using bacterial culture.[57] In the previous years, sequencing of fecal specimens was not accurate due to the loss of 80% of microbes which observed by microscope in the culture.[58] Gut microbiota may influence the degree of poststroke inflammation due to their ability to release neuroactive molecules and modulation of intestinal T-cell trafficking to the meninges of the brain. Mice lacking segmented filamentous bacteria have significantly larger brain infarcts due to the inhibition of Treg expansion.[59] Benakis et al. discovered the negative impact of intestinal flora and meningeal IL-17+ γδ T-cells on ischemic injury. Their study also exhibited reduced ischemic brain injury in the mice with altered intestinal flora.[29] In the mouse model of stroke, Winek et al. raised the importance of microbiota in the protection of acute and severe colitis after cerebral ischemia. Moreover, Singh et al. showed reduced brain lesion volume after transfer of fecal microbiota from naïve animals to the wild-type mice but not to the Rag1-/-mice after MCAO stroke.[7]
Since altered gut microbiome takes place after stroke, fecal microbiota transplantation raises hope to better management of a large amount of microbiota-related disorders including stroke by restoring normal gut microbiota in stroke patients.[60] Wang et al. demonstrated that inhibition of microbial trimethylamine (TMA) lyases with the choline analog 3,3-dimethyl-1-butanol reduces the risk of cardiometabolic disease through the inhibition of microbial metabolite trimethylamine N-oxide produced from the microbial metabolism of TMA-rich foods.[61] Recently, Stanley et al. used phylogenetic investigation of communities by reconstruction of unobserved states and revealed increased abundance of human intestinal mucin degrading bacterium (Akkermansia muciniphila) and clostridial species after stroke.[62] In the mouse cerebral ischemia model, brain injury leads to increased noradrenaline release from the autonomic nervous system into the gut to alter microbial communities in the caecum.[63] These changes may have an impact on the recovery and treatment of patients after stroke,[63] Gram-negative bacteria is a primary source of TLR4 ligand and accelerates activation of the brain endothelial TLR4 receptor to stimulate cerebral cavernous malformation (CCM). Blockade of TLR4 signaling protects mice from the onset of CCM formation and protects the brain from stroke injury.[64] TLRs are mainly expressed in innate immune cells and in nonimmune cells such as fibroblast and epithelial cells.[65,66] Recent studies strongly suggest that gut microbiota, as well as gut inflammatory and immune cells, play a key role in the development of brain injury in a variety of CNS disease conditions, including stroke and TBI.[29,67] Migration of gut inflammatory and immune cells to the CNS may have a significant impact on the brain damage severity and duration after stroke.[29] These studies provide rationales for targeting gut inflammatory and immune response as a novel therapeutic strategy for the treatment of CNS conditions.
Important Issues to Be Solved
GI tract contains >70% of inflammatory and immune cells in the body, and thus should play a key role in inflammatory and immune response after injury in remote organs including the brain. However, the major issue in the field of brain–gut axis research is the lack of complete understanding of the role of gut microbiome, the involvement of gut inflammatory and immune cells and the communications through the PAMPs, humoral, and vagus nerves between the brain and gut after stroke. Several recent promising studies have shown that the communications between the brain and gut may be important therapeutic targets for stroke treatment in animal models. However, whether these discoveries in animal disease models can be translated into the clinical application in humans remain to be studied. Although the composition and distribution of gut inflammatory and immune cells are similar among different animal species and humans, there are significant differences in the microbiome complexity between animals and humans. Development of antibiotic-resistant bacterial strains after prolonged use of antibiotics and augmented chance of infection are among a few key issues to be solved during the treatment of stroke and cardiovascular disease.[68,69]
Innate and adaptive immune cells are responsible for the inflammatory and immune response following stroke. These inflammatory and immune cells may migrate to the brains from different organs including the gut, spleen, or bone marrow at different time points after stroke. On the other hand, stroke leads to damage to the intestinal barrier, resulting in leaky gut and even sepsis, as well as activation of gut inflammatory and immune cells including neutrophils, macrophage or monocytes, dendritic cells, conventional and Treg through brain injury-derived DAMPs, and altered intestinal microbiota. However, the exact mechanisms underlying intestinal barrier damage and activation of inflammatory and immune cells after stroke remain elusive.[70,71] Understanding the origins, time-course, functions, and fates of the inflammatory and immune cells migrating to the brain injury site and the injury surrounding structures such as meanings and the newly identified lymphatic system in the brain, is crucial for the development of novel therapies against inflammatory and immune response after stroke.
Concluding Remarks and Future Directions
The dysfunction of the brain–gut axis after stroke is a promising area of research for identifying novel mechanisms and preventive and treatment strategies against stroke. The brain–gut axis is an important network communicating through a number of pathways such as between the vagus nerve and ENS. After stroke, the brain–gut axis is significantly distressed by injury-induced DAMPs, cytokine release, the BBB changes, altered microbiota or dysbiosis, and leaky gut, resulting in migration of inflammatory and immune cells from gut to the brain. However, the exact molecular mechanisms underlying the changes in the brain–gut axis remain to be further studied. The poststroke BBB changes and gut-elicited inflammatory and immune response as well as poststroke leaky gut and dysbiosis may be among important research subjects to be studied in animal models of stroke. The changes in gut-elicited inflammatory and immune response and poststroke leaky gut and dysbiosis have yet to be carefully studied in human stroke patients.
The changes in the BBB after stroke allow inflammatory and immune cells from the circulation to enter into the brain parenchyma where they interact with innate immune cells in the CNS. However, how and to what extent these inflammatory and immune cells change the disease progression is still incompletely understood. The findings by Benakis et al. show that inhibition of intestinal IL-17 secreting γδ T-cells by Treg cells may alleviate poststroke inflammation in mice. This study provides a rationale for developing a therapy against activation and migration of intestinal IL-17 secreting γδ T-cells. Th17 cells are playing important role in maintaining mucosal barrier, inflammation, and microbial translocation in the gut and have the ability to efficiently breach the BBB to infiltrate in the CNS.[72] Inhibition of Th17 cytokines may be targeted to reduce inflammation. The novel transgenic models may be helpful for the identification of the origin, role, and fate of gut migrating immune cells, and thus for the development of novel therapies targeting the gut inflammatory and immune cells for the treatment of stroke.
Financial support and sponsorship
This work was supported by National Institutes of Health grants: NS40407, NS102815 and NS097875; by Veteran Affair Merit grant: I01BX003926; and by American Heart Association 0940042N-5 to B. R. H.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
Drs. Awadhesh Arya and Bingren Hu wrote the manuscript. Ms. Rose Lee helped with figures and proofreading of the manuscript.
References
- 1.Johnson W, Onuma O, Owolabi M, Sachdev S. Stroke: A global response is needed. Bull World Health Organ. 2016;94:634–A. doi: 10.2471/BLT.16.181636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang Q, Tong X, Schieb L, Vaughan A, Gillespie C, Wiltz JL, et al. Vital signs: Recent trends in stroke death rates-United States, 2000-2015. MMWR Morb Mortal Wkly Rep. 2017;66:933–9. doi: 10.15585/mmwr.mm6635e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28:203–9. [PMC free article] [PubMed] [Google Scholar]
- 4.Wang HX, Wang YP. Gut microbiota-brain axis. Chin Med J (Engl) 2016;129:2373–80. doi: 10.4103/0366-6999.190667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wen SW, Wong CH. An unexplored brain-gut microbiota axis in stroke. Gut Microbes. 2017;8:601–6. doi: 10.1080/19490976.2017.1344809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camara-Lemarroy CR, Ibarra-Yruegas BE, Gongora-Rivera F. Gastrointestinal complications after ischemic stroke. J Neurol Sci. 2014;346:20–5. doi: 10.1016/j.jns.2014.08.027. [DOI] [PubMed] [Google Scholar]
- 7.Singh V, Roth S, Llovera G, Sadler R, Garzetti D, Stecher B, et al. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J Neurosci. 2016;36:7428–40. doi: 10.1523/JNEUROSCI.1114-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winek K, Dirnagl U, Meisel A. The gut microbiome as therapeutic target in central nervous system diseases: Implications for stroke. Neurotherapeutics. 2016;13:762–74. doi: 10.1007/s13311-016-0475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boehme AK, Ranawat P, Luna J, Kamel H, Elkind MS. Risk of acute stroke after hospitalization for sepsis: A case-crossover study. Stroke. 2017;48:574–80. doi: 10.1161/STROKEAHA.116.016162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ritzel RM, Lai YJ, Crapser JD, Patel AR, Schrecengost A, Grenier JM, et al. Aging alters the immunological response to ischemic stroke. Acta Neuropathol. 2018;136:89–110. doi: 10.1007/s00401-018-1859-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sherwin E, Sandhu KV, Dinan TG, Cryan JF. May the force be with you: The light and dark sides of the microbiota-gut-brain axis in neuropsychiatry. CNS Drugs. 2016;30:1019–41. doi: 10.1007/s40263-016-0370-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol. 2012;10:735–42. doi: 10.1038/nrmicro2876. [DOI] [PubMed] [Google Scholar]
- 13.Neunlist M, Van Landeghem L, Mahé MM, Derkinderen P, des Varannes SB, Rolli-Derkinderen M, et al. The digestive neuronal-glial-epithelial unit: A new actor in gut health and disease. Nat Rev Gastroenterol Hepatol. 2013;10:90–100. doi: 10.1038/nrgastro.2012.221. [DOI] [PubMed] [Google Scholar]
- 14.Furness JB. The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol. 2012;9:286–94. doi: 10.1038/nrgastro.2012.32. [DOI] [PubMed] [Google Scholar]
- 15.Matteoli G, Boeckxstaens GE. The vagal innervation of the gut and immune homeostasis. Gut. 2013;62:1214–22. doi: 10.1136/gutjnl-2012-302550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonaz B, Bazin T, Pellissier S. The vagus nerve at the interface of the microbiota-gut-brain axis. Front Neurosci. 2018;12:49. doi: 10.3389/fnins.2018.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Lartigue G, de La Serre CB, Raybould HE. Vagal afferent neurons in high fat diet-induced obesity; intestinal microflora, gut inflammation and cholecystokinin. Physiol Behav. 2011;105:100–5. doi: 10.1016/j.physbeh.2011.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 19.Svensson E, Horváth-Puhó E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, et al. Vagotomy and subsequent risk of Parkinson's disease. Ann Neurol. 2015;78:522–9. doi: 10.1002/ana.24448. [DOI] [PubMed] [Google Scholar]
- 20.Winek K, Meisel A, Dirnagl U. Gut microbiota impact on stroke outcome: Fad or fact? J Cereb Blood Flow Metab. 2016;36:891–8. doi: 10.1177/0271678X16636890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashiro K, Tanaka R, Urabe T, Ueno Y, Yamashiro Y, Nomoto K, et al. Gut dysbiosis is associated with metabolism and systemic inflammation in patients with ischemic stroke. PLoS One. 2017;12:e0171521. doi: 10.1371/journal.pone.0171521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin J, Liao SX, He Y, Wang S, Xia GH, Liu FT, et al. Dysbiosis of gut microbiota with reduced trimethylamine-N-oxide level in patients with large-artery atherosclerotic stroke or transient ischemic attack. J Am Heart Assoc. 2015:4. doi: 10.1161/JAHA.115.002699. pii: e002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stanley D, Mason LJ, Mackin KE, Srikhanta YN, Lyras D, Prakash MD, et al. Translocation and dissemination of commensal bacteria in post-stroke infection. Nat Med. 2016;22:1277–84. doi: 10.1038/nm.4194. [DOI] [PubMed] [Google Scholar]
- 24.Kabat AM, Srinivasan N, Maloy KJ. Modulation of immune development and function by intestinal microbiota. Trends Immunol. 2014;35:507–17. doi: 10.1016/j.it.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol. 2010;28:623–67. doi: 10.1146/annurev-immunol-030409-101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–50. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- 27.Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120:3793–802. doi: 10.1182/blood-2012-02-412726. [DOI] [PubMed] [Google Scholar]
- 28.Liesz A, Hu X, Kleinschnitz C, Offner H. Functional role of regulatory lymphocytes in stroke: Facts and controversies. Stroke. 2015;46:1422–30. doi: 10.1161/STROKEAHA.114.008608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med. 2016;22:516–23. doi: 10.1038/nm.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J Clin Invest. 2015;125:699–714. doi: 10.1172/JCI76210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee N, Kim WU. Microbiota in T-cell homeostasis and inflammatory diseases. Exp Mol Med. 2017;49:e340. doi: 10.1038/emm.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luckheeram RV, Zhou R, Verma AD, Xia B. CD4+ T cells: Differentiation and functions. Clin Dev Immunol 2012. 2012:925135. doi: 10.1155/2012/925135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–52. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 34.Bonneville M, O’Brien RL, Born WK. Gammadelta T cell effector functions: A blend of innate programming and acquired plasticity. Nat Rev Immunol. 2010;10:467–78. doi: 10.1038/nri2781. [DOI] [PubMed] [Google Scholar]
- 35.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–89. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–9. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- 37.Huber S, Gagliani N, Esplugues E, O’Connor W, Jr, Huber FJ, Chaudhry A. Th17 cells express interleukin-10 receptor and are controlled by foxp3− and foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity. 2011;34:554–65. doi: 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park SG, Mathur R, Long M, Hosh N, Hao L, Hayden MS, et al. Tregulatory cells maintain intestinal homeostasis by suppressing γδ T cells. Immunity. 2010;33:791–803. doi: 10.1016/j.immuni.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crapser J, Ritzel R, Verma R, Venna VR, Liu F, Chauhan A, et al. Ischemic stroke induces gut permeability and enhances bacterial translocation leading to sepsis in aged mice. Aging (Albany NY) 2016;8:1049–63. doi: 10.18632/aging.100952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2006;26:797–810. doi: 10.1038/sj.jcbfm.9600229. [DOI] [PubMed] [Google Scholar]
- 41.Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: A double-edged sword. Int J Physiol Pathophysiol Pharmacol. 2013;5:73–90. [PMC free article] [PubMed] [Google Scholar]
- 42.Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–70. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- 43.Stevens SL, Bao J, Hollis J, Lessov NS, Clark WM, Stenzel-Poore MP, et al. The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res. 2002;932:110–9. doi: 10.1016/s0006-8993(02)02292-8. [DOI] [PubMed] [Google Scholar]
- 44.Perez-de-Puig I, Miró-Mur F, Ferrer-Ferrer M, Gelpi E, Pedragosa J, Justicia C, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015;129:239–57. doi: 10.1007/s00401-014-1381-0. [DOI] [PubMed] [Google Scholar]
- 45.Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33:586–92. doi: 10.1161/hs0202.103399. [DOI] [PubMed] [Google Scholar]
- 46.Xie L, Li W, Hersh J, Liu R, Yang SH. Experimental ischemic stroke induces long-term T cell activation in the brain. J Cereb Blood Flow Metab. 2018:271678X18792372. doi: 10.1177/0271678X18792372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stubbe T, Ebner F, Richter D, Engel O, Klehmet J, Royl G, et al. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab. 2013;33:37–47. doi: 10.1038/jcbfm.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahluwalia B, Magnusson MK, Öhman L. Mucosal immune system of the gastrointestinal tract: Maintaining balance between the good and the bad. Scand J Gastroenterol. 2017;52:1185–93. doi: 10.1080/00365521.2017.1349173. [DOI] [PubMed] [Google Scholar]
- 49.Tomasello E, Bedoui S. Intestinal innate immune cells in gut homeostasis and immunosurveillance. Immunol Cell Biol. 2013;91:201–3. doi: 10.1038/icb.2012.85. [DOI] [PubMed] [Google Scholar]
- 50.de Jong PR, González-Navajas JM, Jansen NJ. The digestive tract as the origin of systemic inflammation. Crit Care. 2016;20:279. doi: 10.1186/s13054-016-1458-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chamorro Á, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R, et al. The immunology of acute stroke. Nat Rev Neurol. 2012;8:401–10. doi: 10.1038/nrneurol.2012.98. [DOI] [PubMed] [Google Scholar]
- 52.Neher MD, Weckbach S, Flierl MA, Huber-Lang MS, Stahel PF. Molecular mechanisms of inflammation and tissue injury after major trauma-is complement the “bad guy”? J Biomed Sci. 2011;18:90. doi: 10.1186/1423-0127-18-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boehme AK, Kapoor N, Albright KC, Lyerly MJ, Rawal PV, Bavarsad Shahripour R, et al. Predictors of systemic inflammatory response syndrome in ischemic stroke undergoing systemic thrombolysis with intravenous tissue plasminogen activator. J Stroke Cerebrovasc Dis. 2014;23:e271–6. doi: 10.1016/j.jstrokecerebrovasdis.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dziedzic T. Systemic inflammation as a therapeutic target in acute ischemic stroke. Expert Rev Neurother. 2015;15:523–31. doi: 10.1586/14737175.2015.1035712. [DOI] [PubMed] [Google Scholar]
- 55.Vourc’h M, Roquilly A, Asehnoune K. Trauma-induced damage-associated molecular patterns-mediated remote organ injury and immunosuppression in the acutely ill patient. Front Immunol. 2018;9:1330. doi: 10.3389/fimmu.2018.01330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Absinta M, Ha SK, Nair G, Sati P, Luciano NJ, Palisoc M, et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife. 2017;6:e29738. doi: 10.7554/eLife.29738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lim MY, Song EJ, Kim SH, Lee J, Nam YD. Comparison of DNA extraction methods for human gut microbial community profiling. Syst Appl Microbiol. 2018;41:151–7. doi: 10.1016/j.syapm.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 58.Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65:4799–807. doi: 10.1128/aem.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sadler R, Singh V, Benakis C, Garzetti D, Brea D, Stecher B, et al. Microbiota differences between commercial breeders impacts the post-stroke immune response. Brain Behav Immun. 2017;66:23–30. doi: 10.1016/j.bbi.2017.03.011. [DOI] [PubMed] [Google Scholar]
- 60.Borody T, Fischer M, Mitchell S, Campbell J. Fecal microbiota transplantation in gastrointestinal disease: 2015 update and the road ahead. Expert Rev Gastroenterol Hepatol. 2015;9:1379–91. doi: 10.1586/17474124.2015.1086267. [DOI] [PubMed] [Google Scholar]
- 61.Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, et al. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell. 2015;163:1585–95. doi: 10.1016/j.cell.2015.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stanley D, Moore RJ, Wong CH. An insight into intestinal mucosal microbiota disruption after stroke. Sci Rep. 2018;8:568. doi: 10.1038/s41598-017-18904-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Houlden A, Goldrick M, Brough D, Vizi ES, Lénárt N, Martinecz B, et al. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav Immun. 2016;57:10–20. doi: 10.1016/j.bbi.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang AT, Choi JP, Kotzin JJ, Yang Y, Hong CC, Hobson N, et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature. 2017;545:305–10. doi: 10.1038/nature22075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McGhan LJ, Jaroszewski DE. The role of toll-like receptor-4 in the development of multi-organ failure following traumatic haemorrhagic shock and resuscitation. Injury. 2012;43:129–36. doi: 10.1016/j.injury.2011.05.032. [DOI] [PubMed] [Google Scholar]
- 66.Hörmann N, Brandão I, Jäckel S, Ens N, Lillich M, Walter U, et al. Gut microbial colonization orchestrates TLR2 expression, signaling and epithelial proliferation in the small intestinal mucosa. PLoS One. 2014;9:e113080. doi: 10.1371/journal.pone.0113080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Winek K, Engel O, Koduah P, Heimesaat MM, Fischer A, Bereswill S, et al. Depletion of cultivatable gut microbiota by broad-spectrum antibiotic pretreatment worsens outcome after murine stroke. Stroke. 2016;47:1354–63. doi: 10.1161/STROKEAHA.115.011800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–84. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meisel A, Smith CJ. Prevention of stroke-associated pneumonia: Where next? Lancet. 2015;386:1802–4. doi: 10.1016/S0140-6736(15)00127-0. [DOI] [PubMed] [Google Scholar]
- 70.Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–57. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- 71.Grønberg NV, Johansen FF, Kristiansen U, Hasseldam H. Leukocyte infiltration in experimental stroke. J Neuroinflammation. 2013;10:115. doi: 10.1186/1742-2094-10-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–5. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]