

Abstract
Purpose
Fusions that involve neurotrophic-tropomyosin receptor kinase (NTRK) genes are known drivers of oncogenesis. Therapies that target these ultra-rare, constitutionally active NTRK fusions have been remarkably effective. Herein, we analyze the prevalence of the full array of NTRK alterations—fusions, mutations, copy number alterations, and increased transcript expression—in diverse adult and pediatric tumor types to understand the landscape of NTRK aberrations in cancer.
Methods
We assessed 13,467 samples available from The Cancer Genome Atlas (adult tumors) and the St Jude PeCan database (pediatric tumors) for the prevalence of NTRK fusions, as well as associated genomic and transcriptomic co-aberrations in different tumor types.
Results
NTRK fusions were observed in 0.31% of adult tumors and in 0.34% of pediatric tumors. The most common gene partners were NTRK3 (0.16% of adult tumors) followed by NTRK1 (0.14% of pediatric tumors). NTRK fusions were found more commonly in pediatric melanoma (11.1% of samples), pediatric glioma (3.97%), and adult thyroid cancers (2.34%). Additional genomic and transcriptomic NTRK alterations—mutation, amplification, and mRNA overexpression—occurred in 14.2% of samples, whereas the frequency of alterations that implicated NTRK ligands and the NTRK co-receptor (p75NTR) ranged from 3.8% to 5.4%. Among 31 adult samples carrying NTRK fusions, co-alterations occurred often and usually involved the downstream phosphoinositide-3-kinase signaling pathway, cell-cycle machinery, other tyrosine-kinase receptors, and mitogen-activated protein kinase signals.
Conclusion
Whereas NTRK fusions are exceedingly rare, other NTRK abnormalities affect 14% of patients with cancer. Affecting these alterations has not yet been achievable in cancer. Genomic co-alterations occur frequently with NTRK fusions, but it is not known if co-targeting them can attenuate primary or secondary resistance to NTRK inhibitors.
INTRODUCTION
NTRK1, NTRK2, and NTRK3 genes encode the neurotrophic-tropomyosin receptor tyrosine kinases (NTRKs) TrkA (NTRK1), TrkB (NTRK2), and TrkC (NTRK3). Ligands for the NTRK receptors are called neurotrophins. Nerve growth factor (NGF) binds to NTRK1; brain-derived neurotrophic factor (BDNF) and neurotrophin-4 (NT-4) and NT-5 bind to NTRK2; and NT-3 binds both NTRK1 and NTRK3.1 Binding of neurotrophic factors to their receptors activates the downstream effectors of NTRK: phospholipase C-γ, mitogen-activated protein kinase (MAPK), and phosphatidylinositol-3-kinase (PI3K)/AKT pathways. In addition, neurotrophins also bind to the low-affinity NGF receptor p75NTR. p75NTR is a positive regulator of the NGF/NTRK1 system that reduces ligand-induced receptor ubiquitination and delays receptor internalization and degradation.2
NTRK receptors promote the proliferation and survival of neuronal cells3-8 (Fig 1). Of interest, NTRK alterations induce tumorigenesis in both neurogenic and non-neurogenic cancers and are targets for therapeutic agents.9-11 Although the clinical implications of NTRK single-nucleotide variants or copy number alterations are unclear, several NTRK transcript fusions have been identified. These drive NTRK mRNA and protein overexpression, which further leads to constitutive activation of downstream signaling.12 The prevalence of NTRK fusions is low, but can reach more than 80% in some rare tumors, such as mammary-analog secretory carcinoma of the salivary gland, secretory breast carcinoma, and infantile congenital fibrosarcoma.12-20 NTRK fusions are also found in 40% of pediatric non-brainstem high-grade glioma.21
Fig 1.

Neurotrophic-tropomyosin receptor tyrosine kinase (NTRK) receptor signaling pathway and inhibitors. The ligands nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3), and NT-4 bind to their receptors, namely NTRK1 (tropomyosin receptor kinase A or TrkA), NTRK2 (tropomyosin receptor kinase B or TrkB), and NTRK3 (tropomyosin receptor kinase C or TrkC). These receptors are under the regulation of the co-receptor p75 neurotrophin receptor (p75NTR). The binding of the ligand to the receptor promotes to the dimerization of the receptor and its subsequent intracellular phosphorylation. Several signaling cascades are further activated—phospholipase Cγ (PLC-γ), mitogen-activated protein kinase (MAPK), and phosphoinositide-3-kinase (PI3K) —and are converging to protumorigenic cell processes, such as proliferation, survival invasion, or differentiation. The hyperactivation of the NTRK signaling pathway induced by NTRK alterations—fusions or point mutations—can be overcome by the use of NTRK antagonists (eg, ANA-12 and cyclotraxin B) or small-molecule tyrosine kinase inhibitors (eg, larotrectinib and entrectinib). For now, only small-molecule tyrosine kinase inhibitors are used in the clinic.
Among all alterations in NTRK genes, transcript fusions are currently the best characterized and the most pharmacologically tractable. Nonfusion NTRK alterations—for example, mutation or amplification—have been associated with a lack of response with some NTRK inhibitors.22 Because NTRK fusions are rare, the number of patients who can benefit from drugs that target NTRK receptors is relatively low, but the antitumor activity of such agents is remarkable.23,24 Indeed, larotrectinib, a pan-NTRK inhibitor, demonstrated a response rate of 76% in patients with NTRK fusion–positive tumors (17 cancer types).15,18 Tumor regression has been maintained for more than 1 year in 71% of patients. Entrectinib, an oral pan-NTRK, ROS1, and ALK inhibitor demonstrated a 79% objective response in patients with NTRK, ROS1, or ALK fusions.22
In May 2017, a new precedent was set when an immune checkpoint inhibitor—pembrolizumab—was approved by the US Food and Drug Administration (FDA) for use in a tissue-agnostic fashion on the basis of a genomic biomarker (mismatch repair gene deficiency).25 NTRK-selective inhibitors represent another pharmacology class that has been developed on the sole basis of somatic molecular patterns. Therefore, a comprehensive understanding of individual genomic alterations is becoming crucial.
In the current study, we assessed the landscape of NTRK genomic and transcriptomic alterations, as well as co-alterations in common signaling pathways, using a large cohort of samples available from The Cancer Genome Atlas (TCGA; adult, 33 tumor types) and the St Jude PeCan (pediatric, 17 tumor types).
METHODS
NTRK Receptor Fusions
Adult tumor NTRK-related transcript fusions were retrieved from The Jackson Laboratory Tumor Fusion Gene Data Portal.26 These fusions were defined after an integrated analysis of paired-end RNA sequencing and DNA copy number data from TCGA that corresponded to 9,966 adult tumors (33 different tumor types).
Pediatric tumor NTRK-related transcript fusions were retrieved from the St Jude PeCan Data Portal database.27 These fusions were defined after analysis of RNA sequencing data by the CICERO algorithm (Pediatric Cancer Genome Project) and corresponded to 3,501 pediatric tumors (17 different tumor types).28,29
Genomic and Transcriptomic Alterations in NTRK Receptors, Co-Receptor, and Ligands (beyond fusions)
Adult and pediatric tumor NTRK-related mutations, copy number variations, and mRNA expression for NTRK receptors (NTRK1, NTRK2, and NTRK3), co-receptor (p75NTR), and ligands (NGF, BDNF, NT-3, and NT-4) were retrieved from the UCSC Xena Portal.30 These data include information on 13,467 samples from TCGA (n = 9,966 adults) and St Jude PeCan (n = 3,501 children) pan-cancer cohorts, of which 11,621 (n = 9,966 TCGA and n = 1,655 PeCan) had comprehensive information on fusions, mutations, and copy number alterations. Data were available without restriction of use on the date of February 1, 2018. All data used in this study respected the TCGA’s Human Subjects Protection and Data Access Policies31 and the St Jude Cloud Terms of Use.32
Lists of significant variants were generated using whole-genome somatic mutation data and the MutSig2CV algorithm (http://www.broadinstitute.org/cancer/cga/mutsig), taking into account the somatic background mutation rate for each gene and its neighbor genes.33
Focal copy number variations that correspond to genome-wide single-nucleotide polymorphism array data were normalized and assessed at the gene level using the GISTIC2 protocol,34 where a deep loss was documented by the value (−2), a single-copy loss by the value (−1), a low-level gain by the value (+1), and an amplification by the value (+2). Only NTRK-related gene amplifications were kept for the analysis.
Sequencing-based mRNA expression signals were integrated and normalized for each gene per sample using the RNA-Sequencing by Expectation Maximization protocol. The standard score (z-score) for each gene per sample was calculated using the mean values and standard deviation found in all similar tumors—same tumor type—that are diploid for the said gene. A z-score of ≥ 1.96 standard deviation was used as the threshold of overexpression, whereas a threshold of ≤ −1.96 standard deviation was used to qualify underexpressed genes. Only NTRK-related mRNA overexpression was considered for the analysis.
Genomic and Transcriptomic Co-Alterations Occurring in NTRK Fusion–Positive Adult Tumors (n = 31 patients)
Comprehensive co-alteration data were not available in pediatric tumors. In adults, co-alterations within signaling cascades, such as TP53, MAPK, PI3K, tyrosine kinase receptor, or cell-cycle signaling pathways, were curated from TCGA. All nonsynonymous missense, nonsense, nonstop, deletion/insertions, frameshift, or splicing site mutations within the genes of interest, as well as deep losses or amplifications and mRNA under- or overexpressions, were kept for analysis.
RESULTS
Prevalence of NTRK Fusions in TCGA (adult) and St Jude PeCan (pediatric) Databases
NTRK Fusion Frequency in Adults
Of the 9,966 adult tumor samples in the TCGA database, 0.31% (n = 31 samples) presented an NTRK fusion. This alteration was most common in thyroid cancer (2.34% of samples), colon adenocarcinoma (0.97%), and low-grade glioma (0.94%). Twenty-two adult tumor types had no NTRK fusions. (There were 5,023 patient samples with these 22 NTRK fusion–negative tumor types [samples per tumor type = 36 to 541].) NTRK3 fusions were the most common (n = 16), followed by NTRK1 (n = 9) and NTRK2 (n = 6) fusions in adults (Table 1).
Table 1.
Frequency of NTRK Receptor Transcript Fusions in TCGA (n = 9,966 adult tumor samples) and St Jude Pediatric Cancer Database (n = 3,501 pediatric tumor samples), and Specific Tumors With High Incidence of NTRK Fusions in the Literature

NTRK Fusion Frequency in Children
Of the 3,501 pediatric tumor samples (St Jude PeCan database), 0.34% (n = 12) presented an NTRK fusion. Of interest, NTRK fusions were found in one of nine melanomas. NTRK fusions were also found in glioma (high and low grade [3.97%]) and B-cell acute lymphoblastic leukemia (0.14%). Thirteen pediatric tumor types (n = 2,524 patient samples) had no NTRK fusions (samples per tumor type = 26 to 714). Of 12 pediatric tumor samples with NTRK fusions, the most common partner gene was NTRK1 (n = 5) followed by NTRK2 (n = 4) and NTRK3 (n = 3; Table 1).
Therapeutic or Experimental Molecules With Activity Against NTRK Receptors
Overall, 32 molecules have demonstrated preclinical inhibition activity against one or more NTRK receptors35xref ref-type="bibr" rid="B37"/>-70 (Table 2). Surprisingly, five of these small inhibitors are already approved by the FDA for other indications, namely cabozantinib (Cabometyx; Exelixis, South San Francisco, CA; IC50 against NTRK2, 7 nM), crizotinib (Xalkori; Pfizer, New York, NY; IC50 against NTRK1 and NTRK2, 1 nM), midostaurin (Rydapt; Novartis, Basel Switzerland; IC50 ranging from 11 to 51 nM), nintedanib (Ofev; Boehringer Ingelheim, Ingelheim am Rhein, Germany; IC50 ranging from 17 to 264 nM), and regorafenib (Stivarga; Bayer, Leverkusen, Germany; IC50 against NTRK1, 74 nM). It is not known if these five molecules exhibit clinical activity in patients who harbor NTRK-aberrant tumors. Sixteen molecules are currently being evaluated in clinical trials, with the most advanced being larotrectinib (Loxo Oncology, Stamford, CT; IC50 for NTRK1, NTRK2, and NTRK3 fusions ranging from 4 to 9 nM). The new drug application was submitted to the FDA in December 2017 and granted priority review status on the basis of remarkable clinical activity23 (Table 2).
Table 2.
Target Specificity and IC50 of NTRK-Targeting Inhibitors

Types of NTRK-Related Alterations in Adult and Pediatric Tumors and Sensitivity to NTRK Inhibitors
To understand the potential benefits of selective NTRK inhibitors for the treatment of adult and pediatric patients with cancer, we first aimed to describe the prevalence and type of NTRK-activating pathway alterations, including point mutations, gene copy number amplifications, and mRNA overexpression of NTRK receptors, co-receptor, and ligands, within a large cohort of pan-cancer samples (Figs 1 and 2). The number of samples with comprehensive data for this analysis was 11,621 (9,966 adults and 1,655 children).
Fig 2.
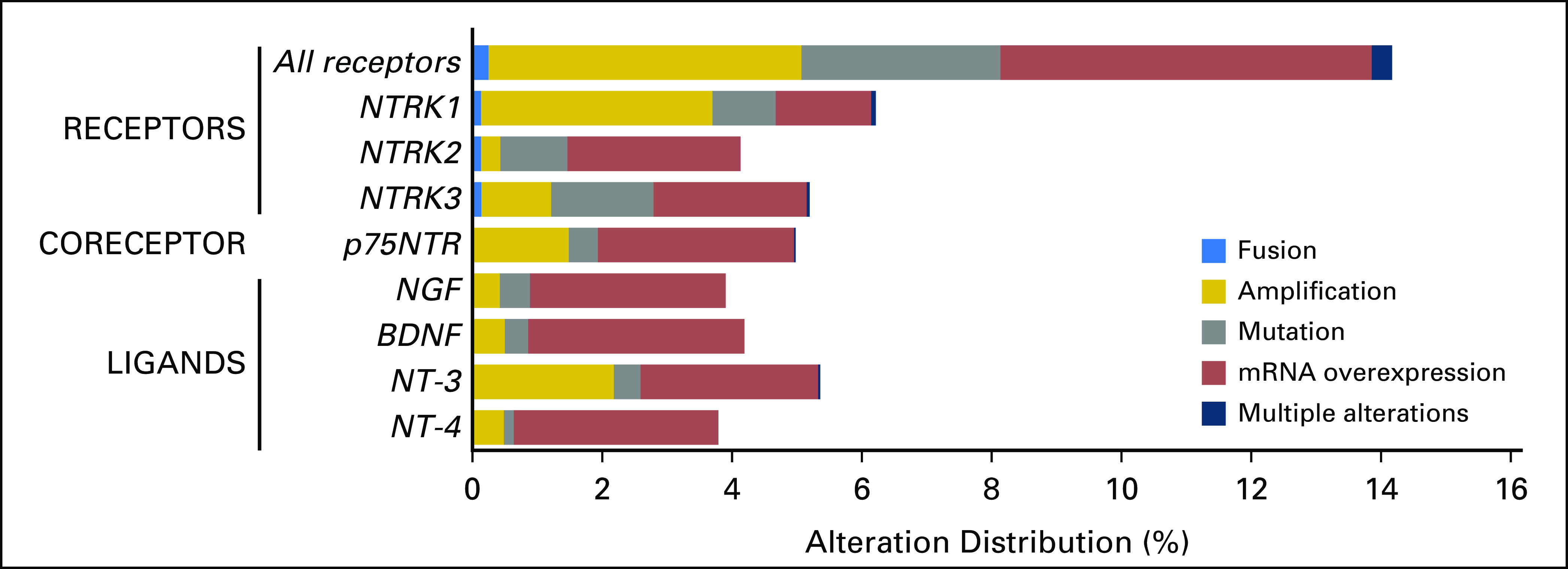
Distribution of molecular alterations leading to the hyperactivation of the neurotrophic-tropomyosin receptor tyrosine kinase (NTRK) signaling pathway in human tumors (N = 11,621 samples with comprehensive molecular data). All samples that presented a nonsilent mutation, gene copy amplification, gene fusion, or mRNA overexpression of NTRK receptors (NTRK1, NTRK2, and NTRK3), co-receptor (p75NTR), or ligands (nerve growth factor [NGF], brain-derived neurotrophic factor [BDNF], neurotrophin 3 [NT-3], and NT-4) were retrieved from a large adult and pediatric pan-cancer cohort (The Cancer Genome Atlas and St Jude’s PeCan databases; N = 11,621samples). Among the NTRK fusion cases (n = 31 from TCGA cohort), four cases had concomitant alteration within the genes that code the NTRK pathway members—ligands, co-receptor, and receptors—as follows: low-grade glioma, NTRK3 fusion plus NTF3 amplification (n = 1); low-grade glioma, NTRK1 fusion plus NTRK1 amplification (n = 1); glioblastoma, NTRK1 fusion plus NTRK1 amplification (n = 1); head and neck squamous cell carcinoma, NTRK3 fusion plus NTRK3 amplification (n = 1).
Alterations in NTRK Receptors and Ligands
Genomic and/or transcriptomic NTRK receptor alterations were found in 14.2% (1,648 of 11,621) of samples, with gene amplification and mRNA overexpression being the most frequent alterations. The three NTRK receptors were equally impacted, with frequencies of alterations ranging from 4.1% to 6.2%. In addition, the co-receptor p75NTR presented one or more presumably activating alteration in almost 5% (579 of 11,621 samples) of tumors. NTRK ligands presented an alteration in 3.8% to 5.4% of samples. Transcript fusions were observed in NTRK receptor genes only, with the exception of two samples that presented one transcript fusion of BDNF ligand and one transcript fusion of p75NTR (positive regulator of the NGF/NTRK1 machinery; Fig 2).
NTRK-Transcript Fusion Types
NTRK-transcript fusions that were observed in the pan-cancer cohort and/or described in the literature are listed in Table 3. The ETV6-NTRK3 rearrangement was the most frequently observed (0.09% of samples). This variant is a known biomarker of sensitivity to larotrectinib and entrectinib.71,72 Variants TPM3-NTRK1 (0.04%), IRF2BP2-NTRK1 (< 0.01%), and SQSTM1-NTRK1 (< 0.01%) are also sensitive to larotrectinib; however, the sensitivity of the remaining 22 unique variants observed in the pan-cancer cohort is not currently known. Nine rearrangements previously described in the literature were not found in the TCGA and St Jude PeCan databases (Table 3).
Table 3.
NTRK Alterations, Frequency in TCGA/St Jude PeCan Databases, and Clinical Response to Illustrative NTRK-Targeting Inhibitors

NTRK Point Mutations
Several point mutations are acquired resistant variants to first-generation NTRK inhibitors (larotrectinib or entrectinib), but not to LOXO-195, specifically designed to overcome secondary resistance. These variants, namely NTRK1 G595R, NTRK1 G667C, NTRK3 G696A, and NTRK3 G623R, were not observed in any of the 13,467 combined adult and pediatric tumors reviewed (treatment-naïve samples; Table 3).
Co-Alterations Observed in NTRK Fusion-Positive Adult Tumor Samples
Among 31 adult tumors presenting NTRK fusions, 61.3% (19 of 31) harbored one or more co-alteration that activated the downstream PI3K signaling pathway; 58.1% (18 of 31) harbored one or more co-alteration within cell-cycle[en]associated genes; 58.1% (18 of 31) harbored one or more co-alteration within other tyrosine kinase receptors; 32.2% (10 of 31) harbored one or more co-alteration within the MAPK signaling pathway; and 35.5% (11 of 31) harbored one or more co-alteration within TP53-associated genes. NF2-activating mutations were associated with NTRK fusions in 42% (13 of 31) of samples, and TP53 (10 of 31), RB1 (six of 31) and CDKN2A (five of 31) occurred in more than 15% of the NTRK fusion–positive samples (Fig 3 and Appendix Table A1). (Adequate data to comprehensively assess co-alteration data in children was not available.) Samples bearing NTRK fusions were significantly associated with NTRK mRNA overexpression compared with samples without the fusion (Appendix Fig A1). Moreover, tumors with NTRK fusions were significantly associated with lower tumor mutational burden compared with the fusion-negative cases (Appendix Fig A2).
Fig 3.

Co-alterations associated with neurotrophic-tropomyosin receptor tyrosine kinase (NTRK) fusions in adult tumors (from The Cancer Genome Atlas). All samples that presented a gene fusion of NTRK receptors—NTRK1, NTRK2, and NTRK3—were retrieved from a large adult pan-cancer cohort (The Cancer Genome Atlas database; n = 9,966 samples). Among 31 patients with NTRK fusions, some patients also harbored co-alterations that can lead to tumorigenesis. Those co-alterations include TP53-associated genes, cell cycle–associated genes, tyrosine kinase families, and mitogen-activated protein kinase (MAPK) and phosphoinositide-3-kinase (PI3K) signaling alterations.
DISCUSSION
Along with recent advances in sequencing technology, a histology-agnostic, matched, targeted approach has emerged as a newer strategy by which to manage malignancies.80-84 Targeting activated gene mutations or amplification/overexpression has demonstrated some remarkable successes—for example, targeting of KIT mutations for GI stromal tumors and targeting of EGFR mutation for non–small-cell lung cancer, BRAF V600E mutation for melanoma and lung cancer, and human epidermal growth factor receptor 2 overexpression for breast cancer—although in some cases the responses may be short lived.85-91 Tumor heterogeneity and co-alterations result in resistance to targeted therapeutics.92 Thus, for many cancers, combination therapy may be necessary.93-96
In some instances, targeting fusions—even with monotherapy—has shown more marked antitumor activity than targeting other alteration types. Examples include the suppression of aberrant Bcr-Abl kinase enzymatic activity that characterizes chronic myeloid leukemia. Exploitation of imatinib, dasatinib, or nilotinib leads to near-universal responses, and life expectancy increases from approximately 5 years before the imatinib era to a near-normal life span currently; however, it is also conceivable that, in this case, the success of Bcr-Abl–targeted agents is attributable to their deployment in patients with newly diagnosed disease, as advanced chronic myeloid leukemia responds poorly to single-agent Bcr-Abl kinase inhibitors.97,98 Conversely, in patients with advanced non–small-cell- lung cancer, targeting ALK fusions demonstrates a median progression-free survival of 25.7 months with an 83% response rate, and targeting the ROS1 fusion demonstrates a median progression-free survival of 19.2 months with an approximate 70% response rate.99,100 In addition, larotrectinib, an NTRK inhibitor, resulted in a 76% response rate in patients with an NTRK fusion.23 These observations indicate that certain fusions act as strong drivers of tumorigenesis in specific cancers that are likely addicted to this type of founder alteration.
We reviewed data from 13,467 tumor samples in the TCGA (adult tumors) and St Jude PeCan (pediatric tumors) databases and found NTRK fusions in 0.3% of pan-cancer tumors (Table 1). Although the prevalence of these alterations is low, NTRK fusions are more often found in specific and rare tumors, such as mammary-analog secretory carcinoma of the salivary gland (93% to 100% of tumors presenting an ETV6-NTRK3 fusion), secretory breast carcinoma (ETV6-NTRK3 fusions in 92% of tumors), infantile congenital fibrosarcoma (ETV6-NTRK3 fusions in 86% to 91% of tumors), and pediatric non-brainstem high-grade glioma14-21 (40% of tumors presenting an NTRK fusion; Table 1).
Of importance, various NTRK inhibitors are in clinical development and have differential activities (Table 2). Drugs with established clinical trial data and the ability to affect NTRK1, NTRK2, and NTRK3 fusions at low IC50 include, but are not limited to, larotrectinib (76% response rate in diverse malignancies bearing NTRK fusions) and entrectinib, which also affects ALK and ROS1 rearrangements (79% response rate), and some of these responses are durable and occur with remarkable speed22,23 (Table 2). Of interest, 32 molecules have demonstrated inhibition activity against one or more NTRK receptor (Table 2). Furthermore, five of these small inhibitors are already approved by the FDA for other indications: cabozantinib (IC50 against NTRK2, 7 nM), crizotinib (IC50 against NTRK1 and NTRK2, 1 nM), midostaurin (IC50 against NTRK1, -2, and -3 ranging from 11 to 51 nM), nintedanib (IC50 against NTRK1, -2, amd -3 ranging from 17 to 264 nM), and regorafenib (IC50 against NTRK1, 74 nM). Even so, it is not known whether these five medications have anti-NTRK activity in patients. Multiple other molecules that target NTRK are also in clinical trials (Table 2).
Resistance to NTRK inhibitors is now emerging. NTRK mutations that are associated with larotrectinib or entrectinib resistance include NTRK1 F589L G595R, G667C, G667S, V573M, and NTRK3 G696A, G623R (Table 3). (These mutations were not detected in TCGA, likely because these patients had not been previously treated with NTRK inhibitors.) The resistant alterations are targetable with LOXO-195, a next-generation, selective NTRK inhibitor with promising preliminary clinical activity50 (Fig 2). Other mechanisms of resistance may include the presence or emergence of genomic co-alterations. In the current study, NTRK-associated co-alterations were commonly discerned in genes that are involved in PI3K signaling (61% of patient samples), tyrosine kinase families (58% of patient samples), cell-cycle machinery (58% of patient samples), and MAPK pathways (32% of patient samples; Fig 3 and Appendix Table A1). Moreover, cases with NTRK fusions were significantly associated with NTRK mRNA overexpression (Appendix Fig A1), which is consistent with a previous report.101 Of interest, in the adult cohort, NTRK fusion–positive samples were significantly associated with a lower mutational burden compared with fusion-negative tumors (P < .001; Appendix Fig A2). This observation echoes a previous report that demonstrated that tumors harboring a driver fusion tend to have a lower number of point mutations.101 In contrast, high microsatellite unstable metastatic colorectal tumors have been shown to preferentially bear ALK, ROS1, or NRTK fusions.102 In our cohort, three NTRK fusion–positive colon cancer samples were observed and two of them presented with microsatellite instability-high status (data not shown). Finally, we found that nonfusion NTRK gene alterations, such as mutation, amplification, and mRNA overexpression, were found in approximately 14% of pan-cancer samples (Fig 2). Nonfusion NTRK alterations have not yet demonstrated druggability.
There are several limitations to the current study. First, clinical correlation with disease outcome among patients with NTRK alterations was not feasible because the data were not fully clinically annotated. Second, the possibility of sample size bias cannot be excluded because the number of tumor cases depended on the number of specimens submitted by investigators. Third, direct comparison between the TCGA and St Jude PeCan databases is challenging as a result of the use of different sequencing methods. Finally, we did not observe NTRK fusions in a number of cancer types, which may be a result of low sample size. Despite these limitations, the current report provides a comprehensive portrait of the genomic landscape of NTRK alterations among pan-cancer tumors using large databases.
In conclusion, NTRK fusions were observed in 0.31% (31 of 9,966) of adult tumors and 0.34% (12 of 3,501) of pediatric cancers, mostly in NTRK3 (0.16% of adult tumors) and NTRK1 (0.14% of pediatric tumors); however, some tumor types had more frequent NTRK fusions (Table 1). Additional genomic and transcriptomic NTRK alterations—mutation, amplification, and mRNA overexpression—occurred in 14.2% of samples. Genomic co-alterations were commonly observed in NTRK fusion–positive cancers, including in genes involved in PI3K signaling, tyrosine kinase families, cell-cycle–associated regulators, and the MAPK pathway (Fig 3). Additional investigation is needed to elucidate whether these genes mediate resistance to NTRK inhibition and if co-targeting them augments anti-NTRK antitumor activity. Furthermore, it would be of interest to determine whether the salutary effects of NTRK inhibitors in patients who harbor cancers with NTRK fusions can be extended via rational compound design to any of the more common NTRK alterations, such as mutation, amplification, and overexpression. Finally, the rarity of NTRK fusions, but their remarkable tractability in multiple cancer types, further expands the paradigm of tissue-agnostic genomic drug development.
Appendix
Fig A1.

Association of neurotrophic-tropomyosin receptor tyrosine kinase (NTRK) fusions and NTRK mRNA overexpression in adult tumors.
Fig A2.

Association of neurotrophic-tropomyosin receptor tyrosine kinase (NTRK) fusions and mutational burden in adult tumors. The mutational burden corresponds to the total number of nonsynonymous mutations detected by whole-exome sequencing in each sample.
Table A1.
Details of Co-Alterations With NTRK Fusions in Adult Tumors From The Cancer Genome Atlas

Footnotes
This work was supported in part by the Joan and Irwin Jacobs Fund, and NIH P30 CA023100 (R.K.). The authors also acknowledge the support of the Jon Schneider Memorial Cancer Research Fund NIH K08 CA168999 and R21 CA192072 (J.K.S.)
AUTHOR CONTRIBUTIONS
Conception and design: Ryosuke Okamura, Amélie Boichard, Razelle Kurzrock
Collection and assembly of data: Ryosuke Okamura, Amélie Boichard
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Ryosuke Okamura
No relationship to disclose
Amélie Boichard
No relationship to disclose
Shumei Kato
No relationship to disclose
Jason K. Sicklick
Consulting or Advisory Role: BioTheranostics, Loxo Pharmaceuticals, Grand Rounds
Research Funding: Novartis, Foundation Medicine, Blueprint Medicines
Lyudmila Bazhenova
Stock and Other Ownership Interests: Epic Sciences
Consulting or Advisory Role: AstraZeneca, Novartis, Ariad Pharmaceuticals, Genentech, Bristol-Myers Squibb
Speakers' Bureau: Genentech, AstraZeneca, Pfizer
Research Funding: BeyondSpring Pharmaceuticals
Razelle Kurzrock
Leadership: CureMatch
Stock and Other Ownership Interests: CureMatch
Consulting or Advisory Role: Sequenom, Actuate Therapeutics, Loxo Pharmaceuticals, NeoMed, Xbiotech
Speakers' Bureau: Roche
Research Funding: Guardant Health (Inst), Sequenom (Inst), Merck Serono (Inst), Genentech (Inst), Pfizer (Inst), Foundation Medicine (Inst), Incyte (Inst), Konica Minolta (Inst)
REFERENCES
- 1.Segal RA. Selectivity in neurotrophin signaling: Theme and variations. Annu Rev Neurosci. 2003;26:299–330. doi: 10.1146/annurev.neuro.26.041002.131421. [DOI] [PubMed] [Google Scholar]
- 2.Arévalo JC, Wu SH. Neurotrophin signaling: Many exciting surprises! Cell Mol Life Sci. 2006;63:1523–1537. doi: 10.1007/s00018-006-6010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 4.Kawamura K, Kawamura N, Fukuda J, et al. Regulation of preimplantation embryo development by brain-derived neurotrophic factor. Dev Biol. 2007;311:147–158. doi: 10.1016/j.ydbio.2007.08.026. [DOI] [PubMed] [Google Scholar]
- 5.Kawamura K, Kawamura N, Kumazawa Y, et al. Brain-derived neurotrophic factor/tyrosine kinase B signaling regulates human trophoblast growth in an in vivo animal model of ectopic pregnancy. Endocrinology. 2011;152:1090–1100. doi: 10.1210/en.2010-1124. [DOI] [PubMed] [Google Scholar]
- 6.Kawamura K, Kawamura N, Sato W, et al. Brain-derived neurotrophic factor promotes implantation and subsequent placental development by stimulating trophoblast cell growth and survival. Endocrinology. 2009;150:3774–3782. doi: 10.1210/en.2009-0213. [DOI] [PubMed] [Google Scholar]
- 7.Klein R, Jing SQ, Nanduri V, et al. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- 8.Loeb DM, Stephens RM, Copeland T, et al. A Trk nerve growth factor (NGF) receptor point mutation affecting interaction with phospholipase C-gamma 1 abolishes NGF-promoted peripherin induction but not neurite outgrowth. J Biol Chem. 1994;269:8901–8910. [PubMed] [Google Scholar]
- 9.Nakagawara A. Trk receptor tyrosine kinases: A bridge between cancer and neural development. Cancer Lett. 2001;169:107–114. doi: 10.1016/s0304-3835(01)00530-4. [DOI] [PubMed] [Google Scholar]
- 10.Skaper SD. The neurotrophin family of neurotrophic factors: An overview. Methods Mol Biol. 2012;846:1–12. doi: 10.1007/978-1-61779-536-7_1. [DOI] [PubMed] [Google Scholar]
- 11.Khotskaya YB, Holla VR, Farago AF, et al. Targeting TRK family proteins in cancer. Pharmacol Ther. 2017;173:58–66. doi: 10.1016/j.pharmthera.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Vaishnavi A, Capelletti M, Le AT, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19:1469–1472. doi: 10.1038/nm.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Créancier L, Vandenberghe I, Gomes B, et al. Chromosomal rearrangements involving the NTRK1 gene in colorectal carcinoma. Cancer Lett. 2015;365:107–111. doi: 10.1016/j.canlet.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 14.Skálová A, Vanecek T, Sima R, et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: A hitherto undescribed salivary gland tumor entity. Am J Surg Pathol. 2010;34:599–608. doi: 10.1097/PAS.0b013e3181d9efcc. [DOI] [PubMed] [Google Scholar]
- 15.Skálová A, Vanecek T, Simpson RH, et al. Mammary analogue secretory carcinoma of salivary glands: Molecular analysis of 25 ETV6 gene rearranged tumors with lack of detection of classical ETV6-NTRK3 fusion transcript by standard RT-PCR—Report of 4 cases harboring ETV6-X gene fusion. Am J Surg Pathol. 2016;40:3–13. doi: 10.1097/PAS.0000000000000537. [DOI] [PubMed] [Google Scholar]
- 16.Bishop JA, Yonescu R, Batista D, et al. Utility of mammaglobin immunohistochemistry as a proxy marker for the ETV6-NTRK3 translocation in the diagnosis of salivary mammary analogue secretory carcinoma. Hum Pathol. 2013;44:1982–1988. doi: 10.1016/j.humpath.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tognon C, Knezevich SR, Huntsman D, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2:367–376. doi: 10.1016/s1535-6108(02)00180-0. [DOI] [PubMed] [Google Scholar]
- 18.Bourgeois JM, Knezevich SR, Mathers JA, et al. Molecular detection of the ETV6-NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. Am J Surg Pathol. 2000;24:937–946. doi: 10.1097/00000478-200007000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Orbach D, Brennan B, De Paoli A, et al. Conservative strategy in infantile fibrosarcoma is possible: The European Paediatric Soft Tissue Sarcoma Study Group experience. Eur J Cancer. 2016;57:1–9. doi: 10.1016/j.ejca.2015.12.028. [DOI] [PubMed] [Google Scholar]
- 20.Rubin BP, Chen CJ, Morgan TW, et al. Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion: Cytogenetic and molecular relationship to congenital (infantile) fibrosarcoma. Am J Pathol. 1998;153:1451–1458. doi: 10.1016/S0002-9440(10)65732-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu G, Diaz AK, Paugh BS, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46:444–450. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drilon A, Siena S, Ou SI, et al. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor entrectinib: Combined results from two phase I trials (ALKA-372-001 and STARTRK-1) Cancer Discov. 2017;7:400–409. doi: 10.1158/2159-8290.CD-16-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378:731–739. doi: 10.1056/NEJMoa1714448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “wild-type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339. doi: 10.1186/s12967-016-1075-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The Jackson Laboratory Tumor fusion gene data portal. http://www.tumorfusions.org
- 27.St Jude Children’s Research Hospital St Jude PeCan data portal. http://pecan.stjude.org/proteinpaint/study/target-tall
- 28.Li Y, Bo T, Rusch M, et al. Detecting complex fusion transcripts in pediatric cancer using a novel assembly-based algorithm CICERO. abstr 1387M Am Soc Human Genet. 2014 [Google Scholar]
- 29.Downing JR, Wilson RK, Zhang J, et al. The pediatric cancer genome project. Nat Genet. 2012;44:619–622. doi: 10.1038/ng.2287. Erratum: Nat Genet 44:1072, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.University of California Santa Cruz UCSC Xena. http://xena.ucsc.edu/
- 31.The Cancer Genome Atlas Human subjects protection and data access policies. https://cancergenome.nih.gov/abouttcga/policies/tcga-human-subjects-data-policies
- 32.St Jude Children’s Research Hospital St Jude Cloud: Terms of use. https://stjude.cloud/terms-of-use.html
- 33.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Broad Institute TCGA copy number portal. http://www.broadinstitute.org/tcga/about
- 35.US Food and Drug Administration Cabozantinib (S)-malate: Pharmacology review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203756Orig1s000PharmR.pdf
- 36.US Food and Drug Administration Crizotinib: Pharmacology review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202570Orig1s000PharmR.pdf
- 37.US Food and Drug Administration Midostaurin: Pharmacology review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/207997Orig1Orig2s000PharmR.pdf
- 38.Nishiyama A, Yamada T, Kita K, et al. Foretinib overcomes entrectinib resistance associated with the NTRK1 G667C mutation in NTRK1 fusion-positive tumor cells in a brain metastasis model. Clin Cancer Res. 2018;24:2357–2369. doi: 10.1158/1078-0432.CCR-17-1623. [DOI] [PubMed] [Google Scholar]
- 39.Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68:4774–4782. doi: 10.1158/0008-5472.CAN-07-6307. [DOI] [PubMed] [Google Scholar]
- 40.US Food and Drug Administration Regorafenib: Pharmacology review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203085Orig1s000PharmR.pdf
- 41.Smith BD, Kaufman MD, Leary CB, et al. Altiratinib inhibits tumor growth, invasion, angiogenesis, and microenvironment-mediated drug resistance via balanced inhibition of MET, TIE2, and VEGFR2. Mol Cancer Ther. 2015;14:2023–2034. doi: 10.1158/1535-7163.MCT-14-1105. [DOI] [PubMed] [Google Scholar]
- 42.Weiss GJ, Sachdev JC, Infante JR, et al. Phase (Ph) 1/2 study of TSR-011, a potent inhibitor of ALK and TRK, including crizotinib-resistant ALK mutations. J Clin Oncol. 2014;32(suppl; abstr e19005) [Google Scholar]
- 43.Carboni JM, Wittman M, Yang Z, et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther. 2009;8:3341–3349. doi: 10.1158/1535-7163.MCT-09-0499. [DOI] [PubMed] [Google Scholar]
- 44.Schroeder GM, An Y, Cai ZW, et al. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J Med Chem. 2009;52:1251–1254. doi: 10.1021/jm801586s. [DOI] [PubMed] [Google Scholar]
- 45.Carpinelli P, Ceruti R, Giorgini ML, et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther. 2007;6:3158–3168. doi: 10.1158/1535-7163.MCT-07-0444. [DOI] [PubMed] [Google Scholar]
- 46.Kiga M, Iwasaki S, Togashi N, et al. Preclinical characterization and antitumor efficacy of DS-6051b, a novel, orally available small molecule tyrosine kinase inhibitor of ROS1 and NTRKs. Eur J Cancer. 2016;69:S35–S36. [Google Scholar]
- 47.Fletcher GC, Brokx RD, Denny TA, et al. ENMD-2076 is an orally active kinase inhibitor with antiangiogenic and antiproliferative mechanisms of action. Mol Cancer Ther. 2011;10:126–137. doi: 10.1158/1535-7163.MCT-10-0574. [DOI] [PubMed] [Google Scholar]
- 48.Braud FGD, Pilla L, Niger M, et al. Phase 1 open label, dose escalation study of RXDX101, an oral pan-trk, ROS1, and ALK inhibitor, in patients with advanced solid tumors with relevant molecular alterations. J Clin Oncol. 2014;32(suppl; abstr 2502) [Google Scholar]
- 49.Rolfo C, Ruiz R, Giovannetti E, et al. Entrectinib: A potent new TRK, ROS1, and ALK inhibitor. Expert Opin Investig Drugs. 2015;24:1493–1500. doi: 10.1517/13543784.2015.1096344. [DOI] [PubMed] [Google Scholar]
- 50.Drilon A, Nagasubramanian R, Blake JF, et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 2017;7:963–972. doi: 10.1158/2159-8290.CD-17-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghilardi JR, Freeman KT, Jimenez-Andrade JM, et al. Administration of a tropomyosin receptor kinase inhibitor attenuates sarcoma-induced nerve sprouting, neuroma formation and bone cancer pain. Mol Pain. 2010;6:87. doi: 10.1186/1744-8069-6-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shabbir M, Stuart R. Lestaurtinib, a multitargeted tyrosine kinase inhibitor: From bench to bedside. Expert Opin Investig Drugs. 2010;19:427–436. doi: 10.1517/13543781003598862. [DOI] [PubMed] [Google Scholar]
- 53.Miknyoczki SJ, Chang H, Klein-Szanto A, et al. The Trk tyrosine kinase inhibitor CEP-701 (KT-5555) exhibits significant antitumor efficacy in preclinical xenograft models of human pancreatic ductal adenocarcinoma. Clin Cancer Res. 1999;5:2205–2212. [PubMed] [Google Scholar]
- 54.Yan SB, Peek VL, Ajamie R, et al. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Invest New Drugs. 2013;31:833–844. doi: 10.1007/s10637-012-9912-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Konicek BW, Bray SM, Capen AR, et al. Merestinib (LY2801653), targeting several oncokinases including NTRK1/2/3, shows potent anti-tumor effect in colorectal cell line-and patient-derived xenograft (PDX) model bearing TPM3-NTRK1 fusion. Cancer Res. 2016;76(suppl; abstr 2647) [Google Scholar]
- 56.Shimomura T, Hasako S, Nakatsuru Y, et al. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther. 2010;9:157–166. doi: 10.1158/1535-7163.MCT-09-0609. [DOI] [PubMed] [Google Scholar]
- 57.Brasca MG, Amboldi N, Ballinari D, et al. Identification of N,1,4,4-tetramethyl-8-[4-(4-methylpiperazin-1-yl)phenyl]amino-4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline-3-carboxamide (PHA-848125), a potent, orally available cyclin dependent kinase inhibitor. J Med Chem. 2009;52:5152–5163. doi: 10.1021/jm9006559. [DOI] [PubMed] [Google Scholar]
- 58.ECMC Network PLX7486 background information October 2015. http://www.ecmcnetwork.org.uk/sites/default/files/PLX7486%20Background%20for%20CRUK%20Combinations%20Alliance%20(Non-CI)%202015-10-08%20final.pdf
- 59.Patwardhan PP, Ivy KS, Musi E, et al. Significant blockade of multiple receptor tyrosine kinases by MGCD516 (Sitravatinib), a novel small molecule inhibitor, shows potent anti-tumor activity in preclinical models of sarcoma. Oncotarget. 2016;7:4093–4109. doi: 10.18632/oncotarget.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ivanov SV, Panaccione A, Brown B, et al. TrkC signaling is activated in adenoid cystic carcinoma and requires NT-3 to stimulate invasive behavior. Oncogene. 2013;32:3698–3710. doi: 10.1038/onc.2012.377. [DOI] [PubMed] [Google Scholar]
- 61.Cazorla M, Jouvenceau A, Rose C, et al. Cyclotraxin-B, the first highly potent and selective TrkB inhibitor, has anxiolytic properties in mice. PLoS One. 2010;5:e9777. doi: 10.1371/journal.pone.0009777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cazorla M, Prémont J, Mann A, et al. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest. 2011;121:1846–1857. doi: 10.1172/JCI43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chong CR, Bahcall M, Capelletti M, et al. Identification of existing drugs that effectively target NTRK1 and ROS1 rearrangements in lung cancer. Clin Cancer Res. 2017;23:204–213. doi: 10.1158/1078-0432.CCR-15-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qian F, Engst S, Yamaguchi K, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009;69:8009–8016. doi: 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- 65.Albaugh P, Fan Y, Mi Y, et al. Discovery of GNF-5837, a selective TRK inhibitor with efficacy in rodent cancer tumor models. ACS Med Chem Lett. 2012;3:140–145. doi: 10.1021/ml200261d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wood ER, Kuyper L, Petrov KG, et al. Discovery and in vitro evaluation of potent TrkA kinase inhibitors: Oxindole and aza-oxindoles. Bioorg Med Chem Lett. 2004;14:953–957. doi: 10.1016/j.bmcl.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 67.Jani JP, Arcari J, Bernardo V, et al. PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol Cancer Ther. 2010;9:883–894. doi: 10.1158/1535-7163.MCT-09-0915. [DOI] [PubMed] [Google Scholar]
- 68.Skerratt SE, Andrews M, Bagal SK, et al. The discovery of a potent, selective, and peripherally restricted pan-Trk inhibitor (PF-06273340) for the treatment of pain. J Med Chem. 2016;59:10084–10099. doi: 10.1021/acs.jmedchem.6b00850. [DOI] [PubMed] [Google Scholar]
- 69.Ignyta Catalyzing precision medicine with integrated RX/DX in oncology. https://www.ignyta.com/PDF/Ignyta%20Presentation_Jan%202014_20140110%20Final.pdf
- 70.Gamo K, Belmont B, Stockett D, et al. SNS-314, a novel small-molecule Aurora kinase inhibitor, induces cell-cycle defects and potently suppresses tumor growth. https://www.sunesis.com/data-pdf/314/SNS-314_Poster.pdf
- 71.Hyman DM, Laetsch TW, Kummar S, et al. The efficacy of larotrectinib (LOXO-101), a selective tropomyosin receptor kinase (TRK) inhibitor, in adult and pediatric TRK fusion cancers. J Clin Oncol. 2017;35(suppl; abstr LBA2501) [Google Scholar]
- 72.Smith KM, Fagan PC, Pomari E, et al. Antitumor activity of entrectinib, a pan-TRK, ROS1, and ALK inhibitor, in ETV6-NTRK3-positive acute myeloid leukemia. Mol Cancer Ther. 2018;17:455–463. doi: 10.1158/1535-7163.MCT-17-0419. [DOI] [PubMed] [Google Scholar]
- 73.Cook PJ, Thomas R, Kannan R, et al. Somatic chromosomal engineering identifies BCAN-NTRK1 as a potent glioma driver and therapeutic target. Nat Commun. 2017;8:15987. doi: 10.1038/ncomms15987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nagasubramanian R, Wei J, Gordon P, et al. Infantile fibrosarcoma with NTRK3-ETV6 fusion successfully treated with the tropomyosin-related kinase inhibitor LOXO-101. Pediatr Blood Cancer. 2016;63:1468–1470. doi: 10.1002/pbc.26026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doebele RC, Davis LE, Vaishnavi A, et al. An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov. 2015;5:1049–1057. doi: 10.1158/2159-8290.CD-15-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Farago AF, Le LP, Zheng Z, et al. Durable clinical response to entrectinib in NTRK1-rearranged non-small cell lung cancer. J Thorac Oncol. 2015;10:1670–1674. doi: 10.1097/01.JTO.0000473485.38553.f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wei G, Ardini E, Patel R, et al. Entrectinib is effective against the gatekeeper and other emerging resistance mutations in NTRK-, ROS1- and ALK- rearranged cancers. Cancer Res. 2016;76(suppl; abstr 2136) [Google Scholar]
- 78.Russo M, Misale S, Wei G, et al. Acquired resistance to the TRK inhibitor entrectinib in colorectal cancer. Cancer Discov. 2016;6:36–44. doi: 10.1158/2159-8290.CD-15-0940. [DOI] [PubMed] [Google Scholar]
- 79.Drilon A, Li G, Dogan S, et al. What hides behind the MASC: Clinical response and acquired resistance to entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC) Ann Oncol. 2016;27:920–926. doi: 10.1093/annonc/mdw042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsimberidou AM, Hong DS, Ye Y, et al. Initiative for molecular profiling and advanced cancer therapy (IMPACT): An MD Anderson precision medicine study. JCO Precis Oncol. doi: 10.1200/PO.17.00002. doi: 10.1200/PO.17.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsimberidou AM, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin Cancer Res. 2012;18:6373–6383. doi: 10.1158/1078-0432.CCR-12-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wheler JJ, Janku F, Naing A, et al. Cancer therapy directed by comprehensive genomic profiling: A single center study. Cancer Res. 2016;76:3690–3701. doi: 10.1158/0008-5472.CAN-15-3043. [DOI] [PubMed] [Google Scholar]
- 83.Schwaederle M, Chattopadhyay R, Kato S, et al. Genomic alterations in circulating tumor DNA from diverse cancer patients identified by next-generation sequencing. Cancer Res. 2017;77:5419–5427. doi: 10.1158/0008-5472.CAN-17-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kato S, Krishnamurthy N, Banks KC, et al. Utility of genomic analysis in circulating tumor DNA from patients with carcinoma of unknown primary. Cancer Res. 2017;77:4238–4246. doi: 10.1158/0008-5472.CAN-17-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 86.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Balduzzi S, Mantarro S, Guarneri V, et al. Trastuzumab-containing regimens for metastatic breast cancer. Cochrane Database Syst Rev. 2014;2014:CD006242. doi: 10.1002/14651858.CD006242.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 89.Catenacci DVT, Tebbutt NC, Davidenko I, et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:1467–1482. doi: 10.1016/S1470-2045(17)30566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Le Tourneau C, Kurzrock R. Targeted therapies: What have we learned from SHIVA? Nat Rev Clin Oncol. 2016;13:719–720. doi: 10.1038/nrclinonc.2016.164. [DOI] [PubMed] [Google Scholar]
- 91.Schwaederle M, Parker BA, Schwab RB, et al. Precision oncology: The UC San Diego Moores Cancer Center PREDICT experience. Mol Cancer Ther. 2016;15:743–752. doi: 10.1158/1535-7163.MCT-15-0795. [DOI] [PubMed] [Google Scholar]
- 92.Farago AF, Piotrowska Z, Sequist LV. Unlocking the mystery of small-cell lung cancer transformations in EGFR mutant adenocarcinoma. J Clin Oncol. 2017;35:2987–2988. doi: 10.1200/JCO.2017.73.5696. [DOI] [PubMed] [Google Scholar]
- 93.Janku F, Hong DS, Fu S, et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Reports. 2014;6:377–387. doi: 10.1016/j.celrep.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Subbiah V, Kurzrock R. Challenging standard-of-care paradigms in the precision oncology era. Trends Cancer. 2018;4:101–109. doi: 10.1016/j.trecan.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kato S, Subbiah V, Kurzrock R. Counterpoint: Successes in the pursuit of precision medicine: Biomarkers take credit. J Natl Compr Canc Netw. 2017;15:863–866. doi: 10.6004/jnccn.2017.0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bieg-Bourne CC, Millis SZ, Piccioni DE, et al. Next-generation sequencing in the clinical setting clarifies patient characteristics and potential actionability. Cancer Res. 2017;77:6313–6320. doi: 10.1158/0008-5472.CAN-17-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 98.Westin JR, Kurzrock R. It’s about time: Lessons for solid tumors from chronic myelogenous leukemia therapy. Mol Cancer Ther. 2012;11:2549–2555. doi: 10.1158/1535-7163.MCT-12-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med. 2017;377:829–838. doi: 10.1056/NEJMoa1704795. [DOI] [PubMed] [Google Scholar]
- 100.Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963–1971. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gao Q, Liang WW, Foltz SM, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Reports. 2018;23:227.e3–238.e3. doi: 10.1016/j.celrep.2018.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pietrantonio F, Di Nicolantonio F, Schrock AB, et al. ALK, ROS1, and NTRK rearrangements in metastatic colorectal cancer. J Natl Cancer Inst. 2017;109:109. doi: 10.1093/jnci/djx089. [DOI] [PubMed] [Google Scholar]