

Abstract
Exosomes are extracellular nanosized vesicles with lipid bilayers encapsulating nucleic acids and proteins, both with and without glycosylation. While exosomal nucleic acids and proteins have previously been explored to identify cancer biomarkers with some promising results, little information has been available concerning their glycoconjugate content. Exosomes were isolated from normal urine samples through multistep differential centrifugation. The isolated exosomes have an average size of 146 nm and a spherical shape, as determined by dynamic light scattering and transmission electron microscopy, respectively. N-Glycans were enzymatically released from the isolated vesicles. After being reduced and permethylated, N-glycans were measured by MALDI mass spectrometry. Paucimannosidic, high-mannose, and complex type glycans were identified and their relative abundances were determined. Some detailed structures of these glycans were revealed through liquid chromatography/tandem mass spectrometry (LC/MS-MS). The reduced N-glycans, without being permethylated, were also separated and analyzed by LC/MS-MS, and their structures were further detailed through isomeric separation on porous graphitized carbon (PGC) packed in long capillaries. Using microfractionation before LC/MS-MS, minor multiantennary N-glycans were preconcentrated as based on hydrophobicity or charge. Preconcentration of the reduced and permethylated glycans on a C18 cartridge revealed numerous large glycans, whereas fractionation of the reduced N-glycans by ion-exchange cartridges facilitated detection of sulfated glycans. After removing N-glycans from the original sample aliquot, O-glycans were chemically released from urinary exosomes and profiled, revealing some unusual structures.
Graphical Abstract
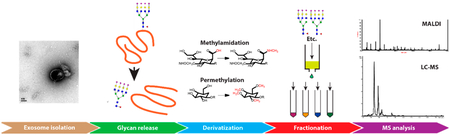
A recent surge of interest in the molecular content of exosomes encountered in biological fluids stems from their diagnostic and therapeutic potential.1 Exosomes are nanosized extracellular entities that appear to reflect directly the status of their parent cells. The intracellular origin, secretion mechanism and numerous intercellular interactions of various exosomes have been reviewed,2 indicating their important roles in intercellular communication and, therefore, their major physiological and pathological importance. While the roles of exosomal vesicles were previously thought to be confined to the mechanisms of “cellular waste management”, such as removal of dangerous or redundant substances, oxidized phospholipids, caspase-3, etc.,3 their signaling function is currently receiving considerable attention. To this end, the exosomes may use proteins, lipids, and RNAs packaged in the vesicles with a lipid bilayer.
There is increasing evidence of the involvement of exosomes in the cancer process: suppression of the immune system;4 transfer of mRNA from tumor to normal cells;5 and drug resistance.6 Melanoma-originated exosomes mediate lymphatic metastasis by cancer cells.7 While the potential scope for analyzing exosome-derived samples and their disease connections is wide-ranging, to isolate, purify and characterize them in their detailed molecular terms will necessitate development of new analytical approaches and methodologies. The protocols for isolation and characterization of exosomes from biological fluids and cellular cultures have been under development for some time,8–10 but the availability of biomedically practical methodologies still represent challenges. The exosomes isolated from different sources can be characterized morphologically, histochemically and immuno-logically, but their molecular content has become of increasing interest as sensitive “omics” methodologies become gradually available. As an outstanding example, a surface proteoglycan, glypican-1, was recently identified to represent cancer exosomes in small volumes of blood sera from early pancreatic cancer patients.11 However, molecular studies of various exosomal fractions have been typically confined to RNAs and proteins.
The present communication concerns the analytical aspects of the isolation and comprehensive glycomic characterization of human urinary exosomes. Since the pioneering study on the isolation and proteomic profiling of urinary exosomes by Pisitkun et al.,12 additional characterization studies have been reported.13–16 While the presence of glycoproteins in urinary exosomes15 and prostatic secretions17,18 has recently been indicated in terms of a few specific N-glycan structures, there is a need to characterize more completely the glycosylation patterns associated with urinary glycoproteins, in general, and biomolecules associated with urinary exosomes as a unique source of potential disease biomarkers,19 in particular.
The isolation of exosomes in small volumes of biological samples has been a challenging task and the limitation to perform thorough structural studies. Urine samples provide an exceptional opportunity for certain exosome studies as a rich source of biochemical information, including glycoproteins. In their recent pioneering work, Saraswat et al.15 partially characterized N-glycopeptides and glycans isolated from 800 mL urine samples. In a different investigation, Staubach et al.16 described differential centrifugation and isolation of exovesicles from 200 mL aliquots of urine from galactosemic patients; their targeted glycoproteins were connected to proteinuria and renal damage associated with this condition. Their previous study14 provided indication of a shift from high-mannose to complex structures due to galactosemia. In this study, we have devised a comprehensive system of in-depth glycomic profiling techniques, on relatively small volumes of urine (40 mL), which permits us to follow concentrations of a wide range of N-glycans as well as O-glycans. High sensitivity of our measurements through MALDI-mass spectrometry (MS) and capillary LC/tandem MS techniques makes it feasible to work with relatively small urine samples as needed in clinical investigations. Moreover, the miniaturized sample preconcentration techniques, based on hydrophobicity and charge, are demonstrated to extend the range of detected and identified glycans to large oligosaccharides that were previously implicated in other biofluids as putative cancer biomarkers.20–25
Finally, the use of long capillary columns packed with small graphitized carbon particles under the conditions of ultrahigh-pressure liquid chromatography (UHPLC) is demonstrated as being effective to resolve glycan isomers prior to their MS characterization.
EXPERIMENTAL SECTION
The subsection of materials, sample preparation for MALDITOF measurements of glycans, MALDI-TOF mass analysis and LC-MS of released N-glycans from urinary exosomes are described in Supporting Information.
Isolation of Urinary Exosomes from Urine.
Forty milliliters of fresh early morning urine were collected from several healthy male individuals. Immediately after the collection, 0.02% NaN3 and 0.5 mL of protease inhibitor cocktail were added as previously described.26 The urine was first centrifuged at 17,000 g for 30 min at room temperature to remove urinary sediment. The pellet was redissolved in 500 μL isolation solution (IS), containing 10 mM triethanolamine, 250 mM sucrose, 0.02% NaN3 at pH 7.6, and 100 mg of D,L-dithiothreitol was added to release the entrapped exosomes from aggregated Tamm–Horsfall protein (THP).27 The redissolved pellet was combined with the supernatant and centrifuged at 200 000g for 1 h at room temperature. The pellet was again redissolved in 500 μL IS, and 100 mg of D,L-dithiothreitol was added. The mixture was incubated for 45 min at 37 °C, vortexed, and centrifuged at 17 000g for 1 min. To the supernatant, 20 mL of IS was added and centrifuged at 200 000g for 1 h at room temperature. The pellet was resuspended in 20 mL of phosphate buffered saline (PBS) and centrifuged at 200 000g, while the pellet was again collected. Finally, the pellet was dissolved in 200 μL of 0.2% sodium dodecyl sulfate (SDS) in 10 mM phosphate buffer pH7.4 and heated for 10 min at 60 °C for glycomic studies or PBS for dynamic light scattering and transmission electron microscopic characterization.
Characterization of Urinary Exosomes by Transmission Electron Microscopy (TEM) and Dynamic Light Scattering.
Purified urinary exosomes in PBS (10 μL) were applied to carbon-coated copper grids and incubated for 5 min at room temperature. Following removal of the excess fluid, 10 μL of 2% uranyl acetate was added to the grid and incubated for 5 min. The excess uranyl acetate was removed, and the grids were allowed to air-dry. Electron micrographs were recorded using a JEOL JEM 1010 transmission electron microscope (TEM) (JEOL USA Inc., Peabody, MA). For the dynamic light-scattering measurements, exosomes of 80 μg/mL in PBS buffer were placed in the Zetasizer Nano S instrument (Malvern, Herrenberg, Germany), and the size distribution was determined by intensity.
Prefractionation of Permethylated N-Glycans with Micro-Spin C18 Column.
The reduced and permethylated N-glycans released from urinary exosomes were separated using a C18 Micro-Spin column. To the dried samples, 200 μL of 50% methanol was added, and the solution was placed on preequilibrated Micro-Spin C18 columns with three passages and eluted with a sequence of 600 μL of 5%, 15%, 35%, 50%, and 75% acetonitrile containing 0.1% TFA. Several fractions containing the released N-linked glycans were collected and dried using the SpeedVac.
Prefractionation of Native Reduced N-Glycans Using Ion-Exchange Chromatography.
Reduced N-glycan mixture released from urinary exosomes was further fractionated with an Oasis max (3 cc) column cartridge (Waters Corporation, Milford, MA) and eluted by gravity. The protocol from Wang et al.28 was modified for this purpose. The column was preconditioned with a stepwise addition of 1 mL of 95% acetonitrile, 2 mL of 100 mM sodium acetate, 6 mL of water, and 6 mL of 95% acetonitrile. The dried glycans were dissolved in 3 mL of 95% acetonitrile and run through the column three times, after which the neutral glycans were eluted with 6 mL of 50% acetonitrile. The acidic glycans were eluted into four fractions by a stepwise addition of the increasing levels of sodium acetate: 3 mL of 10 mM sodium acetate, 3 mL of 20 mM sodium acetate, 3 mL of 50 mM sodium acetate, and 3 mL of 100 mM sodium acetate. The fractions eluted with sodium acetate were desalted with macro-spin charcoal columns (Harvard apparatus, Holliston, MA). Columns were preconditioned three times with 85%/15%/0.1% (acetonitrile/water/TFA) and three times with 5%/95%/0.1% (acetonitrile/water/TFA) by centrifuging for 2 min at 1800 rpm. 150 μL acetonitrile and 3 μL TFA were added to the samples before putting the sample to the column in 1 mL at a time, and centrifuged for 2 min at 1800 rpm. The process was repeated, so that the sample mixture passed through the column twice. Subsequently, the column was washed two-times with 500 μL 5%/95%/0.1% (acetonitrile/water/TFA). To elute the sample, 500 μL of 50%/50%/0.1% (acetonitrile/water/TFA) was added and centrifuged for a few seconds. The column was allowed to equilibrate for about 10 min before centrifuging for1.5 min at 1800 rpm. Another 500 μL of 50%/50%/0.1% (acetonitrile/water/TFA) was added to elute the sample by centrifuging for 1.5 min at 3000 rpm. Samples were finally dried on the CentriVap Centrifugal Vacuum Concentrator.
LC-ESI-MS of Methylamidated N-Glycans.
Nonreduced samples separated through ion-exchange chromatography, as described earlier, were subjected to methylamidation of the sialic residues on the glycans. The procedure was performed as previously described.29,30 Briefly, the samples were dissolved in 5 μL of DMSO containing 2 M methylamine hydrochloride and 1 M 4-methylmorpholine. Five microliters of 100 mM PyAOP in DMSO were added and allowed to react for 2 h at room temperature in darkness. Two hundred forty microliters of 85% acetonitrile/15% water was used to quench the reaction. Sample was purified using Ultra-Micro SpinColumn amino columns. After methylamidation, the samples were reduced as described earlier. Samples were run on LC-MS with the same settings as neutral (unreactive) glycans, except the buffers that were changed to contain 0.1% FA.
O-Glycan Measurements from Urinary Exosomes, Human Blood Serum (HBS), and Fetuin.
O-Glycans were extracted from the lysed and denatured urinary exosomes, HBS and fetuin (used as O-glycan “controls”) based on our previous method with little modification.31 Urinary exosomes (85 μg protein), HBS (2.5 μg) and fetuin (100 μg) were denatured by0.1% SDS and 0.1% ß-mercaptoethanol in 10 mM sodium phosphate (pH 7.5) at 60 °C for 1 h. After the samples were cooled for 10 min at room temperature, 2.5 μL of 10% Nonidet P-40 (NP-40) was added and allowed to equilibrate for 10 min. Subsequently, 1 μL of 0.25 mU of PNGase F was added to each sample and incubated at 37 °C for 21 h. While N-glycans were removed by size exclusion through Amicon Ultra-0.5 (10 kDa), the remaining O-glycoproteins were digested by 5 μg of trypsin at room temperature for 4 h and 20 μg of pronase at 60 °C for 48 h. Subsequently, using amino microcolumn (Harvard Apparatus), the released amino acids were washed out, while the residual O-linked amino acids were subjected to a chemical release of O-linked oligosaccharides during the solid-phase permethylation.31 Their profiles were recorded using MALDITOF/TOF mass spectrometry.
RESULTS AND DISCUSSION
General Considerations.
Much current interest in the isolation and bioanalytical characterization of exosomes has been justified by the hypotheses linking the composition of exosomes to specific cells that determine or influence their biological destination (e.g., their signaling functions or a potential role in cancer metastasis).
The genitourinary tract is lined with a variety of cells that release their respective exosomes into the urine. It is thus reasonable to expect that they are a source of potential glycoconjugate biomarkers of the diseases such as ovarian, prostate or urothelial cancers. Urine collection and chemical analyses represent easy and noninvasive procedures in a clinical setting. As a prelude to glycomic biomarker investigations, detailed characterization of the normal urinary exosome glycome is in order. Herein, we describe a set of powerful analytical techniques, applied in concert, to separate, identify and quantify both N-glycans and O-glycans in 40 mL aliquots of normal male urine. Our additional emphasis has been placed on tracing certain components and high-mass tri- and tetraantennary N-glycans due to their expected diagnostic importance.20–25
Isolation of Urinary Exosomes and Their Characterization.
The urinary exosomes were isolated from urine through multiple ultracentrifugation (scheme in Figure S1), according to a modification of the previously published procedures.27,32 Isolation of exosomes from urine can be complicated because of the presence of THP cross-linked network, which can entrap exosomes. Reducing the disulfide bonds of THP to break down the network was suggested as an effective way of a release of the entrapped exosomes.27 Herein, during the purification, reducing agent DTT was introduced and proven to be effective and reproducible. We also found it convenient to dissolve the purified exosomes in 10 mM phosphate buffer at pH 7.4, containing 0.2% SDS, prior to glycomic studies. Otherwise, a precipitate can appear, possibly due to the aggregation of the exosomes in phosphate-buffered saline (PBS). However, for TEM examination, a diluted exosome solution in PBS seems more appropriate, as the presence of 0.2% SDS would necessarily disrupt the membrane structure of exosomes.
The protein contents of the urinary exosomes isolated from 40 mL urine typically reached about 300 μg or slightly higher levels, depending on how concentrated the urine samples may be during the day and diurnal variations.
As is often performed in the practice of clinical determinations, we prefer acquiring samples as the morning first voided urine aliquots; alternatively creatinine or other normalizing procedures can be employed.33 To test the reproducibility of the purification procedure, exosomes were processed from one batch of urine sample three times, and the average yield based on their protein contents was 322 ± 9 μg per 40 mL of urine. The purified urinary exosomes were characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM). DLS measurement gave the average size of 146 nm for the isolated urinary exosomes by intensity (Figure 1A). TEM images show that the isolated urinary exosomes have a membrane structure (Figure 1B).
Figure 1.

(A) Size distribution of the purified urinary exosomes measured with dynamic light scattering by intensity and (B) the TEM images of purified urinary exosomes.
SDS-PAGE and LC-MS Characterization of Urinary Exosomes.
The exosomes were characterized with SDS-PAGE with Coomassie blue staining (Figure S2). Many protein bands were observed in the exosome sample, the strongest at around 100 kDa, which is the expected size of monomeric uromodulin. In addition, tryptic digests of exosomes were analyzed with nanoLC-MS (Supporting Information). MS/MS spectra were analyzed with search engine MASCOT, which identified 93 proteins with a MudPIT score of 50 or better. The proteins identified were similar to previous reports on human urinary exosomes.12,15 Several previously reported exosomal markers were identified, such as programmed cell death 6-interacting protein (ALIX), ezrin and neprilysin. Four members of the ESCRT-III complex were also identified. The identified proteins indicated that the exosomes may stem from several cell types. The entire proteomic profile will be reported in more detail in a future communication.
Exosomal N-Glycan Profile Determined through MALDI-TOF-MS.
Figure 2 shows a fairly comprehensive MS profile of N-glycans. Since exhaustive permethylation23,34 was utilized as a sample derivatization procedure, both neutral and acidic glycans are readily observed in the same analytical runs. In particular, tetra-antennary glycans became clearly visible and easily quantifiable in our procedure, as seen even more succinctly in the normalized graph (Figure S3). As this region of a profile may have significant meaning vis-à-vis cancer-related N-glycan structures, it represents a significant extension over previously reported studies.14,15 Figure S3, in comparison to a typical N-glycan profile of a human blood serum,20–25 further demonstrates how remarkably different are the urinary exosomal samples from those of other biological fluids. Even more profoundly, larger N-glycans become more visible after preconcentration using a C18 cartridge (Figure 3).
Figure 2.

Profile of N-glycans of urinary exosomes identified by MALDI-TOF/MS after permethylation.
Figure 3.

MALDI-MS of urinary exosomal N-glycans after C18 preconcentration of permethylated “heavy fraction”.
The profiles seen in Figures 2 and S3 start with the paucimannosidic structures and continue with high-mannose and complex-type glycans in a wide range of structures featuring fairly extensive fucosylation and a very high level of sialylation toward the profile ends. The high representation of multiply sialylated structures, unlike with the previous report,15 is likely due to the use of permethylation that effectively stabilizes these multiantennary glycans and provides greater detection sensitivity. The overall profiles are profoundly different from those obtained through MALDI-MS in blood sera,20–25 which are normally dominated by the immunoglobulin-originated N-glycans. However, various bisecting structures are prominently represented in the urinary exosome isolates. As seen in Figure S3, the quantitative proportions of multiantennary glycans is quite high, and it is not clear, at this stage of research, to what extent some of these structures originate from the “contaminant THP” as to their possible content from the exosomes per se. This, in principle, should not impair the future clinical comparative studies, as long as the quantitative profile reproducibility would be maintained.
Interestingly, we have been able to observe paucimannosidic glycans in our urinary exosome fractions. Previously, these glycans have been chiefly reported in invertebrates and plants, which have limited complex glycosylation.35 However, some groups detected more recently these glycans in human specimens. Proteins modified with paucimannose structures were found in human embryonic stem cells36 and human buccal epithelial cells.37 Increases in the paucimannosidic glycans were also found in human colorectal cancer tissues.38 In contrast, reduced paucimannosylation was found in tuberculosis-infected macrophages.39 Recently, paucimannosidic glycans were identified in the azurophilic granules of human neutrophils.40 The paucimannosidic glycans were reported to be trimmed from other types of glycans by distinct glycosidases. Such glycans are usually related to inflammatory conditions including cancers.41
Reduced and permethylated glycans ionize in a more uniform manner than native glycans do, which makes it feasible to perform quantitative evaluation of the abundance of these glycans. The relative abundance of each glycan from the purified urinary exosomes is shown in Figure S3. Permethylated N-glycans became more hydrophobic and can be thus enriched by a C-18 precolumn. Through this step, smaller glycans were eluted with 35% acetonitrile with 0.1% TFA, and the remaining “heavier” glycans were eluted by 50% acetonitrile with 0.1% TFA. Figure 3 demonstrates more distinctly the presence of larger glycans after the C-18 enrichment.
All observed glycans are sialylated and tetra-antennary glycans are abundant. In addition to glycans carrying the Sda determinant, lactosamine-elongated glycans are also observed.
Exosomal N-Glycan Profiles Displayed through LC/ESI-MS.
Permethylated N-glycans released from the exosomes were separated on a C18 column with increasing concentration of acetonitrile prior to detection and automatic tandem MS fragmentation of the most abundant ions in the scan range of m/z 300–2000. The structures found previously by MALDIMS were confirmed through LC-MS/MS, while some additional structures could also be detected. The annotated composite spectrum of sodiated ions from the heavy C18 fraction is shown in Figure 4. All suggested structures are either tri- or tetra-antennary N-glycans that are terminally sialylated and only one of the 17 structures is lacking core fucose. The major ion in the spectrum is the m/z molecular ion 1550.73+, corresponding to a tetra-antennary, sialylated, core fucosylated N-glycan. The tetra-antennary glycans also display variable degree of substitutions with the terminal NeuAc(HexNAc-)Hex Sda-determinant (one Sda: m/z 1632.33+/1230.24+, two Sda: m/z 1714.03+, three Sda: m/z 1796.03+ and four Sda: m/z 1877.74+). The annotated MS2 spectrum of m/z 1877.74+ is shown in Figure 5. The Sda-determinant was previously elucidated on the N-glycans of Tamm-Horsfall glycoprotein in human urine,42 but it is also known to be found with glycoproteins and glycolipids in several organs and blood.43 Sialylated terminal lactosamine structures are also observed on tri- (m/z 1430.23+) and tetra-antennary N-glycans on one (m/z 1281.44+/1700.33+), or two arms (m/z 1850.04+). The LC A buffer had to contain high concentration of sodium ions to avoid peak splitting between protonated and sodiated ions. Interestingly, sodium adducts also served here as a prerequisite for a strong and informative MS2 fragmentation.
Figure 4.

Reverse-phase-LC-MS composite spectrum of permethylated glycans eluted between the retention times of 85–108 min after a C18 preconcentration.
Figure 5.

MS/MS of the permethylated N-glycan ion m/z 1877.74Na+ at retention time of 96 min. MS/MS of the sodiated ion provides informative fragments which were not usually observed for protonated glycans. The small extracted ion chromatogram shows that the ion originated from elution as a single peak during chromatography on the C18 capillary column.
Chromatography on porous graphitized carbon (PGC) of native or reduced glycans offers the advantage of isomeric separation. With such a separation, the MS/MS runs in many cases resolve their structural differences. A comprehensive base chromatogram of human urinary exosome N-glycans run on a 900 × 0.075 mm PGC column displayed a variety of N-glycans, mainly of the high-mannose and complex type structures. Several isomeric compounds can be seen resolved, such as the [M + 2H+]2+ ions at m/z 968.5 and m/z 864.9 (Figure 6). The ions at m/z 968.5 show similar MS/MS spectra with some quantitative differences among fragment peaks. Most likely, this is an indication that the sialic acid is located on different antennae. In the negative-ion mode, the 1–6 linked branch fragmentation was seen as dominant,44 so that a similar case could be possible for the positive-ion mode, but this has not been reported yet to our knowledge. The MS/MS spectra for the two peaks at m/z 864.9 are also similar to each other, but the peak at retention time of 104.9 min reveals a fragment at 1475.3, indicating a NeuAc-Hex-HexNAc-(NeuAc-Hex-Hex-NAc-)Hex fragment. This indicates that the sialylated antenna is linked to the same mannose (data not shown).
Figure 6.

Base–peak chromatogram obtained with a PGC long capillary column. Sample represents total reduced N-glycan fraction from urinary exosomes. Green and purple peaks give examples of isomeric separations with the ions at m/z 968.5 and 864.9, respectively. Under the used chromatographic conditions, most tetra-antennary glycans remain unresolved in spite of a fairly high column efficiency.
To further enhance the number of detectable compounds in the exosome preparation, sample fractionation was carried out. An ion-exchange microcolumn separated the native glycans as based on the number of anionic groups (such as sialic residues and sulfation). Five fractions were eluted with an increasing concentration of sodium acetate and recovered. The first fraction (ion exchange fraction A) was eluted with 1:1 water/acetonitrile. In this fraction, the glycans without sialic acids and sulfate groups were detected (Figure 7). Generally, the same glycans found in the total base-peak chromatogram (Figure 6) could be found in this eluate. One additional glycan was a core-fucosylated isomer to the antennae-fucosylated bisecting structure at m/z 997.0. The next fraction (ion-exchange fraction B) mainly contained N-glycans with one sialic acid, with some minor peaks with two sialic acids, while ion-exchange fraction C displays glycans with two sialic acids (data not shown), some minor peaks with one sialic acid and one sulfated glycan. In the ion-exchange fraction D, glycans were eluted with 50 mM sodium acetate, while many tri- and tetraantennary N-glycans were found (Figure 8A). Most glycans contained three sialic acids, but some were found with one sialic acid and one sulfate group. Finally, the ion-exchange E fraction contained glycans with three or four sialic acids, two sulfate groups or a combination or sulfates and sialic acids (Figure 8B). In total 12 sulfated structures were detected.
Figure 7.

(A) Base-peak chromatogram from PGC column of the ion-exchange fraction A (neutral fraction) with an extracted ion chromatogram of m/z 997.0. (B and C) MS/MS of m/z 997.0 at the retention times (RT) of 67.1 and 84.5 min. The B fragment ion at 511 designates a fucosylated branch for RT 67.1 min, while the peak at RT 84.5 min lacks this ion, indicating a core fucosylation.
Figure 8.

PGC base peak chromatograms of acidic ion-exchange fractions of N-glycans from urinary exosomes preconcentrated through ion-exchange chromatography. Gray circle with a red letter S signifies a sulfate group. (A, B) Reduced N-glycans and (C, D) methylamidated reduced N-glycans.
The separation of N-glycans according to their sialic acid and sulfate composition seems to yield many additional detectable peaks. Their positive identification will necessitate further studies in future. One way this procedure can be used is to enrich for glycans that are not highly abundant, such as the larger glycans that generally contain more sialic acid residues. It can also be used to get more spectra from the simplified chromatograms for an easier analysis and at the same time allowing the detection of more glycans hiding in the spectral background. A good example of this are the sulfated N-glycans that could not be seen in the total base peak chromatogram (Figure 6), while in the ion-exchange fractions they could be identified in fractions C, D, and E. This shows the importance of prefractionation even when the chromatographic resolution is genuinely high, with the long PGC capillaries and high column pressures.
Several isomers could be seen in the total chromatograms and spectra of the native, reduced glycans, but to separate totally glycans on PGC with more than three sialic acids with good resolution proves to be a challenge with our current setting and conditions. Pabst and Altmann45 suggested an increased concentration of formic acid (buffered with ammonium hydroxide in the mobile phase), but this caused problems with the electrospray on our instrument, as well as a reduced peak intensity. As a possible solution to this seemed to be neutralization of the carboxylic moiety on the sialic acids, we tried methylamidation of the glycans in the acidic ion-exchange fractions to yield a better separation.30 The LC-MS recordings and spectra for this procedure indicated that it worked well for the glycans with three to four sialic acids (Figure 8). However, it seems to have less benefit compared to the native reduced glycan separation when the analytes contain up to two sialic acids, or for the solutes containing sulfated structures (data not shown).
Comparison of Methods.
The different analytical methods yielded a variety of glycans with an overlap of detected N-glycans, however additional glycans can be detected in each method. Because of the inability to distinguish isomers, MALDI-TOF-MS detects less glycans compared to the other methods. However, it is quicker and more accurate in quantitative comparisons of nonisomeric glycans. LC-MS with a PGC column detects a higher number of glycans with its ability to separate isomeric compounds. The preconcentration with ion-exchange enabled detection of more glycans, however, the large N-glycans with three of four sialic acids had poor resolution. The methylamidation of the sialic acids yielded an increase in resolution for these glycans, however, the weakness of methylamidation seems to be the loss of sulfated structures. A simple comparison in the numbers of tentative N-glycans found by the different procedures can be seen in Figure S4.
O-Glycan Analysis.
Profiling O-glycans in small biological specimens appears less popular than similar studies involving N-glycomics. This is in spite of the major biological roles that O-glycans play and their significance as disease biomarkers.46,47 Presumably, this is due to, at least in part, the more facile enzymatic hydrolysis of N-glycans and the subsequently developed profiling methodologies. Nevertheless, with the recent advent of better cleavage protocols31,48,49 in mind, it deemed feasible to profile the O-glycan content of an 85-μg protein aliquot of the urinary exosomes isolated in this study. For comparative purposes, we also analyzed 100 μg of bovine fetuin (as a “model glycoprotein” with well-characterized set of O-glycans) and human blood serum through our earlier developed method31 that involves the combination of complex enzymatic degradation with a chemical release during the solid–phase permethylation of O-linked oligosaccharides prior to MALDI-TOF-MS measurements. Consequently, we have profiled 16 mucin type O-glycans, using MALDI-TOF MS/MS analysis and in-house database (Figure 9). The quantification is based on relative abundance of the released glycans, in which the profiled exosome O-glycans were normalized for the sake of comparison with the common glycoprotein source (fetuin) and human blood serum analyzed by a Data Explorer Software (AB Sciex, LLC, Framingham, MA).
Figure 9.

Comparison of the relative abundances of mucin-type O-glycans released from 85-μg protein aliquots of the urinary exosomes, 100-μg bovine fetuin, and 2.5-μg human blood serum (HBS). Certain O-glycan structures appear specific to urinary exosomes (m/z 967.47, 1124.54, 1141.55, 13165, 1328.65, and 1502.76). The scale of the lower panel is expanded.
The results of comparative analyses are shown in Figure 9, demonstrating that Tn(GalNAc), sialylated Tn and extended core 2 Galβ1–3(GlcNAcβ1–6)GalNAc (m/z 1689.82) are common to all three sample types, albeit at different abundances per protein amount. Certain O-glycans appear specific to the exosome samples. Herein, we have established for the first time that (a) there are quantifiable levels of O-glycans in the glycoproteins extracted from urinary exosomes and (b) there may be O-glycans of different structures than those expressed in the major serum glycoproteins. However, any biomedical significance of urinary O-glycans remains to be investigated in future studies.
CONCLUSIONS
Using a multistep differential centrifugation protocol, we have succeeded in isolating a fraction of urinary exosomes that is adequate for performing both N-glycan and O-glycan reproducible profiling from 40 mL urine aliquots. For procedural reasons, small volumes of urinary samples are an essential prerequisite for future clinical investigations. Using a combination of highly sensitive analytical techniques, we have been able to provide the means for a wide-range characterization of glycan structures and reliable measurements. The analytical profiles recorded by MALDI- and ESI-MS from the exosome samples feature paucimannosidic structures, high- mannose and complex-type N-glycans, some with a high degree of fucosylation and sialylation. We have succeeded in partially resolving some sialylated isomers of tetra-antennary glycans, which represent one of the most difficult analytical challenges in glycoscience;50 a combination of the column selectivity and the efficiency of long packed capillary columns will undoubtedly be the key to future advances in this direction. A successful enrichment of trace tri- and tetra-antennary glycans was accomplished in both their permethylated and native forms through the use of appropriate microcolumns (based on hydrophobicity and ion exchange). Our future studies will concentrate on a complete, in-depth analysis of the glycome and glycoproteome of the urinary exosomes. The present study has been a prelude to glycomic profiling of genitourinary tract diseases.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institute of General Medical Sciences, U.S. Department of Health (Grants NIH/NIGMS RO1 GM024349, NIH R21GM118340, and NIH RO1 GM106084). Thanks to Professor J.W. Jorgenson (University of North Carolina) for sharing his knowledge on packing chromatographic columns under high pressure. We also appreciate assistance of Dr. Sungyun Kang (Indiana University) with SDS-PAGE.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.7b00062.
Materials and methods section, scheme of the procedure for the isolation of exosomes from urine, SDS-PAGE of human urinary exosomes, relative abundance of the N-glycans identified from urinary exosomes as compared to a typical blood serum, and the overlap of the detected N-glycans by different methods (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Fais S; O’Driscoll L; Borras FE; Buzas E; Camussi G; Cappello F; Carvalho J; Cordeiro da Silva A; Del Portillo H; El Andaloussi S; Ficko Trček T; Furlan R; Hendrix A; Gursel I; Kralj-Iglic V; Kaeffer B; Kosanovic M; Lekka ME; Lipps G; Logozzi M; Marcilla A; Sammar M; Llorente A; Nazarenko I; Oliveira C; Pocsfalvi G; Rajendran L; Raposo G; Rohde E; Siljander P; van Niel G; Vasconcelos MH; Yáñez-Mó M; Yliperttula ML; Zarovni N; Zavec AB; Giebel B ACS Nano 2016, 10, 3886–3899. [DOI] [PubMed] [Google Scholar]
- (2).Colombo M; Raposo G; Thery C Annu. Rev. Cell Dev. Biol 2014, 30, 255–289. [DOI] [PubMed] [Google Scholar]
- (3).Nieuwland R; Sturk A Thromb. Res 2010, 125 (Suppl 1), S49–S51. [DOI] [PubMed] [Google Scholar]
- (4).Al-Nedawi K; Meehan B; Micallef J; Lhotak V; May L; Guha A; Rak J Nat. Cell Biol 2008, 10, 619–624. [DOI] [PubMed] [Google Scholar]
- (5).Skog J; Wurdinger T; van Rijn S; Meijer DH; Gainche L; Curry WT Jr.; Carter BS; Krichevsky AM; Breakefield XO Nat. Cell Biol 2008, 10, 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Shedden K; Xie XT; Chandaroy P; Chang YT; Rosania GR Cancer Res 2003, 63, 4331–4337. [PubMed] [Google Scholar]
- (7).Hood JL; San RS; Wickline SA Cancer Res 2011, 71, 3792–3801. [DOI] [PubMed] [Google Scholar]
- (8).Thery C; Amigorena S; Raposo G; Clayton A Curr. Protoc Cell Biol 2006, DOI: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
- (9).Cantin R; Diou J; Belanger D; Tremblay AM; Gilbert CJ Immunol. Methods 2008, 338, 21–30. [DOI] [PubMed] [Google Scholar]
- (10).Im H; Shao H; Park YI; Peterson VM; Castro CM; Weissleder R; Lee H Nat. Biotechnol 2014, 32, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Melo SA; Luecke LB; Kahlert C; Fernandez AF; Gammon ST; Kaye J; LeBleu VS; Mittendorf EA; Weitz J; Rahbari N; Reissfelder C; Pilarsky C; Fraga MF; Piwnica-Worms D; Kalluri R Nature 2015, 523, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pisitkun T; Shen RF; Knepper MA Proc. Natl. Acad. Sci. U.S. A 2004, 101, 13368–13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gonzales PA; Pisitkun T; Hoffert JD; Tchapyjnikov D; Star RA; Kleta R; Wang NS; Knepper MA J. Am. Soc. Nephrol 2009, 20, 363–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Staubach S; Schadewaldt P; Wendel U; Nohroudi K; Hanisch FG J. Proteome Res 2012, 11, 906–916. [DOI] [PubMed] [Google Scholar]
- (15).Saraswat M; Joenvaara S; Musante L; Peltoniemi H; Holthofer H; Renkonen R Mol. Cell. Proteomics 2015, 14, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Staubach S; Pekmez M; Hanisch FG J. Proteome Res 2016, 15, 1754–1761. [DOI] [PubMed] [Google Scholar]
- (17).Principe S; Jones EE; Kim Y; Sinha A; Nyalwidhe JO; Brooks J; Semmes OJ; Troyer DA; Lance RS; Kislinger T; Drake RR Proteomics 2013, 13, 1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nyalwidhe JO; Betesh LR; Powers TW; Jones EE; White KY; Burch TC; Brooks J; Watson MT; Lance RS; Troyer DA; Semmes OJ; Mehta A; Drake RR Proteomics: Clin. Appl 2013, 7, 677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nilsson J; Skog J; Nordstrand A; Baranov V; Mincheva-Nilsson L; Breakefield XO; Widmark A Br. J. Cancer 2009, 100, 1603–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kyselova Z; Mechref Y; Al Bataineh MM; Dobrolecki LE; Hickey RJ; Vinson J; Sweeney CJ; Novotny MV J. Proteome Res 2007, 6, 1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kyselova Z; Mechref Y; Kang P; Goetz JA; Dobrolecki LE; Sledge GW; Schnaper L; Hickey RJ; Malkas LH; Novotny MV Clin. Chem 2008, 54, 1166–1175. [DOI] [PubMed] [Google Scholar]
- (22).Goldman R; Ressom HW; Varghese RS; Goldman L; Bascug G; Loffredo CA; Abdel-Hamid M; Gouda I; Ezzat S; Kyselova Z; Mechref Y; Novotny MV Clin. Cancer Res 2009, 15, 1808–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Alley WR Jr.; Vasseur JA; Goetz JA; Svoboda M; Mann BF; Matei DE; Menning N; Hussein A; Mechref Y; Novotny MV J. Proteome Res 2012, 11, 2282–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Snyder CM; Alley WR Jr.; Campos MI; Svoboda M; Goetz JA; Vasseur JA; Jacobson SC; Novotny MV Anal. Chem 2016, 88, 9597–9605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Vasseur JA; Goetz JA; Alley WR Jr.; Novotny MV Glycobiology 2012, 22, 1684–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhou H; Yuen PS; Pisitkun T; Gonzales PA; Yasuda H; Dear JW; Gross P; Knepper MA; Star RA Kidney Int 2006, 69, 1471–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Fernandez-Llama P; Khositseth S; Gonzales PA; Star RA; Pisitkun T; Knepper MA Kidney Int 2010, 77, 736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wang SH; Tsai CM; Lin KI; Khoo KH Glycobiology 2013, 23, 677–689. [DOI] [PubMed] [Google Scholar]
- (29).Mitra I; Snyder CM; Zhou X; Campos MI; Alley WR Jr.; Novotny MV; Jacobson SC Anal. Chem 2016, 88, 8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang Q; Feng X; Li H; Liu BF; Lin Y; Liu X Anal. Chem 2014, 86, 7913–7919. [DOI] [PubMed] [Google Scholar]
- (31).Goetz JA; Novotny MV; Mechref Y Anal. Chem 2009, 81, 9546–9552. [DOI] [PubMed] [Google Scholar]
- (32).Alvarez ML; Khosroheidari M; Kanchi Ravi R; DiStefano JK Kidney Int 2012, 82, 1024–1032. [DOI] [PubMed] [Google Scholar]
- (33).Ginsberg JM; Chang BS; Matarese RA; Garella SN Engl. J. Med 1983, 309, 1543–1546. [DOI] [PubMed] [Google Scholar]
- (34).Kang P; Mechref Y; Klouckova I; Novotny MV Rapid Commun. Mass Spectrom 2005, 19, 3421–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Durocher Y; Butler M Curr. Opin. Biotechnol 2009, 20, 700–707. [DOI] [PubMed] [Google Scholar]
- (36).Satomaa T; Heiskanen A; Mikkola M; Olsson C; Blomqvist M; Tiittanen M; Jaatinen T; Aitio O; Olonen A; Helin J; Hiltunen J; Natunen J; Tuuri T; Otonkoski T; Saarinen J; Laine J BMC Cell Biol 2009, 10, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Everest-Dass AV; Jin D; Thaysen-Andersen M; Nevalainen H; Kolarich D; Packer NH Glycobiology 2012, 22, 1465–1479. [DOI] [PubMed] [Google Scholar]
- (38).Balog CI; Stavenhagen K; Fung WL; Koeleman CA; McDonnell LA; Verhoeven A; Mesker WE; Tollenaar RA; Deelder AM; Wuhrer M Mol. Cell. Proteomics 2012, 11, 571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hare NJ; Lee LY; Loke I; Britton WJ; Saunders BM; Thaysen-Andersen MJ Proteome Res 2017, 16, 247. [DOI] [PubMed] [Google Scholar]
- (40).Thaysen-Andersen M; Venkatakrishnan V; Loke I; Laurini C; Diestel S; Parker BL; Packer NH J. Biol. Chem 2015, 290, 8789–8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Tomiya N; Awaya J; Kurono M; Hanzawa H; Shimada I; Arata Y; Yoshida T; Takahashi NJ Biol. Chem 1993, 268, 113–126. [PubMed] [Google Scholar]
- (42).Donald ASR; Yates AD; Soh CPC; Morgan WTJ; Watkins WM Biochem. Biophys. Res. Commun 1983, 115, 625–631. [DOI] [PubMed] [Google Scholar]
- (43).Morton JA; Pickles MM; Terry AM Vox Sang 1970, 19, 472–482. [DOI] [PubMed] [Google Scholar]
- (44).Chai W; Piskarev V; Lawson AM J. Am. Soc. Mass Spectrom 2002, 13, 670–679. [DOI] [PubMed] [Google Scholar]
- (45).Pabst M; Altmann F Anal. Chem 2008, 80, 7534–7542. [DOI] [PubMed] [Google Scholar]
- (46).Karlsson NG; McGuckin MA Glycobiology 2012, 22, 918–929. [DOI] [PubMed] [Google Scholar]
- (47).Ju T; Wang Y; Aryal RP; Lehoux SD; Ding X; Kudelka MR; Cutler C; Zeng J; Wang J; Sun X; Heimburg-Molinaro J; Smith DF; Cummings RD Proteomics: Clin. Appl 2013, 7, 618–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Gizaw ST; Koda T; Amano M; Kamimura K; Ohashi T; Hinou H; Nishimura S Biochim. Biophys. Acta, Gen. Subj 2015, 1850, 1704–1718. [DOI] [PubMed] [Google Scholar]
- (49).Song X; Ju H; Lasanajak Y; Kudelka MR; Smith DF; Cummings RD Nat. Methods 2016, 13, 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Cummings RD; Pierce JM Chem. Biol 2014, 21, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.