

CONSPECTUS:
Nitrogenase is a complicated two-component enzyme system that uses ATP binding and hydrolysis energy to achieve one of the most difficult chemical reactions in nature, the reduction of N2 to NH3. One component of the Mo-based nitrogenase system, Fe protein, delivers electrons one at a time to the second component, the catalytic MoFe protein. This process occurs through a series of synchronized events collectively called the “Fe protein cycle”. Elucidating details of the events associated with this cycle has constituted an important challenge in understanding the nitrogenase mechanism. Electron delivery is a multistep process involving three metal clusters with intra- and interprotein events. It is proposed that the first electron transfer event is a gated intraprotein transfer of one electron from the MoFe protein P-cluster to the FeMo cofactor. Measurement of the effect of osmotic pressure on the rate of this electron transfer process revealed that it is gated by protein conformational changes. This first electron transfer is activated by binding of the Fe protein containing two bound ATP molecules. The mechanism of how this protein–protein association triggers electron transfer remains unknown. The second electron transfer event is proposed to be a rapid interprotein “backfill” with transfer of one electron from the reduced Fe protein 4Fe–4S cluster to the oxidized P-cluster. In this way, electron delivery can be viewed as a case of “deficit spending”. Such a deficit-spending electron transfer process can be envisioned as a way to achieve one-direction electron flow, limiting the potential for back electron flow. Hydrolysis of two ATP molecules associated with the Fe protein occurs after the electron transfer and therefore is not used to directly drive the electron transfer. Rather, ATP hydrolysis is proposed to contribute to relaxation of the “activated” conformational state associated with the ATP form of the complex, with the free energy from ATP hydrolysis being used to pay back energy associated with component protein association and electron transfer. Release of inorganic phosphate (Pi) and protein–protein dissociation follow electron transfer and ATP hydrolysis. The rate-limiting step for the Fe protein cycle is not dissociation of the two proteins, as previously believed, but rather is release of Pi after ATP hydrolysis, which is then followed by rapid protein–protein complex dissociation. Nitrogenase is composed of two catalytic halves that do not function independently but rather exhibit anticooperative nuclear motion in which electron transfer in one-half of the complex partially inhibits electron transfer and ATP hydrolysis in the other half. Calculations indicated the existence of anticooperative interactions across the entire nitrogenase complex, suggesting a mechanism for the control of events on opposite ends of this large complex. The mechanistic necessity for this anticooperative process remains unknown. This Account presents a working model for how all of these processes work together in the nitrogenase “machine” to transduce the energy from ATP binding and hydrolysis to drive N2 reduction.
Graphical Abstract
INTRODUCTION
The element nitrogen is essential to all life and is the limiting nutrient in many environments.1–3 Atmospheric N2 gas is the predominant source of available N in the biosphere, and it is the reduction of N2 to two molecules of ammonia, called nitrogen fixation, that converts nitrogen into a usable form. This reduction occurs at large scale by two processes: the industrial Haber–Bosch process, which uses H2 as the N2 reductant with a supported Fe catalyst to carry out the reaction,1 and nitrogen fixation by microbes, which uses electrons and protons generated separately and is catalyzed by the enzyme nitrogenase.4 Each of these processes contributes about half of the N2 reduced globally each year.3,5 Although conversion of N2 to ammonia is an exergonic process, both industrial and biological nitrogen fixation require considerable energy input in order to carry out the first step of the reaction, cleavage of the N2 triple bond.2,3 Fossil fuels generate the H2, heat, and pressure required for the Haber–Bosch process, whereas ATP generated from metabolism supplies the energy for microbial nitrogen fixation.
Nitrogenase is found in specialized bacteria and archaea, called diazotrophs, but not in any known eukaryotes.6,7 Three different forms of nitrogenase have been reported, known as the Mo-, V- and Fe-dependent enzymes.8 The first form of nitrogenase to be discovered, and the most widely distributed in nature, is the Mo-based system,9 which is the focus of this Account. The overall architectures of these three nitrogenase forms are similar,10–15 and mechanistic studies on the different nitrogenases support the view that mechanistic insights on one system are generally applicable to all three.8,16,17
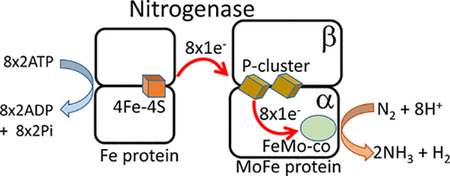
The Mo-dependent nitrogenase comprises two catalytic partners, the MoFe protein and the Fe protein (Figure 1). The MoFe protein is an α2β2 tetramer, with each αβ dimer containing two metal- and sulfur-containing clusters, an 8Fe–7S cluster (P-cluster) associated with electron transfer to the catalytic 7Fe–9S–1Mo–1C homocitrate cofactor, also called the FeMo cofactor (FeMo-co or M). The Fe protein is a homodimer with a single bridging 4Fe–4S cluster (F) and two ATP-binding sites, one located in each subunit.
Figure 1.

Diagram of the nitrogenase proteins and the metal-containing cofactors. (left) Schematic representation of the Fe protein component and the MoFe protein component with metal cofactors. (center) Structures of the 4Fe–4S cluster (F), the P-cluster (P), and the FeMo cofactor (M). Structures are from PDB entry 4WZA. (right) Legend showing representations of the metal clusters and oxidation states.
Building on decades of earlier work,9 recent studies18 have revealed key aspects of the chemistry carried out at the active-site FeMo-co, where N2 is bound and reduced, and why this surprisingly requires the transfer of eight electrons from the Fe protein to the active-site FeMo-co and thus the overall reaction stoichiometry shown in eq 1:
| (1) |
The electrons are transferred one at a time from the Fe protein’s 4Fe–4S cluster as part of a series of events collectively called the “Fe protein cycle”. Elucidating details of the events associated with the Fe protein cycle is one of the grand challenges in understanding the nitrogenase mechanism. This Account describes recent progress in meeting this challenge by revealing how nitrogenase functions as a “chemo-mechanical” machine that utilizes the energy of ATP binding and hydrolysis to achieve substrate reduction.
FE PROTEIN CYCLE
Each electron transfer event occurs during the transient (~200 ms) association of the two proteins, with key steps in the process deduced from a number of kinetic studies, including association of the two proteins, inter- and intramolecular electron transfer, hydrolysis of two ATP molecules, release of two phosphates, and finally, dissociation of the catalytic partners.
Early kinetic studies provided an initial framework for the Fe protein cycle.9 Those studies established that the cycle is initiated by rapid binding of two reduced ATP-bound Fe proteins to the MoFe protein (second-order rate constant ≈ 5 × 107 M−1 s−1) to form a ternary complex having one Fe protein associated with each half of the MoFe protein. Electron transfer from the Fe protein to the MoFe protein (Figure 1) was monitored in the pre-steady state by following absorbance changes associated with the oxidation of the Fe protein 4Fe–4S cluster, revealing an apparent first-order rate constant of 140 s−1. The rate constant for ATP hydrolysis was not directly determined, and thus, the order of electron transfer and ATP hydrolysis was not established. Kinetic measurement of inorganic phosphate (Pi) released after ATP hydrolysis indicated a rate constant of 16 s−1.9 Because the Fe protein 4Fe–4S cluster is inaccessible to reducing agents in the ternary complex but not in the free form, dissociation of the oxidized ADP-bound Fe protein from the one-electron-reduced MoFe protein was measured by absorbance changes associated with the re-reduction of the Fe protein 4Fe–4S cluster by dithionite. Such measurements indicated a protein–protein dissociation rate constant of 6 s−1.
Those early studies established some key features of the Fe protein cycle, but important elements remained to be resolved. Studies over the last 10 years have revealed the involvement of conformational changes in controlling electron transfer, the role of the P-cluster during inter- and intramolecular electron transfer, and the coupling and synchronization of electron transfer and ATP hydrolysis. These studies have led to the formulation of a revised Fe protein cycle that is discussed here.
Conformational Gating of Electron Transfer
To examine the possible involvement of conformational control (gating) of the observed electron transfer from the Fe protein to the MoFe protein, the influences of the viscosity (η) and osmotic pressure of the solution on the rate of electron transfer were examined.19 These two solution properties influence different kinds of conformationally controlled processes. Thus, the rate constant for a reaction controlled by a dynamical conformational transition (controlled by the kinetics of transformation) decreases with increasing viscosity as k(η) ∝ 1/η, where k(η) is the rate constant for electron transfer. In contrast, the rate constant for a conformational change that alters the number of bound waters is modulated by the energetics of water uptake/release through a term proportional to exp [−(Δn/55.6)m], where m is the solute molality and Δn is the number of waters absorbed in the transformation, with Δn ≥ 0 for water binding or Δn ≤ 0 for water release.19
It was found that the rate constant for electron transfer is independent of viscosity but decreases exponentially with solute molality (osmotic pressure), revealing that intracomplex oxidation of Fe protein by the resting-state MoFe protein is gated by a process controlled by the energetics of an uptake of water molecules, with Δn ≥ +80.19 This uptake corresponds to a conformational transition in which ≥800 Å2 of surface becomes exposed to solvent and binds waters (Figure 2). To place this value into context, the “ADP” structure of the Fe protein–MoFe protein complex exposes 2000 Å2 more protein surface than does the “ATP” structure.20 Thus, the conformational changes revealed by these measurements can be plausibly attributed to a large-scale motion of the Fe protein relative to the MoFe protein.
Figure 2.

Gated electron transfer in nitrogenase. Shown is a schematic of gated electron transfer from the Fe protein to half of the MoFe protein. The electron transfer event is initiated by association of the Fe protein with the MoFe protein (left). In this state, the gate for electron transfer is closed (shown as a blue bar). Next follow protein conformational changes in the complex that result in the uptake of about 80 water molecules (center). The electron transfer gate is opened (shown as two blue lines), allowing electron transfer (red arrow) to occur (right).
The finding that the effects of solute on nitrogenase electron transfer are indeed wholly energetic and not dynamical is captured by a model in which the rate-limiting step is preceded by a rapid pre-equilibrium between the energetically favored structure of the ATP-bound form of the complex and a higher-energy structure activated for electron transfer (eq 2):
| (2) |
On the basis of these considerations, it can be further speculated that the nitrogenase complex employs a “compound” conformational gating mechanism that involves other elements of the complex.19 For example, gating motions at the protein–protein interface might be accompanied by as yet unknown conformational changes, also occurring within the MoFe protein, that trigger intramolecular electron transfer from the P-cluster to FeMo-co, ATP hydrolysis, Pi release, and dissociation of the complex. Providing molecular-level details of these processes is a frontier area that requires more study.
Deficit-Spending Electron Transfer: Role of the P-Cluster
Various X-ray structures of the nitrogenase Fe protein–MoFe protein complex place the P-cluster roughly “in line” between the Fe protein [4Fe–4S] cluster and the FeMo-co within the MoFe protein.21,22 Since most electron transfer events between redox cofactors proceed by an electron tunneling mechanism and their rates drop rapidly with increasing distance, the P-cluster is expected to mediate electron transfer from the Fe protein to the FeMo-co. The involvement of the P-cluster in electron transfer was supported by a study showing that the P-cluster is redox-active during turnover.23 Still, the details of how the P-cluster participates remained poorly understood. One important observation is that all of the Fe atoms of the P-cluster are in the ferrous oxidation state in the resting protein. This P-cluster state is often designated as PN in the literature,9 where N indicates the as-isolated “native” state. A model involving the P-cluster as the initial acceptor during primary electron transfer followed by intramolecular electron transfer from the P-cluster to the FeMo-co is therefore counterintuitive because it would involve reduction of Fe atoms below the ferrous state, which would be unprecedented for FeS clusters observed to date. This situation suggests two alternatives: direct electron transfer from the Fe protein to the FeMo-co without involvement of the P-cluster or a two-step “deficit spending” process.24 In the “deficit spending” model, PN first transfers one electron to the “native” resting-state FeMo-co, designated as MN, generating a one-electron deficit within the P-cluster relative to PN plus the FeMo cofactor reduced by one electron, designated as MR. Indeed, such a P-cluster state, designated as P1+, can be experimentally generated by treating the MoFe protein resting state with oxidants. In the second step, the deficit is paid back through re-reduction of the P1+ state to PN by rapid delivery of an electron from reduced Fe protein (Figure 3, steps 2 to 3 and 3 to 4). Evidence supporting the deficit-spending electron transfer mechanism came from examination of pre-steady-state electron transfer using a MoFe protein variant (α-188Ser substituted by α-188Cys), for which 65% of the P-clusters are in the P1+ state in the resting enzyme.24 The availability of such a catalytically competent MoFe protein having P-clusters populated in both the PN and P1+ states permitted individual examination of both inter- and intramolecular electron transfer events either by monitoring optical changes associated with oxidation of the Fe protein [4Fe–4S] cluster or loss of the S = 1/2 electron paramagnetic resonance (EPR) signature associated with the P1+ state of the P-cluster in freeze-quench experiments.
Figure 3.

Updated Fe protein cycle. Shown is the Fe protein with two bound nucleotides and half of the MoFe protein with the P-cluster (P) and the FeMo cofactor (M). The cycle starts at the top left and proceeds clockwise, with the relevant transitions between states and reported rate constants noted on the black arrows. Electron transfers are noted with red arrows. The metal clusters and oxidation states are noted in Figure 1.
Electron delivery from the reduced Fe protein to the restingstate wild-type MoFe protein occurs with the rate constant kobs = 170 s−1. In contrast, Fe protein reduces α-188Cys MoFe protein in two steps: an electron transfer “burst”, with 65% of the oxidation of the Fe protein 4Fe–4S cluster occurring during the instrument dead time (≤2 ms), and a time-resolved phase (35%) having a rate constant essentially the same as that observed for the wild-type system.24 The “burst” electron transfer phase is interpreted to reflect rapid reduction of the P1+ state of the as-isolated α-188Cys MoFe protein fraction captured in the P1+/MN state, having an estimated rate constant k ≥ 1700 s−1. The kinetically resolved phase arises from the “normal” reduction of the α-188Cys MoFe protein population present in the PN/MN state. These observations are consistent with a deficit-spending electron transfer process and indicate that intramolecular transfer from the P-cluster to the FeMo cofactor is the slower of the two electron transfer steps.
Varying the concentration of sucrose as the osmolyte showed that a protein conformational gate for electron transfer acts specifically on the initiating intramolecular electron transfer deficit-spending step (PN → MN), while the rapid Fe-protein-dependent re-reduction of P1+ is not gated.24 Consideration of the X-ray structure of an oxidized MoFe protein (P1+, MN) suggested that conformational activation of the resting-state MoFe protein occurs when an as yet unidentified change in the Fe protein–MoFe protein interface causes the α-188Ser side chain to transiently coordinate to an Fe atom of PN, thereby creating an activated state of the P-cluster (designated as PN*) whose potential is lowered to the point that PN* → MN electron transfer becomes favorable. An earlier study concluded that specific residues on the MoFe protein surface where the Fe protein binds are involved in triggering electron transfer and ATP hydrolysis in the complex, suggesting one possible pathway for activation.25 It is also possible that the desolvation forces mentioned above, arising from the Fe protein–MoFe protein interaction and water exclusion could similarly energize the P-cluster, contributing to the deficit-spending electron transfer reaction.
Electron Transfer Precedes ATP Hydrolysis
The challenge of determining the pre-steady-state rate constant for ATP hydrolysis was recently overcome by using a double-mixing quench flow method employing α32P-ATP.26 In those experiments, ATP hydrolysis was measured by mixing Fe protein and MoFe protein with α32P-ATP for defined times before quenching the reaction and monitoring the formation of α32P-ADP by thin-layer chromatography. These studies established the rate constant for ATP hydrolysis as 45 to 70 s−1, which is significantly lower than the rate constant of 170 s−1 for the gated primary electron transfer.26 Thus, it was concluded that electron transfer occurs before ATP hydrolysis. Conversely, measurement of Pi release revealed that this step occurs with a rate constant of 25 s−1, placing events associated with Pi release well after ATP hydrolysis. These findings redefine the order of the Fe protein cycle, with the key events shown in Figure 3.
Thermodynamics of the Cycle
Because electron transfer precedes ATP hydrolysis, it follows that electron transfer is not driven by the free energy released at the time of ATP hydrolysis. Instead, it is ATP-dependent protein–protein binding that elicits electron transfer. The 4Fe–4S cluster desolvation hypothesis cited above could provide a mechanism for such energy transduction as part of the protein–protein interaction and water exclusion events. Also, because ATP hydrolysis and Pi release occur as temporally separated steps, both of which precede the dissociation of the complex, both hydrolysis and Pi release are required to relax the conformationally activated system, thereby inducing dissociation of the Fe protein from the reduced MoFe protein to complete the cycle.27 In the absence of ATP hydrolysis and phosphate release, the two catalytic partners would otherwise be captured in a “dead-end complex”. A similar sequence of steps was predicted by detailed theoretical thermodynamic analysis of nitrogenase electron transfer and ATP hydrolysis.27
The resulting Fe protein thermodynamic cycle is analogous to the four-step thermodynamic cycle of the ABC (ATP-binding cassette) transmembrane substrate transporters, in particular the exporter subclass.28 These transporters operate by a process dependent on conformational changes initiated by ATP binding, with the system being reset for another cycle by ATP hydrolysis. Thus, the concept of the Fe protein as a “nucleotide switch” that drives nucleotide-dependent “electron transport” must involve toggling of the Fe protein–MoFe protein complex through two conformational states: “activated” for electron transfer with ATP bound and “relaxed” following ATP hydrolysis and Pi release (Figure 3).
Exactly how nucleotide-dependent changes in the conformation of the Fe protein–MoFe protein complex initiate the electron transfer cycle remains an enigma. Neither the structure of the complex that binds the ATP analogue, AMPPCP, nor the complex that binds an analogue of the transition state of ATP hydrolysis, ADP-AlF4, reveal significant perturbations within the MoFe protein relative to isolated MoFe protein.20–22 Further nuances might arise because of the sequential nature of nucleotide binding to each subunit of the Fe protein complex. The two ATP molecules bind with varying affinities and are hydrolyzed sequentially. This was also seen in recent structural studies showing two different nucleotide analogues bound to the subunits of a single Fe protein dimer.20 However, the ABC-transporter-like mechanism does suggest why no perturbations of MoFe protein are seen. ATP hydrolysis and Pi release occur only after electron transfer and therefore contribute only to relaxation of the “activated” conformational state associated with the ATP complex. Consequently, there is no requirement that the transition state for ATP hydrolysis be associated with a high-energy state within the MoFe protein. In addition, as the deficit-spending electron transfer process is conformationally gated, it could be the case that the elusive state in which the FeMo cofactor and its environs are activated for substrate reduction are formed only by transient conformational fluctuations that never exist in high occupancy.
Pi Release Is the Rate-Limiting Step of the Fe Protein Cycle
A number of early kinetic studies reported that the release of the oxidized Fe protein-(2ADP) from the MoFe protein occurs with a rate constant of 6 s−1, clearly placing this step as the final and rate-limiting step of the overall Fe protein cycle.9 However, all of those studies used dithionite (S2O42−) to reduce the oxidized Fe protein following its dissociation from the MoFe protein. Although dithionite is a convenient experimental reagent, the equilibrium concentration of the actual species responsible for reduction (SO2•−) is low, and this species forms slowly in solution, with a rate constant near 10 s−1. This raised the possibility that such kinetic experiments, although expertly performed and confirmed by multiple investigators, were compromised by the slow formation of the active SO2•− reductant.
The issue of the rate-limiting step was resolved by using flavodoxin, a physiological electron donor to the Fe protein.29 Those studies revealed that dissociation of the Fe protein–MoFe protein complex is extremely fast following Pi release. In fact, the dissociation step measured in this way is too fast for a rate constant to be accurately established. Thus, the rate-limiting step in the Fe protein cycle is not dissociation of the Fe protein from the MoFe protein but instead is an event associated with Pi release29 (Figure 3, steps from 5 to 6 to 7). For completeness, we note that recent studies indicate that reduction of the oxidized ADP-bound Fe protein is triggered (by flavodoxin or ferredoxin) prior to exchange of ATP for ADP, thus completing the Fe protein cycle.30
Negative Cooperativity in Nitrogenase
The nitrogenase MoFe protein is a dimer of identical αβ units, with each αβ unit containing one P-cluster, one active-site FeMo-co, and one Fe protein binding site. Previous kinetic studies assumed that the MoFe protein functions as two independent catalytic units.9 However, quantitative studies undertaken to complement the kinetic measurements described above revealed that the two αβ units actually do not function independently.31 Instead, during the pre-steady-state reaction of the ternary [MoFe protein–2Fe protein-(2ATP)] complex, only approximately two ATPs are hydrolyzed per ternary complex, which is approximately half the value of four ATPs per ternary complex that would be anticipated if each half of the complex operated independently, with hydrolysis of the two ATPs bound to its Fe protein. Likewise, only approximately two Pi are released during the pre-steady-state period, not four, and freeze-quench EPR experiments showed that only about half of the FeMo-co is reduced.31 In aggregate, these quantitative measurements suggest that only about one of the two bound Fe proteins in the ternary complex proceeds through the Fe protein cycle in the pre-steady state, while the other Fe protein in the complex does not immediately and independently initiate a separate Fe protein cycle. These studies were consistent with earlier studies suggesting that the two halves of nitrogenase are not equivalent.32
To understand the stoichiometric measurements that gave values that were about half of those expected if each half of the nitrogenase complex worked independently, the experimentally measured progress curves for ATP hydrolysis, electron transfer, and Pi release were globally fit to alternative kinetic models.31 A model having each half of the complex operate independently sharply overestimates the number of ATPs hydrolyzed, while at the other extreme, a model having strictly “half-site” reactivity, with one half completely inactive, sharply underestimates the number of ATPs hydrolyzed.
A “negative cooperativity” kinetic scheme was therefore developed in which electron delivery in one half of the complex induces conformational changes that partially suppresses but do not completely eliminate the Fe protein cycle in the other half. A global fit of this model to all three progress curves (electron transfer, ATP hydrolysis, and Pi release) reproduced the time course of ATP hydrolysis and is equally good at describing the electron transfer and Pi release steps.31
These studies make it clear that the two halves of nitrogenase do not operate independently but rather are mechanically linked via conformational changes that cause them to anticooperate. Computational studies using normal-mode analysis of the large-amplitude motions in the Fe protein–MoFe protein complex provided a possible mechanism by which such motions couple the two halves of the ternary complex, thereby introducing negative cooperativity into electron transfer31 (Figure 4).
Figure 4.

Scheme illustrating negative cooperativity in the nitrogenase ternary complex. Following protein conformational changes, the electron transfer goes forward in the bottom half (step 1), while the electron transfer does not occur in the top half. Once the bottom half completes electron transfer, ATP hydrolysis, and Pi release, the top half then proceeds through electron transfer and ATP hydrolysis (step 2).
Covariance analysis of peptide backbone vibrational displacement, based on a coarse-grained model of the nitrogenase complex, indicates a cross-correlation between the motions of the two Fe proteins (Figure 4), suggesting that the motion of one of the two Fe proteins causes a response in the region between the P-cluster and the FeMo-co. Specifically, the motion of residues β-64Tyr, β-98Tyr, and β-99Phe in the MoFe protein, residues already implicated in participating in intramolecular electron transfer on the basis of amino acid substitution experiments,33 correlates with the motion of the Fe protein.31 The most significant contributor is an out-of-phase rolling motion of the Fe proteins on the surface of the MoFe protein wherein one Fe protein rolls toward the position crystallographically observed to be adopted after ATP hydrolysis,20 whereas the other rolls away from such a conformation. These results further suggest a dynamic coupling between the motions of the Fe protein and the MoFe protein region lying between the P-cluster and the FeMo-co, which may provide clues toward understanding the unique role of the Fe protein in regulating intramolecular electron transfer and substrate activation.
HOW DOES THE NITROGENASE “CHEMO-MECHANICAL MACHINE” FUNCTION?
Gating of the electron transfer step and negative cooperativity during the Fe cycle together show that nitrogenase functions as a complex “machine” in which electron delivery is coupled to nucleotide utilization and modulated by dynamic allosteric coupling between the two halves of the ternary complex. These findings, as augmented by the determination of the sequence of steps in the Fe protein cycle (electron transfer first, ATP hydrolysis second) and as complemented by molecular dynamics computations, are here combined into a first-pass discussion of the mechanism for negative cooperativity. What follows is the outline of a hypothetical model for future testing, as stimulated, in part, by a recent discussion of “half-site” reactivity in the homodimeric enzyme fluoroacetate dehalogenase.34
The model proposes that although the ternary complex is chemically and crystallographically symmetrical, the rocking motion that couples the two halves of the ternary complex (Figure 4) desymmetrizes the complex in a way that activates one side by driving its Fe protein toward the ADP-bound/ ATP-hydrolyzed state (recalling that ATP hydrolysis follows electron transfer) while hyperstabilizing the unactivated ATP-bound structure of the partner Fe protein on the other side of the complex (first gating motion). This activation of the “first” half of the complex involves activation of the P-cluster, for example by attack of β-188Ser-OH on P (second motion of the compound gate), lowering its potential and initiating P → M electron transfer with subsequent rapid backfill from the reduced Fe protein. In parallel, the motion of the other waiting side Fe protein partially stabilizes the environment of the waiting P-cluster against attack by its β-188Ser-OH, thereby lowering the effective rate constant for electron transfer in the second half of the MoFe protein, although not abolishing it. Accompanying these events, a net of approximately 80 waters are bound to protein surfaces uncovered by the motions of the Fe proteins relative to the MoFe protein on both sides of the complex. Although this binding contributes an unfavorable entropy to conformational activation, the overall energy may be favorable, further favoring the activated gating. The negative cooperativity kinetic model described above embodies these elements, both providing a consistent interpretation of the available data and pointing the way to new studies to test it.
SUMMARY AND DIRECTIONS
This Account summarizes the recent reformulation of the Fe protein cycle. The updated cycle involves the following sequence of steps: association of the two proteins, electron transfer, ATP hydrolysis, rate-limiting Pi release, rapid dissociation of oxidized Fe protein, re-reduction of the Fe protein, and nucleotide exchange (as presented in Figure 3). The electron transfer event proceeds by a two-step “deficit spending” process in which conformationally gated intramolecular electron transfer is rate limiting and is rapidly followed by intermolecular electron transfer “backfill”. The energetics of this process is described by a thermodynamic cycle in which ATP is involved in two ways. Initially, the reduced Fe protein, with two ATPs bound, forms a complex with the MoFe protein. This complex is activated to transfer an electron from the Fe protein to the catalytic FeMo cofactor via the P-cluster. After the electron transfer event, the protein–protein complex is weakened by conformational changes driven by ATP hydrolysis, resulting in the dissociation of the two proteins.
Quantitative measurements of electron transfer, ATP hydrolysis, and Pi release show that the two halves of the ternary complex do not function independently. Instead, vibrationally induced coupling between the two halves introduces a negative cooperativity in which electron transfer in one half partially suppresses electron transfer in the other. The conformational gating that controls electron transfer could well be a component of the conformational coupling associated with this negative cooperativity. The “mechanical” element of the Fe cycle, namely, the conformational coupling between the two halves of the ternary complex and the gating of electron transfer, reveal the nitrogenase two-component system to be a remarkable chemo-mechanical machine for delivering the electrons to the catalytic FeMo-co, which uses them to cleave the N2 triple bond and form two NH3.
ACKNOWLEDGMENTS
L.C.S., J.W.P., and D.N.B. were supported by the Biological Electron Transfer and Catalysis (BETCy) Energy Frontiers Research Center (EFRC), funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award DE-SC0012518 to investigate electron transfer and energy coupling in enzymes. E.A., L.C.S., and D.R.D. were funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Awards DE-SC0017866, DE-SC0010834, and DE-SC0010687 to understand the mechanism of dark operative protochlorophyllide reductase and N2 and CO2 activation by nitrogenase. S.R. was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, under Award DE-AC05-76RL01830/FWP66476 to conduct calculations to gain insights into metalloenzyme mechanism. B.M.H. was funded by the National Institutes of Health under Awards HL 63203 and GM 111097 to conduct spectroscopic and kinetic studies to gain mechanistic insights on metalloenzymes. J.W.P. and L.C.S were supported by the National Science Foundation under Award MCB-1330807 for work on structurally characterizing poised redox states of the MoFe protein.
Biography
Lance C. Seefeldt was born in Redlands, California, in 1960. He received a B.S. degree in Chemistry from the University of Redlands and a Ph.D. in Biochemistry from the University of California Riverside. He is currently a Professor of Chemistry and Biochemistry at Utah State University, where his research is focused on elucidating the mechanism of the enzyme nitrogenase.
Brian M. Hoffman was born in Chicago and was an undergraduate at the University of Chicago. He then wandered to the West Coast for a Ph.D. from Caltech under the direction of Harden McConnell and on to the East Coast for a postdoctoral year with Alex Rich at MIT before joining the faculty at Northwestern University, where he is Morrison Professor of Chemistry and Professor of Molecular Biosciences. His research interests beyond metalloenzyme mechanism have included electron transfer within protein complexes and the study of porphyrazine macrocycles. He and his wife Janet are avid collectors of daughters (four total) and grandkids (seven to date).
John W. Peters received his B.S. in Microbiology in 1989 from the University of Oklahoma. He did graduate work in the Department of Anaerobic Microbiology the Anaerobe Lab at Virginia Tech, where he was the recipient of a Pratt Animal Nutrition Fellowship, and postdoctoral work at CalTech, where he was the recipient of an NIH Ruth L. Kirschstein National Research Service Award. He is currently Professor and Director of the Institute of Biological Chemistry at Washington State University. His research interests focus on examining structure–function relationships in enzymes in various aspects of microbial redox catalysis.
Simone Raugei obtained a Ph.D. in Theoretical Chemistry from the University of Florence in 2000. He was an Assistant Professor of Biophysics at the International School for Advanced Studies (SISSA) in Trieste from 2002 to 2009, where he was tenured in 2010. He has been a Chief Scientist at Pacific Northwest National Laboratory (PNNL) since 2010 and Research Professor at Washington State University since 2017. His research deals with computation and modeling of chemical and biochemical processes for energy storage and energy delivery.
David N. Beratan studied chemistry at Duke (B.S., 1980) and Caltech (Ph.D., 1986). He was a National Research Council Postdoctoral Associate and Member of the Technical Staff at NASA’s Jet Propulsion Laboratory before beginning his academic career at the University of Pittsburgh. In 2001, he moved to Duke University, where he is the R. J. Reynolds Professor of Chemistry, Professor of Biochemistry, and Professor of Physics. His studies focus on theoretical aspects of chemical and biochemical processes.
Edwin Antony was born in Chennai, India, in 1976. He received his B.Sc. degree in Zoology and Chemistry from Loyola College, an M.Sc. degree in Biochemistry from St. Joseph’s College, and a Ph.D. in Molecular Biology and Biochemistry from Wesleyan University in Connecticut. He is currently an Assistant Professor at Marquette University, where his research is focused on the mechanism of action of various ATPases.
Dennis R. Dean was born in Indianapolis, Indiana, in 1951. He received a B.A. degree in Biology from Wabash College and a Ph.D. in Molecular Biology from Purdue University. He is currently a University Distinguished Professor, Stroobants Professor of Biotechnology, and Director of the Fralin Life Science Institute at Virginia Tech. His research is focused on genetic manipulation of biological nitrogen fixation and the mechanisms for the assembly of simple and complex iron–sulfur cofactors.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Smil V Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production; The MIT Press: Cambridge, MA, 2004. [Google Scholar]
- (2).Nørskov J; Chen J Sustainable Ammonia Synthesis; U.S. Department of Energy: Washington, DC, 2016; https://science.energy.gov/~/media/bes/pdf/reports/2016/SustainableAmmoniaReport.pdf (accessed May 5, 2018). [Google Scholar]
- (3).Lehnert N; Coruzzi G; Hegg E; Seefeldt L; Stein L Feeding the World in the 21st Century: Grand Challenges in the Nitrogen Cycle; National Science Foundation: Alexandria, VA, 2015; https://www.nsf.gov/mps/che/workshops/nsf_nitrogen_report_int.pdf (accessed May 5, 2018). [Google Scholar]
- (4).Patil BS; Hessel V; Seefeldt LC; Dean DR; Hoffman BM; Cook BJ; Murray LJ Nitrogen Fixation In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH, 2017; DOI: 10.1002/14356007.a17_471.pub2. [DOI] [Google Scholar]
- (5).Chen JG; Crooks RM; Seefeldt LC; Bren KL; Bullock RM; Darensbourg MY; Holland PL; Hoffman B; Janik MJ; Jones AK; Kanatzidis MG; King P; Lancaster KM; Lymar SV; Pfromm P; Schneider WF; Schrock RR Beyond fossil fuel driven nitrogen transformations. Science 2018, 360, eaar6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vicente EJ; Dean DR Keeping the nitrogen-fixation dream alive. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 3009–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Burén S; Rubio LM State of the art in eukaryotic nitrogenase engineering. FEMS Microbiol Lett. 2018, 365, fnx274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Eady RR Structure function relationships of alternative nitrogenases. Chem. Rev 1996, 96, 3013–3030. [DOI] [PubMed] [Google Scholar]
- (9).Burgess BK; Lowe DJ Mechanism of molybdenum nitrogenase. Chem. Rev 1996, 96, 2983–3012. [DOI] [PubMed] [Google Scholar]
- (10).Chan MK; Kim J; Rees DC The nitrogenase FeMo cofactor and P-cluster pair: 2.2 Å resolution structures. Science 1993, 260, 792–794. [DOI] [PubMed] [Google Scholar]
- (11).Kim J; Rees DC Structural models for the metal centers in the nitrogenase molybdenum-iron protein. Science 1992, 257, 1677–1682. [DOI] [PubMed] [Google Scholar]
- (12).Rees DC; Howard JB Structural bioenergetics and energy transduction mechanisms. J. Mol. Biol 1999, 293, 343–350. [DOI] [PubMed] [Google Scholar]
- (13).Kim J; Rees DC Crystallographic structure and functional implications of the nitrogenase molybdenum-iron protein from Azotobacter vinelandii. Nature 1992, 360, 553–560. [DOI] [PubMed] [Google Scholar]
- (14).Georgiadis MM; Komiya H; Chakrabarti P; Woo D; Kornuc JJ; Rees DC Crystallographic structure of the nitrogenase iron protein from Azotobacter vinelandii. Science 1992, 257, 1653–1659. [DOI] [PubMed] [Google Scholar]
- (15).Sippel D; Einsle O The structure of vanadium nitrogenase reveals an unusual bridging ligand. Nat. Chem. Biol 2017, 13, 956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Harris DF; Lukoyanov DA; Shaw S; Compton P; Tokmina-Lukaszewska M; Bothner B; Kelleher N; Dean DR; Hoffman BM; Seefeldt LC Mechanism of N2 reduction catalyzed by Fe-nitrogenase involves reductive elimination of H2. Biochemistry 2018, 57, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hales BJ Alternative nitrogenase. Adv. Inorg. Biochem 1990, 8, 165–198. [PubMed] [Google Scholar]
- (18).Hoffman BM; Lukoyanov D; Yang Z-Y; Dean DR; Seefeldt LC Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem. Rev 2014, 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Danyal K; Mayweather D; Dean DR; Seefeldt LC; Hoffman BM Conformational gating of electron transfer from the nitrogenase Fe protein to MoFe protein. J. Am. Chem. Soc 2010, 132, 6894–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tezcan FA; Kaiser JT; Howard JB; Rees DC Structural evidence for asymmetrical nucleotide interactions in nitrogenase. J. Am. Chem. Soc 2015, 137, 146–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tezcan FA; Kaiser JT; Mustafi D; Walton MY; Howard JB; Rees DC Nitrogenase complexes: multiple docking sites for a nucleotide switch protein. Science 2005, 309, 1377–1380. [DOI] [PubMed] [Google Scholar]
- (22).Owens CP; Katz FEH; Carter CH; Luca MA; Tezcan FA Evidence for functionally relevant encounter complexes in nitrogenase catalysis. J. Am. Chem. Soc 2015, 137, 12704–12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chan JM; Christiansen J; Dean DR; Seefeldt LC Spectroscopic evidence for changes in the redox state of the nitrogenase P-cluster during turnover. Biochemistry 1999, 38, 5779–5785. [DOI] [PubMed] [Google Scholar]
- (24).Danyal K; Dean DR; Hoffman BM; Seefeldt LC Electron transfer within nitrogenase: evidence for a deficit-spending mechanism. Biochemistry 2011, 50, 9255–9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Christiansen J; Chan JM; Seefeldt LC; Dean DR The role of the MoFe protein α-125Phe and β-125Phe residues in Azotobacter vinelandii MoFe protein–Fe protein interaction. J. Inorg. Biochem 2000, 80, 195–204. [DOI] [PubMed] [Google Scholar]
- (26).Duval S; Danyal K; Shaw S; Lytle AK; Dean DR; Hoffman BM; Antony E; Seefeldt LC Electron transfer precedes ATP hydrolysis during nitrogenase catalysis. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 16414–16419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kurnikov IV; Charnley AK; Beratan DN From ATP to electron transfer:electrostatics and free-energy transduction in nitrogenase. J. Phys. Chem. B 2001, 105, 5359–5367. [Google Scholar]
- (28).Locher KP Structure and mechanism of ATP-binding cassette transporters. Philos. Trans. R. Soc., B 2009, 364, 239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yang Z-Y; Ledbetter R; Shaw S; Pence N; Tokmina-Lukaszewska M; Eilers B; Guo Q; Pokhrel N; Cash VL; Dean DR; Antony E; Bothner B; Peters JW; Seefeldt LC Evidence that the Pi release event is the rate-limiting step in the nitrogenase catalytic cycle. Biochemistry 2016, 55, 3625–3635. [DOI] [PubMed] [Google Scholar]
- (30).Pence N; Tokmina-Lukaszewska M; Yang Z-Y; Ledbetter RN; Seefeldt LC; Bothner B; Peters JW Unraveling the interactions of the physiological reductant flavodoxin with the different conformations of the Fe protein in the nitrogenase cycle. J. Biol. Chem 2017, 292, 15661–15669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Danyal K; Shaw S; Page TR; Duval S; Horitani M; Marts AR; Lukoyanov D; Dean DR; Raugei S; Hoffman BM; Seefeldt LC; Antony E Negative cooperativity in the nitrogenase Fe protein electron delivery cycle. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E5783–E5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Maritano S; Fairhurst SA; Eady RR Long-range interactions between the Fe protein binding sites of the MoFe protein of nitrogenase. JBIC, J. Biol. Inorg. Chem 2001, 6, 590–600. [DOI] [PubMed] [Google Scholar]
- (33).Peters JW; Fisher K; Newton WE; Dean DR Involvement of the P cluster in intramolecular electron transfer within the nitrogenase MoFe protein. J. Biol. Chem 1995, 270, 27007–27013. [DOI] [PubMed] [Google Scholar]
- (34).Kim TH; Mehrabi P; Ren Z; Sljoka A; Ing C; Bezginov A; Ye L; Pomès R; Prosser RS; Pai EF The role of dimer̀ asymmetry and protomer dynamics in enzyme catalysis. Science 2017, 355, eaag2355. [DOI] [PubMed] [Google Scholar]