

Abstract
Background and Purpose
The biological significance of the multi‐site phosphorylation of Bcl‐2 at its loop region (T69, S70 and S87) has remained controversial for decades. This is a major obstacle for understanding apoptosis and anti‐tumour drug development.
Experimental Approach
We established a mathematical model into which a phosphorylation and de‐phosphorylation process of Bcl‐2 was integrated. Paclitaxel‐treated breast cancer cells were used as experimental models. Changes in the kinetics of binding with its critical partners, induced by phosphorylation of Bcl‐2 were experimentally obtained by surface plasmon resonance, using a phosphorylation‐mimicking mutant EEE‐Bcl‐2 (T69E, S70E and S87E).
Key Results
Mathematical simulations combined with experimental validation showed that phosphorylation regulates Bcl‐2 with different dynamics depending on the extent of Bcl‐2 phosphorylation and the phosphorylated Bcl‐2‐induced changes in binding kinetics. In response to Bcl‐2 homology 3 (BH3)‐only protein Bmf stress, Bcl‐2 phosphorylation switched from diminishing to enhancing the Bcl‐2 anti‐apoptotic ability with increased phosphorylation of Bcl‐2, and the turning point was 50% Bcl‐2 phosphorylation induced by 0.2 μM paclitaxel treatment. In contrast, Bcl‐2 phosphorylation enhanced the anti‐apoptotic ability of Bcl‐2 towards other BH3‐only proteins Bim, Bad and Puma, throughout the entire phosphorylation procedure.
Conclusions and Implications
The model could accurately predict the effects of anti‐tumour drugs that involve the Bcl‐2 family pathway, as shown with ABT‐199 or etoposide.
Abbreviations
- BH3
Bcl‐2 homology 3
- MAC
mitochondrial apoptosis channel
- MOMP
mitochondrial outer membrane permeabilization
- ODEs
ordinary differential equations
- pBcl‐2
phosphorylated Bcl‐2
- SPR
surface plasmon resonance
Introduction
The Bcl‐2 protein promotes cancer cell accumulation by antagonizing pro‐apoptotic Bcl‐2 family members via the shared Bcl‐2 homology 3 (BH3) domain (Sharpe et al., 2004; Hinds and Day, 2005). It is regulated by phosphorylation to a much greater extent than protein expression because it has a long half‐life of ~20 h (Blagosklonny et al., 1996; Ruvolo et al., 2001). The several phosphorylation sites of the Bcl‐2 protein, T69, S70 and S87, are located in its loop region, which is far away from the BH3 binding site (Ruvolo et al., 2001). To date, there is neither structural information for phosphorylated Bcl‐2 (pBcl‐2) nor measured data on changes in kinetics of binding with its critical partners, induced by phosphorylation of Bcl‐2.
Moreover, there is still controversy over the functional consequence of Bcl‐2 phosphorylation resulting from a great number of cell‐based explorations (May et al., 1994; Ito et al., 1997; Srivastava et al., 1999; Deng et al., 2000, 2004; Mingo‐Sion et al., 2004; Terrano et al., 2010; Barille‐Nion et al., 2012; Chae et al., 2012; Notte et al., 2013). Two opposing models of Bcl‐2 phosphorylation were proposed. The ‘Taxol‐induced’ model implies that phosphorylation inhibits the anti‐apoptotic function of Bcl‐2 (Srivastava et al., 1999; Mingo‐Sion et al., 2004; Terrano et al., 2010; Barille‐Nion et al., 2012; Chae et al., 2012), while the ‘IL‐3‐induced’ model demonstrates that the phosphorylation of Bcl‐2 enhances its anti‐apoptotic function (May et al., 1994; Ito et al., 1997; Deng et al., 2000, 2004). Even in investigations with paclitaxel, an opposing conclusion has been reached by some independent groups that Bcl‐2 phosphorylation enhances its anti‐apoptotic ability (Deng et al., 2004; Notte et al., 2013). This is a major obstacle for understanding apoptosis regulation and anti‐tumour drug development.
The effect of phosphorylation in proteins is manifold. For example, it can induce conformational changes, promote order–disorder transitions and modulate binding (Jiang and McKnight, 2006; Volkman et al., 2001). Recently, our studies on low MW pBcl‐2 inhibitors revealed that Bcl‐2 phosphorylation in the loop region induced changes in the BH3 binding groove and then reshuffled the binding preference of Bcl‐2 with various partners – for example, Bax and Bak favour the phospho‐mimic mutant T69E/S70E/S87E Bcl‐2 (EEE‐Bcl‐2) much more than the non‐phospho‐mimic mutant T69A/S70A/S87A Bcl‐2 (AAA‐Bcl‐2), but Bim disfavours EEE‐Bcl‐2 (Liu et al., 2013; Zhang et al., 2013; Song et al., 2015, 2016). Therefore, it is impossible to identify how phosphorylation alters Bcl‐2 function based on these individual binding properties. We hypothesized that the phosphorylation‐induced functional changes of Bcl‐2 that emanate from multi‐protein interplay should be explored at the Bcl‐2 network level rather than by individual component behaviour.
A well‐established network model using ordinary differential equations (ODEs) and mathematical simulations via Bcl‐2's multi‐protein interaction interplay has been successfully used to understand the dynamic functions of the apoptotic network (Chen et al., 2007; Cui et al., 2008; Lindner et al., 2013; Wurstle et al., 2014; Zhao et al., 2015). For example, Pei et al. utilized ODE model simulation to map mutation‐induced changes in protein interaction kinetics to changes in network dynamics and found cancer‐related mutations (Zhao et al., 2015). Prehn et al. performed a systems analysis of Bcl‐2 protein family interactions using an ODE model to predict the responses to chemotherapy (Lindner et al., 2013). However, these models did not consider the functional processes of Bcl‐2 phosphorylation, the changes in Bcl‐2 interaction kinetics induced by phosphorylation or the function of pBcl‐2 in the changes in network dynamics.
We parameterized the changes in kinetics of binding to proteins, induced by phosphorylation of Bcl‐2 and established a pBcl‐2/Bcl‐2 mathematical model to understand the apoptosis network dynamics under Bcl‐2 phosphorylation. The experimentally validated pBcl‐2/Bcl‐2 model provided a quantitative and unifying mechanism to reconcile the previous conflicting conclusions. The diminishing and enhancing of anti‐apoptotic function following Bcl‐2 phosphorylation occurs sequentially under a given condition. The enhancement of Bcl‐2 anti‐apoptotic function is predominant because diminishing anti‐apoptotic function following Bcl‐2 phosphorylation only occurs under Bmf stress and when pBcl‐2/total Bcl‐2 (TBcl‐2) ratio is less than 50%. Other than that, pBcl‐2 enhances the anti‐apoptotic function with the pBcl‐2/TBcl‐2 ratio increased from 0 to 80% in cells sensitized by other BH3‐only proteins, including Bim, Puma and Bad. Additionally, the pBcl‐2/Bcl‐2 model not only fits the experimental data but also provides an accurate prediction for the effects of anti‐tumour drugs in a broad spectrum that involves the Bcl‐2 family pathway, similar to our prediction of the anti‐tumour effects of ABT‐199 or etoposide.
Methods
Cell lines and culture
MDA‐MB‐231(RRID: CVCL_0062), MCF‐7 (RRID: CVCL_0031), T47D (RRID: CVCL_0553), OCI‐AML2 (CVCL_1619), KG‐1 (CVCL_0374), THP‐1 (CVCL_0006), NB4 (CVCL_0005), OCI‐AML3 (CVCL_1844), LoVo (CVCL_0399), HCT‐116 (CVCL_0291), SW480 (CVCL_0546), DLD1 (CVCL_0248), HT29 (CVCL_0320) were purchased from the Cell Bank of Chinese Academy of Sciences. The identity of each cell line has been validated by STR profiling before providing it to research labs. We used these cell lines within 6 months to avoid contamination. All the cells were maintained in RPMI‐1640 supplemented with 10% fetal calf serum, 100 U mL‐1 penicillin and 100 mg mL‐1 streptomycin at 37°C in 5% CO2.
Computational modelling
Protein production, degradation, phosphorylation, dephosphorylation and interaction were modelled by a pseudo‐reaction network using mass‐action kinetics and were translated into a set of ODEs solved using MATLAB 7.3 (MathWorks, R2007b, RRID: SCR_001622) function ode 15 s. The concentrations of phosphorylated JNK (p‐JNK) and mitochondria‐accumulated Bmf or Puma in the three breast cancer cell lines upon paclitaxel treatment were considered as surrogates of the chemotherapeutic dose and were used as model inputs. The low MW Bcl‐2 inhibitor ABT‐199 (venetoclax) acted by binding to and inhibiting anti‐apoptotic proteins (Table 1).
Table 1.
Binding kinetics of BH3 peptides towards different Bcl‐2 variants by SPR
| Bcl‐1 protein | WT‐Bcl‐2 | AAA‐Bcl‐2 | EEE‐Bcl‐2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| BH3 peptides | K D (nM) | k a (nM−1S−1) | k d (S−1) | K D (nM) | k a (nM−1S−1) | k d (S−1) | K D (nM) | k a (nM−1S−1) | k d (S−1) |
| Bim | 2.5 | 4.48 × 10−5 | 1.12 × 10−4 | 2.9 | 4.28 × 10−5 | 1.24 × 10−4 | 17.6 | 3.25 × 10−5 | 5.72 × 10−4 |
| Puma | 3.2 | 4.91 × 10−5 | 1.57 × 10−4 | 3.6 | 4.53 × 10−5 | 1.63 × 10−4 | 18.2 | 3.16 × 10−5 | 5.75 × 10−4 |
| Bmf | 7.8 | 3.17 × 10−5 | 2.47 × 10−4 | 7.3 | 3.24 × 10−5 | 2.37 × 10−4 | 7.1 | 3.56 × 10−5 | 2.53 × 10−4 |
| Bad | 8.5 | 3.25 × 10−5 | 2.76 × 10−4 | 8.6 | 3.31 × 10−5 | 2.85 × 10−4 | 264.7 | 2.83 × 10−5 | 7.49 × 10−3 |
| Bax | 11.2 | 3.24 × 10−5 | 3.63 × 10−4 | 10.3 | 3.26 × 10−5 | 3.36 × 10−4 | 1.2 | 3.03 × 10−5 | 3.64 × 10−5 |
| Bak | 36.3 | 2.57 × 10−5 | 9.33 × 10−4 | 32.5 | 2.45 × 10−5 | 7.96 × 10−4 | 4.1 | 2.26 × 10−5 | 9.27 × 10−5 |
| ABT‐199 | 2.6 | 3.88 × 10−5 | 1.01 × 10−4 | 3.2 | 3.95 × 10−5 | 1.26 × 10−4 | 283 | 2.15 × 10−5 | 6.08 × 10–3 |
| Noxa | >1000 | ND | ND | >5000 | ND | ND | >1000 | ND | ND |
Kinetic parameters for the interaction between BH3 peptides and three forms of Bcl‐2, obtained from SPR assays. Although the binding profile of AAA‐Bcl‐2 to the BH3 peptides was almost the same as that of WT‐Bcl‐2, the binding profile of EEE‐Bcl‐2 showed clear differences from that of both WT and AAA‐Bcl‐2. Particularly, EEE‐Bcl‐2 showed greater KD values for Bim, Puma, Bad and ABT‐199 and lower values for Bax and Bak. Note that none of the three variants of Bcl‐2 bound the BH3 protein Noxa. Data shown are means from three assays. ND, not determined.
Model conception: available prior information and assumptions concerning the network
We constructed a simplified apoptotic network focused on the regulation of mitochondrial outer membrane permeabilization (MOMP). MOMP leads to irreversible cell death by releasing pro‐apoptotic factors such as cytochrome c (Cyt c) and Smac from the mitochondria to cytosolic, and the releasing channel, mitochondrial apoptosis channel (MAC), is composed of Bax/Bak. MOMP is controlled primarily through interactions among proteins of the Bcl‐2 family. Bcl‐2 family members can be classified into two groups: pro‐ and anti‐apoptotic factors. The former contains the following three subtypes: the MOMP effectors Bax and Bak; the select BH3‐only proteins termed ‘activators’ (Bim and Puma); and the other BH3‐only proteins, such as Bmf and Bad. Anti‐apoptotic members, such as Bcl‐2, Bcl‐xL and Mcl‐1, inhibit MOMP by binding these pro‐apoptotic members.
In our present experiments, using surface plasmon resonance (SPR) assay in vitro and co‐immunoprecipitation (co‐IP) assay in cells, we found that pBcl‐2 formed complexes with Bim, Bax, Bad, Bmf and Puma proteins, similar to those formed by Bcl‐2, but with different interaction affinities and kinetics. For these reasons, the model makes the assumption that pBcl‐2 did not alter the network topology, the production or degradation of proteins but acted though changing the protein interactions. Thus, we obtained a pBcl‐2/Bcl‐2 model with the proposed pBcl‐2 mechanism to further simulation and analysis (Figure 2A).
Figure 2.
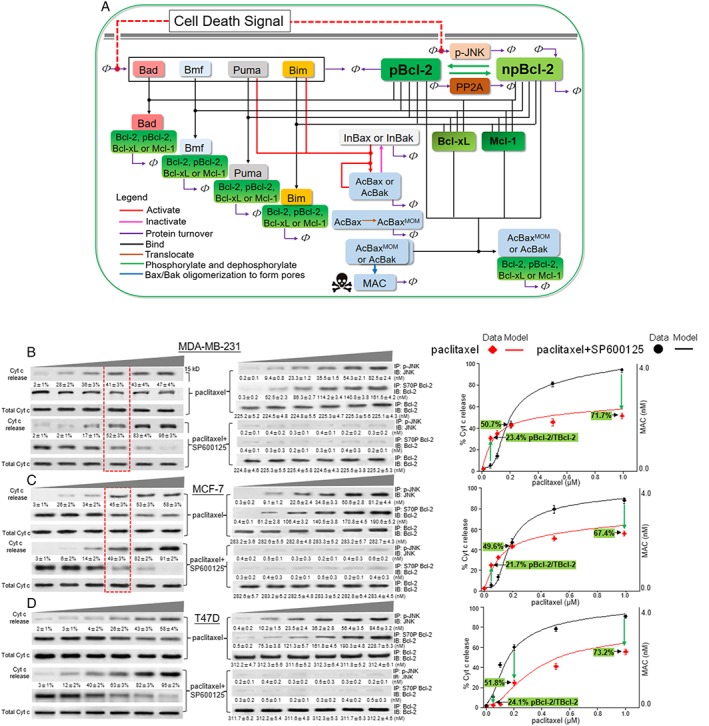
The pBcl‐2/Bcl‐2 model predicts MOMP in response to paclitaxel. (A) Mathematical model of Bcl‐2‐controlled MOMP that was formulated in terms of Bcl‐2 protein interactions. The interactions considered were (see key) Bax and Bak activation, inactivation, protein turnover, binding, Bax translocation, Bcl‐2 phosphorylation/de‐phosphorylation and Bax and Bak dimerization to form pores (MAC). All the corresponding equations are shown in Supporting Information Tables S1–S8. The red, dashed line indicates activation of Bcl‐2 kinases by the cell death signal, for example, paclitaxel. The death signal produced a model input by inducing BH3‐only proteins Bmf, Puma, Bad or Bim activated and accumulated on the mitochondrial outer membrane. MAC served as the model output. Bax and Bak are represented by Bax. (B–D), left panel: MDA‐MB‐231, MCF‐7 and T47D cells were treated with graded concentrations of paclitaxel (0.05, 0.1, 0.2, 0.5 and 1 μM) alone or in combination with SP600125 (20 μM) for 24 h, and then Cyt c release was analysed by immunoblotting. Total Cyt c was used as a loading control. Middle panel: After treatment with paclitaxel alone or in combination with SP600125, cells were counted, collected and lysed, and then quantitative immunoblotting assays for p‐JNK, pBcl‐2 and TBcl‐2 were performed as described in the Supporting Information . Right panel: Simulation of the MAC levels are shown by red and black lines. The experimental curve of Cyt c release (mean ± SD, n = 5,) is compared to the simulated MAC levels. The experimentally detected ratio of pBcl‐2 to TBcl‐2 as a function of paclitaxel concentration is labelled.
Finally, the model assumptions are supported by the good fit with the experimental data with paclitaxel, overexpression of BH3‐only proteins, effects of ABT‐199 and etoposide accompanied with the assay on protein stability for Bax, Bak, Bim and Puma.
Computational modelling of Bcl‐2 protein reactions
The signalling pathway of the Bcl‐2 family proteins was modelled by a pseudo‐reaction network. Using mass‐action kinetics, this network was subsequently translated into a set of ODEs which describe the changes in concentration of a single protein or a single protein complex. We translated the modelled protein–protein interaction from a general reaction notation to ODEs by a detailed guideline listed in Supporting Information Table S1. ODEs for npBcl‐2 protein interaction, pBcl‐2 protein interaction, Bcl‐2 phosphorylation/de‐phosphorylation and concentration of Bax, Bim and Bcl‐2 were parameterized by experiments (Supporting Information Tables S3–S6). Proteins that sustain turnover were modelled to reach equilibrium concentrations that were given by the interplay of protein production and degradation (kpro/kdeg). Based on this, the production rate was calculated by multiplying the actual protein concentration by the degradation constant rate. The other parameters for chemical reactions were derived from literature or fitted by experimental data (Supporting Information Table S8). Subsequently, ODEs were solved using MATLAB 7.3 (MathWorks, R2007b) function ode 15 s at time point t = 300 min.
Materials
Paclitaxel, ABT‐199 and etoposide were purchased from Selleck Chemicals (S1150, S8048 and S1225, Houston, TX, USA). SP600125 (S5567) was purchased from Sigma (St. Louis, MO, USA). The 36‐mer Bax BH3 (residues 49–84), Bak BH3 (residues 62–97), Bim BH3 (residues 136–171), Puma BH3 (residues 127–162), Bad BH3 (residues 137–172) and Bmf BH3 (residues 210–245) were synthesized at HD Biosciences (CL‐1655, CL‐1734, CL‐2123, CL‐1453, CL‐2248, CL‐1216, ShangHai, China).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
Bcl‐2 phosphorylation induces changes in the parameters of the kinetics of protein interactions
EEE‐Bcl‐2 has been confirmed previously to reproduce the function of native multi‐site pBcl‐2 (Deng et al., 2004). Here, we explored the changes in kinetics of protein interactions, induced by phosphorylation of Bcl‐2 using EEE‐Bcl‐2.
AAA‐Bcl‐2 and EEE‐Bcl‐2 were transfected into MCF‐7 cells in which endogenous Bcl‐2 was silenced (Supporting Information Figure S1A). Apoptosis signal‐regulating kinase 1 (ASK1) and JNK1 proteins were overexpressed in the MCF‐7 cell line to produce endogenous pBcl‐2 in mitochondria (Supporting Information Figure S1B) (Yamamoto et al., 1999). As shown in Figure 1A, Bcl‐2 was phosphorylated at T69, S70 and S87 in ASK1/JNK1‐transfected cells, as shown by immunoblotting assays with specific antibodies. TBcl‐2 could be completely pulled down in the co‐IP assays using the S70P Bcl‐2 antibody (Supporting Information Figure S1C), suggesting that almost all the Bcl‐2 was phosphorylated in ASK1/JNK1‐transfected cells. Using the TBcl‐2 antibody, which can detect both the phosphorylated and non‐phosphorylated forms of Bcl‐2 (npBcl‐2), and the S70P Bcl‐2 antibody, we performed co‐IP assays and observed similar amounts of Bax as well as other partners in the complexes (Figure 1A, right column), confirming complete Bcl‐2 phosphorylation in ASK1/JNK1‐transfected cells. As shown in Figure 1B, pBcl‐2 bound to more Bax and Bak and less to Bim, Puma and Bad in ASK1/JNK1‐transfected cells, than npBcl‐2 in cells transfected with empty vectors. Next, the changes in interaction strength were entirely reproduced in cells transfected with EEE‐Bcl‐2 compared with those transfected with wild‐type Bcl‐2 (WT‐Bcl‐2) or AAA‐Bcl‐2 (Figure 1C). Taking these data together, we concluded that EEE‐Bcl‐2 mimicked the activity of native pBcl‐2, in terms of changing the parameters of interaction with protein partners.
Figure 1.
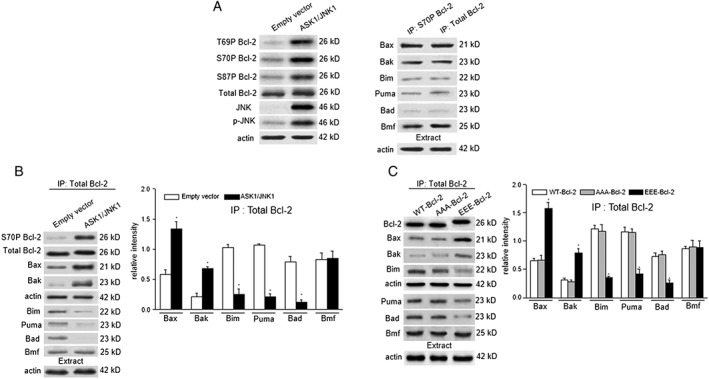
EEE‐Bcl‐2 reproduces the activity of native pBcl‐2 in changing the interaction parameters with partners. (A) Empty vector‐transfected control MCF‐7 cells or ASK1/JNK1‐cotransfected MCF‐7 cells were collected and lysed followed by immunoblotting analysis with T69P, S70P or S87P Bcl‐2 antibody as well as TBcl‐2, JNK or p‐JNK antibody. In ASK1/JNK1‐transfected MCF‐7 cells, S70P Bcl‐2 and TBcl‐2 immunoprecipitation (IP) assays were performed, and the immunoprecipitated fractions were analysed by immunoblotting for the indicated proteins. Actin was blotted in 10% cell lysates from each sample before immunoprecipitation to confirm equal loading. (A, C) In empty vector‐transfected or ASK1/JNK1‐transfected MCF‐7 cells (B) and WT‐Bcl‐2, AAA‐Bcl‐2 or EEE‐Bcl‐2‐transfected MCF‐7 cells in which endogenous Bcl‐2 was silenced (C), TBcl‐2 immunoprecipitations were performed, and the immunoprecipitated fractions were analysed by immunoblotting for the indicated proteins. Relative protein quantification is shown by the intensities of individual immunoblots normalized to that of actin. Data are the mean ± SD (n = 5). The levels of significance were evaluated by two‐tailed t‐test (*P < 0.05 significantly different from empty vector (B) or, **P < 0.01).
To quantitatively measure the changes in kinetics of protein binding, induced by phosphorylation of Bcl‐2, we performed SPR assays to compare the binding affinity constant (K D) and the association (k a) and dissociation (k d) rates of EEE‐Bcl‐2 and WT‐Bcl‐2 (full length with loop region, 1–206) for BH3 peptides from Bax, Bak, Bim, Puma, Bmf, Bad and Noxa (Supporting Information Figure S2 and Table 1). The increased and decreased binding affinities with different partners of EEE‐Bcl‐2 were found in agreement with the co‐IP results described above. Specifically, we determined that EEE‐Bcl‐2 binds Bax BH3 (1.2 nM) and Bak BH3 (4.1 nM) with approximately nine‐fold higher affinities compared with the WT‐Bcl‐2 (11.2 and 36.3 nM, respectively). In contrast, approximately six‐ to seven‐fold lower affinities were found for Bim BH3 (17.6 nM) and Puma BH3 (18.2 nM) towards EEE‐Bcl‐2 than those towards WT‐Bcl‐2 (2.5 and 3.2 nM, respectively), and the affinity loss increased to 31‐fold for Bad BH3 (264.7 nM vs. 8.5 nM). For the Bmf BH3 peptides, EEE‐Bcl‐2 displays a binding affinity similar to that of WT‐Bcl‐2 (7.1 vs. 7.8 nM). Both EEE‐Bcl‐2 and WT‐Bcl‐2 could not bind Noxa BH3. The changed pBcl‐2 interaction kinetics with other partners are given in Table 1. We also performed fluorescent polarization assays to compare the binding affinity constanst (K D) (Supporting Information Figure S3), which are in good agreement with the SPR data. Additionally, AAA‐Bcl‐2 exhibited similar binding affinities and kinetics to those of WT‐Bcl‐2, suggesting that the effect of glutamate substitution is dependent on the generation of phosphoepitopes at the three phosphorylation sites rather than destroying their original interactions with other residues in Bcl‐2.
Bcl‐2 phosphorylation regulates the Bcl‐2 network
A mixture of increasing and decreasing interaction strength changes induced by Bcl‐2 phosphorylation makes it difficult to identify its significance of enhancing or diminishing the anti‐apoptotic function of Bcl‐2. To understand the significance of pBcl‐2 from a network perspective, a mathematical model was conducted using ODEs (Supporting Information Tables S1–S8). This is a well‐established method based on the commonly accepted topology of the Bcl‐2 interaction network (Chen et al., 2007; Cui et al., 2008). Specifically, based on the pBcl‐2 interaction kinetics given in Table 1, especially the six sets of parameters for kon and koff values of pBcl‐2 interactions with partners (Supporting Information Table S6), we developed a pBcl‐2/Bcl‐2 model focused on the changes in kinetics induced by phosphorylation of Bcl‐2 (Figure 2A, for model details, see Supporting Information). The model integrates Bcl‐2 phosphorylation and de‐phosphorylation by kinases (JNK, ERK or PKC), here exemplified by JNK and phosphatases (PP2A).
The model was implemented in the MATLAB software. For details on the model conception, the kinetic reaction parameters, and the calibration of the model, see Supporting Information . The estimation of five unknown parameters was based on experimental data of overexpressing BH3‐only proteins to induce MOMP, showing that Cyt c release fits the modelled concentration of oligomerized pores (MAC) (Supporting Information Figure S4). To estimate the kinetic parameter values of kphos and kdephos, we used time course data for Bcl‐2 phosphorylation which was mediated by the active p‐JNK (Supporting Information Figure S5).
The model is applied to further simulation and analysis of apoptosis under conditions where Bcl‐2 is continuously phosphorylated. We established tumour cells exposed to paclitaxel as a case study because conflicting evidence regarding the anti‐apoptotic role of pBcl‐2 following paclitaxel treatment has been reported for decades.
We identified that paclitaxel induced JNK activation and the phosphorylation of Bcl‐2 (Thr69, Ser70 and Ser87) in the mitochondria of MDA‐MB‐231, MCF‐7 and T47D cells as previously reported (Srivastava et al., 1999) (Figure 2B–D and Supporting Information Figure S6). These three cell lines were treated with paclitaxel in a dose range of 0.05–1.0 μM. Immunoblotting assays showed that paclitaxel induced a dose‐dependent induction of p‐JNK and Bcl‐2 phosphorylation accompanied by apoptosis in these cells, and the JNK inhibitor SP600125 could inhibit Bcl‐2 phosphorylation and affect apoptosis (Figure 2B–D and Supporting Information Figure S7).
In MDA‐MB‐231 and MCF‐7 cells, we compared the dynamics of Cyt c release following paclitaxel treatment and paclitaxel plus SP600125 treatment. We found a turning point at 0.2 μM paclitaxel (Figure 2B, C, red box). Paclitaxel at concentrations of 0.05–0.2 μM induced less than half of pBcl‐2 in TBcl‐2, and pBcl‐2 acted in favour of apoptosis, as evidenced by a down‐regulation of Cyt c release when combined with SP600125 (Figure 2B,C, left and middle panel). However, paclitaxel at higher concentrations (0.2–1 μM) induced phosphorylation in more than half of the Bcl‐2 and, under these conditions, pBcl‐2 strongly inhibited apoptosis as shown by a marked up‐regulation of Cyt c release when combined with SP600125. When Bcl‐2 was mostly phosphorylated, only half of the total Cyt c could be released by paclitaxel compared with nearly complete Cyt c release with npBcl‐2 resulting from SP600125 (0.47 vs. 0.96 in MDA‐MB‐231 cells, and 0.58 vs. 0.91 in MCF‐7 cells). The percentage of Cyt c release was plotted as a function of paclitaxel concentration in paclitaxel‐treated cells (Figure 2B,C, right panel, red rhombus) and paclitaxel plus SP600125‐treated cells (black circles), respectively, to obtain experimental curves.
The apoptotic network involving pBcl‐2 interaction kinetics was then simulated with our pBcl‐2/Bcl‐2 model using the production rate of Bmf as one model input because Bmf was determined as the BH3‐only protein that connected paclitaxel‐induced microtubule perturbation to MOMP in MDA‐MB‐231 and MCF‐7 cells (Kutuk and Letai, 2010) and was comfirmed by us (Supporting Information Figure S8). Meanwhile, paclitaxel induced p‐JNK in a dose‐dependent manner (Figure 2B,C, middle panel). Thus, the concentrations of Bmf and p‐JNK were considered as surrogates of the chemotherapeutic dose and were used as model inputs (for mathematical details, see Supporting Information ). The simulation results showed excellent agreement with the cell‐based experiments in that the shape of the experimental curve and the turning point at 0.2 μM paclitaxel were correctly reproduced by our model (Figure 2B,C, right panel). The tuning point was also detected in MCF‐7 cells transfected with WT‐Bcl‐2 and AAA‐Bcl‐2 respectively, while EEE‐Bcl‐2 transfected cells showed the highest resistance to paclitaxel (Supporting Information Figure S9).
Thus far, we reconciled the conflicting opinions regarding the enhancement or inactivation of the anti‐apoptotic function of Bcl‐2 by phosphorylation. In 1999, Srivastava et al. applied paclitaxel in MDA‐MB‐231 cells and showed the anti‐apoptotic function of Bcl‐2 to be inactivated by phosphorylation at a concentration of 0.1 μM (Srivastava et al., 1999). In contrast, results from other groups conflict with the pBcl‐2‐inactivating mode in the same cell line using the same stimuli but at different concentrations. For example, Michiels et al. found that, in MDA‐MB‐231 cells, inhibition of Bcl‐2 phosphorylation by JNK siRNA promoted paclitaxel‐induced apoptosis at a concentration of 50 μM, thus implying that pBcl‐2 enhanced anti‐apoptotic function (Notte et al., 2013). When integrating the modelling with experimental validation, we found that the two apparently conflicting phenomena indeed occurred sequentially. A turning point emerged at a concentration of 0.2 μM paclitaxel, below which less than 50% of the TBcl‐2 is phosphorylated in MDA‐MB‐231 cells, and pBcl‐2 acts to weaken the anti‐apoptotic function of the Bcl‐2 network. However, when the concentration exceeds 0.2 μM, more than 50% of the TBcl‐2 is phosphorylated, and now pBcl‐2 acts to significantly block apoptosis. There is a consensus that pBcl‐2 enhances the anti‐apoptotic ability of Bcl‐2 in response to >1 μM paclitaxel (Deng et al., 2004).
In T47D cells, a different curve was obtained. As shown in Figure 2D (left and middle panels), throughout the concentration range of paclitaxel from 0.05 to 1 μM, which induces increasing Bcl‐2 phosphorylation (from 20 to 70%), pBcl‐2 functions to inhibit paclitaxel‐induced Cyt c release. The experimental curves could also be reproduced by the pBcl‐2/Bcl‐2 model when Puma is simulated as the input (Figure 2D, right panel) because Puma served as the BH3‐only protein that connected paclitaxel to MOMP in T47D cells (Kutuk and Letai, 2010), a finding that was verified by us (Supporting Information Figure S8). The consensus modelling and experimental results showed that pBcl‐2 enhanced the anti‐apoptosis function of Bcl‐2 towards Puma throughout the entire range of phosphorylation. Trajectories of the other model species were shown in Supporting Information Figure S10.
The extent of Bcl‐2 phosphorylation and changes in kinetics of protein‐protein binding, induced by phosphorylation determine the dynamics of the apoptotic network
To further investigate the availability of the turning point for paclitaxel‐induced apoptosis in different cells with variability in protein concentrations, we investigated the minimum dose of BH3 ligands necessary to induce MOMP (referred to as dose ω hereafter) by numerical simulations of the MAC level, where a sudden increase in MAC level will occur if the stimuli increases beyond the dose ω. The location of ω reflects the death threshold.
The level of p‐JNK determines the extent of Bcl‐2 phosphorylation. By varying kpro_p‐JNK, the model simulation in response to Bmf showed that p‐JNK levels that were lower than 36 nM shifted the dose ω to the left from the original non‐phosphorylated location, suggesting that phosphorylation diminished the anti‐apoptosis function of Bcl‐2 against Bmf (Figure 3A, left panel). However, when p‐JNK level was increased above 36 nM, a significant rightward shift of the dose ω occurred. Under these conditions, approximately three‐fold higher production of Bmf was required to switch on apoptosis when p‐JNK (80 nM) led to an 80% ratio of pBcl‐2/TBcl‐2 (Figure 3A, left panel). Importantly, the p‐JNK level (36 nM) that makes the turning point between diminishing and enhancing the anti‐apoptosis function remained unchanged when the Bcl‐2 protein levels were varied at 100, 200 and 300 nM (Supporting Information Figure S11), confirming the availability of the turning point in different cells. The p‐JNK level at 36 nM led to a 50% ratio of pBcl‐2/TBcl‐2, which correctly reproduced the experimental data on the turning point at 0.2 μM paclitaxel, under which the ratio of pBcl‐2/TBcl‐2 was also 50%, although the levels of Bcl‐2 or pBcl‐2 were different in both MDA‐MB‐231 and MCF‐7 cells (Figure 2B,C, middle panel). In conclusion, the proportion of Bcl‐2 that is phosphorylated, rather than the levels of Bcl‐2 or pBcl‐2, determines the turning point from diminishing to enhancing anti‐apoptotic function. A low proportion of Bcl‐2 phosphorylation facilitates cell death induced by Bmf, but cell apoptosis was strongly blocked when pBcl‐2 was a higher proportion (>50%) of TBcl‐2.
Figure 3.
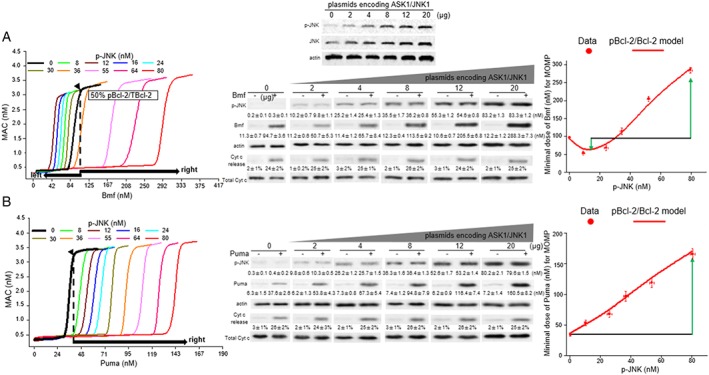
The extent of Bcl‐2 phosphorylation, which is regulated by changes in the kinetics of interactions between p‐JNK and pBcl‐2, determines the dynamics of apoptosis. (A, B) Left panel: The diagram for MAC levels using the levels of Bmf (A) or Puma (B) as the model inputs. Different concentrations of p‐JNK determines the extent of Bcl‐2 phosphorylation. Each curve represents the variation in dose ω with Bmf or Puma concentration, at a given concentration of p‐JNK. Middle panel: The experimentally determined dose of Bmf or Puma under different p‐JNK level conditions to induce Cyt c release. A series of MCF‐7 cells stably expressing different levels of JNK1 and p‐JNK were derived from each clone of cells transfected with increasing concentrations of plasmids encoding ASK1/JNK1 (up to 20 μg of plasmids). Then, these cells were transiently transfected with increasing concentrations of plasmids encoding Bmf (up to 20 μg of plasmids) or Puma (up to 10 μg of plasmids) for 24 h, after which Cyt c release was assayed by immunoblotting (Figure S13). The beginning of Cyt c release after Bmf or Puma expression were specifically shown. Equal loading was confirmed by stripping the immunoblot and reprobing it for actin. Right panel: The experimentally derived values of BH3‐only protein concentration required for MOMP (dose ω; mean ± SD, n = 5) are compared to the curves derived from the simulation.
In contrast to the biphasic changes in dose ω triggered by Bmf, when Puma, Bim and Bad were the model inputs, Bcl‐2 phosphorylation resulted in the rightward shift of dose ω, from the very beginning of Bcl‐2 phosphorylation (Figure 3B, left panel, and Supporting Information Figure S12), a finding that was consistent with the paclitaxel experiments.
To evaluate whether our model was able to reproduce the experimental data, we co‐transfected MCF‐7 cells with increasing concentrations of plasmids encoding each of Bmf, Puma, Bim and Bad with ASK1/JNK1 and then determined the concentration of individual BH3‐only proteins required to induce Cyt c release under different p‐JNK levels (Figure 3A,B, middle panel, and Supporting Information Figure S13). The experimental results showed excellent agreement with the model predictions that Bmf triggered a biphasic regulation of apoptosis whereas, with the other three BH3‐only stress contexts, pBcl‐2 always enhanced the network against apoptosis (Figure 3A,B, right panel, and Supporting Information Figure S12). The result of Puma stress is consistent with the report that pBcl‐2 inhibited etoposide‐induced apoptosis, which is mediated through p53‐activated Puma (Ito et al., 1997; Dai et al., 2013; Jamil et al., 2015), and the ‘IL‐3‐induced model’ supposed by Deng et al. (Ito et al., 1997; Deng et al., 2000, 2004; Ekert et al., 2006).
Next, a single‐parameter sensitivity analysis was performed. By increasing or decreasing each of the model parameters by 10% and recording the percentage change of dose ω, a sensitivity spectrum of all parameters was achieved. Twenty sensitive parameters that led to a dose ω shift above 10% are shown in Supporting Information Figure S14. Although the protein half‐life of Bax, Bak, Puma, Bim, Bcl‐2, Bcl‐xL and Mcl‐1 was supposed to be sensitive for the network, they had no influence on the pBcl‐2‐induced network because the half‐life of Bax, Bak, Puma and Bim before and after Bcl‐2 phosphorylation in paclitaxel‐induced apoptosis experiments showed that pBcl‐2 has little influence on their stability (Supporting Information Figure S15A,B). In addition, paclitaxel treatment did not influence the expression levels of Bcl‐2, Bcl‐xL and Mcl‐1, at least in the time period of the experiment (0 to 24 h) (Supporting Information Figure S15C). As such, only the changes in kinetics, induced by phosphorylation of Bcl‐2, in binding of activated Bax/Bcl‐2, Bak/Bcl‐2, Bim/Bcl‐2, Puma/Bcl‐2, Bmf/Bcl‐2 and Bad/Bcl‐2 (kon and koff value in Supporting Information Table S6) affected the dynamic properties of the pBcl‐2/Bcl‐2 network. The changes in interaction kinetics of Bcl‐xL and Mcl‐1 were other sensitive parameters and not changed in the pBcl‐2/Bcl‐2 model.
To illustrate how Bcl‐2 phosphorylation‐induced changes in the parameters of protein interaction kinetics can account for the biphasic effects on dose ω, under Bmf stress, we tried to input apparent binding affinities of Bax/Bcl‐2 and Bim/Bcl‐2 which is continuously changed as the proportion of pBcl‐2 increased in TBcl‐2 (Supporting Information Figure S16, left panel) to model the Bcl‐2 interactions. Indeed, when blindly varying apparent binding affinity of Bcl‐2/Bax and Bcl‐2/Bim in the mode during Bmf stress, a mixed increase and decrease in dose ω for MOMP was identified (Supporting Information Figure S16, right panel). For example, when Kapp_Bcl‐2/Bax is increased from 0.1 to 0.2, the dose ω required for MOMP is raised between the outputs with the same Bcl‐2/Bim affinity (shown as two black squares between the 1st and 2nd columns, Supporting Information Figure S16, right panel). However, when Kapp_Bcl‐2/Bim was decreased (in the order of black, red, blue, pink and green), the dose ω was also decreased. Thus, the dose ω found with an apparent affinities combination corresponding to 20% pBcl‐2/TBcl‐2, was lower than that appearing at 0% pBcl‐2/TBcl‐2. In addition, compared to 0% pBcl‐2/TBcl‐2, a similar dose ω appears under apparent affinities combination corresponding to 50% pBcl‐2/TBcl‐2. Then, dose ω under an apparent affinities combination corresponding to 70% pBcl‐2/TBcl‐2 is raised to a higher value. Thus, the biochemical terms at the level of interactions change induced by Bcl‐2 phosphorylation could account for the turning point under Bmf stress, and the quantified point is determined by a combination of apparent affinities, which is decided by the pBcl‐2/TBcl‐2 ratio.
Thus far, we identified that the extent of Bcl‐2 phosphorylation and phosphorylation‐induced protein–protein interaction kinetics underlies the dynamics of the apoptosis network.
The model correctly predicts ABT‐199 and etoposide
Having established that the pBcl‐2/Bcl‐2 model fits the experimental data, we used it to predict the effects of combining paclitaxel with ABT‐199, a selective Bcl‐2 inhibitor that has been recently approved by the FDA (Souers et al., 2013; Deeks, 2016). The kinetics of the interactions of ABT‐199 with WT‐Bcl‐2 and with EEE‐Bcl‐2 were assayed by SPR, and the data showed an approximately 100‐fold decreased binding affinity towards Bcl‐2 by the phosphorylation‐mimicking mutant (Table 1), consistent with our previous report (Song et al., 2016).
Using the concentration of ABT‐199 alone or plus paclitaxel at different ratios as the model input, the dose ω that represents a surrogate of the ABT‐199 alone or in combination required for MOMP was predicted. The degree of synergism between ABT‐199 and paclitaxel was determined by the combination index (CI), which were calculated based on either predicted or experimentally assayed dose of ABT‐199 alone, paclitaxel alone and the combination of the two drugs at different ratios (for mathematical and experimental details, see Supporting Information ). As shown in Figure 4A,B, no synergism (CI > 1) at any ratio of the two drugs was accurately predicted by the pBcl‐2/Bcl‐2 model in MDA‐MB‐231 and T47D cells (r = 0.83 and 0.78, respectively). If pBcl‐2 was not included in the model, a synergistic effect of paclitaxel/ABT‐199 combination was predicted (CI < 1). Consistent with the model simulation, experimental measurements on paclitaxel/ABT‐199 combination showed synergism when adding SP600125 to inhibit pBcl‐2 (CI < 1). These results demonstrated that pBcl‐2 enhances anti‐apoptotic ability against the combination of paclitaxel and ABT‐199.
Figure 4.
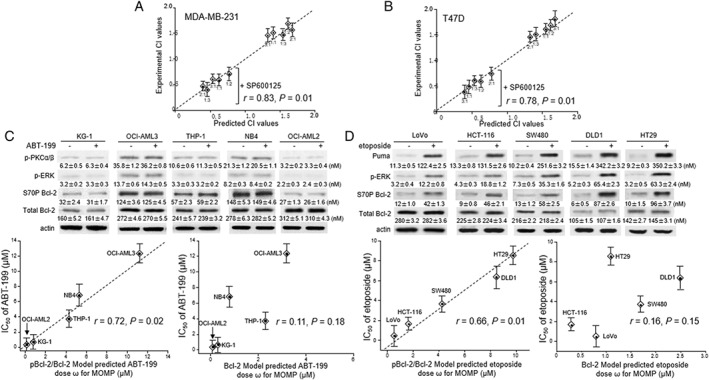
The pBcl‐2/Bcl‐2 model predicts the MOMP in response to ABT‐199 or etoposide. (A, B) The experimentally derived CI values for the ABT‐199/paclitaxel combination were plotted against those predicted from the pBcl‐2/Bcl‐2 model, using MDA‐MB‐231 (A) and T49D cells (B), with and without SP600125. Data are the mean ± SD (n = 5). Spearman's correlation (r) and P values are shown. (C, D) Upper panel: Expression of p‐PKCα/β, p‐ERK and Bcl‐2 proteins in the five AML cell lines (A) before and after ABT‐199 treatment (0.2 μM for OCI‐AML2 and KG‐1; 4 μM for THP‐1 and NB4; 10 μM for OCI‐AML3; 24 h) and in the five colorectal cancer cell lines (B) before or after etoposide treatment (1 μM for LoVo and HCT‐116; 2 μM for SW480; 4 μM for DLD1 and HT29; 24 h). Equal loading was confirmed by stripping the immunoblot and reprobing it for actin. Lower panel: The experimental IC50 values for apoptosis induced by ABT‐199 or etoposide treatment after 48 h were plotted against the dose ω predicted by the pBcl‐2/Bcl‐2 model or the Bcl‐2 model Correlations (r values shown) between the experimental IC50 values and the predicted dose ω predicted by pBcl‐2/Bcl‐2 model were statistically significant whereas those using the Bcl‐2 model were not significant .
Then, we evaluated the applicability of the model to predict drug effects against cancer cells expressing various levels of Bcl‐xL, Mcl‐1, Bcl‐2 and pBcl‐2 under different physiological conditions. Five AML cell lines were detected with varied expression of activated p‐PKC and p‐ERK, which produced pBcl‐2 (Figure 4C, top panel). We compared the accuracy of prediction between the pBcl‐2/Bcl‐2 model including the two Bcl‐2 kinases as model species and the Bcl‐2 model without considering Bcl‐2 phosphorylation (for mathematical details, see Supporting Information ). The predicted dose ω that represents a surrogate of the ABT‐199 dose required for MOMP was calculated. The experimental assayed IC50 value for ABT‐199‐induced apoptosis is shown in comparison with the predicted dose ω (Figure 4C, middle panel). Values of dose ω predicted by the pBcl‐2/Bcl‐2 model showed a positive correlation with the experimental IC50 value (r = 0.72, P = 0.02; Figure 4C, bottom panel). However, if we did not include Bcl‐2 phosphorylation in the model, the predictions showed no correlation with the experimental data (r = 0.11, P = 0.18).
Further, we used the pBcl‐2/Bcl‐2 model to predict a genotoxic drug, etoposide (Karpinich et al., 2002; Stefanelli et al., 2002; Wang et al., 2007). In five p53‐competent colorectal cancer cell lines, Puma was confirmed as connecting etoposide with mitochondrial apoptosis, as previously reported (Wang et al., 2007). Also. etoposide induced activation of ERK1/2, one of pBcl‐2 kinases (Stefanelli et al., 2002). Consistent with these earlier data, we detected increased p‐ERK and pBcl‐2 upon etoposide treatment (Figure 4D, top panel). The predicted dose ω , using the concentration of Puma and p‐ERK as a surrogate of etoposide, showed a positive correlation with the IC50 value (r = 0.66, P = 0.01; Figure 4D, middle and bottom panel). The predictions failed when Bcl‐2 phosphorylation was not considered in the model (r = 0.16, P = 0.15).
To examine which interactions are properly accounted for in the pBcl‐2/Bcl‐2 model but not the Bcl‐2 model to better explain the cellular response to ABT‐199 and etoposide, we performed Bcl‐2 co‐IP experiments in NB4 and HT‐29 cells respectively. The disruption of Bcl‐2/Bax complex was assessed by measuring the cellular response to 5 μM ABT‐199, and formation of the Bcl‐2/Puma complex was assessed by measuring the cellular response to 10 μM etoposide, which is accurately predicted by pBcl‐2/Bcl‐2 (Supporting Information Figure S17). These results further confirmed the accuracy of our modelling involving Bcl‐2 phosphorylation and showed the significance and importance of considering Bcl‐2 phosphorylation when predicting the effects of anti‐tumour drugs.
Discussion
We successfully elucidated the outcomes of Bcl‐2 phosphorylation by integrating mathematical modelling and experimental validation. Our investigation of an entire Bcl‐2 phosphorylation procedure explored the dynamics of the apoptotic network, rather than adopting a local focus. We provided a precise, quantitative, predictive and unifying mechanism of an apoptosis network regulated by Bcl‐2 phosphorylation that reconciles conflicting opinions that have persisted for decades. We identified a turning point for the first time. When the exposure to paclitaxel was was lower than 0.2 μM in those breast cancer cell lines where Bmf is the sensitizer, pBcl‐2 showed an inactivated anti‐apoptotic function, relative to Bcl‐2. When the concentration was higher than 0.2 μM, pBcl‐2 exhibited enhanced anti‐apoptotic function in these cells, similar to that in other cell lines, and was sensitized by the other BH3‐only proteins Bim, Puma or Bad. The existence of such a turning point is the root of the controversy.
Notably, 0.2 μM is the IC50 value for paclitaxel in most breast cancer cell lines. However, paclitaxel administration to patients with breast cancer results in a peak plasma concentration of 4.5 μM (Fogli et al., 2002). As shown by six commonly used chemotherapeutic drugs (cisplatin, carboplatin, 5‐FU, MTX, bleomycin and vincristine), the peak plasma concentration is at least threefold higher than the IC50 values in cell‐based experiments (Schuler et al., 2002). As such, it is more meaningful to focus on in vivo drug concentrations higher than the in vitro IC50 values when we evaluate the drug effects. Therefore, in the clinical setting, pBcl‐2 definitely plays an enhanced anti‐apoptotic role.
The pBcl‐2/Bcl‐2 ratio simulation and parameter sensitivity analysis, combined with experimental validation, showed that the changes in Bcl‐2 interaction kinetics, induced by phosphorylation, are the major factors affecting the dynamics of the Bcl‐2 network. We suspected that pBcl‐2 functions differently in binding BH3‐only proteins or Bax and Bak due to the alterations in the BH3 groove following phosphorylation. Our previous study has identified the first low MW pBcl‐2 inhibitor, which is structurally slightly different from the non‐pBcl‐2 (npBcl‐2) inhibitor (Song et al., 2015). It is the most convincing evidence so far to illustrate that the structure of the BH3 groove changes upon phosphorylation. Based on the semi‐quantitative evidence that EEE‐Bcl‐2 reproduces the binding kinetics of pBcl‐2, by cellular co‐IP assays in this study, quantitative binding kinetics of k a, and k d values were obtained for EEE‐Bcl‐2 to replace those of native pBcl‐2. Particularly, we found that the effects of the phospho‐mimicking mutation on K D were due primarily to the change in k d. Without any information on the structure of pBcl‐2, it is difficult to infer k d from the K D by analysing short‐range (related to k d) and long‐range interactions (related to k a) (Schreiber, 2002). Additionally, it is difficult to calculate it by molecular dynamics simulation. Here, we established that EEE‐Bcl‐2 could quantitatively reproduce the pBcl‐2 binding kinetics, providing a convenient method for other agents to obtain their binding kinetics towards pBcl‐2. It is worth noting that the mutants are not natural and thus may behave differently from native pBcl‐2 in other aspects. The biological effects of EEE‐Bcl‐2 should interpreted with caution.
The observation of decreased affinity of pBcl‐2 to Bim was not consistent with a previous study by Dai et al. (2013). Their SPR results showed that Bim increased its binding affinity towards both the non‐phosphorylated mimicking Bcl‐2 mutant (S70A‐Bcl‐2) and the phosphorylated mimicking Bcl‐2 mutant (S70E‐Bcl‐2) to a similar extent. These data are in contrast with the present studies by using non‐phosphorylated mimicking Bcl‐2 mutant, which showed that the partner proteins bound to S70A‐Bcl‐2 were less than those bound to S70E‐Bcl‐2, as detected by co‐IP (Deng et al., 2000, 2004; Bassik et al., 2004; Wang et al., 2005; Konopleva et al., 2006). However, our binding parameters of AAA‐Bcl‐2 and EEE‐Bcl‐2 are convincing because of the consistency in the K D values from the three methods, FP, ITC and SPR, that we were able to obtain.
Phosphorylation‐induced Bcl‐2 interaction kinetics determine not only the significance of pBcl‐2 but also the drug lethality, either as a single agent or in combination. Our model allows three types of prediction to be made about mechanisms involving Bcl‐2.
(i) Prediction of the Bcl‐2 inhibitors and chemotherapeutic drugs (e.g. cisplatin, paclitaxel and etoposide) alone or in combination. To calculate the effective dose of Bcl‐2 inhibitor as monotherapy, its binding kinetics towards pBcl‐2 and npBcl‐2 need to be parameterized, as we have done for ABT‐199 in this study. Then, the concentrations of Bcl‐2 inhibitor are used as the model input to calculate the dose of Bcl‐2 inhibitor (given alone) needed to trigger MOMP. Because most of the chemotherapeutic drugs have been identified to trigger MOMP by activating certain BH3‐only proteins (Vo and Letai, 2010; Elkholi et al., 2011), for example, Puma in the context of DNA damage drugs, using a quantitative curve of drug‐induced Puma concentration as the model input, the dose of the chemotherapeutic drug (given alone) to trigger MOMP could be calculated. After the first Bcl‐2 inhibitor ABT‐199 was approved, its combination with other available chemotherapeutic drugs has been of great interest. To calculate the combination doses that trigger MOMP, both concentrations of chemotherapeutic drug‐induced BH3‐only proteins and Bcl‐2 inhibitors at fixed ratios are used as the model input. Moreover, Bcl‐2 phosphorylation has been shown to occur under various chemotherapeutic drugs – for example etoposide, retinoic acid and paclitaxel (Hu et al., 1998; Makin and Dive, 2001). Thus, although the previous Bcl‐2 network modelling cannot accurately predict the drug lethality,our pBcl‐2/Bcl‐2 mathematical modelling could be used.
(ii) Prediction of the kinase inhibitors alone or in combination. Kinase inhibitors are the best developed target anti‐tumour drugs and more of them are becoming first‐line anti‐tumour drugs (Wu et al., 2015). The prediction of kinase inhibitors is important for precision medicine. Based on the available equations (Yang et al., 2005), a single dose of kinase inhibitors that trigger MOMP could be calculated by a curve with decreased p‐JNK, p‐ERK or p‐PKC concentrations as the model input.
(iii) Prediction of the anti‐tumour drugs targeting the extrinsic apoptosis pathway (e.g. TRAIL) (Holoch and Griffith, 2009). Because caspase 8 activates tBid, a BH3‐only activator, it is the only known species that connects extrinsic apoptosis stimuli with the intrinsic apoptosis pathway. Thus, a quantitative curve of caspase 8 activation and tBid cleavage after TRAIL treatment is needed as the model input. Also, using model‐merging methods – for example, SBMLmerge – our pBcl‐2/Bcl‐2 modelling could be merged with a mass‐action‐based, extrinsic apoptosis pathway to predict the anti‐tumour effects (Krause et al., 2010).
Because the phosphorylation is embedded inside the apoptosis network and induces many changes in protein binding kinetics that disturb the network, the response to different stimuli is non‐uniform, leading to a mis‐understanding of pBcl‐2 functionalities when the focus is a linear pathway instead of the entire network. The validation of our pBcl‐2/Bcl‐2 modelling identified that the significance of Bcl‐2 phosphorylation must be explored at the network and system levels. The clarification that the enhanced anti‐apoptotic function through Bcl‐2 phosphorylation is predominant and depends on pBcl‐2 binding kinetics resolved a long‐standing controversy. This modelling could provide predictions for agents damaging DNA, Bcl‐2 inhibitors, paclitaxel and other anti‐tumour drugs involved in the Bcl‐2 pathway.
It has been reported that Bcl‐2 phosphorylation can retard the G1/S transition. We have not included this possible outcome in our pBcl‐2/Bcl‐2 model (Deng et al., 2003). We do not expect that such an effect would influence the dynamics of the anti‐apoptotic effect of pBcl‐2 in mitochondria. However, cells can generally withstand stress better in G1/G0 than during DNA replication. Therefore, cell cycle retardation and the anti‐apoptotic effects of Bcl‐2 phosphorylation may provide a cooperative function for Bcl‐2 in managing cell stress.
Author contributions
Z.Z. conceived the project. T.S. and Z.Z. designed the experiments. T.S., P.W., X.Y., G.C. and Y.F. performed the experiments and analysed the data. T.S., P.W. and A.W. performed the computational modelling. Z.Z. and T.S. wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Overexpression of different Bcl‐2 variants in Bcl‐2‐silenced MCF‐7 cells, and ASK1/JNK1 proteins to phosphorylate endogenous Bcl‐2.
Figure S2 Kinetics characteristics of BH3 peptides binding to WT‐Bcl‐2 and EEE‐Bcl‐2 as measured by SPR.
Figure S3 Binding affinities of BH3 peptides to WT‐Bcl‐2 and EEE‐Bcl‐2 as measured by FPAs.
Figure S4 Statistical analysis of the best‐fit parameter set.
Figure S5 Experimental‐based parameter estimations for phosphorylation and de‐phosphorylation kinetics.
Figure S6 Paclitaxel induced Bcl‐2 phosphorylation at three phosphorylation sites in mitochondrialby active (phosphorylated) form of JNK but not ERK or PKC.
Figure S7 The apoptosis inducing effect of paclitaxel alone or in combination with SP600125 in the three selected breast cancer.
Figure S8 Paclitaxel induced mitochondrial apoptosis via Bmf in MDA‐MB‐231 and MCF‐7 cells, and via Puma in T47D cells.
Figure S9 The apoptosis inducing effect of paclitaxel in MCF‐7 cells transfected with WT‐Bcl‐2, AAA‐Bcl‐2 and EEE‐Bcl‐2 respectively.
Figure S10 Trajectories of model species for the three selected breast cancer cell lines.
Figure S11 The p‐JNK controlled extent of Bcl‐2 phosphorylation, rather than the levels of Bcl‐2 proteins, determines the tuning points from diminishing to enhancing antiapoptotic ability of Bcl‐2 by phosphorylation.
Figure S12 Bcl‐2 phosphorylation caused a right shift of dose ω induced by Bim or Bad stress, which could reproduce experimental data.
Figure S13 Different p‐JNK level influenced the dose of Bmf or Puma to induce cytochrome c release.
Figure S14 Percentage change of the dose ω in response to a 10% increase or decrease of each parameter.
Figure S15 Paclitaxel did not influence the half‐life of Puma, Bim, Bax and Bak and the expression level of Bcl‐2, Bcl‐xL and Mcl‐1.
Figure S16 Apparent affinity of Bcl‐2/Bax and Bcl‐2/Bim complex as a function of pBcl‐2/TBcl‐2 ratio and the effects on model of MOMP in response to Bmf.
Figure S17 Changes of Bcl‐2 interactions in NB4 and HT29 cells upon ABT‐199 or etoposide treatment.
Figure S18 Quantification of protein levels of Bcl‐2, Bim, Bax and Bak in MDA‐MB‐231, MCF‐7 and T47D cells.
Table S1 Translation of protein interactions into Ordinary Differential Equations Protein reactions were grouped into different classes and translated into a mathematical form. (A) Irreversible Bax or Bak activation by BH3‐only activator protein (Bim and Puma) or by auto‐activation. BH3‐only activator protein were recycled after one activation so that the concentration of activator was unchanged in this chemical reaction. Changes of Bax or Bak concentration over time were proportional to the product of reactant concentrations. (B) Protein turnover of a R1was modelled to consist of constant protein production and degradation. The protein degradation rate of R2was modelled to be proportional to the actual protein concentration. (C) Irreversible Bax or Bak inactivation. Changes of Bax or Bak concentration over time were proportional to the product of reactant concentrations. (D) Translocation of activated cytosolic Bax to mitochondrial membrane was modelled by a first order reaction. The translocated protein was considered as different model entity “AcBaxMOM”. (E) Reversible reactions of two proteins R1and R2forming a complex R1/R2. Change of protein concentration was given by balancing binding on and off reactions. (F) Reversible reactions of two Bax or Bak forming pores (MAC). Change of protein concentration was given by balancing forward and backward reactions. (G) Irreversible Bcl‐2 phosphorylation by kinases (p‐JNK, ERK or PKC) and Bcl‐2 dephosphorization by PP2A. The concentration of kinases and PP2A was unchanged in this chemical reaction. Changes of npBcl‐2 or pBcl‐2 concentration over time were proportional to the product of reactant concentrations.
Table S2 Bax and Bak activation kinetics by BH3‐only activator protein and auto‐activation. According to the literature report on that Bax and Bak conformational change in each activation case is equivalent, we assumed the same kinetic constants for Bax and Bak activation. Activation kinetic parameters of Bax and Bak by Bim or Puma were fitted by data set I‐IV in Figure S4.
Table S3 Degradation rates as used in in the model. Proteins and protein complexes that were modelled to be subjected to degradation are depicted. Based on the experimental data, as shown in Figure S12C, in which the protein level of Bcl‐2 was not changed following phosphorylation, we considered for pBcl‐2the same half‐life time as for Bcl‐2 (1200 min). In addition, we considered for Bcl‐xL the same half‐life time as for Bcl‐2 due to the similar amino acid sequence (EMBOSS Needle alignment, similarity 53%). The half‐life times of Bim, Puma Bax andBak were assayed as shown in Figure S12 Aand B. The half‐life times of other proteins were derived from the literature. Half‐life times of most complexes of anti‐and pro‐apoptotic proteins were modelled to be the same as the half‐life time of the Bcl‐2/tBid complex (75 min). The degradation constants kdegwere calculated from the half‐life time by the following equation: kdeg = ln (2) / (60*t1/2).
Table S4 Kinetics for Bax and Bak inactivation. According to the literature report on equivalent function of Bax and Bak, the same kinetic constants for Bax and Bak inactivation were used. The parameter was 25 fitted by data set I‐IV in Figure S4.
Table S5 Kinetics for translocation of activated cytosolic Bax to mitochondrial membrane. Activated cytosolic Bax was modelled to translocate with a half‐life time 1/10 min (k = 1.16E‐01 s‐1) to the mitochondrial membrane.
Table S6 Kinetics for inhibition of BH3‐only proteins and effectors Bax and Bak by anti‐apoptotic proteins. The association and dissociation constants for npBcl‐2 and pBcl‐2 binding BH3‐only proteins and effectors were taken from SPR experiment data in Table 1. The association and dissociation constants for Bcl‐xL and Mcl‐1 were taken from literature as indicated. No binding of Mcl‐1 to Bax and Mcl‐1 to Bad were detected.
Table S7 Kinetics for Bax and Bak homo‐or hetero‐dimerization Activated Bax and Bak were modelled to homo‐or hetero‐dimerize in the mitochondrial outer membrane. Given on the equivalent function of Bax and Bak, the same kinetic constants for them were assumed. The reaction constant k+/k‐was fitted by data set I‐IV in Figure S4. We fixed k‐value of 0.02, and k + was obtained by k + = k+/k‐ × k‐.
Table S8 Kinetics for Bcl‐2 phosphorylation and de‐phosphorylation Activated p‐JNK, p‐ERK or p‐PKC were modelled to catalyse Bcl‐2 phosphorylation in the mitochondrial outer membrane. PP2A was modelled to catalyse Bcl‐2 de‐phosphorylation. The reaction constantskphosby p‐JNK and kdephosby PP2A were fitted by data set V in Figure S5. We assumed the same kinetic constants for p‐JNK, p‐ERK or p‐PKC.
Acknowledgements
This research was supported by the Natural Science Foundation of China (81570129, 21502015 and 81430083). This work was supported by the Fundamental Research Funds for the Central Universities (DUT17LK32).
Song, T. , Wang, P. , Yu, X. , Wang, A. , Chai, G. , Fan, Y. , and Zhang, Z. (2019) Systems analysis of phosphorylation‐regulated Bcl‐2 interactions establishes a model to reconcile the controversy over the significance of Bcl‐2 phosphorylation. British Journal of Pharmacology, 176: 491–504. 10.1111/bph.14555.
References
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ (2004). Phosphorylation of BCL‐2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J 23: 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barille‐Nion S, Bah N, Vequaud E, Juln P (2012). Regulation of cancer cell survival by Bcl‐2 family members upon prolonged mitotic arrest: opportunities for anticancer therapy. Anticancer Res 32: 4225–4233. [PubMed] [Google Scholar]
- Blagosklonny MV, Alvarez M, Fojo A, Neckers LM (1996). Bcl‐2 protein downregulation is not required for differentiation of multidrug resistant HL60 leukemia cells. Leuk Res 20: 101–107. [DOI] [PubMed] [Google Scholar]
- Chen C, Cui J, Zhang W, Shen P (2007). Robustness analysis identifies the plausible model of the Bcl‐2 apoptotic switch. FEBS Lett 581: 5143–5150. [DOI] [PubMed] [Google Scholar]
- Chae S, Kim YB, Lee JS, Cho H (2012). Resistance to paclitaxel in hepatoma cells is related to static JNK activation and prohibition into entry of mitosis. Am J Physiol Gastrointest Live Physiol 302: G1016–G1024. [DOI] [PubMed] [Google Scholar]
- Cui J, Chen C, Lu H, Sun T, Shen P (2008). Two independent positive feedbacks and bistability in the Bcl‐2 apoptotic switch. Plos One 3: e1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Ding H, Meng XW, Lee SH, Schneider PA, Kaufmann SH (2013). Contribution of Bcl‐2 phosphorylation to Bak binding and drug resistance. Cancer Res 73: 6998–7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks ED (2016). Venetoclax: first global approval. Drugs 76: 979–987. [DOI] [PubMed] [Google Scholar]
- Deng X, Ruvolo P, Carr B, May WS Jr (2000). Survival function of ERK1/2 as IL‐3‐activated, staurosporine‐resistant Bcl‐2 kinases. Proc Natl Acad Sci U S A 97: 1578–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Gao F, Flagg T, May WS Jr (2004). Mono‐ and multisite phosphorylation enhances Bcl2's antiapoptotic function and inhibition of cell cycle entry functions. Proc Natl Acad Sci U S A 101: 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Gao F, May WS Jr (2003). Bcl2 retards G1/S cell cycle transition by regulating intracellular ROS. Blood 102: 3179–3185. [DOI] [PubMed] [Google Scholar]
- Ekert PG, Jabbour AM, Manoharan A, Heraud JE, Yu J, Pakusch M et al (2006). Cell death provoked by loss of interleukin‐3 signaling is independent of Bad, Bim, and PI3 kinase, but depends in part on Puma. Blood 108: 1461–1468. [DOI] [PubMed] [Google Scholar]
- Elkholi R, Floros KV, Chipuk JE (2011). The role of BH3‐only proteins in tumor cell development, signaling, and treatment. Genes Cancer 2: 523–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogli S, Danesi R, Gennari A, Donati S, Conte P, Del Tacca M (2002). Gemcitabine, epirubicin and paclitaxel: pharmacokinetic and pharmacodynamics interactions in advanced breat cancer. Ann Oncol 13: 919–927. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZB, Minden MD, McCulloch EA (1998). Phosphorylation of Bcl‐2 after exposure of human leukemic cells to retinoic acid. Blood 92: 1768–1775. [PubMed] [Google Scholar]
- Holoch PA, Griffith TS (2009). TNF‐related apoptosis‐inducing ligand (TRAIL): a new path to anti‐cancer therapies. Eur J Pharmaco 625: 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds MG, Day CL (2005). Regulation of apoptosis: uncovering the binding determinants. Curr Opin Struct Biol 15: 690–699. [DOI] [PubMed] [Google Scholar]
- Ito T, Deng X, Carr B, May WS Jr (1997). Bcl‐2 phosphorylation required for anti‐apoptosis function. J Biol Chem 272: 11671–11673. [DOI] [PubMed] [Google Scholar]
- Jamil S, Lam I, Majd M, Tsai SH, Duronio V (2015). Etoposide induces cell death via mitochondrial‐dependent actions of p53. Cancer Cell Int 15: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang ZG, McKnight CJ (2006). A phosphorylation‐induced conformation change in dematin headpiece. Structure 14: 379–387. [DOI] [PubMed] [Google Scholar]
- Karpinich NO, Tafani M, Rothman RJ, Russo MA, Farber JL (2002). The course of etoposide‐induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochrome c. J Biol Chem 277: 16547–16552. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S et al (2006). Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT‐737 in acute myeloid leukemia. Cancer Cell 10: 375–388. [DOI] [PubMed] [Google Scholar]
- Krause F, Uhlendorf J, Lubitz T, Schulz M, Klipp E, Liebermeister W (2010). Annotation and merging of SBML models with semanticSBML. Bioinformatics 26: 421–422. [DOI] [PubMed] [Google Scholar]
- Kutuk O, Letai A (2010). Displacement of Bim by Bmf and Puma rather than increase in Bim level mediates paclitaxel‐induced apoptosis in breast cancer cells. Cell Death Differ 17: 1624–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner AU, Concannon CG, Boukes GJ, Cannon MD, Liambi F, Ryan D et al (2013). Systems analysis of Bcl‐2 protein family interactions establishes a model to predict responses to chemotherapy. Cancer Res 73: 519–528. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhang Z, Song T, Liang F, Xie M, Sheng H (2013). Resistance to BH3 mimetic S1 in SCLC cells that up‐regulate and phosphorylate Bcl‐2 through ERK1/2. Br J Pharmacol 169: 1612–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makin G, Dive C (2001). Modulating sensitivity to drug‐induced apoptosis: the future for chemotherapy? Breast Cancer Res 3: 150–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May WS, Tyler PG, Ito T, Armstrong DK, Qatsha KA, Davidson NE (1994). Interleukin‐3 and bryostatin‐1 mediate hyperphosphorylation of Bcl‐2a in association with suppression of apoptosis. J Biol Chem 269: 26865–26870. [PubMed] [Google Scholar]
- Mingo‐Sion AM, Marietta PM, Koller E, Wolf DM, Van Den Berg CL (2004). Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breat cancer cells. Oncogene 23: 596–604. [DOI] [PubMed] [Google Scholar]
- Notte A, Ninane N, Arnould T, Michiels C (2013). Hypoxia counteracts taxol‐induced apoptosis in MDA‐MB‐231 breat cancer cells: rold of autophagy and JNK activation. Cell Death Dis 4: e638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvolo PP, Deng X, May WS (2001). Phosphorylation of Bcl‐2 and regulation of apoptosis. Leukemia 15: 515–522. [DOI] [PubMed] [Google Scholar]
- Schuler PJ, Trellakis S, Greve J, Bas M, Bergmann C, Bolke E et al (2002). In vitro chemosensitivity of head and neck cancer cell lines. Eur J Med Res 15: 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G (2002). Kinetic studies of protein‐protein interactions. Curr Opin Struct Biol 12: 41–47. [DOI] [PubMed] [Google Scholar]
- Sharpe JC, Arnoult D, Yould RJ (2004). Control of mitochondrial permeability by Bcl‐2 family members. Biochim Biophys Acta 1644: 107–113. [DOI] [PubMed] [Google Scholar]
- Song T, Chai G, Liu Y, Yu X, Wang Z, Zhang Z (2016). Bcl‐2 phosphorylation confers resistance on chronic lymphocytic leukaemia cells to the BH3 mimetics ABT‐737, ABT‐263 and ABT‐199 by impeding direct binding. Br J Pharmacol 173: 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song T, Yu X, Liu Y, Li X, Chai G, Zhang Z (2015). Discovery of a small‐molecule pBcl‐2 inhibitor that overcomes pBcl‐2‐mediated resistance to apoptosis. Chembiochem 16: 757–765. [DOI] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J et al (2013). ABT‐199, a potent and selective Bcl‐2 inhibitors achieves antitumor activity while sparing platelets. Nat Med 19: 202–208. [DOI] [PubMed] [Google Scholar]
- Srivastava RK, Mi QS, Hardwick JM, Longo DL (1999). Deletion of the loop region of Bcl‐2 completely blocks paclitaxel‐induced apoptosis. Proc Natl Acad Sci U S A 96: 3775–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanelli C, Tantini B, Fattori M, Stanic I, Pignatti C, Clo C et al (2002). Caspase activation in etoposide‐treated fibroblasts is correlated to ERK phosphorylation and both events are blocked by polyamine depletion. FEBS Lett 527: 223–228. [DOI] [PubMed] [Google Scholar]
- Terrano DT, Upreti M, Chambers TC (2010). Cyclin‐dependent kinase 1‐mediated Bcl‐xL/Bcl‐2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol Cell Biol 30: 640–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman BF, Lipson D, Wemmer DE, Kern D (2001). Two‐state allosteric behaviour in a single‐domain signalling protein. Science 291: 2429–2433. [DOI] [PubMed] [Google Scholar]
- Vo TT, Letai A (2010). BH3‐only proteins and their effects on cancer. Adv Exp Med Biol 687: 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Yu J, Zhang L (2007). The nuclear function of p53 is required for PUMA‐mediated apoptosis induced by DNA damage. Proc Natl Acad Sci U S A 104: 4054–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Chen L, Benincosa J, Fortney J, Gibson LF (2005). VEGF‐induced phosphorylation of Bcl‐2 influences B lineage leukemic cell response to apoptotic stimuli. Leukemia 19: 344–353. [DOI] [PubMed] [Google Scholar]
- Wurstle ML, Zink E, Prehn JH, Rehm M (2014). From computational modelling of the intrinsic apoptosis pathway to a systems‐based analysis of chemotherapy resistance: achievements, perspectives and challenges in systems medicine. Cell Death Dis 5: e1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Nielsen TE, Clausen MH (2015). FDA‐approved small‐molecule kinase inhibitors. Trends Pharmacol Sci 36: 422–439. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Ichijo H, Korsmeyer SJ (1999). Bcl‐2 is phosphorylated and inactivated by an ASK1/Jun N‐terminal protein kinase pathway normally activated at G (2)/M. Mol Cell Biol 19: 8469–8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Shapiro BE, Mjolsness ED, Hatfield GW (2005). An enzyme mechanism language for the mathematical modeling of metabolic pathways. Bioinformatics 21: 774–780. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Liu Y, Song T, Xue Z, Shen X, Liang F et al (2013). An antiapoptotic Bcl‐2 family protein index predicts the response of leukaemic cells to the pan‐Bcl‐2 inhibitor S1. Br J Cancer 108: 1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Sun T, Pei J, Ouyang Q (2015). Mutation‐induced protein interaction kinetics changes affect apoptotic network dynamic properties and facilitate oncogenesis. Proc Natl Acad Sci U S A 112: E4045–E4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Overexpression of different Bcl‐2 variants in Bcl‐2‐silenced MCF‐7 cells, and ASK1/JNK1 proteins to phosphorylate endogenous Bcl‐2.
Figure S2 Kinetics characteristics of BH3 peptides binding to WT‐Bcl‐2 and EEE‐Bcl‐2 as measured by SPR.
Figure S3 Binding affinities of BH3 peptides to WT‐Bcl‐2 and EEE‐Bcl‐2 as measured by FPAs.
Figure S4 Statistical analysis of the best‐fit parameter set.
Figure S5 Experimental‐based parameter estimations for phosphorylation and de‐phosphorylation kinetics.
Figure S6 Paclitaxel induced Bcl‐2 phosphorylation at three phosphorylation sites in mitochondrialby active (phosphorylated) form of JNK but not ERK or PKC.
Figure S7 The apoptosis inducing effect of paclitaxel alone or in combination with SP600125 in the three selected breast cancer.
Figure S8 Paclitaxel induced mitochondrial apoptosis via Bmf in MDA‐MB‐231 and MCF‐7 cells, and via Puma in T47D cells.
Figure S9 The apoptosis inducing effect of paclitaxel in MCF‐7 cells transfected with WT‐Bcl‐2, AAA‐Bcl‐2 and EEE‐Bcl‐2 respectively.
Figure S10 Trajectories of model species for the three selected breast cancer cell lines.
Figure S11 The p‐JNK controlled extent of Bcl‐2 phosphorylation, rather than the levels of Bcl‐2 proteins, determines the tuning points from diminishing to enhancing antiapoptotic ability of Bcl‐2 by phosphorylation.
Figure S12 Bcl‐2 phosphorylation caused a right shift of dose ω induced by Bim or Bad stress, which could reproduce experimental data.
Figure S13 Different p‐JNK level influenced the dose of Bmf or Puma to induce cytochrome c release.
Figure S14 Percentage change of the dose ω in response to a 10% increase or decrease of each parameter.
Figure S15 Paclitaxel did not influence the half‐life of Puma, Bim, Bax and Bak and the expression level of Bcl‐2, Bcl‐xL and Mcl‐1.
Figure S16 Apparent affinity of Bcl‐2/Bax and Bcl‐2/Bim complex as a function of pBcl‐2/TBcl‐2 ratio and the effects on model of MOMP in response to Bmf.
Figure S17 Changes of Bcl‐2 interactions in NB4 and HT29 cells upon ABT‐199 or etoposide treatment.
Figure S18 Quantification of protein levels of Bcl‐2, Bim, Bax and Bak in MDA‐MB‐231, MCF‐7 and T47D cells.
Table S1 Translation of protein interactions into Ordinary Differential Equations Protein reactions were grouped into different classes and translated into a mathematical form. (A) Irreversible Bax or Bak activation by BH3‐only activator protein (Bim and Puma) or by auto‐activation. BH3‐only activator protein were recycled after one activation so that the concentration of activator was unchanged in this chemical reaction. Changes of Bax or Bak concentration over time were proportional to the product of reactant concentrations. (B) Protein turnover of a R1was modelled to consist of constant protein production and degradation. The protein degradation rate of R2was modelled to be proportional to the actual protein concentration. (C) Irreversible Bax or Bak inactivation. Changes of Bax or Bak concentration over time were proportional to the product of reactant concentrations. (D) Translocation of activated cytosolic Bax to mitochondrial membrane was modelled by a first order reaction. The translocated protein was considered as different model entity “AcBaxMOM”. (E) Reversible reactions of two proteins R1and R2forming a complex R1/R2. Change of protein concentration was given by balancing binding on and off reactions. (F) Reversible reactions of two Bax or Bak forming pores (MAC). Change of protein concentration was given by balancing forward and backward reactions. (G) Irreversible Bcl‐2 phosphorylation by kinases (p‐JNK, ERK or PKC) and Bcl‐2 dephosphorization by PP2A. The concentration of kinases and PP2A was unchanged in this chemical reaction. Changes of npBcl‐2 or pBcl‐2 concentration over time were proportional to the product of reactant concentrations.
Table S2 Bax and Bak activation kinetics by BH3‐only activator protein and auto‐activation. According to the literature report on that Bax and Bak conformational change in each activation case is equivalent, we assumed the same kinetic constants for Bax and Bak activation. Activation kinetic parameters of Bax and Bak by Bim or Puma were fitted by data set I‐IV in Figure S4.
Table S3 Degradation rates as used in in the model. Proteins and protein complexes that were modelled to be subjected to degradation are depicted. Based on the experimental data, as shown in Figure S12C, in which the protein level of Bcl‐2 was not changed following phosphorylation, we considered for pBcl‐2the same half‐life time as for Bcl‐2 (1200 min). In addition, we considered for Bcl‐xL the same half‐life time as for Bcl‐2 due to the similar amino acid sequence (EMBOSS Needle alignment, similarity 53%). The half‐life times of Bim, Puma Bax andBak were assayed as shown in Figure S12 Aand B. The half‐life times of other proteins were derived from the literature. Half‐life times of most complexes of anti‐and pro‐apoptotic proteins were modelled to be the same as the half‐life time of the Bcl‐2/tBid complex (75 min). The degradation constants kdegwere calculated from the half‐life time by the following equation: kdeg = ln (2) / (60*t1/2).
Table S4 Kinetics for Bax and Bak inactivation. According to the literature report on equivalent function of Bax and Bak, the same kinetic constants for Bax and Bak inactivation were used. The parameter was 25 fitted by data set I‐IV in Figure S4.
Table S5 Kinetics for translocation of activated cytosolic Bax to mitochondrial membrane. Activated cytosolic Bax was modelled to translocate with a half‐life time 1/10 min (k = 1.16E‐01 s‐1) to the mitochondrial membrane.
Table S6 Kinetics for inhibition of BH3‐only proteins and effectors Bax and Bak by anti‐apoptotic proteins. The association and dissociation constants for npBcl‐2 and pBcl‐2 binding BH3‐only proteins and effectors were taken from SPR experiment data in Table 1. The association and dissociation constants for Bcl‐xL and Mcl‐1 were taken from literature as indicated. No binding of Mcl‐1 to Bax and Mcl‐1 to Bad were detected.
Table S7 Kinetics for Bax and Bak homo‐or hetero‐dimerization Activated Bax and Bak were modelled to homo‐or hetero‐dimerize in the mitochondrial outer membrane. Given on the equivalent function of Bax and Bak, the same kinetic constants for them were assumed. The reaction constant k+/k‐was fitted by data set I‐IV in Figure S4. We fixed k‐value of 0.02, and k + was obtained by k + = k+/k‐ × k‐.
Table S8 Kinetics for Bcl‐2 phosphorylation and de‐phosphorylation Activated p‐JNK, p‐ERK or p‐PKC were modelled to catalyse Bcl‐2 phosphorylation in the mitochondrial outer membrane. PP2A was modelled to catalyse Bcl‐2 de‐phosphorylation. The reaction constantskphosby p‐JNK and kdephosby PP2A were fitted by data set V in Figure S5. We assumed the same kinetic constants for p‐JNK, p‐ERK or p‐PKC.