

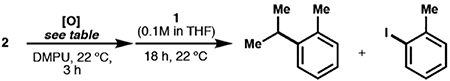
Abstract
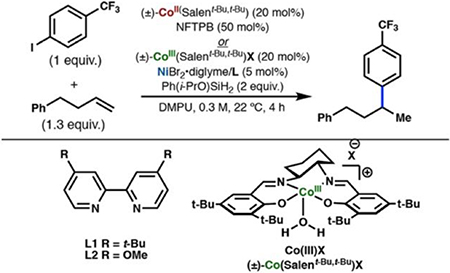
Cobalt/nickel-dual catalyzed hydroarylation of terminal olefins with iodoarenes builds complexity from readily available materials, with a high preference for the Markovnikov (branched) product. Here, we advance a mechanistic model of this reaction through the use of reaction progress kinetic analysis (RPKA), radical clock experiments, and stoichiometric studies. Through exclusion of competing hypotheses, we conclude that the reaction proceeds through an unprecedented alkylcobalt to nickel direct transmetalation. Demonstration of catalytic alkene prefunctionalization, via spectroscopic observation of an organocobalt species, distinguishes this Csp2–Csp3 cross-coupling method from a conventional transmetalation process, which employs a stoichiometric organometallic nucleophile, and from a bimetallic oxidative addition of an organohalide across nickel, described by radical scission and subsequent alkyl radical capture at a second nickel center. A refined understanding of the reaction leads to an optimized hydroarylation procedure that excludes exogenous oxidant, demonstrating that the transmetalation is net redox neutral. Catalytic alkene prefunctionalization by cobalt and engagement with nickel catalytic cycles through direct transmetalation provides a new platform to merge these two rich areas of chemistry in preparatively useful ways.
INTRODUCTION
Transition-metal-catalyzed cross-coupling is an indispensable tool for constructing carbon−carbon bonds. Seminal examples, such as the Stille, Suzuki−Miyaura, Kumada−Corriu, and Negishi reactions, use stoichiometric organometallic nucleophiles (M = Sn, B, Mg, Zn) to transfer an organic ligand to a catalytically generated metal species (such as Pd or Ni) in a transmetalation step.1 Formation of an organometallic (or metalloid) complex serves to prefunctionalize the substrate, enabling the predictable formation of new carbon−carbon bonds catalyzed at a single metal site. A complementary approach to classical cross-coupling methods is dual catalysis, in which a nucleophile, which is catalytically generated in situ, undergoes transmetalation to a second metal catalyst, which undergoes a canonical catalytic cycle (Figure 1A).2,3 Dual catalysis addresses some of the practical limitations of single-site catalysis using stoichiometric nucleophiles, including requisite substrate prefunctionalization, potential air and water sensitivity, and substrate functional group incompatibility. While dual catalysis can directly address these drawbacks, the rational design of novel dual catalytic systems is limited by our identification and understanding of efficient and selective transmetalations.
Figure 1.

Electron-neutral alkenes and electron-deficient aryl iodides couple efficiently in Co/Ni-catalyzed cross-coupling.
Our group recently described a cobalt/nickel-dual catalyzed iodoarene-olefin cross-coupling that results in highly branch-selective (Markovnikov) hydroarylation (Figure 1B).4 This reaction originated from our prior interest in Drago− Mukaiyama-type radical hydrofunctionalization methods.5
To adapt these reactions to a cross-coupling platform, we envisaged that the intermediate carbon-centered radical could be intercepted by a catalytically generated organonickel species,6 whereas past iterations of Drago−Mukaiyama reactions used stoichiometric, classic radical electrophiles. Merging these metal−hydride hydrogen atom transfer (MHAT) reactions with nickel catalysis would benefit from improved atom economy as compared to existing nickel-catalyzed radical cross-coupling reactions, which rely on cleavage of a high-formula weight radical precursor and ablation of stereochemical information. In contrast, MHAT/ nickel dual catalysis forms two new bonds from a pro-chiral alkene via sequential C−H and C−C bond formation in one step.
In our initial report, mechanistic details of the reaction were absent, especially regarding intersection of the two catalytic cycles. Two separate experiments, small ring opening and cyclization onto an alkene, indicated that an alkyl radical was generated in the reaction, but the kinetically relevant reaction pathway of that radical remained unknown.
Initial insights were drawn from our recent connection between Drago−Mukaiyama hydrofunctionalization reactions catalyzed by cobalt, manganese, and iron hydrides to the paradigm of MHAT explored by Simandi and Nagy,7 Kwiatek,8 Jackman,9 Halpern,10 Norton,11 and others.12
The MHAT mechanism postulates the reversible formation of a caged carbon-radical/metal pair. Subsequent reactivity proceeds via three available pathways: (1) alkyl radical solvent cage escape and interception by a suitable radical trap; (2) isomerization via M• abstraction of an adjacent C−H bond; or (3) cage collapse to form an organometallic complex (Scheme 1).13 The intermediacy of a carbon-centered radical leads to the high Markovnikov selectivity observed for olefin hydro-functionalizations on account of increased stabilization of 3° > 2° > 1° carbon radicals. The metal center, ligand, and substrate can influence the preference for pathways 1−3 by altering the relative stabilities of organic, metallic, and organometallic intermediates.11f,g In our bimetallic system, it was unclear which Co−H HAT pathway, formation of a solvent-separated radical (Scheme 1, pathway 1) or discrete organometallic (Scheme 1, pathway 3), was relevant to the catalytic cycle and could interact productively with a catalytically generated nickel species.
Scheme 1.

Observed Reactivity of Metal–Hydrides (M = Co, Mn, Fe) with Unactivated Olefins
Through a combination of reaction kinetics, radical clock experiments, and stoichiometric reactions, the mechanism not excluded by our data describes a rate-determining organo-cobalt to nickel transmetalation. Furthermore, we propose that the mechanism proceeds through a “cage rebound” process described by an electron-transfer/ligand-transfer step between a Co(III)/Ni(III) organometallic pair within a solvent cage, supporting the redox neutrality of both cycles.
RESULTS
The following hypotheses, formulated by analogy to established reactivity, guided our inquiry into the mechanism of this reaction (Scheme 2A–D):
Scheme 2.

Possible Mechanisms of the Cobalt/Nickel-Dual Catalyzed Hydroarylation
(A) A nickel-catalyzed reductive Heck was suggested by a referee of our initial communication and evaluated here for completeness. The rate is expected to be first order in nickel and zero order in cobalt.
(B) Another possibility is generation of a radical caged pair via Co−H HAT reaction with the alkene. Radical cage escape (Scheme 1, pathway 1) would generate a low concentration of a short-lived radical that could be captured by a much larger concentration of an arylnickel(II) intermediate. The resultant dioorganonickel(III) intermediate would undergo reductive elimination to form coupled product and nickel(I).14 Excess 1-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate (NFTPB) would reoxidize cobalt(II) to cobalt(III) to turn over the cobalt cycle.15 In this mechanistic scenario, the rate is expected to be first order in cobalt and zero order in nickel.
(C) An alternative mechanism would involve collapse of the alkyl radical to a discrete organocobalt(III) species (Scheme 1, pathway 3). A nickel(I) species could reduce the organocobalt-(III) complex to an unstable organocobalt(II) anionic intermediate,16,17 which would homolyze to release an alkyl radical and cobalt(I) anion. A separate arylnickel(II) species would capture the alkyl radical, and the resultant dioorganonickel(III) species would reductively eliminate product and regenerate nickel(I). The cobalt(I) species could be oxidized to the active cobalt(III) catalyst by excess NFTPB or adventitious molecular oxygen. The radical capture step would occur at a nickel center different from that of radical initiation, constituting a radical chain process in which Ni(I) serves as a chain carrier in the propagation step; similar mechanisms have been described by Weix’s cross-electrophile coupling18 or the bimetallic oxidative addition processes detailed by Hu19 and Fu.20
(D) A final possibility is the direct transfer of an alkyl ligand from cobalt to nickel. In this mechanism, the cobalt−carbon and nickel−carbon bonds are broken and formed in the same step, reminiscent of a conventional transmetalation found in traditional cross-coupling methods. Few literature precedents exist for this type of transformation between cobalt and nickel, and so our understanding of this potential process was limited at the beginning of this study. Various oxidation states for both metals were considered and will be discussed (vide infra).
Exclusion of a Cage Escape Mechanism.
Our first foray into mechanistic inquiry involved reaction progress kinetic analysis (RPKA) of the Co/Ni-catalyzed cross-coupling between 4-phenyl-1-butene and 4-iodobenzotrifluoride (Scheme 3). RPKA provides a unique advantage of gaining kinetic information by monitoring a reaction under relevant reaction conditions, rather than artificially high excesses of reagents to mimic pseudoconstant concentrations.21
Scheme 3.

Standard Hydroarylation Conditions
Although the principles of RPKA are derived algebraically from rate laws, the use of a graphical rate equation makes interpretation of the data straightforward. Different excess experiments were used to establish any dependence of the reaction rate on reagents and reactants (see the Supporting Information).
Different initial concentrations of silane, iodoarene, and olefin produced no variation in the rate as observed by the overlay of product concentration versus time (Figure 2).
Figure 2.

Different excess experiments varying initial concentrations of iodoarenes, olefin, and silane. Standard condition concentrations are [ArI]0 = 0.32 M, [olefin]0 = 0.42 M, and [silane]0 = 0.64 M.
Because of the large excess of these stoichiometric reactants relative to the catalysts (at 5 and 20 mol %), saturation kinetics does not exclude these substrates from participating in steps prior to formation of the catalyst resting states of cobalt and nickel. The same excess experiments were performed to corroborate the use of RPKA analysis (see the Supporting Information). Catalyst deactivation did not occur until high conversions of starting material, and there was no apparent product inhibition.
At different initial concentrations of cobalt or nickel, the graphs no longer overlay when plotted directly as a function of time, but instead displayed a direct relationship between rate and catalyst loading. We decided to use a normalized time scale to plot the concentration of product directly against t•[cat]n, in which n is the order of the catalyst, in accordance with Bureś ’ graphical rate law analysis (Figure 3).22 For both catalysts, the three different catalyst loadings overlay when n = 1, indicating that the reaction is first order in cobalt and nickel, and a mixed second-order reaction overall.23 Different excess experiments with 1-fluoro-2,4,6-trimethyl-pyridinium tetrafluoroborate (NFTPB) led us to conclude the oxidant is not involved in the rate-determining step (Figure 4).
Figure 3.

Burés graphical rate analysis for (A) cobalt and (B) nickel.
Figure 4.

Different excess experiments with NFTPB. An induction period is observed at 20 mol % loading, but the rate is parallel to the standard conditions (a line has been added as a visual aid). Higher loadings of oxidant led to deleterious effects on the yield and rate, possibly due to off-pathway reactions with the nickel catalyst.
In the design of this catalyst system, NFTPB was chosen, in accordance with the literature, as a compatible oxidant in cobalt(III)−hydride-mediated alkene hydrofunctionalization reactions.24 After initial oxidization of the cobalt(II) precatalyst to the cobalt(III) oxidation state,25 subsequent reaction with Ph(i-PrO)SiH2 forms the cobalt(III)−hydride catalyst.26 Because the oxidant is involved in formation of the active cobalt(III)−hydride catalyst, the presence of an induction period was unsur prising at a lower oxidant loading (20 mol %). However, after the induction period, it appears that the rates of both the 20 and 50 mol % loading of NFTPB are equivalent (line has been added as a visual aid). Therefore, it was reasonable to conclude that NFTPB was not involved in the rate-determining step. Interestingly, increasing the oxidant loading (75 mol %) led to a deleterious effect on the reaction yield and rate. This result indicated that the oxidant could exhibit off-pathway reactivity with the nickel catalyst, which would inhibit the cross-coupling. At this point, the kinetic profile of NFTPB offered no definitive answers for its role in the reaction, although we speculated that at 50 mol % loading or lower, it was not involved in the reaction beyond initial cobalt(II) precatalyst oxidation. Further experimentation would provide a clear answer on the role of the oxidant, which we will explore in a later section.
Taken together, these graphical rate analyses showed a rate law that included both transition metal catalysts, but not their substrates. The oxidant was implicated in the initial cobalt(II) to cobalt(III) oxidation, but not in the rate-determining step. The observed rate law excluded a nickel-only-catalyzed reductive Heck (Scheme 2A), and a cage escape mechanism (Scheme 2B), and were consistent with, but did not differentiate between, a radical chain (Scheme 2C) or direct transmetalation mechanism (Scheme 2D).
Evidence for the Formation of an Organocobalt Species.
The potential intermediacy of an organocobalt complex in the hydroarylation also informed our initial reaction design.4 Previously explored olefin hydrofunctionalization reactions utilizing a cobalt salen catalyst, hydrosilane, and oxidant have explicitly suggested the intermediacy of an organocobalt complex,24,26,27 although its presence in a catalytic reaction could not be confirmed spectroscopically.27a
During our previous studies of olefin isomerization, also employing a cobalt salen catalyst and phenylsilane, we hypothesized the off-cycle formation of an organocobalt species during the isomerization of terminal olefins.5d Whereas 1,1-disubstituted alkenes isomerized rapidly at room temperature, monosubstituted alkenes required elevated temperatures (>60 °C). A competition experiment between a 1,1-disubstituted alkene and monosubstituted alkene led to isomerization only upon heating; the room temperature isomerization was completely suppressed (Figure 5A). This observation was consistent with our hypothesis that reactivity was arrested by catalyst sequestration through secondary organocobalt formation, rather than a higher kinetic barrier of MHAT to monosubstituted alkenes as compared to 1,1-disubstituted alkenes. Even gentle heating leads to homolysis of the cobalt−carbon bond, followed by M• abstraction of the internal C−H bond and subsequent isomerization to the more thermodynamically stable internal alkene.28
Figure 5.

(A) Evidence for stability of sec-alkyl-Co(Salt-Bu,t-Bu) organometallic complexes at room temperature: the active catalyst appears to be sequestered;5d and (B) observation of two diastereotopic methyls at –0.4 and –0.5 ppm from Co(I) SN2 and Co(III)/PhSiH3/alkene combination.
The stability of organocobalt complexes (Scheme 1, pathway 3) is largely influenced by steric interaction between the cobalt ligand and the carbon substituent, as well as the identity of the trans-axial ligand.29 The synthesis and characterization of secondary organocobalt complexes has precedent in the literature, while the isolation of tertiary alkyl cobalt species has not been possible, lending further support to our hypothesis for organocobalt formation in the isomerization reaction of monosubstituted olefins.30 Despite the lack of spectroscopic characterization of sec-alkyl cobalt salen complexes, we set out to observe the formation of an organocobalt in the related isomerization reaction explored by our lab5d via 1H NMR spectroscopy, because it appeared relevant to the rate-determining step in the hydroarylation. When 4-phenyl-1-butene was subjected to MHAT isomerization conditions in d6-benzene, no isomerization took place at room temperature, as observed previously, but a new diamagnetic alkylcobalt complex could be observed by 1H NMR in d6-DMSO. The identity of this organocobalt complex was confirmed by independent synthesis from NaCo(I)salen and (3-bromobutyl)benzene (Figure 5B). Both reaction products showed identical peaks in the far-upfield region of their 1H NMR spectra: a distinctive pair of doublets at δ −0.43 and −0.54 ppm corresponding to diastereotopic methyls.31 The methine proton could also be visualized as a multiplet at δ 5.33 ppm. The observation of an organocobalt complex was consistent with the observed rate law of the catalytic hydroarylation in which an organocobalt complex described the cobalt catalyst resting state and could act as either a stable reservoir of alkyl radicals or a reactive intermediate.
Although we had spectroscopic evidence suggesting formation of an organocobalt complex under relevant isomerization conditions, we wondered if its formation was reversible and more accurately described as an equilibrium favoring starting alkene and cobalt(III)−hydride (“back HAT”). A rapid, reversible MHAT equilibrium would be kinetically indistinguishable from a stable organometallic catalyst resting state as it comes before the rate-determining step in the hydroarylation. We therefore ran the hydroarylation reaction with a d2-alkene isomer to probe reversibility through isotopic exchange. The hydroarylation product was isolated from the catalytic reaction with a slight increase in hydrogen incorporation at the terminal methyl group (13% increase in hydrogen incorporation as compared to that expected) (Figure 6). This experiment agreed with cage-pair formation followed by collapse to a metastable organocobalt complex: the cobalt resting state before rate-determining transmetalation.
Figure 6.

Hydroarylation with d2-alkene indicates an irreversible HAT under catalytic conditions.
We then began to consider how alkyl ligand transfer between an organocobalt and nickel could proceed. The most relevant literature stems from studies of the bacterial Wood− Ljungdahl pathway32 for conversion of CO2 to acetyl-CoA via transmetalation of methylcobalamine(III) to a multimeric cysteine−nickel complex.33
This transmetalation has been recapitulated using MeCo(dmgBF2)2L, which reacts with inorganic Ni(I) and Ni(0) complexes by radical and SN2 pathways, respectively (Scheme 4).34 There are additional examples of alkylcobalt transmetalation to other metals (i.e., Cr, Pd) in the literature, which have suggested both radical chain processes or direct group transfer mechanisms.35 These previously explored mechanisms informed our evaluation of two potential mechanisms, a radical chain mechanism (Scheme 2C) and a direct transmetalation (Scheme 2D).
Scheme 4.

Literature Precedent for Alkyl Ligand Transfer from Organocobalt to Nickel Chelates Relevant to the Wood–Ljungdahl Pathway
Exclusion of Radical Chain Mechanism via Radical Clock Cyclization Experiments.
Mechanisms C and D both involve generation of carbon-centered radicals, implicated by ring formation and ring cleavage in our original report,4 but are distinct in whether the nickel species that affects cobalt−carbon bond homolysis and captures that radical are different (radical chain) or the same (cage rebound) (Scheme 5). Recent mechanistic studies of similar nickel-catalyzed reactions answered this same question with a “radical clock” substrate under catalytic conditions to distinguish both mechanisms.18–20,36 These studies identified a radical chain mechanism in which a nickel(I) complex initiates carbon radical formation via reduction of an alkyl halide. A second arylnickel(II) intermediate then captures this radical to form an unstable dioorganonickel(III) species, which reductively eliminates coupled product, thereby regenerating Ni(I), and propagating the chain. This radical chain, in which one nickel species effects radical formation and a second captures the radical, was reflected in the effect of nickel concentration on the ratio of unrearranged and rearranged products from either 5-hexenyl iodide18 or 3-(2-bromoethoxy)prop-1-ene.19 The alkene-tethered radicals in these studies were formed from C− X bonds. Our study required radical formation from an alkene in the presence of another alkene. Fortunately, the steric demand of the Salt-Bu,t-Bu ligand differentiates between substituted alkenes: monosubstituted alkenes undergo hydroarylation, but 1,2-disubtituted alkenes do not.4,5d
Scheme 5.

Coupling Reaction with a Radical Clock Substrate to Differentiate Mechanisms C and D
The simplest substrate to fulfill these criteria, (E)-1,6-octadiene,37 was subjected to our standard catalytic hydroarylation conditions (Figure 7A). The presence of four rearranged diastereomeric products eliminated the possibility that cyclized products arose from a nickel-only coordinated Heck-type pathway, which would yield only two diastereomers.38
Figure 7.

(A) The coupling reaction with radical clock substrate (E)-1,6-octadiene and varying concentrations of nickel can indicate whether a radical chain mechanism is operative. (B) A comparative graph of relative U/R increase as a function of relative nickel precatalyst loading in nickel-radical cross-coupling mechanistic studies (see refs 18 and 19). The lack of a direct relationship in our work effectively excludes a radical chian mechanism (mechanism C).
After alkyl radical formation, two reaction pathways are available: the alkyl radical can react (1) intramolecularly and generate a different, rearranged alkyl radical species, according to a fast, unimolecular rate constant (k1), or (2) bimolecularly with a suitable radical trap, described in this experiment by k2[Ni]. These competitive pathways will give rise to two products, the rearranged hydroarylation product (R) and the unrearranged hydroarylation product (U), respectively. The rearranged alkyl radical intermediate also reacts bimolecularly (k2[Ni]), but this rate would have no impact on the ratio of products.
If a radical chain mechanism were operative, the ratio of unrearranged to rearranged products would be reflected by k2[Ni]/k1, the relative rates of the competitive pathways after the initial alkyl radical formation. Because this ratio is directly proportional to nickel concentration, there should be a direct, linear relationship between concentration of nickel precatalyst and unrearranged to rearranged (U/R) ratio.
To our surprise, the ratio of rearranged and unrearranged isomers was insensitive to changes in nickel precatalyst concentration across five concentrations (Figure 7B). Correlation of isomer ratios (0.8−1.1) to nickel concentration (15−45 mM) generated a best-fit line slope of only 0.0093 (R2 = 0.91), far from the directly proportional relationship we would expect if a radical chain were present. In comparison, nickel catalytic cycles that separate radical generation from radical capture have yielded non-normalized slopes of 0.4218 and 0.519 (see the Supporting Information).
The independence of the U/R ratio on nickel concentration is inconsistent with a radical chain mechanism (Scheme 2C), leaving a cage rebound mechanism (Scheme 2D) as a plausible mechanistic scenario.39 How then would the rearranged hydroarylation product arise?
Whereas radical cyclization in a radical chain mechanism18,19 is easy to understand, rearrangement in a cage rebound mechanism is not. Running the same experiment, but varying cobalt precatalyst concentration, provided some insight.
Unlike the nickel variation experiment, a positive correlation between cobalt(II) precatalyst loading and U/R ratio is present (Figure 8A). Whereas a 200% increase in nickel concentration led to only a 37% increase in product ratio, the same increase in cobalt concentration led to a 383% increase. At the outset of this experiment, we had assumed that nickel was acting as a radical trap, but we had not accounted for the high propensity for cobalt(II)40 and alkylcobalt(III)41 complexes to trap alkyl radicals.
Figure 8.

(A) An alkyl radical is generated and captured by cobalt, not nickel, in the reaction. The unrearranged and rearranged alkyl cobalt species directly transfer an alkyl ligand to nickel. The ratio of product displays a positive, albeit nonlinear, relationship to the concentration of salenCo(II) precatalyst. (B) Alkyl radical chain mechanisms using primary hexenyl-Co(salen) complexes elucidated by Kochi and coworkers.42
Therefore, the direct relationship between cobalt(II) precatalyst loading and U/R ratio could reflect the following scenario. Initiation of a radical chain can occur by (a) cage escape after initial HAT; (b) homolysis of the carbon−cobalt bond at ambient temperature; or (c) competitive cage escape in the cage rebound mechanism. Option (b) seems unlikely given the extremely low rate of isomerization at room temperature (trace isomerization observed after 14 h in DMPU) as compared to the hydroarylation reaction rate (>70% after 3 h).
Options (a) and (c), however, merely require an imperfect carbon radical−metal collapse to allow solvent cage escape and a radical chain initiation. We considered that NFTPB could be initiating the alkyl radical chain by alkylcobalt(III) oxidation, but this was subsequently ruled out after further experimentation (see the Supporting Information).
Radical capture by cobalt(II) inhibits a radical chain, while SH2 at another alkylcobalt(III) center propagates alkyl radical formation. This alkyl radical chain capture/propagation proposal is consistent with Kochi’s previous work on the molecular rearrangement of 1-hexenyl Co(salen) to cyclopentylmethyl Co(salen) complexes (Figure 8B).42 At higher cobalt loadings, there is a higher concentration of cobalt(II) (likely due to incomplete cobalt(II) precatalyst oxidation), and we observe a higher U/R ratio. Both unrearranged and rearranged alkylcobalt(III) complexes directly engage nickel in an alkyl ligand transfer step (Figure 8A), giving rise to both unrearranged and rearranged products, while still consistent with a cage rebound mechanism (Scheme 2D).
At this point, a mechanism that could explain the presence of all four diastereomers of the rearranged product became apparent. An alkyl radical formed by any of the pathways mentioned above could rearrange and/or propagate alkyl radical formation by SH2 at another organocobalt(III) center. The cyclization arises via a radical pathway but is captured by an alkylcobalt(III) or a cobalt(II) species, not a nickel intermediate. A radical chain was, in fact, present, but did not describe the transfer of an alkyl ligand from cobalt to nickel.
Stoichiometric Experiments.
We decided that probing a direct transmetalation with stoichiometric experiments between an alkylcobalt complex and an isolable nickel(II) oxidative addition intermediate could provide additional mechanistic insights into the reaction.43 We had shown that sec-alkylcobalt complexes could be observed spectroscopically (Figure 5B), giving credibility to their preparative synthesis.
As mentioned in a previous section, extensive literature precedent details the synthesis and characterization of primary alkyl organocobalt complexes, but fewer examples were found describing the synthesis of secondary alkyl organocobalt species.30 In certain cases, instability was attributed to air- and moisture-sensitivity.44 Moreover, a representative example, (i-C3H7)Co(salen), with an ethylene backbone, was reported to possess a much lower bond dissociation energy (BDE) relative to an analogous primary alkylcobalt, C2H5Co(salen) (19 vs 30 kcal/mol).44a To circumvent potential problems due to instability, we synthesized i-Pr-Co(Salt-Bu,t-Bu)(pyr) (1) in a glovebox as a solution in tetrahydrofuran. To our knowledge, this is the first example of a spectroscopically (1H NMR) characterized sec-alkyl cobalt salen species.
When 1 and dtbbpyNi(o-Tol)I (2) were allowed to stir for 18 h in THF:DMPU (1:1), the expected cross-coupled product, o-cymene, was observed in 10% yield (Scheme 6). The low yield in the stoichiometric reaction suggested that a nickel(II) aryl species was not the transmetalating species. The formation of 2-iodotoluene and 2,2′-dimethyl-1-1′-biphenyl indicated that low-valent nickel was present over the course of the stoichiometric experiment, but clearly did not initiate quantitative radical formation and capture; this observation provides additional evidence against a radical chain mechanism.45 At this point, we began to further interrogate the role of the oxidant, NFTPB. During our initial RPKA studies, we had observed a deleterious effect on the reaction yield and rate when a high excess of NFTPB was present. This observation led us to question the effect of the oxidant on the nickel catalytic cycle in addition to its role in the initial oxidation of the cobalt(II) precatalyst.
Scheme 6. Stoichiometric Transmetalation Experiments.

a The reaction of i-Pr-Co(Salt-Bu,tBu)(pyr) (1) and dtbbpyNi(o-Tol)I (2) led to coupled product, o-cymene, in only 10% yield. 2-iodotoluene and biaryl (2,2’-dimethyl-1,1’-biphenyl) were also observed
To test the potential role of NFTPB in the transmetalation, we returned to the stoichiometric experiment. Stirring 1 equiv of NFTPB with 2 led to an immediate color change from red to green. After 3 h at room temperature, a solution of 1 was added and the reaction was stirred for 18 h. We observed mostly 2-iodotoluene (78% yield), and a slightly higher yield of o-cymene (17%) (Table 1, entry 2). We reasoned that the high yield of 2-iodotoluene stemmed from reductive elimination from a Ni(III) or Ni(IV) intermediate formed by oxidation of 2, consistent with similar observations for C−C bond formation using nickel(II) organometallic complexes and NFTPT (1-fluoro-2,4–6-trimethylpyridinium triflate) in the literature.46
Table 1.
Stoichiometric Cross-Coupling Studiesa
 | |||
|---|---|---|---|
| entrya | oxidant | o-cymene (%) | 2-iodotoluene (%) |
| 1c | none | 10 | 25 |
| 2b | NFTPB (1 equiv) | 17 | 78 |
| 3c | AcFcBF4 (1 equiv) | 18 | 18 |
| 4 | AcFcBF4 (2 equiv) | 41 | 44 |
Reactions run on 10 μmol scale. Average of two trials. Yield determined by GC-FID with internal standard.
No fluorinated products observed.
2,2’-Dimethyl-1,1’-biphenyl also observed.
The high yield of 2-iodotoluene observed argued against the kinetic relevance of an arylnickel(II) intermediate in the catalytic reaction, as it would be competitively consumed by excess NFTPB present under our catalytic reaction conditions. Furthermore, the slight increase in o-cymene (7% increase) was interesting but did not convince us that NFTPB was required for the transmetalation by oxidation of an arylnickel(II) intermediate. Altogether, the stoichiometric studies and the kinetic profile of the oxidant (vide supra) indicated that an arylnickel(II) intermediate was not participating in the rate-determining transmetalation within the catalytic reaction.
However, we wondered if the slight increase in o-cymene yield (Table 1, entry 2) meant that a high-valent (Ni(III) or Ni(IV)) arylnickel species, accessible by oxidative addition of the aryl halide across a Ni(I), or possibly, Ni(II) intermediate, did describe the nickel transmetalating species. To test this question, we ran the stoichiometric reaction between 1 and 2 in the presence of acetylferrocenium tetrafluoroborate (AcFcBF4), an established one-electron outer-sphere oxidant for organometallic nickel(II) species.47 Repeating the reaction using 1 equiv of AcFcBF4 led to similar yields in coupled product (18%) and 2-iodotoluene (18%) (Table 1, entry 3). Two equivalents of AcFcBF4 led to an increase in coupled product (41%), along with 2-iodotoluene (44%) (Table 1, entry 4). While we do not presume to know the identity of a higher-valent arylnickel species that could be formed in this experiment, the result led us to consider that an arylnickel(III) or arylnickel(IV) species may participate in the transmetalation under catalytic conditions. To be clear, this species did not have to be formed by the oxidation of an arylnickel(II) intermediate by NFTPB.
Role of NFTPB Oxidant in the Catalytic Reaction.
Both the kinetic profile of the oxidant and the stoichiometric transmetalation experiments indicated that NFTPB may only serve to oxidize Co(II) to the requisite Co(III) oxidation state for catalysis. While the NFTPB oxidant has been used with similar catalytic systems (CoSalen/phenylsilane), the product of the cobalt(II) oxidation has only been speculated.24
We decided to characterize the product of oxidation through X-ray crystallography and NMR studies. We were unable to obtain an adequate crystal structure of the oxidized cobalt(III) complex when using NFTPB (tetrafluoroborate counterion). Fortunately, when repeating the reaction with NFTPT (triflate counterion), also a competent oxidant in the catalytic reaction, albeit lower yielding (47% yield), we obtained a suitable crystal. The crystal structure of the cobalt(III) oxidized product shows two water molecules coordinated to a cationic cobalt(III) center, with an outer-sphere triflate counteranion (Figure 9). No cobalt−fluoride bond was observed in the X-ray crystal structure, and only one signal was observed in the 19F NMR (δ −76.48 ppm) corresponding to the triflate counteranion.48 The observed crystal structure is consistent with observed structures of other cobalt(III) salen complexes with outer-sphere counteranions. These complexes have been demonstrated to contain significant spin delocalization through the salen ligands, which may facilitate through-ligand single electron transfer.56a,b
Figure 9.

(A) Oxidation of CoII(Salt-Bu,t-Bu) by NFTPT. (B) X-ray crystal structure of oxidized cobalt(III) product shown with 50% thermal ellipsoids. Two molecules of DMPU stabilize a coordinated water molecule.
In an effort to further identify the role of NFTPB in the reaction, we decided to synthesize the CoIII(Salt-Bu,t-Bu)BF4 (Co(III)BF4) complex directly and use it as the precatalyst in the catalytic reaction.49 When the catalytic reaction was run using Co(III)BF4 as the precatalyst with no added oxidant, the product was observed in 55% yield (Table 2, entry 1), indicating that the oxidant was not required for turnover of the cobalt catalytic cycle. Switching to a 4,4′-dimethoxy-2,2′-bipyridyl nickel ligand provided slightly higher yields (Table 2, entry 2). When the reaction was run in the glovebox with degassed reagents, the yield was unaffected, indicating that turnover of the cobalt catalyst is not dependent on adventitious oxygen (Table 2, entry 3). Interestingly, we found that using the analogous Co(III)Cl precatalyst resulted in lower yields in all cases.
Table 2.
Oxidant Not Necessary for Cobalt Turnoveraa
 | ||||
|---|---|---|---|---|
| entrya | variations from above | Co(ll)+NFTPB | Co(III) BF4 | Co(III) CI |
| 1 | LI | 79 | 55 | 44 |
| 2 | L2 | 82 | 56 | 42 |
| 3 | L2, in the glovebox | 82 | 56 | 41 |
| 4 | L2, 2 equiv of Mn° | 76 | 57 | |
| 5 | L2, NFTPB (0.3 equiv) | 70 | 53 | |
| 6b | dtbbpyNi(o-ToI)l (2) as | 68 | 49 | |
Reactions run on 50 μmol scale. Yield determined by GC-FID with internal standard. A1-Methyl-2-(4-phenylbutan-2-yl)benzene observed in <1% overall yield (maximum yield is 10%).
1-Methyl-2-(4-phenylbutan-2-yl)benzene observed in <1% overall yield (maximum yield is 10%).
While the yield is lower with Co(III)BF4, we attributed the decrease in yield to potential formation of inactive cobalt and/ or nickel catalysts via oxidation or reduction to catalytically inactive species. Nickel degradation could be oxidative (Ni(II) as an off-pathway intermediate), and thereby rescued by reductants such as Mn0 (Table 2, entry 4), whereas cobalt degradation could be reductive (Co(II) as an inactive MHAT catalyst) and rescued by excess NFTPB oxidant added (Table 2, entry 5). One decomposition pathway for the cobalt catalyst could be reduction to an inactive Co(II) species (by nickel(I)) or via disproportionation of two cobalt(III)−hydride species and H2 loss). Excess oxidant may serve to reoxidize Co(II) to the active Co(III) oxidation state. Taken together, there is no essential role of the NFTPB oxidant, except that it is required for precatalyst oxidation when using a Co(II) precatalyst or oxidation of inactive Co(II) formed in the reaction.
Using dtbbpyNi(o-Tol)I (2) as the nickel pre-catalyst in the catalytic reaction with Co(III)BF4, under otherwise standard catalytic conditions, led to a similar yield of hydroarylation product (49%) (Table 2, entry 6). The only products we observed from the “pre-catalyst” were 2-iodotoluene (5% yield) and the o-tolyl hydroarylation product (1-methyl-2-(4-phenylbutan-2-yl)benzene), present in 0.3% yield (theoretical yield is 10%) (Figure 10).50 We did not observe biaryl byproducts (mixed or 2,2′-dimethyl-1,1′-biphenyl), although those products have been invoked in the in situ generation of Ni(0) from similar Ni(II)(aryl)chloride pre-catalysts.51 The low yield of the o-tolyl hydroarylation product was consistent with the stoichiometric experiments (vide supra) in which we ruled out an arylnickel(II) species as the active transmetalating species.
Figure 10.

Low yield of o-tolyl hydroarylation product observed in catalytic reaction when using 2 as the nickel precatalyst.
DISCUSSION
The results of our mechanistic study systematically excluded competing mechanistic hypotheses (Scheme 2) described by (A) a nickel-catalyzed reductive Heck; (B) a radical cage escape mechanism; and (C) a radical chain mechanism analogous to previously explored nickel-catalyzed radical cross-coupling methods in the literature.18–20 At this point, the only mechanistic hypothesis not excluded by data describes a direct alkyl ligand transfer from an organocobalt to the nickel catalyst (Figure 11).
Figure 11.

Dual catalytic cycles of cobalt/nickel-catalyzed branch-selective hydroarylation.52
We hypothesize that the transmetalation proceeds by a “cage rebound” mechanism. Mainly four observations led us to consider that the transmetalation could be described by sequential SET/alkyl ligand transfer steps within the solvent cage, according to eq 1 and eq 2:
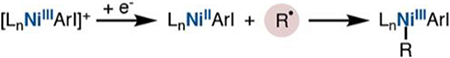 |
(1) |
| (2) |
These observations are as follows: (1) an increase in yield when oxidant was added to the stoichiometric nickel(II) aryl transmetalation experiment (Table 1); (2) the presence of a radical chain dependent on cobalt concentration only, reminiscent of Kochi’s organocobalt radical chain (Figure 8A);42 (3) the ability to use Co(III)BF4 as a suitable cobalt precatalyst with no exogenous oxidant added; and (4) the extremely high rate of carbon radical capture by aryl nickel(II) species, which has a bimolecular rate constant on the order of 108 mol−1 s−1 according to Weix’s radical clock data.53 Any carbon radical formed in close proximity should be captured immediately.
The transmetalation proposed is redox neutral, akin to a conventional polar transmetalation, but is dependent on the ability of both metals to participate in single-electron transfer processes (Figure 12). Both cobalt(III)salen and diimine nickel species are paramagnetic complexes, which exhibit ligand participation in spin delocalization, making a single-electron transfer between both metals plausible.17c,d,54 Electron transfer could be described by oxidation of Co-INT1 by Ni-INT1 to the unstable Co-INT2. Homolysis of the cobalt− carbon of Co-INT2 bond generates Co-INT3 and an alkyl radical that is immediately captured by the resultant Ni-INT2 species. The ensuing Ni-INT3 complex reductively eliminates product and regenerates a nickel(I) species, turning over the catalytic cycle. Co-INT3 may form a cobalt(III) hydride upon reaction with isopropoxy(phenyl)silane to turn over the cobalt cycle.
Figure 12.

Proposed mechanism of direct transmetalation between an alkyl cobalt and aryl nickel species (L = DMPU).
Although the proposed mechanism has no direct mechanistic precedent, the concept of a “cage rebound” mechanism is rooted in the literature. Isolated organocobalt species have been shown to be susceptible to single-electron oxidation by outer-sphere chemical oxidants, followed by subsequent cobalt−carbon bond cleavage.55 A high-valent nickel species, such as Ni-INT1, could be invoked as a suitable outer-sphere oxidant of a metastable organocobalt species. While we cannot directly observe an aryl nickel(III) intermediate under our reaction conditions, analogies to the literature provided a frame of reference from which to consider Ni-INT1 as an oxidant and transmetalating species.
Kochi has implicated transiently formed arylnickel(III) intermediates as reactive paramagnetic species in the nickel-catalyzed biaryl formation from aryl halides. These nickel(III)-(aryl)halide intermediates, accessible via aryl halide oxidative addition across Ni(I), form the requisite diorganonickel(III) species through group transfer of a bridging halide or aryl ligand to an aryl nickel(II) species.56 In these studies, the isolation of a paramagnetic nickel(III) intermediate for stoichiometric studies is precluded by the instability of such a species, as is the case for our system.
Electrochemical oxidations of nickel(II)(aryl)halide complexes result in irreversible oxidation waves ranging from +0.18 to +0.8 V (vs SCE),43d,58 making it difficult to directly compare redox potentials to the reported oxidation potentials of organocobalt(III) complexes in the literature (+0.49 V vs SCE).55f Studies of isolated nickel(III)aryl organometallic complexes require stabilization with electron-rich amine ligands, resulting in quasi-reversible redox Ni(III)/Ni(II) redox waves ranging from onset potentials of −1.1 to −0.40 V (vs Fc/Fc+) in the literature.47,58 Arguably, these electron-rich complexes are harder to reduce than Ni-INT1 due to the electron-donating character of the ligands. Perhaps more informative is looking at the study of (bpy)3Ni(III) inorganic complexes, which are stronger one-electron chemical oxidants (E = 1.6 V vs SCE) than analogous Fe(III) complexes and similar in redox potential to Ce(IV).59 As organocobalt complexes can be chemically oxidized by Fe(III) and Ce(IV) complexes,55 it is conceivable that a nickel(III)(aryl)halide could oxidize an organocobalt(III) intermediate. However, further study and characterization of organometallic complexes similar to our proposed intermediates could help elucidate the feasibility of this proposal.
The aforementioned studies of nickel(III) organometallic complexes highlight the propensity for nickel to undergo single electron transfer reactions without necessarily decomposing via unproductive pathways. In fact, this single electron transfer activity may underlie the catalytic activity of certain nickel-catalyzed reactions. In nickel-catalyzed Kharasch reactions using diaminoarylnickel(II) “pincer” complexes, it is proposed that concomitant electron transfer/halide ligand transfer from a Ni(III) species to a caged alkyl radical generates monomeric atom-transfer radical addition (ATRA) products and regenerates the Ni(II) catalyst.60 Complexes that did not exhibit a reversible Ni(II)/Ni(III) redox potential did not display any catalytic activity.61 In an entirely separate account of an alkyl− aryl Kumada coupling catalyzed by a two-coordinate nickel-(II)-bis(amido) complex, Ni(III) mono alkyl species are proposed to react only as single electron transfer reagents, by oxidation of nickel(I) anionic species generated after reductive elimination from dioorganonickel(III) intermediates, and not participate in any bond-forming or -breaking steps.62 These studies link the ability of nickel(III) organometallic species to act as single electron oxidants in catalytic reactions. While nickel(I) diimine complexes have been evaluated and implicated as single electron reductants in many catalytic reactions,17 the comparative ability for nickel(III) organometallic species to react as single electron oxidants in catalytic reactions warrants greater investigation.
Analogies to more recent cross-coupling literature also implicate formation of Ni-INT1 under our reaction conditions. For example, exclusion of an arylnickel(II) species as a kinetically relevant intermediate strengthens the argument that a Ni(I)/Ni(III) cycle is operative.57,63 In computational studies, Molander and co-workers found that oxidative addition was more energetically favorable across a diimine nickel(I) alkyl species, generated via alkyl radical capture by Ni(0), as compared to the oxidative addition across Ni(0) followed by alkyl radical capture.64 In a different study by Schoenebeck and co-workers, nickel diimine catalysts were shown to favor Ni(I)/Ni(III) catalytic cycles due to the lower calculated barrier of iodoarene oxidative addition to Ni(I) intermediates.65 While the former study evaluates dual nickel/ photoredox catalysis and the latter reaction concerns the trifluoromethylthiolation of aryl halides, the use of similar bipyridine ligands implies a lower barrier in our system for oxidative addition of iodoarenes to a Ni(I) intermediate. This reasoning would be consistent with the higher yields exhibited in the catalytic reaction when using electron-withdrawn aryl iodides, as well as the decrease in yields when using aryl bromides and even more significantly when using aryl chlorides (see the Supporting Information).
Within this mechanistic proposal, an analogy can be drawn to the radical cross-couplings in which an arylnickel(II) species traps an alkyl radical, generating a dioorganonickel(III) species that reductively eliminates coupled product.6,18–20 However, a major distinction lies in that an arylnickel(II) intermediate is not the resting state of the nickel catalyst that accepts a distally generated radical. Instead, generation of the alkyl radical by Ni-INT1 leads to formation of Ni-INT2, which can accept the radical generated by homolysis of the cobalt−carbon bond, all within the solvent cage. Overall, the transmetalation is redox neutral; yet electron transfer is necessary to spur radical formation and subsequent capture at the same nickel center.
We cannot, at this time, definitively rule out a mechanism in which a transmetalation between an organocobalt and nickel(I) species occurs before oxidative addition of the aryl iodide. As this process would also have to be redox neutral, a possible mechanism could involve σ bond metathesis to form an alkylnickel(I) species and reform cobalt(III). However, we can find no analogous transformations in the literature. The effect of oxidant on the stoichiometric transmetalation experiments argues against this inorganic Ni(I) mechanism (Table 1). While studies to replicate stoichiometric transmetalation with an isolable aryl nickel(III) complex would be useful, significant changes to the ligand set would be required to stabilize a Ni(III) complex relevant to the catalytic system at hand.
CONCLUSION
We have demonstrated that the cobalt-/nickel-catalyzed hydroarylation of terminal olefins proceeds via a direct and unprecedented organocobalt to nickel transmetalation. Reaction progress kinetic analysis (RPKA) and variable catalyst concentration radical clock experiments differentiate this transmetalation from the cage-escape/nickel-capture mechanism demonstrated by Riordan to occur with MeCo-(dmgBF2)2L,34a as well as the bimetallic oxidative addition mechanisms elucidated in other nickel-catalyzed radical cross-coupling reactions.18–20 Stoichiometric experiments and removal of exogenous oxidant (NFTPB) from the reaction indicate a net redox-neutral transmetalation. Such a process could be reasonably depicted as a cage-rebound process, in which a single electron transfer from cobalt to nickel and alkyl radical ligand transfer occurs within the solvent cage, by analogy to related complexes.61 To our knowledge, this is the only example of a redox-neutral alkylcobalt to nickel transmetalation. A better understanding of this dual-catalytic system has streamlined Co/Ni-dual catalyzed branch-selective hydroarylation, and we hope will aid in the refinement of related reactions. Furthermore, we hope that an understanding of this novel transmetalation mechanism will provide a template from which to rationally design new catalytic reactions that can only be achieved through dual-catalysis.
Supplementary Material
ACKNOWLEDGEMENTS
Financial support for this work was provided by the NIH (R35 GM122606). We thank Dr. Milan Gembicky, Dr. Curtis Moore, and Professor Arnold L. Rheingold for X-ray crystallographic analysis and ORTEP rendering. We also thank Professor Donna G. Blackmond for helpful discussions and Professor Keary Engle for use of his glovebox. Finally, we thank Steven W. M. Crossley, Dr. Francis Barabé, Samantha A. Green, Jeishla L. M. Matos, Dr. Akiko Yagi, and Dr. Suhelen Vaśquez-Ceśpedes for helpful discussions.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
* The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b06458.
REFERENCES
- (1).Miyaura N; Suzuki A Chem. Rev 1995, 95, 2457–2483. [Google Scholar]
- (2).For reviews on bimetallic or dual catalysis: (a) Pye DR; Mankad NP Chem.Sci 2017, 8, 1705–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Peŕez-Temprano MH; Casares JA; Espinet P Chem.-Eur. J 2012, 18, 1864–1884. [DOI] [PubMed] [Google Scholar]; (c) Hirner JJ; Shi Y; Blum SA Acc. Chem. Res 2011, 44, 603–613. [DOI] [PubMed] [Google Scholar]
- (3).For seminal reports on dual-catalytic transmetalations, see Cu/Pd: (a) Sonogashira K; Tohda Y; Hagihara N Tetrahedron Lett 1975, 16, 4467–4470. [Google Scholar]; (b) Gooßen LJ; Deng G; Levy LM Science 2006, 313, 662–664. Cu/Ni: [DOI] [PubMed] [Google Scholar]; (c) Semba K; Ohtagaki Y; Nakao Y Org. Lett 2016, 18, 3956–3959. Pd/Ni: [DOI] [PubMed] [Google Scholar]; (d) Ackerman LKG; Lovell MM; Weix DJ Nature 2015, 524, 454–457. Rh/Pd: [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sawamura M; Sudoh M; Ito YJ Am. Chem. Soc 1996, 118, 3309–3310. Au/Pd: [Google Scholar]; (f) Shi Y; Roth KE; Ramgren SD; Blum SA J. Am. Chem. Soc. 2009, 131, 18022–18023. [DOI] [PubMed] [Google Scholar]; (g) García-Dominguez P; Nevado CJ Am. Chem. Soc 2016, 138, 3266–3269. Fe/Pd: [DOI] [PubMed] [Google Scholar]; (h) Chen B; Ma S Chem. - Eur. J 2011, 17, 754–757. Ni/Cr: [DOI] [PubMed] [Google Scholar]; (i) Fürstner A; Shi NJ Am. Chem. Soc 1996, 118, 2533–2534. V/Pd: [Google Scholar]; (j) Trost BM; Luan XJ Am. Chem. Soc 2011, 133, 1706–1709. [DOI] [PubMed] [Google Scholar]
- (4).Green SA; Matos JLM; Yagi A; Shenvi RA J. Am. Chem. Soc 2016, 138, 12779–12782. [DOI] [PubMed] [Google Scholar]
- (5).(a) Iwasaki K; Wan KK; Oppedisano A; Crossley SWM; Shenvi RA J. Am. Chem. Soc 2014, 136, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crossley SWM; Martinez RM; Guevera-Zuluaga S; Shenvi RA Org. Lett 2016, 18, 2620–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Obradors C; Martinez RM; Shenvi RA J. Am. Chem. Soc 2016, 138, 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Crossley SWM; Barabe,´ F.; Shenvi, R. A. J. Am. Chem. Soc 2014, 136, 16788–16791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).To the best of our knowledge, this is the first nickel cross-coupling whose radical precursor is an aliphatic alkene as opposed to an alkyl (pseudo)halide, boronate, amide, oxirane, alkylpyridinium, or carboxylate. For examples, see: (a) Zhou J; Fu GC J. Am. Chem. Soc 2003, 125, 14726. [DOI] [PubMed] [Google Scholar]; (b) Zhao Y; Weix DJ J. Am. Chem. Soc 2014, 136, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tellis JC; Primer DN; Molander GA Science 2014, 345, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Science, 2014, 345, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ackerman LKG; Anka-Lufford LL; Naodovic M; Weix DJ Chem. Sci 2015, 6, 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhao Y; Weix DJ J. Am. Chem. Soc 2015, 137, 3237. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DW C. Science 2016, 352, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS Science 2016, 352, 801. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Huihui KMM; Caputo JA; Melchor Z; Olivares AM; Spiewak AM; Johnson KA; DiBenedetto TA; Kim S; Ackerman LKG; Weix DJ J. Am. Chem. Soc 2016, 138, 5016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Basch CH; Liao J; Xu J; Piane JJ; Watson MP J. Am. Chem. Soc 2017, 139, 5313–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Simandi L; Nagy F Magy. Kem. Foly 1965, 71, 6. [Google Scholar]; (b) Simandi L; Nagy F Acta Chim. Acad. Sci. Hung 1965, 46, 137. [Google Scholar]
- (8).Kwiatek J Catal. Rev.: Sci. Eng 1967, 1, 37. [Google Scholar]
- (9).Jackman LM; Hamilton JA; Lawlor JM J. Am. Chem. Soc 1968, 90, 1914. [Google Scholar]
- (10).Sweany RL; Halpern JJ Am. Chem. Soc 1977, 99, 8335–8337. [Google Scholar]
- (11).(a) Hartung J; Pulling ME; Smith DM; Yang DX Tetrahedron 2008, 64, 11822–11830. [Google Scholar]; (b) Tang L; Papish ET; Abramo GP; Norton JR; Baik M-H; Friesner RA; Rappe AJ Am. Chem. Soc 2003, 125, 10093–10102. [DOI] [PubMed] [Google Scholar]; (c) Estes DP; Grills DC; Norton JR J. Am. Chem. Soc 2014, 136, 17362–17365. [DOI] [PubMed] [Google Scholar]; (d) Choi J; Tang L; Norton JR J. Am. Chem. Soc 2012, 129, 234–240. [DOI] [PubMed] [Google Scholar]; (e) Hartung J; Norton JR Catalysis Without Precious Metals 2010, 1–24. [Google Scholar]; (f) Li G; Han A; Pulling ME; Estes DP; Norton JR J. Am. Chem. Soc 2012, 134, 14662–14665. [DOI] [PubMed] [Google Scholar]; (g) Li G; Kuo JL; Han A; Abuyan JM; Young LC; Norton JR; Palmer JH J. Am. Chem. Soc 2016, 138, 7698–7704. [DOI] [PubMed] [Google Scholar]
- (12).Crossley SWM; Obradors C; Martinez RM; Shenvi RA Chem. Rev 2016, 116, 8912–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Eisenberg DC; Norton JR Isr. J. Chem 1991, 31, 55–66. [Google Scholar]
- (14).A similar mechanism was invoked in (a) ref 6e [Google Scholar]; (b) Hofstra JL; Cherney AH; Ordner CM; Reisman SE J. Am. Chem. Soc 2018, 140, 139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).To account for an 80% yield with only 0.5 equiv of oxidant relative to starting material, we hypothesized that the oxidant could act as a two-electron SET oxidant, or Co(II), a known O2 carrier, could bring molecular oxygen into the reaction.
- (16).Similar mechanism posited in: Komeyama K; Ohata R; Shinnosuke K; Osaka I Chem. Commun 2017, 53, 6401–6404. [DOI] [PubMed] [Google Scholar]
- (17).For seminal reports on radical coupling in which a Ni(I) species is implicated as a reductant in catalytic cross-coupling. With alkyl halides: (a) Wilsily A; Tramutola F; Owston NA; Fu GC J. Am. Chem. Soc 2012, 134, 5794–5797. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zultanski SL; Fu GC J. Am. Chem. Soc 2013, 135, 624–627. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jones GD; McFarland C; Anderson TJ; Vicic DA Chem. Commun 2005, 4211–4213. [DOI] [PubMed] [Google Scholar]; (d) Jones GD; Martin JL; McFarland C; Allen OR; Hall RE; Haley AD; Brandon RJ; Konovalova T; Desrochers PJ; Pulay P; Vicic DA J. Am. Chem. Soc 2006, 128, 13175–13183. With redox-active esters [DOI] [PubMed] [Google Scholar]; (e) Cornella J; Edwards JT; Qin T; Kawamura S; Wang J; Pan C-M; Gianatassio R; Schmidt M; Eastgate MD; Baran PS J. Am. Chem. Soc 2016, 138, 2174–2177. With alkylpyridinium salts [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Basch CH; Liao J; Xu J; Piane JJ; Watson MP J. Am. Chem. Soc 2017, 139, 5313–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Biswas S; Weix DJ J. Am. Chem. Soc 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Breitenfeld J; Ruiz J; Wodrich MD; Hu XJ Am. Chem. Soc 2013, 135, 12004–12012. [DOI] [PubMed] [Google Scholar]
- (20).Schley ND; Fu GC J. Am. Chem. Soc 2014, 136, 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Blackmond DG Angew. Chem., Int. Ed 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]
- (22).Bureś J Angew. Chem., Int. Ed 2016, 55, 2028–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).The orders for nickel and cobalt were also evaluated by initial rates analysis to be 1.0 and 0.8, respectively. See the Supporting Information for more details.
- (24) (a).Shigehisa H; Aoki T; Yamaguchi S; Shimizu N; Hiroya K J. Am. Chem. Soc 2013, 135, 10306–10309. [DOI] [PubMed] [Google Scholar]; (b) Shigehisa H; Ano T; Honma H; Ebisawa K; Hiroya K Org. Lett 2016, 18, 3622–3625. [DOI] [PubMed] [Google Scholar]; (c) Shigehisa H; Nishi E; Fujisawa M; Hiroya K Org. Lett 2013, 15, 5158–5161. [DOI] [PubMed] [Google Scholar]
- (25).For explanations on the reactivity of the oxidant (NFTPB), see: (a) Kiselyov AS Chem. Soc. Rev 2005, 34, 1031–1037. [DOI] [PubMed] [Google Scholar]; (b) Oliver EW; Evans DJ J. Electroanal. Chem 1999, 474, 1–8. [Google Scholar]; (c) Adachi K; Takahashi I; Harada M; Umemoto TJ Fluorine Chem 2016, 183, 100–111. [Google Scholar]
- (26).Tokuyasu T; Kunikawa S; Masayuma A; Nojima M Org. Lett 2002, 4, 3595–3598. [DOI] [PubMed] [Google Scholar]
- (27) (a).Waser J; Gaspar B; Nambu H; Carreira EM J. Am. Chem. Soc 2006, 128, 11693–11712. [DOI] [PubMed] [Google Scholar]; (b) Gaspar B; Carreira EM Angew. Chem., Int. Ed 2008, 47, 5758–5760. [DOI] [PubMed] [Google Scholar]; (c) Gaspar B; Carreira EM J. Am. Chem. Soc 2009, 131, 13214–13215. [DOI] [PubMed] [Google Scholar]
- (28).Li S; de Bruin B; Peng C-H; Fryd M; Wayland BB J. Am. Chem. Soc 2008, 130, 13373–13381. [DOI] [PubMed] [Google Scholar]
- (29) (a).Schrauzer GN; Grate JH J. Am. Chem. Soc 1981, 103, 541–546. [Google Scholar]; (b) Tsou T-T; Loots M; Halpern JJ Am. Chem. Soc 1982, 104, 623–624. [Google Scholar]; (c) Ng FTT; Rempel GL; Mancuso C; Halpern J Organometallics 1990, 9, 2762–2772. [Google Scholar]
- (30) (a).Costa G; Mestroni G; Stefani L J. Organomet. Chem 1967, 7, 493–501. [Google Scholar]; (b) Costa G; Mestroni G; Pellizer GJ Organomet. Chem 1968, 11, 333–340. [Google Scholar]; (c) McAllister RM; Weber JH J. Organomet. Chem 1974, 77, 91–105. [Google Scholar]; (d) Li G; Zhang FF; Chen H; Yin HF; Chen HL; Zhang SYJ Chem. Soc., Dalton Trans 2002, 105–110. [Google Scholar]
- (31).The reported 1H NMR spectrum of n-C2H5Co(salen) assigned diastereotopic−CH3 peaks at δ −0.20 and −0.33 ppm. See ref 30c and the Supporting Information for more details.
- (32).Ragsdale SW Crit. Rev. Biochem. Mol. Biol 2004, 39, 165. [DOI] [PubMed] [Google Scholar]
- (33) (a).Darnault C; Volbeda A; Kim EJ; Legrand P; Vernede X; Lindahl PA; Fontecilla-Camps JC Nat. Struct. Mol. Biol 2003, 10, 271. [DOI] [PubMed] [Google Scholar]; (b) A copper-containing heterometallic cluster has also been described: Doukov TI; Iverson T; Seravalli J; Ragsdale SW; Drennan CL Science 2002, 298, 567. [DOI] [PubMed] [Google Scholar]
- (34).For Ni(I) studies: (a) Ram MS; Riordan CG; Yap GPA; Liable-Sands L; Rheingold AL; Marchaj A; Norton JR J. Am. Chem. Soc 1997, 119, 1648. [Google Scholar]; For Ni(0) studies: (b) Eckert NA; Dougherty WG; Yap GPA; Riordan CG J. Am. Chem. Soc 2007, 129, 9286. [DOI] [PubMed] [Google Scholar]
- (35).Organocobalt transmetalation to PdII salts: Vol’pin ME; Volkova LG; Levitin IY; Boronina NN; Yurkevich AM J. Chem. Soc. D 1971, 894–850. [Google Scholar]; (a) To CrII: Bakac A; Espenson JHJ Am. Chem. Soc 1984, 106, 5197–5202. [Google Scholar]; (b) Espenson JH; Sellers TD Jr J. Am. Chem. Soc 1974, 96, 94–97. [Google Scholar]
- (36).These radical clock substrates contain C−X bonds as alkyl radical precursors, whereas an alkene serves as the radical precursor in this study. (a) Kinney RJ; Jones WD; Bergman RG J. Am. Chem. Soc 1978, 100, 635–637. [Google Scholar]; (b) Kinney RJ; Jones WD; Bergman RG J. Am. Chem. Soc 1978, 100, 7902–7915. [Google Scholar]; (c) Ash CE; Hurd PW; Darensbourg MY; Newcomb MJ Am. Chem. Soc 1987, 109, 3313–3317. [Google Scholar]; (d) Peŕalez E; Neǵrel J-C; Chanon M Tetrahedron 1995, 51, 12601–12610. [Google Scholar]; (e) Newcomb M Tetrahedron 1993, 49, 1151–1176. [Google Scholar]
- (37).For calibration of a similar secondary alkyl radical clock generated from an alkyl halide: (a) Lusztyk J; Maillard B; Deycard S; Lindsay A; Ingold KU J. Org. Chem 1987, 52, 3509. [Google Scholar]
- (38).For a proposed mechanism in which Ni inserts into remote olefins to produce a cyclized substrate: Hegedus LS; Thompson DHP J. Am. Chem. Soc 1985, 107, 5663–5669. [Google Scholar]; Photoinduced C−N Ullman coupling shown to proceed via a radical pathway due to the appearance of diastereomers: Creutz SE; Lotito KJ; Fu GC; Peters JC Science 2012, 338, 647–651. [DOI] [PubMed] [Google Scholar]
- (39).A small slope would also result if acyclic products arose from a nickel-only pathway. However, such reductive Heck mechanisms generate linear, not branched, products. Such a pathway is not supported by our data.
- (40).Garr CD; Finke RG Inorg. Chem 1993, 32, 4414–4421. [Google Scholar]
- (41).Stolzenberg AM; Cao YJ Am. Chem. Soc 2001, 123, 9078–9090. [DOI] [PubMed] [Google Scholar]
- (42).Samsel EG; Kochi JK J. Am. Chem. Soc 1986, 108, 4790–4804. [Google Scholar]
- (43).For mechanistic studies utilizing these intermediates, see (a) refs 18 and 20. [Google Scholar]; (b) Primer DN; Molander GA J. Am. Chem. Soc 2017, 139, 9847–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shields BJ; Doyle AGJ Am. Chem. Soc 2016, 138, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]; For synthesis and properties of similar complexes: (d) Klein A; Kaiser A; Wielandt W; Belaj F; Wendel E; Bertagnolli H; Zaĺis S Inorg. Chem 2008, 47, 11324–11333. [DOI] [PubMed] [Google Scholar]
- (44).For determination of Co−C BDE: (a) Li G; Zhang FF; Chen H; Yin HF; Chen HL; Zhang SY J. Chem. Soc., Dalton Trans 2002, 105–110. [Google Scholar]; (b) Synthesis of salenCoR: Costa G J. Organomet. Chem 1967, 7, 493–501. [Google Scholar]; Identification of (salophen) Co(i-Pr) instability: (c) McAllister RM; Weber JH J. Organomet. Chem 1974, 77, 91–105. [Google Scholar]
- (45).In mechanistic studies that propose a radical chain mechanism, radical initiation in stoichiometric experiments using aryl nickel(II) species is proposed to stem from the formation of small amounts of low-valent nickel. See refs 18 and 20.
- (46) (a).Higgs AT; Zinn PJ; Sanford MS Organometallics 2010, 29, 5446–5449. [Google Scholar]; (b) Camasso NM; Sanford MS Science 2015, 347, 1218–1220. [DOI] [PubMed] [Google Scholar]
- (47) (a).Camasso NM; Canty AJ; Ariafard A; Sanford MS Organometallics 2017, 36, 4382–4393. [Google Scholar]; (b) Zheng B; Tang F; Luo J; Schultz JW; Rath NP; Mirica LM J. Am. Chem. Soc 2014, 136, 6499–6504. [DOI] [PubMed] [Google Scholar]; (c) Schultz JW; Fuchigama K; Zheng B; Rath NP; Mirica LM J. Am. Chem. Soc 2016, 138, 12929–12934. [DOI] [PubMed] [Google Scholar]
- (48).Hayashida T; Kondo H; Terasawa J; Kirchner K; Sunada Y; Nagashima HJ Organomet. Chem 2007, 692, 382–394. [Google Scholar]
- (49).See the Supporting Information.
- (50).A competition experiment between 4-iodobenzotrifluoride (1.0 equiv) and 2-iodotoluene (0.1 equiv) led to no observable o-tolyl hydroarylation product. The yield of the o-tolyl hydroarylation product from 2-iodotoluene (1.0 equiv) in the catalytic reaction is 25% (see Supporting Information).
- (51) (a).Standley EA; Jamison TF J. Am. Chem. Soc 2013, 135, 1585–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shields JD; Gray EE; Doyle AG Org. Lett 2015, 17, 2166–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Oxidative addition could be reversible: (a) Tsou TT; Kochi JK J. Am. Chem. Soc 1979, 101, 7547–7560. [Google Scholar]; (b) See ref 63f. [Google Scholar]
- (53).In the aforementioned radical clock studies by Weix (ref 18), the 5-hexenyl iodide radical clock has an intramolecular cyclization rate constant of k = 2.3 × 105 s−1; yet when the concentration of nickel is 50 mM, the ratio of unrearranged to rearranged product is 22 to 1. If we assume that the concentration of LnNiIIArI (catalyst resting state) is approximately 50 mM, then the rate constant of the alkyl radical trapped by nickel is around k = 1.0 × 108 mol−1 s−1.
- (54).Evaluation of LCo(III)/L•Co(II) character: (a) Kochem A; Kanso H; Baptiste B; Arora H; Philouze C; Jarjayes O; Vezin H; Luneau D; Orio M; Thomas F Inorg. Chem 2012, 51, 10557–10571. [DOI] [PubMed] [Google Scholar]; (b) Kurahashi T; Fujii H Inorg. Chem 2013, 52, 3908–3919. [DOI] [PubMed] [Google Scholar]; Ligand-radical delocalization in pyridine(diimine) cobalt hydrogenation catalysts: Chirik PJ Acc. Chem. Res 2015, 48, 1687–1695. [DOI] [PubMed] [Google Scholar]; Redox-active nickel α-diimine hydrosilylation catalysts: Pappas I; Treacy S; Chirik PJ ACS Catal. 2016, 6, 4105–4109 [Google Scholar]; Nickel(I) terpyridine complexes display ligand radical character: see refs 17c and 17d. [Google Scholar]
- (55).For oxidation of (salen)CoR by ferrocenium salts, see ref 42. Oxidation by (dmgH)2CoR by [IrCl6]2−: (a) Topich J; Halpern, J. Inorg. Chem 1979, 18, 1339–1343. [Google Scholar]; (b) Abley P; Dockal ER; Halpern JJ Am. Chem. Soc. 1972, 94, 659–660. [DOI] [PubMed] [Google Scholar]; (c) Anderson SN; Ballard DH; Chrastowski JZ; Dodd D; Johnson MDJ Chem. Soc., Chem. Commun 1972, 685–686. [Google Scholar]; Oxidation of (dmgH)2CoR by Ce(IV): (d) Halpern J; Chan MS; Hanson J; Roche TS; Topich JA J. Am. Chem. Soc 1975, 97, 1606–1608. [Google Scholar]; (e) Magnuson RY; Halpern JJ Chem. Soc., Chem. Commun 1978, 44–46. [Google Scholar]; Electrochemical oxidation of CoSalen-alkyl complexes: (f) Levitin I; Sigan AL; Vol’pin ME J. Chem. Soc., Chem. Commun 1975, 469–470. [Google Scholar]; (g) Costa G; Puxeddu A; Reisenhofer EJ Chem. Soc., Dalton Trans 1972, 1519–1523. [Google Scholar]; Oxidation of (TPP)CoR(pyr) by Fe3+ salts: (h) Fukuzumi S; Miyamoto K; Suenobu T; Caemelbecke EV; Kadish KM J. Am. Chem. Soc 1998, 120, 2880–2889. [Google Scholar]; The nature of LCoIV−carbon bond cleavage, heterolysis or homolysis, is dependent on the nature of the cobalt and alkyl ligands: (i) Vol’pin ME; Levitin IY; Sigan AL Inorg. Chim. Acta 1980, 41, 271–277. [Google Scholar]
- (56) (a).Kochi JK Pure Appl. Chem 1980, 52, 571–605. [Google Scholar]; (b) A similar mechanism was also proposed in the allylation of organic halides, catalyzed by nickel: Hegedus LS; Thompson DHP J. Am. Chem. Soc 1985, 107, 5663–5669. [Google Scholar]
- (57).Zhang C-P; Wang H; Klein A; Biewer C; Stirnat K; Yamaguchi Y; Xu L; Gomez-Benitez V; Vicic DA J. Am. Chem. Soc 2013, 135, 8141–8144. [DOI] [PubMed] [Google Scholar]
- (58) (a).Grove DM; van Koten G; Mul P; Zoet R; van der Linden JGM; Legters J; Schmitz JEJ; Murrall NW; Welch AJ Inorg. Chem 1988, 27, 2466–2413. [Google Scholar]; (b) The oxidation of the isolated Ni(II) bis(imido) methyl complex was reversible: Lipschutz MI; Yang X; Chatterjee R; Tilley TD J. Am. Chem. Soc 2013, 135, 15298–1530. [DOI] [PubMed] [Google Scholar]
- (59).Brodovitch JC; Haines RI; McAuley A Can. J. Chem 1981, 59, 1610–1614. [Google Scholar]
- (60).Gossage RA; van de Kuil LA; van Koten G Acc. Chem. Res 1998, 31, 423–431. [Google Scholar]
- (61).Stohl M; Snelders DJM; Godbole MD; Havenith RWA; Haddleton D; Clarkson G; Lutz M; Spek AL; van Klink GPM; van Koten G Organometallics 2007, 26, 3985–3994. [Google Scholar]
- (62).Lipshultz MI; Tilley TD Angew. Chem., Int. Ed 2014, 53, 7290–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63) (a).Lin C-Y; Power PP Chem. Soc. Rev. 2017, 46, 5347–5399. [DOI] [PubMed] [Google Scholar]; (b) Hu X Chem. Sci. 2011, 2, 1867–1886. [Google Scholar]; (c) Anderson TJ; Jones GD; Vicic DA J. Am. Chem. Soc. 2004, 126, 8100–8101. [DOI] [PubMed] [Google Scholar]; (d) Bakac A; Espenson JH J. Am. Chem. Soc. 1986, 108, 719–723. [DOI] [PubMed] [Google Scholar]; (e) Cornella J; Goḿez-Bengoa E; Martin R J. Am. Chem. Soc 2013, 135, 1997–2009. [DOI] [PubMed] [Google Scholar]; (f) Lin X; Sun J; Xi Y; Lin D Organometallics 2011, 30, 3284–3292. [Google Scholar]; (g) Zhang K; Conda-Sheridan M; Cooke SR; Louie J Organometallics 2011, 30, 2546–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Lin X; Phillips DL J. Org. Chem 2008, 73, 3680–3688. [DOI] [PubMed] [Google Scholar]; (i) Zultanski SL; Fu GC J. Am. Chem. Soc 2011, 133, 15362–15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kalvet I; Guo Q; Tizzard GJ; Schoenebeck F ACS Catal 2017, 7, 2126–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.