

Abstract
Complex interplay between cardiac tissues is crucial for their integrity. The flow responsive transcription factor KLF2, which is expressed in the endocardium, is vital for cardiovascular development but its exact role remains to be defined. To this end, we mutated both klf2 paralogues in zebrafish, and while single mutants exhibit no obvious phenotype, double mutants display a novel phenotype of cardiomyocyte extrusion towards the abluminal side. This extrusion requires cardiac contractility and correlates with the mislocalization of N-cadherin from the lateral to the apical side of cardiomyocytes. Transgenic rescue data show that klf2 expression in endothelium, but not myocardium, prevents this cardiomyocyte extrusion phenotype. Transcriptome analysis of klf2 mutant hearts reveals that Fgf signaling is affected, and accordingly, we find that inhibition of Fgf signaling in wild-type animals can lead to abluminal cardiomyocyte extrusion. These studies provide new insights into how Klf2 regulates cardiovascular development and specifically myocardial wall integrity.
Research organism: Zebrafish
Introduction
The embryonic heart must undergo a series of complex cellular and molecular processes to form a mature pumping organ (Auman and Yelon, 2004; Bakkers et al., 2009; Staudt and Stainier, 2012; Milgrom-Hoffman et al., 2014; Dietrich et al., 2014; Staudt et al., 2014; Wilsbacher and McNally, 2016; Del Monte-Nieto et al., 2018; Miao et al., 2018). Defects in any of these processes can lead to congenital heart malformations, which have estimated occurrences ranging from 4 to 50 cases per 1000 live births, and represent the most prevalent category of congenital anomalies (Hoffman and Kaplan, 2002).
Cardiac development and function strongly rely on myocardial wall integrity, which itself is modulated by cardiac tissue interactions as well as connections between cardiomyocytes (Wu et al., 2009; Tian and Morrisey, 2012; Drechsler et al., 2013; Sojka et al., 2014). Several studies have investigated how cardiac tissues including the endocardium, myocardium and epicardium communicate with each other to generate a correctly patterned and functioning organ (Lee et al., 1995; Stainier et al., 1996; de la Pompa and Epstein, 2012; Ruiz-Villalba and Pérez-Pomares, 2012; Bressan et al., 2014; Wang et al., 2015; Rasouli and Stainier, 2017; Del Monte-Nieto et al., 2018). While many signaling pathways including Notch (Rones et al., 2000; High et al., 2009; D'Amato et al., 2016), Wnt (Marvin et al., 2001; Schneider and Mercola, 2001), Retinoic acid (Stainier and Fishman, 1992; Keegan et al., 2005), Hedgehog (Washington Smoak et al., 2005; Wang et al., 2015), Neuregulin/ErbB2 (Lee et al., 1995; Meyer and Birchmeier, 1995; Crone et al., 2002; Lai et al., 2010; Rasouli and Stainier, 2017), FGF (Lavine et al., 2005), and Angiopoietin-1/Tie2 (Tachibana et al., 2005; Kim et al., 2018) have been implicated in cardiac development, it is not clear how they interact to orchestrate this process, or how genes affected by cardiac function modulate cardiac formation.
Shear stress generated by blood flow can regulate hemodynamic responses that are vital for cardiovascular morphogenesis including blood vessel maturation (Ehling et al., 2013; Nakajima and Mochizuki, 2017a; Nakajima et al., 2017b), atrioventricular valve leaflet formation (Slough et al., 2008; Vermot et al., 2009; Heckel et al., 2015; Steed et al., 2016a; Goddard et al., 2017), and trabeculation (Peshkovsky et al., 2011; Staudt et al., 2014; Samsa et al., 2015; Rasouli and Stainier, 2017). However, the cellular and molecular mechanisms underlying this regulation are poorly understood.
Krüppel-like factors (KLFs) are transcriptional regulators that modulate cell growth and differentiation as well as tissue development. Some members of the KLF family, including KLF2, 4, 6, 10 and 15, have been implicated in cardiovascular development and function (Lee et al., 2006; McConnell and Yang, 2010; Nayak et al., 2011; Dietrich et al., 2014; Renz et al., 2015). Klf2, a flow responsive gene, is a well-known member of this family, and it is expressed in endothelial cells and lymphocytes (Dekker, 2002; Cao et al., 2010; Wang et al., 2010). Klf2 -/- mice die between E12.5 and E14.5 due to hemorrhaging and cardiovascular failure (Lee et al., 2006; Wu et al., 2008). Since loss of Klf2 function in mouse results in embryonic lethality, a detailed understanding of its role in cardiovascular development is still lacking. In order to complement studies in mouse and gain additional insights into the role of klf2 in cardiovascular development, we used the zebrafish (Danio rerio), a model organism ideally suited for developmental genetic studies, especially those of the cardiovascular system (Stainier and Fishman, 1994; Stainier et al., 1996; Beis et al., 2005; Chi et al., 2008; Staudt and Stainier, 2012; Collins and Stainier, 2016).
Zebrafish klf2a and klf2b are known to be collectively orthologous to mammalian Klf2 (Oates et al., 2001). Endothelial klf2a expression has been shown to be flow-dependent (Parmar et al., 2006), and its knockdown by antisense morpholinos was reported to cause pericardial edema, dysfunctional cardiac valves (Vermot et al., 2009) and defective haematopoietic stem cell development (Wang et al., 2011). However, unlike the aforementioned klf2a morpholino-induced cardiovascular defects, no obvious morphological phenotypes were observed in klf2ash317 mutant embryos (Novodvorsky et al., 2015), suggesting possible compensation (Rossi et al., 2015) by klf2b and/or other genes.
Taking a loss-of-function approach, we find that Klf2 is essential for myocardial wall integrity as loss of both klf2a and klf2b leads to a novel phenotype of cardiomyocyte extrusion to the abluminal side. Using high-resolution microscopy, we show that this extrusion is correlated with the mislocalization of N-Cadherin in klf2 mutant cardiomyocytes. Taking a tissue-specific rescue approach, we find that endothelial Klf2 can maintain myocardial wall integrity and rescue klf2 mutants to adulthood. Transcriptome analysis of klf2 WT and mutant hearts led us to analyze Fgf signaling and we found that inhibition of Fgf signaling in wild-type (WT) animals could recapitulate the cardiomyocyte extrusion phenotype observed in klf2 mutants. Overall, these data provide new insights into how an endocardially expressed gene can modulate myocardial wall integrity, and therefore further our knowledge about the complex endocardial-myocardial interactions underlying myocardial wall maturation.
Results
klf2 mutants exhibit a cardiomyocyte extrusion phenotype
Given the role of Klf2 in cardiovascular development in mouse (Lee et al., 2006), we used the Transcription Activator-Like Effector Nuclease (TALEN) technology (Cermak et al., 2011) to mutate both klf2 paralogues in zebrafish (Figure 1A–B and Figure 1—figure supplement 1A–B), and identified a klf2a Δ10 allele (bns11) and a klf2b Δ8 allele (bns12) (Kwon et al., 2016). Notably, the predicted truncated Klf2a bns11 protein lacks the transrepression and zinc finger domains (Figure 1A), and the predicted Klf2b bns12 protein contains only part of the transactivation domain (Figure 1B). Surprisingly, unlike the klf2a morphants which have been reported to exhibit pericardial edema and cardiac defects (Vermot et al., 2009), neither klf2abns11 nor klf2bbns12 mutants exhibit gross morphological defects (Figure 1—figure supplement 2A–C). Close examination revealed no obvious cardiovascular defects in klf2a or klf2b single mutants (Figure 1—figure supplement 2D–P) which can survive to become fertile adults. Quantitative PCR (qPCR) analysis of klf2a, klf2b and klf4a mRNA levels shows that klf2a transcript levels are significantly reduced in klf2abns11 mutants compared to their WT siblings, suggesting nonsense-mediated decay. In addition, the levels of klf2b and klf4a mRNA are increased in klf2abns11 mutants compared to their WT siblings (Figure 1—figure supplement 2Q), indicating that they could be compensating for the loss of Klf2a function. Similarly, klf2a and klf4a mRNA levels are increased in klf2bbns12 mutants compared to their WT siblings (Figure 1—figure supplement 2R). We thus decided to generate klf2abns11; klf2bbns12 double mutants (hereafter referred to as klf2 mutants). Unlike the single mutants, most double mutants exhibit pericardial edema by 96 hours post fertilization (hpf) (Figure 1C–D and Figure 1—figure supplement 3A). Abnormal atrioventricular (AV) canal elongation was observed at 48 hpf in approximately 10% of klf2 mutants (3/26). By 80 hpf, AV valve formation was clearly impaired in approximately 50% of klf2 mutants (17/36) as the canal appeared more elongated than in WT, and the multi-layering process leading to leaflet formation was also compromised in these animals (Figure 1—figure supplement 3B–E), consistent with previous observations in klf2a morphants (Vermot et al., 2009). Although the vascular network appeared unaffected in klf2 mutants, most of them died by 2 weeks post-fertilization and none survived to adulthood (Figure 1—figure supplement 3F–H). Upon closer examination of their hearts at 96 hpf, we found that approximately 90% of klf2 mutants exhibit a previously unreported phenotype of cardiomyocyte extrusion toward the abluminal side (69/75 mutant hearts) (Figure 1E–J). This phenotype first appears around 82 hpf in approximately 15% of klf2 mutants (11/67 mutant hearts), and the number of extruding cardiomyocytes, which are mostly located in the outer curvature of the ventricle, is variable and increases over time (Figure 1—figure supplement 4A–B). We also found that the size and circularity of klf2 mutant cardiomyocytes was significantly affected starting at 96 hpf (Figure 1—figure supplement 4C–D), possibly suggesting that the abnormally shaped cardiomyocytes represent an intermediate population on their way to extrusion.
Figure 1. klf2 mutants exhibit a cardiomyocyte extrusion phenotype.
(A–B) Partial sequence alignment of klf2abns11 (A) and klf2bbns12 (B) alleles with WT and schematics of their predicted protein products. (C–D) Representative brightfield images of a WT (C) and a klf2abns11/bns11; klf2bbns12/bns12 double mutant (hereafter referred to as klf2 mutant) at 96 hpf (D); lateral views, anterior to the left; arrowhead points to pericardial edema. (E–F’) Maximum intensity projections of 96 hpf klf2 WT (E–E’) and mutant (F–F’) hearts; (G–H’) Two-dimensional (2D) confocal images of 96 hpf klf2 WT (G–G’) and mutant (H–H’) hearts; ventricular outer curvature (dashed boxes) in (E, F, G and H) magnified in (E’, F’, G’ and H’), respectively. (I–J) Three-dimensional reconstructions from confocal images of 96 hpf klf2 mutant ventricular wall. Arrows point to extruding cardiomyocytes; V: ventricle; At: atrium; scale bars: 0.5 mm (C–D), 50 µm (E–H), 20 µm (I–J).

Figure 1—figure supplement 1. Identifying the klf2abns11 and klf2bbns12 mutant alleles in the absence or presence of the klf2-p2A-tdTomato transgene.

Figure 1—figure supplement 2. Single klf2a or klf2b mutants do not exhibit any obvious phenotypes.

Figure 1—figure supplement 3. Characterizing additional cardiac phenotypes in klf2 mutants.

Figure 1—figure supplement 4. Additional quantification of the cardiomyocyte extrusion phenotype in klf2 mutants.

Cardiac contractility is required for cardiomyocyte extrusion
Cardiac contractility/blood flow is vital for cardiac morphogenesis and function (Sehnert et al., 2002; Berdougo et al., 2003; Peshkovsky et al., 2011; Samsa et al., 2015; Collins and Stainier, 2016; Rasouli and Stainier, 2017). We found that around 60 percent of klf2 mutants exhibit impaired cardiac contractility at 96 hpf. In order to test whether the cardiomyocyte extrusion phenotype was secondary to impaired contractility, we first checked whether it could be induced in WT animals by modulating contractility with amhc morpholinos (MOs) which completely inhibit atrial contractility and cause secondary effects on ventricular contractility (Berdougo et al., 2003), as well as tnnt2a MOs which block both atrial and ventricular contractility (Sehnert et al., 2002). We observed that impaired contractility could not induce cardiomyocyte extrusion in WT animals (Figure 2—figure supplement 1), indicating that this phenotype is not secondary to impaired contractility. However, injecting tnnt2a MOs into klf2 mutants at the one-cell stage blocked cardiomyocyte extrusion (Figure 2A). In addition, short-term inhibition of cardiac contractility by BDM treatment from 100 to 102 hpf could also block cardiomyocyte extrusion (4/5 hearts) (Figure 2B). Overall, these data indicate that cardiac contractility is required for cardiomyocyte extrusion in klf2 mutants, and that this extrusion is reversible.
Figure 2. Cardiac contractility is required for cardiomyocyte extrusion.
(A) Maximum intensity projections of confocal z-stacks of 96 hpf klf2 WT and mutant hearts, non-injected or injected with tnnt2a MO at the one-cell stage; part of ventricular outer curvature (dashed boxes) magnified on the right side of each panel. (B) Time-lapse 2D confocal images of a klf2 mutant heart during BDM treatment. Arrows point to an extruding cardiomyocyte returning to the compact layer upon inhibiting contraction; V: ventricle; At: atrium; scale bars: 50 µm (A), 10 µm (B).

Figure 2—figure supplement 1. Cardiomyocyte extrusion from the compact layer is not caused by impaired contractility.

Cardiomyocyte extrusion correlates with N-cadherin mislocalization but not with cardiomyocyte death or proliferation
Cell death, crowding and loss of cell adhesion molecules have been implicated in epithelial cell extrusion (Rosenblatt et al., 2001; Semenza, 2008; Chien et al., 2008; Eisenhoffer et al., 2012; Marinari et al., 2012; Kuipers et al., 2014). In order to investigate the cause of the cardiomyocyte extrusion phenotype in klf2 mutants, we first performed Acridine Orange staining and found that the extruding cardiomyocytes in klf2 mutants did not appear to be dying (Figure 3A–D). To investigate cardiomyocyte proliferation, we used the Tg(myl7:mVenus-gmnn) line (Jiménez-Amilburu et al., 2016; Uribe et al., 2018) which allows one to visualize actively proliferating cardiomyocytes in green, and found that while the rate of atrial cardiomyocyte proliferation appeared unchanged in klf2 mutants compared to their WT siblings, the levels of ventricular cardiomyocyte proliferation were considerably reduced (Figure 3E–I), suggesting that crowding is not the cause of the cardiomyocyte extrusion phenotype in klf2 mutants. These observations are consistent with recent reports that inhibition of cardiomyocyte proliferation in zebrafish can reduce the total number of cardiomyocytes while not leading to their extrusion (Liu et al., 2010; Uribe et al., 2018).
Figure 3. Cardiomyocyte extrusion correlates with N-cadherin mislocalization but not with cardiomyocyte death or proliferation.
(A–D) 2D (mid-sagittal sections) (A and C) and maximum intensity projections of confocal z-stacks (B and D) of 82 hpf klf2 WT (A–B) and mutant (C–D) hearts stained with Acridine Orange to visualize cell death; arrows point to extruding cardiomyocytes. (E–H) Confocal images of 96 hpf klf2 WT (E and G) and mutant (F and H) hearts to visualize cardiomyocyte proliferation. (I) Number of mVenus-gmnn positive ventricular and atrial cardiomyocytes in 96 hpf klf2 WT and mutant hearts; dots represent individual hearts; values represent means ±SEM; ***p≤0.001, ns (not significant), by Student’s t-test. (J–K’’) Mid-sagittal confocal sections of 96 hpf klf2 WT (J) and mutant (K) hearts. Higher magnification images of the outer curvature of the ventricular wall (white dashed boxes) in (J) and K) are shown in (J’), (J’’), (K’) and (K’’); arrows point to ectopic accumulation of Cdh2-EGFP proteins on the apical side of cardiomyocytes. (L–M’) 2D confocal views of 96 hpf klf2 WT (L) and mutant (M) hearts. Magnified images of dashed boxes in (L) and (M) are shown in (L’) and (M’), respectively. Arrows point to mislocalized Cdh2-GFP on the apical side of cardiomyocytes; V: ventricle, At: atrium; scale bars, 50 µm.

Figure 3—figure supplement 1. Downregulation of cdh2 leads to cardiomyocyte extrusion.

Figure 3—figure supplement 2. The extruding cardiomyocytes appear polarized in the apicobasal axis.

Figure 3—figure supplement 3. Reduction of the cardiac jelly is not obviously affected in klf2 mutants.

Figure 3—figure supplement 4. The epicardium is affected in klf2 mutants.

It has been reported that N-cadherin (Cdh2), an adhesion molecule, is highly enriched in mouse and zebrafish cardiomyocytes (Kostetskii et al., 2001; Bagatto et al., 2006; Cherian et al., 2016). We thus examined the distribution of Cdh2-EGFP molecules using the TgBAC(cdh2:cdh2-EGFP) line (Revenu et al., 2014), and found that while they are highly enriched on the lateral sides of WT cardiomyocytes at 96 hpf, they have become mislocalized from the lateral to the apical side of cardiomyocytes in klf2 mutants (Figure 3J–M’), suggesting that cardiomyocyte adhesion is affected. In addition, knockdown of N-cadherin by injecting cdh2 MO at the one-cell stage can also cause a cardiomyocyte extrusion phenotype in WT hearts (Figure 3—figure supplement 1). Overall, these data suggest that cardiomyocyte extrusion correlates with the mislocalization of N-cadherin on their membranes but not with cardiomyocyte cell death or proliferation.
The extruding cardiomyocytes appear polarized in the apicobasal axis
It has recently been shown that cardiomyocytes in the compact layer are polarized in their apicobasal axis whereas during the trabeculation process, they lose this polarity as they delaminate (Jiménez-Amilburu et al., 2016). In order to further investigate the extruding cardiomyocytes, we checked their apicobasal polarity by generating a myocardial-specific apical transgenic line, Tg(−0.2myl7:tdtomato-podxl), in which the apical protein podocalyxin is fused with tdTomato, and found that at 96 hpf, extruding cardiomyocytes do not lose their apical polarity (Figure 3—figure supplement 2).
Reduction of the cardiac jelly is not obviously affected in klf2 mutants
Reduction of the cardiac jelly, a gelatinous matrix between the endocardial and myocardial layers, is an important process during cardiac morphogenesis (Brutsaert et al., 1996; Stankunas et al., 2008; Bowers and Baudino, 2010; Tian and Morrisey, 2012; Rasouli and Stainier, 2017; Grassini et al., 2018). In order to investigate whether reduction of the cardiac jelly was affected in klf2 mutants, we used the TgBAC(etv2:EGFP) (Proulx et al., 2010) and Tg(myl7:mCherry-CAAX) (Uribe et al., 2018) lines to mark the endocardium and myocardium in green and red, respectively. Utilizing high-resolution confocal microscopy at 96 hpf, we observed that there was no significant difference in terms of cardiac jelly thickness between klf2 WT and mutant animals (Figure 3—figure supplement 3).
The epicardium is affected in klf2 mutants
The epicardium, the outer layer of vertebrate hearts, is known to be involved in the nourishment of the underlying myocardium (Peralta et al., 2014), the formation of coronaries (Ruiz-Villalba and Pérez-Pomares, 2012; González-Rosa et al., 2012), and tissue regeneration after cardiac injury (Schlueter and Brand, 2012; González-Rosa et al., 2012; Wang et al., 2015). In order to investigate the behavior of epicardial cells during early zebrafish heart development, we used high-resolution microscopy to image hearts in Tg(myl7:mCherry-CAAX);TgBAC(tcf21:NLS-EGFP) (Wang et al., 2015) animals, in which myocardial and epicardial cells are labeled in red and green, respectively. At 55 hpf, we observed a few tcf21-positive cells attaching to the abluminal side of the ventricle, which was fully covered by 96 hpf (Figure 3—figure supplement 4A–H), in agreement with previous studies (Virágh and Challice, 1981; Peralta et al., 2013; Peralta et al., 2014; Wang et al., 2015). At 96 hpf, we observed fewer tcf21-positive cells covering the klf2 mutant hearts compared to their WT siblings, and also a sparser distribution (Figure 3—figure supplement 4I–M), suggesting defective epicardial-myocardial interactions in the absence of Klf2 function. In order to investigate whether incomplete epicardial coverage could contribute to the cardiomyocyte extrusion phenotype observed in klf2 mutants, we used a genetic ablation approach to remove epicardial cells in Tg(myl7:EGFP-Has.HRAS) (D'Amico et al., 2007); TgBAC(tcf21:mCherry-NTR) (Wang et al., 2015) animals using metronidazole (MTZ) treatment from 48 to 96 hpf. We found that this epicardial ablation approach could also induce cardiomyocyte extrusion; however, the number of extruding cardiomyocytes after epicardial ablation was far lower than that observed in klf2 mutants (Figure 3—figure supplement 4N–R).
Klf2 functions cell non-autonomously to maintain the integrity of the myocardial wall
To investigate where Klf2 function is required to maintain the integrity of the myocardial wall, we generated chimeric hearts by cell transplantation and examined the behavior of WT cardiomyocytes in klf2 mutant hearts, and vice versa (Figure 4). We first transplanted Tg(myl7:MKATE-CAAX) WT cells into Tg(myl7:LIFEACT-GFP); klf2+/?or klf2-/- hosts. Using confocal microscopy, we found that at 96 hpf some WT donor-derived cardiomyocytes were extruding from klf2-/- but not klf2+/? hearts (Figure 4A–E; n = 7 hearts). We also performed the converse experiment by transplanting Tg(myl7:LIFEACT-GFP); klf2+/?or klf2-/- cells into WT hosts and observed no extruding klf2-/- cardiomyocytes (Figure 4F–J; n = 9 hearts). These data indicate that Klf2 function is not required in cardiomyocytes to maintain the integrity of the myocardial wall. klf2b overexpression in endothelial cells can rescue the klf2 mutant cardiomyocyte extrusion phenotype
Figure 4. Klf2 functions cell non-autonomously to maintain the integrity of the myocardial wall.

(A) Schematic representation of the experiment shown in (B–E). (B–E) Transplantation of Tg(myl7:MKATE-CAAX); klf2+/+ donor cells into Tg(myl7:LIFEACT-GFP); klf2+/? (B and D) or klf2-/- (C and E) hosts shown at 96 hpf; white arrows point to klf2-/- extruding cardiomyocytes in klf2-/- heart, orange arrows point to klf2+/+ extruding cardiomyocytes in klf2-/- hearts; maximum intensity projections of confocal z-stacks of hearts in (B) and (C) are shown in (D) and (E), respectively. (F) Schematic representation of the experiment shown in (G-J). (G–J) Transplantation of Tg(myl7:LIFEACT-GFP); klf2+/? (G and I) or klf2-/- (H and J) donor cells into Tg(myl7:MKATE-CAAX); klf2+/+ hosts shown at 96 hpf; maximum intensity projections of confocal z-stacks of hearts in (G) and (H) are shown in (I) and (J), respectively. V: ventricle, At: atrium; scale bars, 50 µm.
In zebrafish, as in mouse, klf2 is expressed in the endocardium (Lee et al., 2006; Vermot et al., 2009). Similar to klf2a, klf2b also becomes expressed in the zebrafish heart by 36 hpf (Figure 5—figure supplement 1). To investigate where Klf2 function is required to prevent the cardiomyocyte extrusion phenotype observed in klf2 mutants (Figure 5A–B and and E–F), we generated a Tg(fli1a:klf2b-p2A-tdTomato) line to specifically overexpress klf2b in all endothelial cells including the endocardium (Figure 5C–D and and G–H). We also generated Tg(myl7:klf2b-p2A-tdTomato) and Tg(myl7:klf2a-p2A-tdTomato) lines to overexpress klf2a and klf2b in cardiomyocytes (Figure 5I–P and Figure 1—figure supplement 1C–D). Unlike endothelial klf2b overexpression, myocardial klf2a and klf2b overexpression caused pericardial edema and impaired trabeculation in about forty percent of the F1 and F2 animals when observed at 96 hpf. These cardiac defects became milder beyond the F2 generation. Notably, we observed that endothelial, but not myocardial, klf2b overexpression rescued the pericardial edema as well as the cardiomyocyte extrusion phenotype observed in klf2 mutant hearts (Figure 5A–P), indicating the importance of endothelial/endocardial klf2 expression in maintaining myocardial wall integrity. We also quantified the number of proliferative cardiomyocytes in klf2 WT and mutant animals overexpressing klf2b in their endothelium and found that the reduction of ventricular cardiomyocyte proliferation observed in klf2 mutants was rescued by endothelial klf2b overexpression (Figure 5—figure supplement 2). Interestingly, endothelial klf2b overexpression also rescued the epicardial defects observed in klf2 mutants (Figure 5—figure supplement 3). Overall, these data indicate that endothelial/endocardial klf2 is required for myocardial wall integrity by regulating endocardial-myocardial-epicardial interactions. Although all klf2 mutant larvae die, we were able to rescue almost sixty percent of them to become adults using this endothelial rescue approach (Figure 5—figure supplement 4A–B). Dissecting the adult hearts, we did not observe any significant differences between klf2 WT and rescued mutant hearts, further suggesting the importance of endothelial klf2 expression in maintaining myocardial wall integrity (Figure 5Q–R’ and Figure 5—figure supplement 4C–F).
Figure 5. klf2b overexpression in endothelial cells can rescue the klf2 mutant cardiomyocyte extrusion phenotype.
(A–P) Endothelial- and myocardial-specific overexpression of klf2a or klf2b in klf2 WT and mutant hearts. Endothelial overexpression of klf2b (C–D, G–H). Myocardial overexpression of klf2a (K–L, O–P) or klf2b (I–J, M–N); maximum intensity projections of hearts in (A–D) and (I–L) are shown in (E–H) and (M–P), respectively. (Q–R’) Immunostaining of adult klf2 WT (Q–Q’) and rescued mutant (R–R’) heart sections for Caveolin1 to label epicardial cells and phalloidin for overall myocardial structure; magnified images of dashed boxes in (Q) and (R) are shown in (Q’) and (R’), respectively; arrows point to extruding cardiomyocytes; V: ventricle, At: atrium; scale bars: 50 µm (A–P), 300 µm (Q–R’).

Figure 5—figure supplement 1. klf2b expression at early developmental stages .

Figure 5—figure supplement 2. The cardiomyocyte proliferation defect in klf2 mutants is rescued by endothelial klf2b overexpression.

Figure 5—figure supplement 3. The epicardial defect in klf2 mutants is rescued by endothelial klf2b overexpression.

Figure 5—figure supplement 4. klf2 mutants rescued by endothelial klf2b overexpression can survive to adulthood.

Increased Retinoic Acid signaling or inhibition of Hedgehog signaling does not cause a cardiomyocyte extrusion phenotype
To further understand how endothelial cells regulate myocardial wall integrity, we performed microarray analysis of 96 hpf klf2 WT and mutant hearts. Our transcriptome data show that aldh1a2 and cyp26b1, the highest expressing cyp26 gene in 96 hpf hearts, are respectively up- and down-regulated in klf2 mutant hearts (Figure 6—source data 1), suggesting an increase in retinoic acid (RA) levels. It has recently been reported that excess levels of RA in cyp26a1 and cyp26c1 morphants lead to the failure of second heart field progenitors to join the outflow tract as well as the extrusion of first heart field-derived cardiomyocytes from the ventricle at 48 hpf (Rydeen and Waxman, 2016). In order to test whether increased RA signaling causes cardiomyocyte extrusion at later stages, we treated WT zebrafish larvae with RA from 74 to 96 hpf but did not observe any extruding cardiomyocytes (Figure 6—figure supplement 1). The later onset of RA treatment used in our experiments likely explains the different outcome from that observed by Rydeen and Waxman, 2016. We also found that Hedgehog (Hh) signaling was downregulated in klf2 mutant hearts (Figure 6—figure supplement 2, Supplementary file 2 and Figure 6—source data 2); however, inhibition of this pathway in WT zebrafish from 75 to 96 hpf using Cyclopamine, a Hh signaling pathway inhibitor, did not cause cardiomyocyte extrusion (Figure 6—figure supplement 3). Together, these data indicate that by itself increased RA signaling or inhibition of Hh signaling does not cause the cardiomyocyte extrusion phenotype observed in klf2 mutants.
Inhibition of Fgfr signaling can lead to cardiomyocyte extrusion in WT animals
Fgf signaling has been implicated in the formation of the compact myocardial layer as well as in cardiomyocyte differentiation and proliferation (Reifers et al., 2000; Lavine et al., 2005; Marques et al., 2008; Tirosh-Finkel et al., 2010; Pradhan et al., 2017). Transcriptomic analysis of klf2 WT and mutant hearts shows that Fgf signaling is affected in klf2 mutants (Figure 6—figure supplement 2, Figure 6—source data 3 and Supplementary file 2); follow up qPCR analysis shows that several fgf receptor and ligand genes are expressed at reduced levels in klf2 mutant compared to WT hearts (Figure 6—figure supplement 4). To test the role of Fgf signaling in myocardial wall integrity, we treated Tg(myl7:EGFP-Has.HRAS) larvae from 75 to 96 hpf with SU5402, a FGF receptor inhibitor, and observed cardiomyocyte extrusion (7/9 hearts; Figure 6A–D), as well as mislocalization of Cdh2-EGFP from the lateral to the apical side of cardiomyocytes (10/11 hearts; Figure 6—figure supplement 5). In addition, global overexpression of dominant negative Fgfr1, using the Tg(hsp70:dn-fgfr1-EGFP) line (Lee et al., 2005), could also cause cardiomyocyte extrusion (9/13 hearts; Figure 6E–H); however, the number of extruding cardiomyocytes in these animals was less than that observed in klf2 mutants or SU5402 treated animals (Figure 6—figure supplement 6A). While, this cardiomyocyte extrusion phenotype appeared in only a few SU5402 treated hearts by 82 hpf (7/36 hearts), the number of extruding cardiomyocytes increased over time, as in klf2 mutants (Figure 6—figure supplement 6B–D). These data indicate that inhibition of Fgfr signaling can cause cardiomyocyte extrusion, as in klf2 mutants. We also treated WT and klf2a+/-; klf2b-/- larvae with DMSO or increasing concentrations of SU5402 and found that klf2a+/-; klf2b-/- animals were more sensitive to Fgfr signaling inhibition, that is more prone to exhibit cardiomyocyte extrusion compared to WT (Figure 6I). We further treated WT larvae with SU5402 from 75 to 96 hpf and then stained their hearts with a pERK antibody. Our data show that Fgfr inhibition reduced pERK immunostaining (Figure 6—figure supplement 7), in agreement with previous reports (Shinya et al., 2001; Calmont et al., 2006). We also checked pERK immunostaining after dnfgfr1 overexpression and observed a reduction, although milder than that observed in SU5402-treated hearts (Figure 6—figure supplement 7). We further found that pERK immunostaining was significantly reduced in endocardial cells of klf2 mutants compared to WT (Figure 6J and K), and that it was rescued by endothelial klf2b overexpression (Figure 6L and M). Overall, these data suggest that Fgf signaling is required downstream of Klf2 to maintain myocardial wall integrity. By examining single-cell RNA sequencing data of early zebrafish embryos (https://kleintools.hms.harvard.edu/paper_websites/wagner_zebrafish_timecourse2018/mainpage.html; Wagner et al., 2018), we found that most fgf receptor genes and many fgf ligand genes are expressed in the zebrafish heart (Figure 6—figure supplement 8). Because the level of expression of most of these genes is rather low, and possibly challenging to detect by in situ hybridization, we performed qPCR analysis in cloche/npas4l mutant hearts which lack all endocardial cells (Stainier et al., 1995; Reischauer et al., 2016). As expected, myl7 expression levels were unaffected in npas4l mutant hearts compared to WT while fli1a levels were significantly reduced (Figure 6—figure supplement 9A). Expression levels of all fgf receptor genes were slightly increased in npas4l mutants (Figure 6—figure supplement 9B), suggesting that they are not expressed in the endocardium. Analyzing Fgf ligand genes which are clearly downregulated in klf2 mutants, we observed a reduction in fgf14 expression in npas4l mutant hearts (Figure 6—figure supplement 9C), suggesting its endocardial enrichment. We also observed an upregulation of fgf3 expression in npas4l mutant hearts (Figure 6—figure supplement 9C), suggesting that it is not expressed in the endocardium. Additional studies will be required to identify and analyze the precise role of the fgf receptor and ligand genes that potentially function downstream of Klf2 to maintain myocardial wall integrity. Transcriptomic analysis of klf2 WT and mutant hearts also indicates that genes associated with adherens junctions and focal adhesions are affected in klf2 mutants, further suggesting the importance of Klf2 in myocardial wall integrity (Figure 6—figure supplement 2). Overall, our data suggest that endocardial klf2 regulates myocardial wall integrity at least in part by modulating myocardial Fgfr signaling.
Figure 6. Inhibition of Fgfr signaling can lead to cardiomyocyte extrusion in WT animals.
(A–D) Confocal images of 96 hpf hearts; WT animals treated with DMSO as a control or FGFR inhibitor (SU5402) from 75 to 96 hpf; maximum intensity projections of hearts in (A) and (D) are shown in (C) and (D) respectively; arrows point to extruding cardiomyocytes. (E–H) 75 hpf Tg(myl7: mCherry-CAAX) (E–F) or Tg(hsp70:dn-fgfr1-EGFP);Tg(myl7: mCherry-CAAX) (G–H) animals were heat-stressed at 39°C for 1 hr (F and H) and their hearts imaged at 96 hpf; arrow in (H) points to an extruding cardiomyocyte (n = 9/13 hearts). (I) klf2a+/-; klf2b-/- animals are more likely than WT siblings to exhibit cardiomyocyte extrusion upon Fgfr inhibition; number of treated larvae for each condition is shown above the individual columns. (J–K) Hearts of 96 hpf Tg(myl7: mCherry-CAAX); klf2 +/+ or klf2 -/- animals immunostained for pERK. (L–M) Hearts of 96 hpf Tg(fli1a:klf2b-p2A-tdTomato);Tg(myl7:EGFP-Hsa.HRAS); klf2 +/+ or klf2 -/- animals immunostained for pERK. Arrows and arrowheads point to extruding cardiomyocytes and pERK positive endocardial cells, respectively; V: ventricle, At: atrium; scale bars, 50 µm.

Figure 6—figure supplement 1. Increased Retinoic Acid signaling does not cause cardiomyocyte extrusion.

Figure 6—figure supplement 2. Broad GSEA enrichment plots of selected down-regulated gene sets.

Figure 6—figure supplement 3. Inhibition of Hedgehog signaling does not cause cardiomyocyte extrusion.

Figure 6—figure supplement 4. mRNA levels of fgf ligand and receptor genes in WT and klf2 mutant hearts.
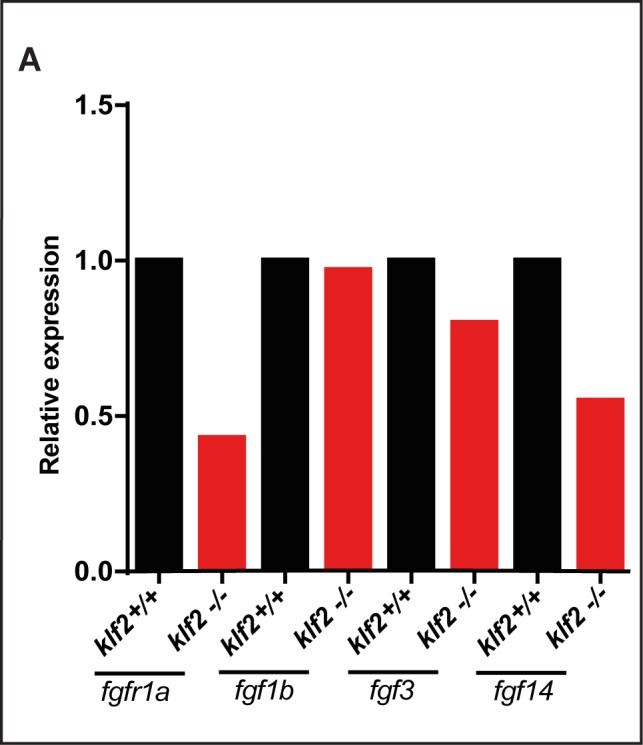
Figure 6—figure supplement 5. Inhibition of Fgfr signaling can lead to Cdh2-GFP mislocalization in cardiomyocytes.

Figure 6—figure supplement 6. Additional quantification of the cardiomyocyte extrusion phenotype upon Fgfr inhibition.

Figure 6—figure supplement 7. Fgf signaling is required for ERK phosphorylation in endocardial cells.

Figure 6—figure supplement 8. Single cell graphs of fgf receptor and ligand genes expressed in zebrafish embryonic endothelium and heart.

Figure 6—figure supplement 9. mRNA levels of fgf ligand and receptor genes in WT and npas4l mutant hearts.

Discussion
KLF2, a well-known molecular transducer of fluid shear stress, is vital for cardiovascular development (Lee et al., 2006; Wu et al., 2008; Goddard et al., 2017). Given the embryonic lethality of mouse Klf2 mutants (Lee et al., 2006; Wu et al., 2008), we mutated both klf2a and klf2b genes in zebrafish. Although klf2a knockdown by antisense morpholino has been reported to cause impaired valvulogenesis (Vermot et al., 2009), single klf2abns11 or klf2bbns12 mutants did not exhibit any gross morphological or cardiovascular defects, consistent with previous observations of klf2ash317 mutants and klf2b morphants (Novodvorsky et al., 2015). In contrast to these findings, a recent study reported that around 80% of 96 hpf klf2aig4 mutants exhibit a range of valvular defects, although milder than those in klf2a morphants (Vermot et al., 2009; Steed et al., 2016b). Additional analyses will be required to examine the potential difference between the various klf2a mutant alleles. Along these lines, since klf2abns11 and/or klf2bbns12 might be hypomorphic alleles, we further examined these loci. If the first ATG downstream of the klf2a bns11 or klf2b bns12 deletion was used to reinitiate translation, a protein lacking the transactivation domain and part of the transrepression domain would be made, and thus such a protein would be lacking at least some of its critical domains. We also found a significant reduction in klf2a mRNA levels in klf2abns11 mutants compared to WT, indicating mRNA decay. klf2b and klf4a mRNA levels are increased in klf2abns11 mutants compared to WT, suggesting that they could be compensating for the loss of Klf2a function (Rossi et al., 2015). Altogether, these data suggest that klf2abns11 and klf2bbns12 are strong alleles, although it might be informative to generate promoter-less alleles to further study the role of these genes (El-Brolosy et al., 2018).
Interestingly, we found that klf2 double mutants exhibit a cardiomyocyte extrusion phenotype towards the abluminal side and that this phenotype was dependent on cardiac contractility. Although cardiomyocytes are not considered classical epithelial cells, they display several epithelial features including morphology, polarity, (Liu et al., 2010; Staudt et al., 2014; Jiménez-Amilburu et al., 2016), and N-cadherin distribution (Cherian et al., 2016). Cell extrusion is a crucial process that epithelial layers use to maintain barrier properties (Marinari et al., 2012; Kuipers et al., 2014). This process can be induced by cell death, crowding or loss of cell adhesion molecules (Rosenblatt et al., 2001; Semenza, 2008; Eisenhoffer et al., 2012; Marinari et al., 2012; Kuipers et al., 2014). Using high-resolution microscopy, we show that, despite the maintenance of apicobasal polarity, cardiomyocyte extrusion in klf2 mutants correlates with the mislocalization of N-cadherin from the lateral to the apical side of cardiomyocytes. We also observed cardiomyocyte extrusion in cdh2 morphants, further suggesting the importance of N-cadherin in myocardial wall integrity. These findings are consistent with studies where N-cadherin-negative cardiomyocytes become rounded and loosely aggregated in zebrafish (Bagatto et al., 2006), and extruded from chimeric hearts in mouse (Kostetskii et al., 2001). A noticeable reduction in ventricular wall thickness was observed in global and endothelial-specific Klf2 mouse mutants, albeit no significant changes in cardiomyocyte proliferation or death were reported (Lee et al., 2006). Therefore, it will be interesting to test whether similar cardiomyocyte extrusion or mislocalization of N-cadherin also occurs in mouse Klf2 mutants. KLF2 has also been implicated in maintaining endothelial barrier integrity, as KLF2 loss- or gain-of-function respectively enhances or prevents endothelial leakage in HUVECs upon thrombin treatment (Lin et al., 2010). Thus, it will also be interesting to investigate how Klf2 modulates endocardial cell adhesion and barrier integrity.
Cardiac jelly remodeling has been reported to be involved in the maturation of vertebrate hearts (Nakamura and Manasek, 1981; Ramasubramanian et al., 2013; Rasouli and Stainier, 2017; Grassini et al., 2018; Del Monte-Nieto et al., 2018). Although cardiac jelly thickness appears unaffected in klf2 mutants, our study does not exclude the potential role of ECM components in the appearance of the extruding cardiomyocytes. In addition, we observed that the epicardium is affected in klf2 mutant hearts. Detachment of the epicardium from the myocardial surface was also reported in Klf2 mutant hearts in mouse (Lee et al., 2006). Therefore, it will be interesting to investigate how the lack of Klf2 function results in impaired epicardial coverage in both species. One possibility is that Klf2 cell-autonomously affects the behavior of epicardial cells, or it could be an indirect effect resulting from the morphological changes of the myocardial wall as the epicardial defect in klf2 mutants was rescued by endothelial/endocardial klf2b overexpression, or a combination of both. New tools are required to test these hypotheses in zebrafish.
Taking cell transplantation and gain-of-function approaches, we found that Klf2 cell non-autonomously maintains the integrity of the myocardial wall as endothelial, but not myocardial, klf2b overexpression can rescue the extruding cardiomyocyte phenotype in klf2 mutants, and allow them to survive to adulthood. We also tried to generate an endothelial specific klf2a overexpression transgenic line but were unable to do so as the injected fish did not survive. One possibility is that klf2a overexpression in endothelial cells leads to their dysfunction or death.
Transcriptome analysis of klf2 mutant hearts revealed changes in multiple signaling pathways including RA, Hh and Fgf. While increasing RA signaling or decreasing Hh signaling did not cause cardiomyocyte extrusion, decreasing Fgf signaling did. Notably, Fgfr inhibition also led to the mislocalization of N-cadherin from the lateral to the apical side of cardiomyocytes. Interestingly, it has been reported that inhibition of Fgf signaling in mouse embryos, by mosaic overexpression of Sprouty2, causes a cell-shedding phenotype in the hepatic epithelium (Calmont et al., 2006). Transcriptome analysis of klf2 mutant hearts also indicated adherens junction and focal adhesion defects. Fgf signaling can have cardioprotective effects by modulating the phosphorylation of the gap junction protein connexin 43 (Srisakuldee et al., 2006), which itself can interact with N-cadherin in cell-cell adhesion (Wei et al., 2005). Interestingly, we also found a downregulation in klf2 mutant hearts of cdon, which is highly expressed in early zebrafish hearts based on single-cell sequencing data (Wagner et al., 2018) and encodes a cell adhesion molecule implicated in myoblast and neuronal differentiation via binding to N-cadherin (Lu and Krauss, 2010); Cdon-mediated N-cadherin localization has also been reported to be crucial in neural crest cell migration (Powell et al., 2015). Cdon expression is induced by KLF2 in vitro (Boon, 2008). Thus, it will be interesting to investigate whether Klf2 regulates N-cadherin localization by modulating cdon expression. We further found that egr1, a flow responsive transcription factor gene involved in FGF-dependent angiogenesis during neo-vascularization and tumor growth (Khachigian et al., 1997; Khachigian, 2006), is downregulated in klf2 mutant hearts. Therefore, it will also be interesting to investigate whether klf2 modulates Fgf signaling by regulating egr1 expression. Altogether, our data suggest that both Klf2 and Fgf signaling are required to maintain myocardial wall integrity, and it will be important to further investigate how endothelial Klf2, and Fgf signaling, modulate this process. Since cardiomyocyte extrusion is a complex phenotype, it is likely that additional signaling pathways function downstream of Klf2 to maintain myocardial wall integrity.
In summary, this study reveals a novel function for endothelial Klf2 in regulating cardiomyocyte behavior, which in turn modulates myocardial wall integrity. It will be important to further investigate endocardial-myocardial-epicardial interactions which are vital for cardiac wall maturation.
Materials and methods
Key resources table.
| Reagent type (species) or resource |
Designation | Source or reference |
Identifiers | Additional information |
|---|---|---|---|---|
| Genetic reagent (Danio rerio) |
klf2abns11 | (Kwon et al., 2016) | ||
| Genetic reagent (Danio rerio) |
klf2bbns12 | (Kwon et al., 2016) | ||
| Genetic reagent (Danio rerio) |
Tg(myl7:
LIFEACT- GFP)s974 |
(Reischauer et al., 2014) | ZFIN ID: ZDB-ALT-150203–1 |
|
| Genetic reagent (Danio rerio) |
Tg(myl7:
mCherry- CAAX)bns7 |
(Uribe et al., 2018) | ZFIN ID: ZDB-ALT-181102–5 |
|
| Genetic reagent (Danio rerio) |
Tg(myl7-
MKATE- CAAX)sd11 |
(Lin et al., 2012) | ZFIN ID: ZDB-ALT-120320–1 |
|
| Genetic reagent (Danio rerio) |
Tg(myl7:EGFP-
Hsa.HRAS)s883 |
(D'Amico et al., 2007) | ZFIN ID: ZDB-ALT-070309–1 |
|
| Genetic reagent (Danio rerio) |
TgBAC
(etv2-EGFP) ci1 |
(Proulx et al., 2010) | ZFIN ID: ZDB-ALT-110131–53 |
|
| Genetic reagent (Danio rerio) |
Tg(−0.8myl7:
nlsDsRed Express)hsc4 |
(Takeuchi et al., 2011) | ZFIN ID: ZDB-ALT-110222–3 |
|
| Genetic reagent (Danio rerio) |
Tg(myl7:
mVenus- gmnn)ncv43 |
(Jiménez-Amilburu et al., 2016) | ZFIN ID: ZDB-ALT-170131–3 |
|
| Genetic reagent (Danio rerio) |
TgBAC(cdh2:cdh2-
EGFP,crybb1: ECFP)zf517 |
(Revenu et al., 2014) | ZFIN ID: ZDB-ALT-141218–5 |
|
| Genetic reagent (Danio rerio) |
Tg(hsp70:dn-
fgfr1-EGFP)pd1 |
(Lee et al., 2005) | ||
| Genetic reagent (Danio rerio) |
Tg(fli1a:klf2b-
p2a-td Tomato)bns235 |
This paper | Details in Materials and methods |
|
| Genetic reagent (Danio rerio) |
Tg(myl7:klf2a-
p2a-td Tomato)bns200 |
This paper | Details in Materials and methods |
|
| Genetic reagent (Danio rerio) |
Tg(myl7:klf2b-
p2a-td Tomato)bns234 |
This paper | Details in Materials and methods |
|
| Genetic reagent (Danio rerio) |
Tg(−0.2myl7:
tdTomato- podxl)bns197 |
This paper | Details in Materials and methods |
|
| Genetic reagent (Danio rerio) |
TgBAC(tcf21:
NLS-EGFP)pd41 |
(Wang et al., 2015) | ZFIN ID: ZDB-ALT-110914–2 |
|
| Genetic reagent (Danio rerio) |
TgBAC(tcf21:
mCherry- NTR) pd108 |
(Wang et al., 2015) | ZFIN ID: ZDB-ALT-150904–1 |
|
| Sequence-based reagent |
tnnt2a
morpholino |
(Sehnert et al., 2002) | ZFIN ID: ZDB- MRPHLNO-060317–4 |
|
| Sequence-based reagent |
amhc
morpholino |
(Berdougo et al., 2003) | ZFIN ID: ZDB- MRPHLNO-061110–1 |
|
| Sequence-based reagent |
cdh2
morpholino |
(Lele et al., 2002) | ZFIN ID: ZDB- MRPHLNO-060815–1 |
|
| Commercial assay or kit |
RNA Clean and Concentrator kit |
Zymo Research | ||
| Software | ZEN Blue 2012 | Zeiss, Germany | ||
| Software | ZEN Black 2012 | Zeiss, Germany |
Zebrafish lines and constructs
Zebrafish were raised and maintained under standard conditions (Westerfield, 2000). All animal experiments were done according to German Animal Protection Laws approved by the local governmental animal protection committee.
Zebrafish mutant and transgenic lines
We used the following mutant and transgenic lines: klf2abns11 and klf2bbns12 (Kwon et al., 2016), Tg(myl7:LIFEACT-GFP)s974 (Reischauer et al., 2014), Tg(myl7:mCherry-CAAX)bns7 (Uribe et al., 2018),Tg(myl7-MKATE-CAAX)sd11 (Lin et al., 2012), Tg(myl7:EGFP-Hsa.HRAS)s883 (D'Amico et al., 2007), TgBAC(etv2-EGFP)ci1 (Proulx et al., 2010), Tg(−0.8myl7:nlsDsRedExpress)hsc4 (Takeuchi et al., 2011), Tg(myl7:mVenus-gmnn)ncv43 (Jiménez-Amilburu et al., 2016), TgBAC(cdh2:cdh2-EGFP,crybb1:ECFP)zf517 (Revenu et al., 2014), Tg(hsp70:dn-fgfr1-EGFP)pd1 (Lee et al., 2005), Tg(fli1a:klf2b-p2a-tdTomato)bns235, Tg(myl7:klf2a-p2a-tdTomato)bns200, Tg(myl7:klf2b-p2a-tdTomato)bns234, Tg(−0.2myl7:tdTomato-podxl)bns197, TgBAC(tcf21:NLS-EGFP)pd41 and TgBAC(tcf21:mCherry-NTR) pd108 (Wang et al., 2015).
Generation of klf2abns11 and klf2bbns12 mutants klf2abns11 and klf2bbns12 mutants were generated by TALEN mutagenesis targeting exon 2. The following TALEN arms were constructed and assembled using the Golden Gate method (Cermak et al., 2011).
For klf2abns11:
TALEN arm1: NN NI HD NI HD HD NG NI HD NG NN HD HD NI NI HD HD NN NG HD NG
TALEN arm2: NN NN NN NN NI NI NI NN HD NI NN NN HD HD NG NN NI HD NG
For klf2bbns12:
TALEN arm1: NN NN HD NI HD NG NN NI NI HD NI HD NI NN NI HD NI HD
TALEN arm2: NG NN HD NG NN NI NN NI NG HD HD NG HD NN NG HD NI NG HD HD NG
100 pg of total TALEN RNA and 50 pg of GFP RNA (used to monitor injection efficiency) were co-injected into the cell at the one-cell stage. The klf2abns11 and klf2bbns12 alleles were genotyped using high-resolution melt analysis (HRMA) with an Eco Real-Time PCR System (Illumina). The HRMA primers are listed in Supplementary file 1.
Generation of transgenic lines
To generate a myocardial specific klf2a or klf2b overexpressing construct, the coding sequence of klf2a or klf2b fused with a self-cleaving peptide 2A and tdTomato, was cloned in a mini tol2 plasmid harbouring a myl7 promoter using the Cold Fusion Cloning Kit (System Bioscience; MC101A-1-SB). We used the same cloning approach to generate an endothelial-specific klf2b overexpression line by putting klf2b-p2a-tdTomato under control of a fli1a promoter.
To generate the Tg(−0.2myl7:tdTomato-podxl) line, we constructed −0.2myl7:tdTomato-podxl by tagging the signal peptide of zebrafish podocalyxin with tdTomato and then cloned it into a Tol2 enabled vector with the −0.2myl7 promoter using the Cold Fusion Cloning Kit.
A total of 15 pg of each aforementioned DNA construct was co-injected with 10 ng of Tol2 RNA into the cell at the one-cell stage. To establish the Tg(fli1a:klf2b-p2a-tdTomato)bns235, Tg(myl7:klf2a-p2a-tdTomato)bns200, Tg(myl7:klf2b-p2a-tdTomato)bns234, and Tg(−0.2myl7:tdTomato-podxl)bns197 lines, injected larvae (F0) were screened for tdTomato fluorescence and raised to adulthood.
Quantitative PCR analysis
qPCR for cardiac expression of fgf ligand and receptor genes was performed in a CFX Connect Real-Time System (Biorad). RNA was isolated using TRIzol (Life Technologies) followed by phenol-chloroform extraction. In brief, 25 75 hpf hearts were lysed and homogenized in TRIzol using the NextAdvance Bullet Blender Homogenizer (Scientific instrument services). Chloroform was then added and phase separation was obtained following vortexing and centrifugation. The top (aqueous) phase (containing RNA) was isolated and purified using a Clean and Concentrator kit (Zymo). Purified RNA was then used for reverse transcription using the Maxima First Strand cDNA synthesis kit (Thermo). All reactions were performed in at least technical duplicates and the results represent biological triplicates. Gene expression values were normalized using the zebrafish rpl13 gene. The qPCR primers are listed in Supplementary file 1. Ct and dCt values are listed in Supplementary file 3. qPCR analysis for global expression of klf2a, klf2b, and klf4a was performed on cDNA obtained from 24 hpf klf2a WT and mutants and 72 hpf klf2b WT and mutants. RNA was extracted using a RNeasy Micro kit (QIAGEN). Gene expression values were normalized using the zebrafish 18S ribosomal RNA and gapdh genes. All reactions were performed in three technical replicates, and the results represent three independent biological samples (25 embryos were pooled for each sample). The qPCR primers are listed in Supplementary file 1. Ct and dCt values are listed in Supplementary file 3.
Morpholinos
1 nl in total volume of the following morpholinos (MOs) were injected at the one-cell stage: tnnt2a MO - 0.5 ng (5’-CATGTTTGCTCTGATCTGACACGCA-3’) (Sehnert et al., 2002) amhc MO - 1 ng (5′-ACTCTGCCATTAAAGCATCACCCAT-3′) (Berdougo et al., 2003) cdh2 MO - 1.5 ng (5’-TCTGTATAAAGAAACCGATAGAGTT-3’) (Lele et al., 2002).
Chemical treatments
To prevent contractility for 2 hr, 100 hpf larvae were treated with 20 mM BDM (Higuchi and Takemori, 1989). To block Fgf and Hh signalling, zebrafish larvae were respectively exposed to SU5402 (15 µM) and Cyclopamine (5 and 10 µM) (Sigma-Aldrich) from 75 to 96 hpf. To increase retinoic acid signaling, dechorionated embryos were treated with 0.5 and 0.75 µM all-trans-retinoic acid (Sigma-Aldrich) from 74 to 96 hpf.
Heat-shock treatments
For heat-shock treatments, 75 hpf larvae were heat-shocked at 39°C for 1 hr. After treatment, the larvae were incubated at 28°C and imaged at 96 hpf.
Acridine orange staining
96 hpf larvae were stained with acridine orange (Sigma) by placing them in a solution of the dye in E3 medium (2 mg/mL) for 30 min. The larvae were then washed carefully with E3 medium three times and imaged using confocal microscopy.
Heart isolation, RNA extraction and microarray profiling
500 hearts from 96 hpf klf2 WT and mutant larvae were extracted, pooled in 1.5 mL and frozen in −80°C as described (Burns and MacRae, 2006). RNA was isolated using Qiazol solution (Qiagen), followed by phenol-chloroform extraction. Briefly, pooled hearts from 96 hpf larvae were first mechanically dissociated and homogenized in Qiazol using the NextAdvance Bullet Blender Homogenizer (Scientific instrument services, inc.). Chloroform was then added and phase separation was obtained following vortexing and centrifugation. The top (aqueous) phase (containing RNA) was isolated and purified using a miRNeasy micro kit (Qiagen).
Microarray analysis was performed by Oak Labs (Germany). The Low Input QuickAmp Labeling Kit (Agilent Technologies) was used to generate fluorescent cRNA (complementary RNA) following manufacturer’s protocol. For 1 st strand synthesis, oligo-dT primer or a random primer/oligo-dT primer mixture (WT primer) was used. After 2nd strand synthesis, an in vitro transcription for synthesis of cRNA labelled with cyanine 3-CTP was performed. The Agilent Gene Expression Hybridisation Kit (Agilent Technologies) was used following manufacturer’s protocol. cRNA was hybridised on a 8 × 60K microarray at 65°C for 17 hr using Agilent‘s recommended hybridisation chamber and oven. Finally, the microarray was washed once with the Agilent Gene Expression Wash Buffer one for one minute at ambient temperature followed by a second wash with preheated (37°C) Gene Expression Wash Buffer two for 1 min. Fluorescence signals on microarrays were detected by the SureScan Microarray Scanner G2600D (Agilent Technologies) at a resolution of 3 micron for SurePrint G3 Gene Expression Microarrays, generating a 20 bit TIFF file. The scanned images were analyzed with Feature Extraction Software 11.5.1.1 (Agilent) using default parameters to obtain background subtracted and spatially detrended processed signal intensities. The processed signal values were quantile normalized. In order to allow analyses at the gene level, the normalized probe values were collapsed to the isoform showing the highest mean signal considering all samples per gene.
Microarray gene set enrichment analysis (GSEA)
The Broad GSEA tool version 3 (Subramanian et al., 2005) was used to identify up- and down-regulated gene sets. The analysis was based on a pre-ranked list of genes scored by the log2 fold change multiplied with the mean normalized signal value. 1000 permutations were performed with a maximum gene set size of 500 and classic weighting versus hallmark and curated gene sets. Downregulated pathways in klf2 mutants are listed in Supplementary file 2.
Whole mount in situ hybridization
Whole mount in situ hybridization was performed as described before (Thisse and Thisse, 2008). 36, 48 and 72 hpf animals were fixed in 4% paraformaldehyde overnight at 4°C and subsequently dehydrated in 100% methanol at −20°C. Samples were rehydrated with 1X phosphate buffered saline (PBS) and permeabilized with proteinase K (10 µg/mL) at RT for 30 min. After washing with 1X PBT (1X PBS, 0.1% Tween 20 (vol/vol)), larvae were hybridized with 150 ng of klf2b antisense DIG-labeled RNA probe overnight at 70°C. The hybridized probes were then detected with alkaline phosphatase conjugated anti-Digoxigenin antibody (Roche, dilution 1:1,000) for 3 hr at RT, and the signal was visualized with BM purple (Roche). The probe for klf2b was amplified from 48 hpf heart cDNA using klf2b forward 5’-TGGCTTTACCTTGCCTTTTG −3’ and reverse 5’- CCGTGTGTTTACGGAAGTGA −3’ primers. The PCR fragment was then subcloned into pGEM-T.
Larval heart immunostaining
Right after stopping heart contraction with 0.2 percent tricaine, larvae were fixed in fish fix (4% paraformaldehyde (PFA)) overnight at 4°C. Fixed larvae were then washed three times with PBS/0.1% Tween. Washed larvae were then deyolked with forceps and permeablized with Proteinase K solution (3 µg/mL) in 1 mL PBS/Tween for 3 hr at RT. Further, the larvae were washed with PBDT (PBS/1% BSA/1% DMSO/0.5% Triton-X 100) and incubated in 2 mL of blocking solution (1.62 mL PBS/0.1% Tween +200 uL sheep serum +80 uL 20% Triton-X +100 µL 20% BSA) for 3 hr at RT. Next, the larvae were incubated in primary antibody overnight at 4°C. After washing with PBDT (four times), the larvae were incubated in secondary antibody for 3 hr at RT. For DAPI staining, larvae were incubated with DAPI (0.01 mg/mL in PBS, Sigma) and washed carefully with PBS/Tween (five times). After washing, larvae were mounted in 2% low melting agarose for imaging. Primary antibodies used for immunofluorescence were anti-GFP at 1:500 (Chicken; Aves Labs), anti-mCherry at 1:100 (mouse; Clontech), and anti-pERK (p44/42 MAPK (Erk1/2)) at 1:100 (rabbit, Cell Signaling). Secondary antibodies were used at 1:500 (Life Technologies).
Adult heart histology and immunostaining
This protocol was adapted from Mateus et al. (2015) with the following modification: after 1 hr fixation with 4% PFA, adult hearts were saturated overnight in 30% sucrose (in PBS). Embedding was done in 7.5% gelatin in 15% sucrose (in PBS), followed by freezing in isopentane cooled in liquid nitrogen. Using a Leica Microtome, the hearts were sectioned at 10 µm and maintained at −20°C until further use. Prior to immunostaining, sections were thawed for 10 min at room temperature and washed twice in PBS at 37°C for 10 min to remove gelatin. Subsequently, sections were washed once in 0.1M glycine for 10 min and permeabilized in acetone for 7 min at −20°C. Sections were washed in 1% Bovine Serum Albumin (BSA, Sigma), 1% DMSO (Sigma) and 0.1% Triton-X100 in 0.5xPBS (PBDX), followed by 2 hr of blocking in PBDX with 5% goat serum. The samples were incubated overnight with phalloidin (1:200, conjugated with Alexa Fluor 488, Invitrogen) and anti-Caveolin one antibody (BD Biosciences, 1:200) at 4°C and counterstained with DAPI (0.001 mg/mL in PBS, Sigma). Secondary antibodies were used at 1:500 (Life Technologies). Slides were mounted with DAKO Fluorescence Mounting Media and imaged using a Zeiss LSM800 confocal microscope.
For Hematoxylin and Eosin staining (H and E), cryosections were stained for 10 min with acidic hemalum (Wladeck), washed in running tap water until sections turned blue and rinsed in deionized water. Sections were subsequently stained for 6 min with an eosin solution (Waldeck), dehydrated in ethanol and cleared in roti-histol prior to mounting in Entellan (Merck). Sections were imaged using a Nikon SMZ25 and the NIS Elements software, and processed using FIJI.
Imaging, quantification and data processing
Zebrafish embryos and larvae were mounted and anesthetized in 2% low-melt agarose (Sigma) containing 0.2% tricaine on glass-bottom dishes. While mounting, samples were manually oriented towards the microscope lens to enhance optical access to the heart. Images were captured with Zeiss LSM780, LSM800, LSM 880 and spinning disk (CSU-X1 Yokogawa) confocal microscopes at 40X magnification. The confocal data were then processed with the ZEN 2012 software (black and blue edition). Adult fish and dissected hearts were imaged using a Nikon SMZ25 and the NIS Elements software and processed using FIJI software.
Cardiomyocyte size and circularity
Size and circularity of compact layer cardiomyocytes in the outer curvature of the ventricle were measured with ImageJ.
Cell transplantation
WT embryos were obtained from Tg(myl7:MKATE-CAAX) incrosses. Mutant and heterozygous embryos were obtained by incrossing Tg(myl7:LIFEACT-GFP); klf2+/- animals. All embryos were dechorionated in pronase (1 mg/mL) for 5 min at 28°C and then incubated in agarose-coated dishes with 1/3 Ringer solution supplemented with penicillin (50 U/mL)/streptomycin (50 μg/mL). Donor cells were taken from embryos at mid-blastula stages and transplanted into age-matched host embryos. Using confocal microscopy, the chimeric hearts were then imaged at 96 hpf. Donors or hosts obtained from incrossing Tg(myl7:LIFEACT-GFP); klf2+/- animals were used for genotyping.
Statistical analysis
Data were analyzed with the Prism six software. Values are presented as mean ±s.e. P values (*p≤0.05, **p≤0.01, ***p≤0.001) were calculated by two-tailed Student’s t test.
Acknowledgements
We acknowledge Albert Wang for critical reading of the manuscript, Radhan Ramadass for confocal imaging and technical support, Oliver Stone, Vanesa Jimenez-Amilburu, Jason Lai, Sri Teja Mullapudi, Michele Marass, Sven Reischauer, Arica Beisaw and Carol Yang for sharing reagents and helpful discussions, Sharon Meaney-Gardian and Simon Perathoner for excellent assistance, as well as Jean-Francois Matuszek, Rita Retzloff and Martin Laszczyk for zebrafish care. PG thanks Thomas Braun for support. We would also like to thank the reviewing editor and the reviewers for their constructive feedback. This work was supported in part by the Max Planck Society and the Leducq Foundation. SJR is a graduate student registered with the Faculty of Biological Sciences at Goethe University, Frankfurt am Main.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Didier Y Stainier, Email: Didier.Stainier@mpi-bn.mpg.de.
Deborah Yelon, University of California, San Diego, United States.
Anna Akhmanova, Utrecht University, Netherlands.
Funding Information
This paper was supported by the following grants:
Fondation Leducq to Didier Y Stainier.
Max-Planck-Gesellschaft Open-access funding to Didier Y Stainier.
Additional information
Competing interests
Senior editor, eLife.
No competing interests declared.
Author contributions
Conceptualization, Data curation, Formal analysis, Validation, Investigation, Visualization, Methodology, Writing—original draft, Writing—review and editing, Performed heart extraction, larval and adult immunostaining, Performed whole mount in situ hybridization and qPCR experiments, Analysed microarray data and performed the rest of the experiments.
Conceptualization, Formal analysis, Validation, Methodology, Writing—review and editing, Performed whole mount in situ hybridization and qPCR experiments.
Conceptualization, Methodology, Writing—review and editing, Perfomed heart extraction.
Conceptualization, Formal analysis, Investigation, Methodology, Writing—review and editing, Performed adult immunostaining.
Conceptualization, Formal analysis, Investigation, Methodology, Writing—review and editing, Performed heart extraction and larval immunostaining.
Conceptualization, Investigation, Performed the transplantation experiments.
Conceptualization, Software, Formal analysis, Validation, Writing—review and editing, Analyzed microarray data.
Conceptualization, Supervision, Funding acquisition, Validation, Writing—original draft, Writing—review and editing.
Ethics
Animal experimentation: All animal experiments were done in accordance with institutional (MPG) and national ethical and animal welfare guidelines approved by the ethics committee for animal experiments at the Regierungspräsidium Darmstadt, Germany (permit numbers B2/1017, B2/1041, B2/1138 and B2/Anz. 1007).
Additional files
Data availability
The authors declare that all data supporting the findings of this study are available within the article and its supplementary information files. Microarray data have been deposited in GEO under accession number GSE122137.
The following dataset was generated:
Rasouli SJ, El-Brolosy M, Taddese Tsedeke A, Bensimon-Brito A. 2018. Microarray data from The flow responsive transcription factor Klf2 is required for myocardial wall integrity by modulating Fgf signaling. NCBI Gene Expression Omnibus. GSE122137
References
- Auman HJ, Yelon D. Vertebrate organogenesis: getting the heart into shape. Current Biology. 2004;14:R152–R153. doi: 10.1016/j.cub.2004.01.044. [DOI] [PubMed] [Google Scholar]
- Bagatto B, Francl J, Liu B, Liu Q. Cadherin2 (N-cadherin) plays an essential role in zebrafish cardiovascular development. BMC Developmental Biology. 2006;6:23. doi: 10.1186/1471-213X-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkers J, Verhoeven MC, Abdelilah-Seyfried S. Shaping the zebrafish heart: from left-right axis specification to epithelial tissue morphogenesis. Developmental Biology. 2009;330:213–220. doi: 10.1016/j.ydbio.2009.04.011. [DOI] [PubMed] [Google Scholar]
- Beis D, Bartman T, Jin S-W, Scott IC, D'Amico LA, Ober EA. Genetic and cellular analyses of zebrafish atrioventricular cushion and valve development. Development. 2005;132:4193–4204. doi: 10.1242/dev.01970. [DOI] [PubMed] [Google Scholar]
- Berdougo E, Coleman H, Lee DH, Stainier DY, Yelon D. Mutation of weak atrium/atrial myosin heavy chain disrupts atrial function and influences ventricular morphogenesis in zebrafish. Development. 2003;130:6121–6129. doi: 10.1242/dev.00838. [DOI] [PubMed] [Google Scholar]
- Boon RA. Universiteit van Amsterdam; 2008. The transcription factor KLF2 in vascular biology.http://hdl.handle.net/11245/1.385648 [Google Scholar]
- Bowers SLK, Baudino TA. Laying the groundwork for growth: Cell-cell and cell-ECM interactions in cardiovascular development. Birth Defects Research Part C: Embryo Today: Reviews. 2010;90:1–7. doi: 10.1002/bdrc.20168. [DOI] [PubMed] [Google Scholar]
- Bressan M, Yang PB, Louie JD, Navetta AM, Garriock RJ, Mikawa T. Reciprocal myocardial-endocardial interactions pattern the delay in atrioventricular junction conduction. Development. 2014;141:4149–4157. doi: 10.1242/dev.110007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brutsaert DL, De Keulenaer GW, Fransen P, Mohan P, Kaluza GL, Andries LJ, Rouleau JL, Sys SU. The cardiac endothelium: functional morphology, development, and physiology. Progress in Cardiovascular Diseases. 1996;39:239–262. doi: 10.1016/S0033-0620(96)80004-1. [DOI] [PubMed] [Google Scholar]
- Burns CG, MacRae CA. Purification of hearts from zebrafish embryos. BioTechniques. 2006;40:274. [PubMed] [Google Scholar]
- Calmont A, Wandzioch E, Tremblay KD, Minowada G, Kaestner KH, Martin GR, Zaret KS. An FGF response pathway that mediates hepatic gene induction in embryonic endoderm cells. Developmental Cell. 2006;11:339–348. doi: 10.1016/j.devcel.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Cao Z, Sun X, Icli B, Wara AK, Feinberg MW. Role of Kruppel-like factors in leukocyte development, function, and disease. Blood. 2010;116:4404–4414. doi: 10.1182/blood-2010-05-285353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Research. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian AV, Fukuda R, Augustine SM, Maischein HM, Stainier DY. N-cadherin relocalization during cardiac trabeculation. PNAS. 2016;113:7569–7574. doi: 10.1073/pnas.1606385113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi NC, Shaw RM, Jungblut B, Huisken J, Ferrer T, Arnaout R, Scott I, Beis D, Xiao T, Baier H, Jan LY, Tristani-Firouzi M, Stainier DY. Genetic and physiologic dissection of the vertebrate cardiac conduction system. PLOS Biology. 2008;6:e109. doi: 10.1371/journal.pbio.0060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien CW, Lin SC, Lai YY, Lin BW, Lin SC, Lee JC, Tsai SJ. Regulation of CD151 by hypoxia controls cell adhesion and metastasis in colorectal cancer. Clinical Cancer Research. 2008;14:8043–8051. doi: 10.1158/1078-0432.CCR-08-1651. [DOI] [PubMed] [Google Scholar]
- Collins MM, Stainier DY. Organ function as a modulator of organ formation: lessons from zebrafish. Current Topics in Developmental Biology. 2016;117:417–433. doi: 10.1016/bs.ctdb.2015.10.017. [DOI] [PubMed] [Google Scholar]
- Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, Liu Y, Peterson KL, Chen J, Kahn R, Condorelli G, Ross J, Chien KR, Lee KF. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nature Medicine. 2002;8:459–465. doi: 10.1038/nm0502-459. [DOI] [PubMed] [Google Scholar]
- D'Amato G, Luxán G, del Monte-Nieto G, Martínez-Poveda B, Torroja C, Walter W, Bochter MS, Benedito R, Cole S, Martinez F, Hadjantonakis AK, Uemura A, Jiménez-Borreguero LJ, de la Pompa JL. Sequential Notch activation regulates ventricular chamber development. Nature Cell Biology. 2016;18:7–20. doi: 10.1038/ncb3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amico L, Scott IC, Jungblut B, Stainier DYR. A Mutation in zebrafish hmgcr1b reveals a role for isoprenoids in vertebrate heart-tube formation. Current Biology. 2007;17:252–259. doi: 10.1016/j.cub.2006.12.023. [DOI] [PubMed] [Google Scholar]
- de la Pompa JL, Epstein JA. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Developmental Cell. 2012;22:244–254. doi: 10.1016/j.devcel.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker RJ. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2) Blood. 2002;100:1689–1698. doi: 10.1182/blood-2002-01-0046. [DOI] [PubMed] [Google Scholar]
- Del Monte-Nieto G, Ramialison M, Adam AAS, Wu B, Aharonov A, D'Uva G, Bourke LM, Pitulescu ME, Chen H, de la Pompa JL, Shou W, Adams RH, Harten SK, Tzahor E, Zhou B, Harvey RP. Control of cardiac jelly dynamics by NOTCH1 and NRG1 defines the building plan for trabeculation. Nature. 2018;557:439–445. doi: 10.1038/s41586-018-0110-6. [DOI] [PubMed] [Google Scholar]
- Dietrich AC, Lombardo VA, Veerkamp J, Priller F, Abdelilah-Seyfried S. Blood flow and Bmp signaling control endocardial chamber morphogenesis. Developmental Cell. 2014;30:367–377. doi: 10.1016/j.devcel.2014.06.020. [DOI] [PubMed] [Google Scholar]
- Drechsler M, Schmidt AC, Meyer H, Paululat A. The conserved ADAMTS-like protein lonely heart mediates matrix formation and cardiac tissue integrity. PLOS Genetics. 2013;9:e1003616. doi: 10.1371/journal.pgen.1003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehling M, Adams S, Benedito R, Adams RH. Notch controls retinal blood vessel maturation and quiescence. Development. 2013;140:3051–3061. doi: 10.1242/dev.093351. [DOI] [PubMed] [Google Scholar]
- Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien CB, Morcos PA, Rosenblatt J. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature. 2012;484:546–549. doi: 10.1038/nature10999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Brolosy M, Rossi A, Kontarakis Z, Kuenne C, Guenther S, Fukuda N. Genetic compensation is triggered by mutant mRNA degradation. bioRxiv. 2018 doi: 10.1101/328153. [DOI] [PMC free article] [PubMed]
- Goddard LM, Duchemin AL, Ramalingan H, Wu B, Chen M, Bamezai S, Yang J, Li L, Morley MP, Wang T, Scherrer-Crosbie M, Frank DB, Engleka KA, Jameson SC, Morrisey EE, Carroll TJ, Zhou B, Vermot J, Kahn ML. Hemodynamic forces sculpt developing heart valves through a KLF2-WNT9B Paracrine signaling axis. Developmental Cell. 2017;43:274–289. doi: 10.1016/j.devcel.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Rosa JM, Peralta M, Mercader N. Pan-epicardial lineage tracing reveals that epicardium derived cells give rise to myofibroblasts and perivascular cells during zebrafish heart regeneration. Developmental Biology. 2012;370:173–186. doi: 10.1016/j.ydbio.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Grassini DR, Lagendijk AK, De Angelis JE, Da Silva J, Jeanes A, Zettler N, Bower NI, Hogan BM, Smith KA. Nppa and Nppb act redundantly during zebrafish cardiac development to confine AVC marker expression and reduce cardiac jelly volume. Development. 2018;145:dev160739. doi: 10.1242/dev.160739. [DOI] [PubMed] [Google Scholar]
- Heckel E, Boselli F, Roth S, Krudewig A, Belting HG, Charvin G, Vermot J. Oscillatory flow modulates mechanosensitive klf2a expression through trpv4 and trpp2 during heart valve development. Current Biology. 2015;25:1354–1361. doi: 10.1016/j.cub.2015.03.038. [DOI] [PubMed] [Google Scholar]
- High FA, Jain R, Stoller JZ, Antonucci NB, Lu MM, Loomes KM, Kaestner KH, Pear WS, Epstein JA. Murine Jagged1/Notch signaling in the second heart field orchestrates Fgf8 expression and tissue-tissue interactions during outflow tract development. Journal of Clinical Investigation. 2009;128:1986–1996. doi: 10.1172/JCI38922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi H, Takemori S. Butanedione monoxime suppresses contraction and ATPase activity of rabbit skeletal muscle1. The Journal of Biochemistry. 1989;105:638–643. doi: 10.1093/oxfordjournals.jbchem.a122717. [DOI] [PubMed] [Google Scholar]
- Hoffman JI, Kaplan S. The incidence of congenital heart disease. Journal of the American College of Cardiology. 2002;39:1890–1900. doi: 10.1016/S0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- Jiménez-Amilburu V, Rasouli SJ, Staudt DW, Nakajima H, Chiba A, Mochizuki N, Stainier DYR. in vivo visualization of cardiomyocyte apicobasal polarity reveals epithelial to mesenchymal-like transition during cardiac trabeculation. Cell Reports. 2016;17:2687–2699. doi: 10.1016/j.celrep.2016.11.023. [DOI] [PubMed] [Google Scholar]
- Keegan BR, Feldman JL, Begemann G, Ingham PW, Yelon D. Retinoic acid signaling restricts the cardiac progenitor pool. Science. 2005;307:247–249. doi: 10.1126/science.1101573. [DOI] [PubMed] [Google Scholar]
- Khachigian LM, Anderson KR, Halnon NJ, Gimbrone MA, Resnick N, Collins T. Egr-1 is activated in endothelial cells exposed to fluid shear stress and interacts with a novel shear-stress-response element in the PDGF A-chain promoter. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17:2280–2286. doi: 10.1161/01.ATV.17.10.2280. [DOI] [PubMed] [Google Scholar]
- Khachigian LM. Early growth response-1 in cardiovascular pathobiology. Circulation Research. 2006;98:186–191. doi: 10.1161/01.RES.0000200177.53882.c3. [DOI] [PubMed] [Google Scholar]
- Kim KH, Nakaoka Y, Augustin HG, Koh GY. Myocardial angiopoietin-1 controls atrial chamber morphogenesis by spatiotemporal degradation of cardiac jelly. Cell Reports. 2018;23:2455–2466. doi: 10.1016/j.celrep.2018.04.080. [DOI] [PubMed] [Google Scholar]
- Kostetskii I, Moore R, Kemler R, Radice GL. Differential adhesion leads to segregation and exclusion of N-cadherin-deficient cells in chimeric embryos. Developmental Biology. 2001;234:72–79. doi: 10.1006/dbio.2001.0250. [DOI] [PubMed] [Google Scholar]
- Kuipers D, Mehonic A, Kajita M, Peter L, Fujita Y, Duke T, Charras G, Gale JE. Epithelial repair is a two-stage process driven first by dying cells and then by their neighbours. Journal of Cell Science. 2014;127:1229–1241. doi: 10.1242/jcs.138289. [DOI] [PubMed] [Google Scholar]
- Kwon HB, Wang S, Helker CS, Rasouli SJ, Maischein HM, Offermanns S, Herzog W, Stainier DY. in vivo modulation of endothelial polarization by apelin receptor signalling. Nature Communications. 2016;7:11805. doi: 10.1038/ncomms11805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai D, Liu X, Forrai A, Wolstein O, Michalicek J, Ahmed I, Garratt AN, Birchmeier C, Zhou M, Hartley L, Robb L, Feneley MP, Fatkin D, Harvey RP. Neuregulin 1 sustains the gene regulatory network in both trabecular and nontrabecular myocardium. Circulation Research. 2010;107:715–727. doi: 10.1161/CIRCRESAHA.110.218693. [DOI] [PubMed] [Google Scholar]
- Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J, Ornitz DM. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Developmental Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature. 1995;378:394–398. doi: 10.1038/378394a0. [DOI] [PubMed] [Google Scholar]
- Lee Y, Grill S, Sanchez A, Murphy-Ryan M, Poss KD. Fgf signaling instructs position-dependent growth rate during zebrafish fin regeneration. Development. 2005;132:5173–5183. doi: 10.1242/dev.02101. [DOI] [PubMed] [Google Scholar]
- Lee JS, Yu Q, Shin JT, Sebzda E, Bertozzi C, Chen M, Mericko P, Stadtfeld M, Zhou D, Cheng L, Graf T, MacRae CA, Lepore JJ, Lo CW, Kahn ML. Klf2 is an essential regulator of vascular hemodynamic forces in vivo. Developmental Cell. 2006;11:845–857. doi: 10.1016/j.devcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Lele Z, Folchert A, Concha M, Rauch GJ, Geisler R, Rosa F, Wilson SW, Hammerschmidt M, Bally-Cuif L. Parachute/n-cadherin is required for morphogenesis and maintained integrity of the zebrafish neural tube. Development. 2002;129:3281–3294. doi: 10.1242/dev.129.14.3281. [DOI] [PubMed] [Google Scholar]
- Lin Z, Natesan V, Shi H, Dong F, Kawanami D, Mahabeleshwar GH, Atkins GB, Nayak L, Cui Y, Finigan JH, Jain MK. Kruppel-like factor 2 regulates endothelial barrier function. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30:1952–1959. doi: 10.1161/ATVBAHA.110.211474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YF, Swinburne I, Yelon D. Multiple influences of blood flow on cardiomyocyte hypertrophy in the embryonic zebrafish heart. Developmental Biology. 2012;362:242–253. doi: 10.1016/j.ydbio.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Bressan M, Hassel D, Huisken J, Staudt D, Kikuchi K, Poss KD, Mikawa T, Stainier DY. A dual role for ErbB2 signaling in cardiac trabeculation. Development. 2010;137:3867–3875. doi: 10.1242/dev.053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Krauss RS. N-cadherin ligation, but not Sonic hedgehog binding, initiates Cdo-dependent p38alpha/beta MAPK signaling in skeletal myoblasts. PNAS. 2010;107:4212–4217. doi: 10.1073/pnas.0908883107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinari E, Mehonic A, Curran S, Gale J, Duke T, Baum B. Live-cell delamination counterbalances epithelial growth to limit tissue overcrowding. Nature. 2012;484:542–545. doi: 10.1038/nature10984. [DOI] [PubMed] [Google Scholar]
- Marques SR, Lee Y, Poss KD, Yelon D. Reiterative roles for FGF signaling in the establishment of size and proportion of the zebrafish heart. Developmental Biology. 2008;321:397–406. doi: 10.1016/j.ydbio.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin MJ, Di Rocco G, Gardiner A, Bush SM, Lassar AB. Inhibition of Wnt activity induces heart formation from posterior mesoderm. Genes & Development. 2001;15:316–327. doi: 10.1101/gad.855501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateus R, Lourenco R, Fang Y, Brito G, Farinho A, Valerio F, Jacinto A. Control of tissue growth by yap relies on cell density and F-actin in zebrafish fin regeneration. Development. 2015;142:2752–2763. doi: 10.1242/dev.119701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell BB, Yang VW. Mammalian Krüppel-like factors in health and diseases. Physiological Reviews. 2010;90:1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–390. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- Miao L, Li J, Li J, Tian X, Lu Y, Hu S, Shieh D, Kanai R, Zhou BY, Zhou B, Liu J, Firulli AB, Martin JF, Singer H, Zhou B, Xin H, Wu M. Notch signaling regulates Hey2 expression in a spatiotemporal dependent manner during cardiac morphogenesis and trabecular specification. Scientific Reports. 2018;8:2678. doi: 10.1038/s41598-018-20917-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milgrom-Hoffman M, Michailovici I, Ferrara N, Zelzer E, Tzahor E. Endothelial cells regulate neural crest and second heart field morphogenesis. Biology Open. 2014;3:679–688. doi: 10.1242/bio.20148078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Mochizuki N. Flow pattern-dependent endothelial cell responses through transcriptional regulation. Cell Cycle. 2017a;16:1893–1901. doi: 10.1080/15384101.2017.1364324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Yamamoto K, Agarwala S, Terai K, Fukui H, Fukuhara S, Ando K, Miyazaki T, Yokota Y, Schmelzer E, Belting HG, Affolter M, Lecaudey V, Mochizuki N. Flow-Dependent Endothelial YAP Regulation Contributes to Vessel Maintenance. Developmental Cell. 2017b;40:523–536. doi: 10.1016/j.devcel.2017.02.019. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Manasek FJ. An experimental study of the relation of cardiac jelly to the shape of the early chick embryonic heart. Development. 1981;65:235–256. [PubMed] [Google Scholar]
- Nayak L, Lin Z, Jain MK. "Go with the flow": how Krüppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxidants & Redox Signaling. 2011;15:1449–1461. doi: 10.1089/ars.2010.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novodvorsky P, Watson O, Gray C, Wilkinson RN, Reeve S, Smythe C, Beniston R, Plant K, Maguire R, M K Rothman A, Elworthy S, van Eeden FJ, Chico TJ. klf2ash317 mutant zebrafish do not recapitulate Morpholino-Induced vascular and haematopoietic phenotypes. PLOS ONE. 2015;10:e0141611. doi: 10.1371/journal.pone.0141611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oates AC, Pratt SJ, Vail B, Yan Y-l, Ho RK, Johnson SL. The zebrafish klf gene family. Blood. 2001;98:1792–1801. doi: 10.1182/blood.V98.6.1792. [DOI] [PubMed] [Google Scholar]
- Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, García-Cardeña G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. Journal of Clinical Investigation. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta M, Steed E, Harlepp S, González-Rosa JM, Monduc F, Ariza-Cosano A, Cortés A, Rayón T, Gómez-Skarmeta JL, Zapata A, Vermot J, Mercader N. Heartbeat-driven pericardiac fluid forces contribute to epicardium morphogenesis. Current Biology. 2013;23:1726–1735. doi: 10.1016/j.cub.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Peralta M, González-Rosa J, Marques I, Mercader N. The epicardium in the embryonic and adult zebrafish. Journal of Developmental Biology. 2014;2:101–116. doi: 10.3390/jdb2020101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peshkovsky C, Totong R, Yelon D. Dependence of cardiac trabeculation on neuregulin signaling and blood flow in zebrafish. Developmental Dynamics. 2011;240:446–456. doi: 10.1002/dvdy.22526. [DOI] [PubMed] [Google Scholar]
- Powell DR, Williams JS, Hernandez-Lagunas L, Salcedo E, O'Brien JH, Artinger KB. Cdon promotes neural crest migration by regulating N-cadherin localization. Developmental Biology. 2015;407:289–299. doi: 10.1016/j.ydbio.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan A, Zeng XI, Sidhwani P, Marques SR, George V, Targoff KL, Chi NC, Yelon D. FGF signaling enforces cardiac chamber identity in the developing ventricle. Development. 2017;144:1328–1338. doi: 10.1242/dev.143719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proulx K, Lu A, Sumanas S. Cranial vasculature in zebrafish forms by angioblast cluster-derived angiogenesis. Developmental Biology. 2010;348:34–46. doi: 10.1016/j.ydbio.2010.08.036. [DOI] [PubMed] [Google Scholar]
- Ramasubramanian A, Chu-Lagraff QB, Buma T, Chico KT, Carnes ME, Burnett KR, Bradner SA, Gordon SS. On the role of intrinsic and extrinsic forces in early cardiac S-looping. Developmental Dynamics. 2013;242:801–816. doi: 10.1002/dvdy.23968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasouli SJ, Stainier DYR. Regulation of cardiomyocyte behavior in zebrafish trabeculation by Neuregulin 2a signaling. Nature Communications. 2017;8:15281. doi: 10.1038/ncomms15281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifers F, Walsh EC, Léger S, Stainier D, Brand M. Induction and differentiation of the zebrafish heart requires fibroblast growth factor 8 (fgf8/acerebellar) Development. 2000;127:225–235. doi: 10.1242/dev.127.2.225. [DOI] [PubMed] [Google Scholar]
- Reischauer S, Arnaout R, Ramadass R, Stainier DY. Actin binding GFP allows 4D in vivo imaging of myofilament dynamics in the zebrafish heart and the identification of Erbb2 signaling as a remodeling factor of myofibril architecture. Circulation Research. 2014;115:845–856. doi: 10.1161/CIRCRESAHA.115.304356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reischauer S, Stone OA, Villasenor A, Chi N, Jin SW, Martin M, Lee MT, Fukuda N, Marass M, Witty A, Fiddes I, Kuo T, Chung WS, Salek S, Lerrigo R, Alsiö J, Luo S, Tworus D, Augustine SM, Mucenieks S, Nystedt B, Giraldez AJ, Schroth GP, Andersson O, Stainier DY. Cloche is a bHLH-PAS transcription factor that drives haemato-vascular specification. Nature. 2016;535:294–298. doi: 10.1038/nature18614. [DOI] [PubMed] [Google Scholar]
- Renz M, Otten C, Faurobert E, Rudolph F, Zhu Y, Boulday G, Duchene J, Mickoleit M, Dietrich AC, Ramspacher C, Steed E, Manet-Dupé S, Benz A, Hassel D, Vermot J, Huisken J, Tournier-Lasserve E, Felbor U, Sure U, Albiges-Rizo C, Abdelilah-Seyfried S. Regulation of β1 integrin-Klf2-mediated angiogenesis by CCM proteins. Developmental Cell. 2015;32:181–190. doi: 10.1016/j.devcel.2014.12.016. [DOI] [PubMed] [Google Scholar]
- Revenu C, Streichan S, Donà E, Lecaudey V, Hufnagel L, Gilmour D. Quantitative cell polarity imaging defines leader-to-follower transitions during collective migration and the key role of microtubule-dependent adherens junction formation. Development. 2014;141:1282–1291. doi: 10.1242/dev.101675. [DOI] [PubMed] [Google Scholar]
- Rones MS, McLaughlin KA, Raffin M, Mercola M. Serrate and Notch specify cell fates in the heart field by suppressing cardiomyogenesis. Development. 2000;127:3865–3876. doi: 10.1242/dev.127.17.3865. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Raff MC, Cramer LP. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Current Biology. 2001;11:1847–1857. doi: 10.1016/S0960-9822(01)00587-5. [DOI] [PubMed] [Google Scholar]
- Rossi A, Kontarakis Z, Gerri C, Nolte H, Hölper S, Krüger M, Stainier DY. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524:230–233. doi: 10.1038/nature14580. [DOI] [PubMed] [Google Scholar]
- Ruiz-Villalba A, Pérez-Pomares JM. The expanding role of the epicardium and epicardial-derived cells in cardiac development and disease. Current Opinion in Pediatrics. 2012;24:569–576. doi: 10.1097/MOP.0b013e328357a532. [DOI] [PubMed] [Google Scholar]
- Rydeen AB, Waxman JS. Cyp26 enzymes facilitate second heart field progenitor addition and maintenance of ventricular integrity. PLOS Biology. 2016;14:e2000504. doi: 10.1371/journal.pbio.2000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsa LA, Givens C, Tzima E, Stainier DYR, Qian L, Liu J. Cardiac contraction activates endocardial Notch signaling to modulate chamber maturation in zebrafish. Development. 2015;142:4080–4091. doi: 10.1242/dev.125724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlueter J, Brand T. Epicardial progenitor cells in cardiac development and regeneration. Journal of Cardiovascular Translational Research. 2012;5:641–653. doi: 10.1007/s12265-012-9377-4. [DOI] [PubMed] [Google Scholar]
- Schneider VA, Mercola M. Wnt antagonism initiates cardiogenesis in Xenopus laevis. Genes & Development. 2001;15:304–315. doi: 10.1101/gad.855601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehnert AJ, Huq A, Weinstein BM, Walker C, Fishman M, Stainier DY. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nature Genetics. 2002;31:106–110. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Does loss of CD151 expression promote the metastasis of hypoxic colon cancer cells? Clinical Cancer Research. 2008;14:7969–7970. doi: 10.1158/1078-0432.CCR-08-2417. [DOI] [PubMed] [Google Scholar]
- Shinya M, Koshida S, Sawada A, Kuroiwa A, Takeda H. Fgf signalling through MAPK cascade is required for development of the subpallial telencephalon in zebrafish embryos. Development. 2001;128:4153–4164. doi: 10.1242/dev.128.21.4153. [DOI] [PubMed] [Google Scholar]
- Slough J, Cooney L, Brueckner M. Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis. Developmental Dynamics. 2008;237:2304–2314. doi: 10.1002/dvdy.21669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sojka S, Amin NM, Gibbs D, Christine KS, Charpentier MS, Conlon FL. Congenital heart disease protein 5 associates with CASZ1 to maintain myocardial tissue integrity. Development. 2014;141:3040–3049. doi: 10.1242/dev.106518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srisakuldee W, Nickel BE, Fandrich RR, Jiang ZS, Kardami E. Administration of FGF-2 to the heart stimulates connexin-43 phosphorylation at protein kinase C target sites. Cell Communication & Adhesion. 2006;13:13–19. doi: 10.1080/15419060600631326. [DOI] [PubMed] [Google Scholar]
- Stainier DY, Fishman MC. Patterning the zebrafish heart tube: acquisition of anteroposterior polarity. Developmental Biology. 1992;153:91–101. doi: 10.1016/0012-1606(92)90094-W. [DOI] [PubMed] [Google Scholar]
- Stainier DY, Weinstein BM, Detrich HW, Zon LI, Fishman MC. Cloche, an early acting zebrafish gene, is required by both the endothelial and hematopoietic lineages. Development. 1995;121:3141–3150. doi: 10.1242/dev.121.10.3141. [DOI] [PubMed] [Google Scholar]
- Stainier DY, Fouquet B, Chen J-N, Warren KS, Weinstein BM, Meiler SE. Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development. 1996;123:285–292. doi: 10.1242/dev.123.1.285. [DOI] [PubMed] [Google Scholar]
- Stainier DYR, Fishman MC. The zebrafish as a model system to study cardiovascular development. Trends in Cardiovascular Medicine. 1994;4:207–212. doi: 10.1016/1050-1738(94)90036-1. [DOI] [PubMed] [Google Scholar]
- Stankunas K, Hang CT, Tsun ZY, Chen H, Lee NV, Wu JI, Shang C, Bayle JH, Shou W, Iruela-Arispe ML, Chang CP. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Developmental Cell. 2008;14:298–311. doi: 10.1016/j.devcel.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt D, Stainier D. Uncovering the molecular and cellular mechanisms of heart development using the zebrafish. Annual Review of Genetics. 2012;46:397–418. doi: 10.1146/annurev-genet-110711-155646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt DW, Liu J, Thorn KS, Stuurman N, Liebling M, Stainier DY. High-resolution imaging of cardiomyocyte behavior reveals two distinct steps in ventricular trabeculation. Development. 2014;141:585–593. doi: 10.1242/dev.098632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steed E, Boselli F, Vermot J. Hemodynamics driven cardiac valve morphogenesis. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2016a;1863:1760–1766. doi: 10.1016/j.bbamcr.2015.11.014. [DOI] [PubMed] [Google Scholar]
- Steed E, Faggianelli N, Roth S, Ramspacher C, Concordet JP, Vermot J. klf2a couples mechanotransduction and zebrafish valve morphogenesis through fibronectin synthesis. Nature Communications. 2016b;7:11646. doi: 10.1038/ncomms11646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana K, Jones N, Dumont DJ, Puri MC, Bernstein A. Selective role of a distinct tyrosine residue on Tie2 in heart development and early hematopoiesis. Molecular and Cellular Biology. 2005;25:4693–4702. doi: 10.1128/MCB.25.11.4693-4702.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi JK, Lou X, Alexander JM, Sugizaki H, Delgado-Olguín P, Holloway AK, Mori AD, Wylie JN, Munson C, Zhu Y, Zhou YQ, Yeh RF, Henkelman RM, Harvey RP, Metzger D, Chambon P, Stainier DY, Pollard KS, Scott IC, Bruneau BG. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nature Communications. 2011;2:187. doi: 10.1038/ncomms1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nature Protocols. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- Tian Y, Morrisey EE. Importance of myocyte-nonmyocyte interactions in cardiac development and disease. Circulation Research. 2012;110:1023–1034. doi: 10.1161/CIRCRESAHA.111.243899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh-Finkel L, Zeisel A, Brodt-Ivenshitz M, Shamai A, Yao Z, Seger R, Domany E, Tzahor E. BMP-mediated inhibition of FGF signaling promotes cardiomyocyte differentiation of anterior heart field progenitors. Development. 2010;137:2989–3000. doi: 10.1242/dev.051649. [DOI] [PubMed] [Google Scholar]
- Uribe V, Ramadass R, Dogra D, Rasouli SJ, Gunawan F, Nakajima H, Chiba A, Reischauer S, Mochizuki N, Stainier DYR. In vivo analysis of cardiomyocyte proliferation during trabeculation. Development. 2018;145:dev164194. doi: 10.1242/dev.164194. [DOI] [PubMed] [Google Scholar]
- Vermot J, Forouhar AS, Liebling M, Wu D, Plummer D, Gharib M, Fraser SE. Reversing blood flows act through klf2a to ensure normal valvulogenesis in the developing heart. PLOS Biology. 2009;7:e1000246. doi: 10.1371/journal.pbio.1000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virágh S, Challice CE. The origin of the epicardium and the embryonic myocardial circulation in the mouse. The Anatomical Record. 1981;201:157–168. doi: 10.1002/ar.1092010117. [DOI] [PubMed] [Google Scholar]
- Wagner DE, Weinreb C, Collins ZM, Briggs JA, Megason SG, Klein AM. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science. 2018;360:981–987. doi: 10.1126/science.aar4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Ha CH, Jhun BS, Wong C, Jain MK, Jin ZG. Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood. 2010;115:2971–2979. doi: 10.1182/blood-2009-05-224824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhang P, Wei Y, Gao Y, Patient R, Liu F. A blood flow-dependent klf2a-NO signaling cascade is required for stabilization of hematopoietic stem cell programming in zebrafish embryos. Blood. 2011;118:4102–4110. doi: 10.1182/blood-2011-05-353235. [DOI] [PubMed] [Google Scholar]
- Wang J, Cao J, Dickson AL, Poss KD. Epicardial regeneration is guided by cardiac outflow tract and Hedgehog signalling. Nature. 2015;522:226–230. doi: 10.1038/nature14325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington Smoak I, Byrd NA, Abu-Issa R, Goddeeris MM, Anderson R, Morris J, Yamamura K, Klingensmith J, Meyers EN. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Developmental Biology. 2005;283:357–372. doi: 10.1016/j.ydbio.2005.04.029. [DOI] [PubMed] [Google Scholar]
- Wei CJ, Francis R, Xu X, Lo CW. Connexin43 associated with an N-cadherin-containing multiprotein complex is required for gap junction formation in NIH3T3 cells. Journal of Biological Chemistry. 2005;280:19925–19936. doi: 10.1074/jbc.M412921200. [DOI] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish. Eugene, United States: University of Oregon Press; 2000. [Google Scholar]
- Wilsbacher L, McNally EM. Genetics of cardiac developmental disorders: cardiomyocyte proliferation and growth and relevance to heart failure. Annual Review of Pathology: Mechanisms of Disease. 2016;11:395–419. doi: 10.1146/annurev-pathol-012615-044336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Bohanan CS, Neumann JC, Lingrel JB. KLF2 transcription factor modulates blood vessel maturation through smooth muscle cell migration. Journal of Biological Chemistry. 2008;283:3942–3950. doi: 10.1074/jbc.M707882200. [DOI] [PubMed] [Google Scholar]
- Wu MT, Su MY, Huang YL, Chiou KR, Yang P, Pan HB, Reese TG, Wedeen VJ, Tseng WY. Sequential changes of myocardial microstructure in patients postmyocardial infarction by diffusion-tensor cardiac MR: correlation with left ventricular structure and function. Circulation. Cardiovascular Imaging. 2009;2:32–40. doi: 10.1161/CIRCIMAGING.108.778902. [DOI] [PubMed] [Google Scholar]