

Abstract
Background and Purpose
The aim of this study was to characterize the human cytochrome P450s (CYPs) involved in oxidative bioactivation of flucloxacillin to 5‐hydroxymethyl flucloxacillin, a metabolite with high cytotoxicity towards biliary epithelial cells.
Experimental Approach
The CYPs involved in hydroxylation of flucloxacillin were characterized using recombinant human CYPs, pooled liver microsomes in the presence of CYP‐specific inhibitors and by correlation analysis using a panel of liver microsomes from 16 donors.
Key Results
Recombinant CYPs showing the highest specific activity were CYP3A4, CYP3A7 and to lower extent CYP2C9 and CTP2C8. Michaelis–Menten enzyme kinetics were determined for pooled human liver microsomes, recombinant CYP3A4, CYP3A7 and CYP2C9. Surprisingly, sulfaphenazole appeared to be a potent inhibitor of 5′‐hydroxylation of flucloxacillin by both recombinant CYP3A4 and CYP3A7.
Conclusions and Implications
The combined results show that the 5′‐hydroxylation of flucloxacillin is primarily catalysed by CYP3A4, CYP3A7 and CYP2C9. The large variability of the hepatic expression of these enzymes could affect the formation of 5′‐hydroxymethyl flucloxacillin, which may determine the differences in susceptibility to flucloxacillin‐induced liver injury. Additionally, the strong inhibition in CYP3A‐catalysed flucloxacillin metabolism by sulfaphenazole suggests that unanticipated drug–drug interactions could occur with coadministered drugs.
Abbreviations
- 5′‐HM‐FLX
5′‐hydroxymethyl flucloxacillin
- CYP
cytochrome P450
- DILI
drug‐induced liver injury
- GWAS
genome‐wide association studies
- HLM
human liver microsomes
- NRS
NADPH‐regenerating system
What is known about this subject
There are large interindividual differences in plasma concentrations of flucloxacillin and 5′‐hydroxymethyl flucloxacillin.
5′‐Hydroxymethyl flucloxacillin is much more toxic to biliary epithelial cells than flucloxacillin.
A previous study on four human CYPs showed that CYP3A4 exerted the highest activity of flucloxacillin hydroxylation. However, only a single substrate concentration was used, without quantification of specific activity due to lack of reference metabolite.
What this study adds
Quantification of specific activities and enzyme kinetics of flucloxacillin hydroxylation by HLM and 14 recombinant human CYPs.
Next to CYP3A4, CYP3A7 and CYP2C9 may contribute significantly to flucloxacillin hydroxylation.
Sulfaphenazole, considered as a selective CYP2C9 inhibitor, strongly inhibits CYP3A‐mediated flucloxacillin hydroxylation.
What is the clinical significance
Interindividual variability and drug‐drug interactions at the level of CYP3A4, CYP3A7 and CYP2C9 may be important factors determining the risk of flucloxacillin‐induced liver injury.
Introduction
The isoxazolyl‐penicillin flucloxacillin is a narrow spectrum β‐lactam antibiotic which is active against Gram‐positive bacteria, such as methicillin‐sensitive Staphylococcus aureus (Sutherland et al., 1970; Leder et al., 1999). Although the antibiotic is generally well tolerated at daily doses as high as 12 g, it is known to cause drug‐induced liver injury (DILI) of a cholesteric nature in susceptible patients (Derby et al., 1993; Eckstein et al., 1993; Fairley et al., 1993; Koek et al., 1994; De Abajo et al., 2004; Russmann et al., 2005; Andrews and Daly, 2008). Although the risk for flucloxacillin induced DILI is in the region of 9 in 100 000 patients (Russmann et al., 2005), the fact that it is one of the most prescribed β‐lactam antibiotics makes flucloxacillin one of the drugs most often involved in DILI (De Abajo et al., 2004). The mechanisms underlying flucloxacillin‐induced cholestasis are still incompletely understood. Association studies have identified several risk factors, such as prolonged treatment with high daily doses and an age over 55 years (Fairley et al., 1993; Russmann et al., 2005). Furthermore, a genome‐wide association study (GWAS) identified the HLA B*57:01 allele as a strong risk factor for flucloxacillin‐induced DILI, with an odds ratio of 80.6, indicative for an important role of the immune system in the mechanism of liver toxicity (Daly et al., 2009). Many forms of idiosyncratic DILI are believed to have an immunological basis, in which covalent binding of drugs and/or metabolites to proteins may play an important role by generation of a hapten and by causing cellular stress, leading to the release of so‐called danger signals which can stimulate the immune response (Uetrecht, 2007; Tailor et al., 2015). Like many other penicillin‐like antibiotics, the reactive β‐lactam‐rings of flucloxacillin and its active metabolite 5′‐hydroxymethyl‐flucloxacillin (5′‐HM‐FLX, Figure 1) have been shown to react with lysine‐residues of proteins thereby forming potential haptens (Jenkins et al., 2009). Interestingly, cytotoxic (CD8+) T‐cells were only activated by flucloxacillin‐exposed antigen‐presenting cells in cells obtained from patients and volunteers with the HLA B*57:01 genotype, supporting the role of this genotype as risk factor for flucloxacillin‐induced DILI (Monshi et al., 2013; Wuillemin et al., 2013, 2014). However, although the association with HLA B*57:01 is one of the strongest ever reported, still only 1 in 1000 patients with the high‐risk allele eventually develops flucloxacillin‐induced DILI. Therefore, non‐genetic risk factors, which escape detection by GWAS‐studies, may also determine the sensitivity for flucloxacillin‐induced cholestasis. Because 5′‐HM‐FLX was shown to be highly cytotoxic to isolated biliary epithelial cells, whereas flucloxacillin was without effect, a high internal exposure to 5′‐HM‐FLX may be considered as a potentially important risk factor (Lakehal et al., 2001).
Figure 1.
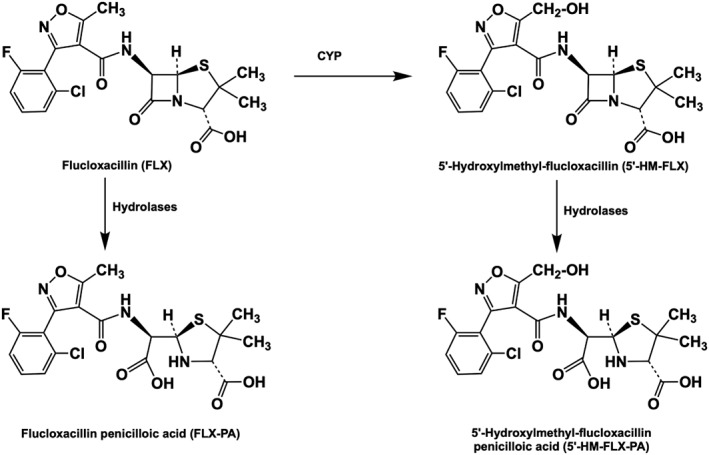
Oxidative metabolism of flucloxacillin (FLX) to 5′‐HM‐FLX and hydrolysis of flucloxacillin and 5′‐HM‐FLX to their corresponding penicilloic acids.
Many studies have investigated the pharmacokinetics of flucloxacillin in groups of healthy volunteers or patients (Thijssen and Wolters, 1982; Adam et al., 1983; Gath et al., 1995; Røder et al., 1995; Landersdorfer et al., 2007; Dijkmans et al., 2012; Maier‐Salamon et al., 2017). Dependent on the dose regimen and route of administration (by i.v. infusion and/or orally), the maximal plasma concentration (Cmax) of flucloxacillin can range 50 to 790 μM. Systemic clearance of flucloxacillin appears to be mainly by renal clearance (65–71%) (Landersdorfer et al., 2007; Maier‐Salamon et al., 2017). Non‐renal clearance of flucloxacillin occurs predominantly by cytochrome P450‐mediated hydroxylation of flucloxacillin to 5′‐HM‐FLX and partly by hydrolysis to the corresponding penicilloic acid (Figure 1). In healthy volunteers, plasma concentrations of 5′‐HM‐FLX are on average approximately 10% of those of FLX, whereas their half‐lives are comparable (Thijssen and Wolters, 1982). Because of the high contribution of renal excretion of flucloxacillin and 5′‐HM‐FLX, renal impairment was shown to lead to a significant increase in the AUCs which may increase the risk for flucloxacillin‐induced DILI (Gath et al., 1995).
The different pharmacokinetics studies have demonstrated that metabolism of flucloxacillin shows significant interindividual variability, which may be a reflection of the large interindividual variability of the enzymes involved in hydroxylation of flucloxacillin and/or the mammalian dipeptidase involved in hydrolysis of flucloxacillin and 5′‐HM‐FLX to their corresponding penicilloic acids. The study of Lakehal et al.(2001), showed that CYP3A4 was able to catalyse the 5′‐hydroxylation of flucloxacillin. However, the involvement of only four CYP isoforms was studied, and flucloxacillin hydroxylation was measured at only one concentration. Another study showed that flucloxacillin induces CYP3A4 in cell lines exposed to 200–400 μM flucloxacillin (Huwyler et al., 2006). This may explain the decrease of plasma concentrations of cyclosporine A, quinidine, voriconazole and warfarin when co‐administered together with flucloxacillin (Cynke et al., 1999; Comuth et al., 2012; Muilwijk et al., 2017; Chaudhuri and Wade, 2018). Whether this induction resulted in increased formation of 5′‐HM‐FLX was not studied, however. Because a recent meta‐analysis of Achour et al. (2014) showed that the hepatic expression levels of all drug metabolizing human CYPs vary considerably, due to genetic polymorphism, enzyme induction and epigenetic factors, it is critically important to fully characterize the CYPs involved in drug metabolism in order to predict the interindividual variability in pharmacokinetics and the risk of drug–drug interaction. Furthermore, a major involvement of CYP3A4 in drug metabolism complicates prediction of pharmacokinetics based on enzyme kinetics because of the occurrence of atypical enzyme kinetics, such as substrate activation (Houston and Kenworthy, 2000; Ekins et al., 2003; Atkins, 2005; Houston and Galetin, 2005). Furthermore, inhibition of CYP3A4 by drugs appears to be substrate‐dependent, further complicating prediction of drug–drug interactions (Stresser et al., 2000).
The aim of the present study was therefore to characterize the bioactivation of flucloxacillin to 5′‐HM‐FLX in more detail by using a larger set of recombinant CYPs, by inhibition studies using pooled human liver microsomes (HLM) and by correlation analysis using a panel of HLM from 16 donors. Furthermore, for HLM and the most active CYPs, the enzyme kinetics of flucloxacillin hydroxylation were determined.
Methods
Enzyme sources
Gentest Supersomes containing recombinant human CYPs were obtained from Corning BV life sciences (Amsterdam, the Netherlands). The enzymes used were CYP1A1 (Lot No. 456211), CYP1A2 (Lot No. 456203), CYP2A6 (Lot No. 456254), CYP2B6 (Lot No. 456255), CYP2C8 (Lot No. 456252), CYP2C9*1(Arg144) (Lot No. 456258), CYP2C18 (Lot No. 456222), CYP2C19 (Lot No. 456259), CYP2D6*1 (Lot No. 456217), CYP2E1 (Lot No. 456206), CYP2J2 (Lot No. 456264), CYP3A4 (Lot No. 456207), CYP3A5 (Lot No. 456256) and CYP3A7 (Lot No. 456237).
Pooled HLM (50 donors, 20 mg·mL−1, Lot No. 0710619) were obtained from Xenotech (Lenexa, USA). A panel of HLM from single donors were prepared from livers kindly provided by Kalycell (Plobsheim, France) (donors: S1399T, S1449T, S1352T, S1342T, B1327T, S1405T, S1336T, S1446T, S1329T, S1339T, S1343T, S1442T, S1344T, S1441T, S1334T and R1341T).
Preparation of 5′‐HM‐FLX
5′‐HM‐FLX was obtained biosynthetically by hydroxylation of flucloxacillin using a drug metabolizing mutant of CYP102A1 from Bacillus megaterium, M11 L437E, which contains mutations R47L, E64G, F81I, F87V, E143G, L188Q, E267V, G415S and L437E compared to wild‐type CYP102A1. This mutant appeared to selectively convert flucloxacillin to 5′‐HM‐FLX (Luirink et al., 2018); 1 mL of a solution of 100 mM flucloxacillin in DMSO was added to 49 mL 100 mM potassium phosphate buffer pH 7.4 and incubated in the presence of 1 μM of M11 L437E and a NADPH‐regenerating system (NRS) for 20 h at room temperature with constant stirring. The NRS was consisted of 100 μM NADPH, 10 mM glucose‐6‐phosphate and 0.5 units·mL−1 glucose‐6‐phosphate dehydrogenase (final concentrations). The reaction was terminated by the addition of an equal volume ice‐cold methanol containing 2% acetic acid. The mixture was centrifuged at 3630× g for 2 h at 4°C after which the supernatant was filtered through a 0.2 μm Phenex RC membrane filter from Phenomenex (Utrecht, the Netherlands). 5′‐HM‐FLX was purified by preparative HPLC using a Xbridge Prep C18‐MS (5 μm; 10 × 55 mm) column. Samples of 1.5 mL were injected and eluted with a flow rate of 3 mL·min−1. A gradient was constructed using eluent A (99% water/0.9% acetonitrile/0.1% formic acid) and eluent B (99% acetonitrile/0.9% water/0.1% formic acid) and programmed from 25% B to 50% B in 11 min after which it returned to 25% B in half a minute. After re‐equilibrating for 4.5 min, the next sample was injected. A fraction collector was triggered to collect peaks appearing at UV detection at 272 nm. The fractions containing 5′‐HM‐FLX were combined and evaporated to dryness and stored at −80°C under N2. The identity of 5′‐HM‐FLX was confirmed by 1H NMR and 19F NMR using a Bruker Avance 250 (Fallanden, Switzerland) equipped with a cryoprobe operating at 250.1 MHz. The metabolite had a purity of 99% based on HPLC analysis.
Oxidative metabolism of flucloxacillin by HLM and recombinant human CYPs
Flucloxacillin was initially incubated at concentrations of 10 and 100 μM in the presence of 100 nM of 14 Gentest Supersomes in order to identify the most active human CYPs. Reactions were initiated in the presence of NRS (see above). Incubations were performed in a 100 mM potassium phosphate buffer pH 7.4 supplemented with 5 mM MgCl2 and 2 mM EDTA and at a total volume of 50 μL. The incubations were started by the addition of NRS and incubated for 10 min at 37°C, during which metabolite production was linear (data not shown). The reactions were terminated as described above. The samples were subsequently centrifuged at 20 800× g for 20 min using a table top centrifuge. The supernatant was filtered through 0.2 μm Phenex RC membrane filters from Phenomenex (Utrecht, the Netherlands) and subsequently analysed by LC–MS, as described below. Because these incubations were only performed to identify the most active enzymes, duplicate experiments were deemed sufficient since reproducibility of incubations with recombinant enzymes from the same source was always within 5%,
For the most active Gentest Supersomes and pooled HLM, the enzyme kinetics of the formation of 5′‐HM‐FLX were determined by incubating with 11 concentrations of flucloxacillin, ranging from 0 to 500 μM. Each flucloxacillin concentration was incubated in duplicate for 10 min with 0.5 mg·mL−1 pooled HLM, 100 nM CYP3A4, CYP3A7 and CYP2C9 Supersomes in 100 mM potassium phosphate buffer, pH 7.4 supplemented with 5 mM MgCl2 and 2 mM EDTA. The reactions were started, stopped and analysed as described above.
Effect of specific inhibitors of CYPs on oxidative metabolism of flucloxacillin by pooled HLM
To study the involvement of individual human CYPs in the hydroxylation of flucloxacillin in incubations with pooled HLM from 50 individuals, the effect of selective P450 inhibitors was investigated. Flucloxacillin was incubated at substrate concentrations of 10 and 100 μM with 0.5 mg·mL−1 HLM, as described above, and in the absence or presence of the following inhibitors: 10 μM α‐naphtoflavone (CYP1A2), 3 μM ticlopidine (CYP2B6), 15 μM quercetin (CYP2C8), 10 μM sulfaphenazole (CYP2C9), 10 μM fluvoxamine (CYP2C9/CYP1A2), 1 μM (+)‐N‐3‐benzyl‐nirvanol (CYP2C19), 2 μM quinidine (CYP2D6), 20 μM diethylthiocarbamate (CYP2E1), 3 μM troleandomycin (CYP3A4); 2 μM miconazole (CYP3A4/CYP2C9) and 0.1 μM (CYP3A4) and 1 μM (CYP3A) ketoconazole respectively. At these concentrations, the inhibitors are considered to be selective for the indicated P450 (Kumar et al., 2006; Niwa et al., 2009; Khojasteh et al., 2011). The mechanism‐based inhibitors ticlopidine and troleandomycin were pre‐incubated with HLM and NRS for 10 min before the addition of flucloxacillin. The inhibition experiment was performed three times, with each condition incubated in duplicate.
Because the CYP2C9‐specific inhibitor sulfaphenazole appeared to almost completely block formation of 5′‐HM‐FLX by pooled HLM, it was also added at different concentrations to incubations of CYP3A4 and CYP3A7 Supersomes with flucloxacillin (10 μM) and testosterone (50 μM).
Variability of oxidative metabolism of flucloxacillin by individual HLM and correlation analysis
To determine the variability of 5′‐hydroxylation of flucloxacillin, the substrate was incubated (10 and 100 μM) with a panel of individual HLM fractions from 16 donors (0.5 mg·mL−1) which were phenotyped with respect to activities of CYP1A2 (phenacetin O‐deethylation), CYP2A6 (coumarin 7‐hydroxylation), CYP2B6 (bupropion hydroxylation), CYP2C8 (amodiaquine N‐deethylation), CYP2C9 (diclofenac 4′‐hydroxylation), CYP2C19 (mephenytoin 4‐hydroxylation), CYP2D6 (bufuralol 1‐hydroxylation), CYP2E1 (chlorzoxazone 6‐hydroxylation) and CYP3A4 (midazolam 1′‐hydroxylation and testosterone 6β‐hydroxylation) using the concentrations described previously (den Braver‐Sewradj et al., 2018).
Analytical methods
Flucloxacillin and its metabolites were measured using an Agilent TOF 6230 mass spectrometer with electrospray ionization source coupled with an Agilent 1200 rapid resolution LC system. The mass spectrometer was operated under the following conditions: a capillary voltage of 3500 V, 10 L·min−1 nitrogen drying gas and 50 psig nitrogen nebulizing gas at 350°C and operating in positive ion mode. Chromatographic separation of flucloxacillin and its metabolites was achieved by reversed‐phase LC using a Phenomenex Luna C18‐column (5 μm; 4.6 × 150 mm). A gradient was constructed using 1% acetonitrile in 0.1% formic acid (A) and 99% acetonitrile in 0.1% formic acid (B) as eluents. The gradient was programmed from 40% B to 99% B in 23.5 min after which the percentage went back to 40% B in 0.5 min. Next, the column was re‐equilibrated in 40% B for 11 min. The flow rate was 0.4 mL·min−1.
The chromatograms were analysed using Agilent Masshunter Qualitative Analysis software (RRID:SCR_016657). Quantification was performed using a standard curve of 5′‐HM‐FLX ranging from 1 nM to 6 μM. The lower limit of quantification of 5′‐HM‐FLX was 8 nM.
Data and statistical analysis
Enzyme kinetic parameters were determined using GraphPad Prism 5.00 for Windows, GraphPad Software (San Diego, USA; RRID:SCR_002798) and fitted to the Michaelis–Menten equation. IC50 was calculated using the log (inhibitor) versus response function of GraphPad Prism 5.00 for Windows, GraphPad Software (San Diego, USA). The Pearson correlation coefficient was calculated for the specific probe substrates and flucloxacillin 5′‐hydroxylation using GraphPad Prism 5.00 for Windows, GraphPad Software (San Diego, USA). A two‐tailed P‐value was used with a confidence interval of 95%. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). However, randomization of samples was deemed unnecessary since no systematic bias was observed related to the sample position or in the order of sample analysis by LC–MS. Blinding was not undertaken since prior knowledge of sample content was not expected to introduce bias of the measurements. Also, data acquisition and data analysis were performed by different individuals.
Materials
Flucloxacillin (CAS: 1847‐24‐1) was a kind gift of Dr Bayliss from the University of Liverpool. β‐Nicotinamide adenine dinucleotide phosphate disodium salt (NADP; CAS: 24292‐60‐2), glucose‐6‐phosphate (G‐6‐P; CAS: 54010‐71‐8), G‐6‐P dehydrogenase from baker's yeast (Saccharomyces cerevisiae; CAS: 9001‐40‐5), (+)‐N‐3‐benzyl‐nirvanol (CAS: 790676‐40‐3), bufuralol (CAS: 60398‐91‐6), chlorzoxazone (CAS: 95‐25‐0), diethyldithiocarbamate (CAS: 20624‐25‐3), fluvoxamine maleate (CAS: 22916‐47‐8), ketoconazole (CAS: 65277‐42‐1), (S)‐mephenytoin (CAS: 70989‐04‐7), miconazole (CAS: 22916‐47‐8), α‐naphtoflavone (CAS: 604‐59‐1), quercetin (CAS: 6151‐25‐3), quinidine (CAS: 56‐54‐2), sulfaphenazole (CAS: 526‐08‐9), testosterone (CAS: 58‐22‐0), ticlopidine (CAS: 53885‐35‐1) and troleandomycin (CAS: 2751‐09‐9) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Midazolam (CAS: 59467‐96‐8) was from Duchefa Farma (Haarlem, the Netherlands). All other chemicals were of analytical grade purity and obtained from standard suppliers.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Oxidative metabolism of flucloxacillin by HLM and recombinant CYPs
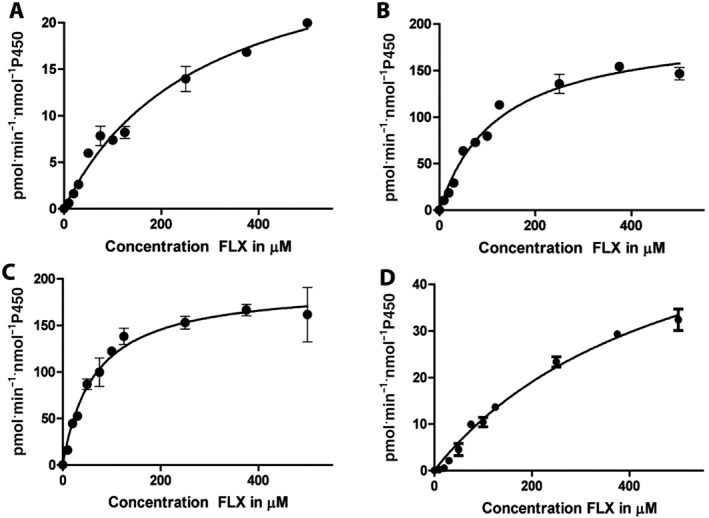
As shown in Figure 2, initial incubations of 10 μM flucloxacillin for 60 min with pooled HLM, in the presence or absence of NRS, resulted in the formation of only one NRS‐dependent metabolite eluting at 11.6 min and with m/z ([M + H]+) of 470.052, which corresponded to the retention time and m/z‐value of biosynthetic 5′‐HM‐FLX. The product eluting at 9.3 min has an m/z of 472.078 and was also formed in the absence of NRS. The mass increase of 18 relative to flucloxacillin (m/z 454.059) indicates that this product most likely corresponds to the penicilloic acid formed by hydrolysis of the β‐lactam ring of flucloxacillin which is not dependent on CYP‐activity. The penicilloic acid of 5′‐HM‐FLX, which was identified in urine (Maier‐Salamon et al., 2017), was not detectable under the incubation conditions used. After checking the linearity of product formation with time, the enzyme kinetics of 5′‐HM‐FLX formation by pooled HLM was studied in 10 min incubations by varying the concentration of flucloxacillin from 10 to 500 μM. As shown in Figure 3A, a hyperbolic curve was obtained consistent with Michaelis–Menten kinetics. Enzyme kinetic parameters obtained by non‐linear regression were K M 284 ± 38 μM and Vmax 30 ± 2 pmol·min−1·mg−1 protein.
Figure 2.
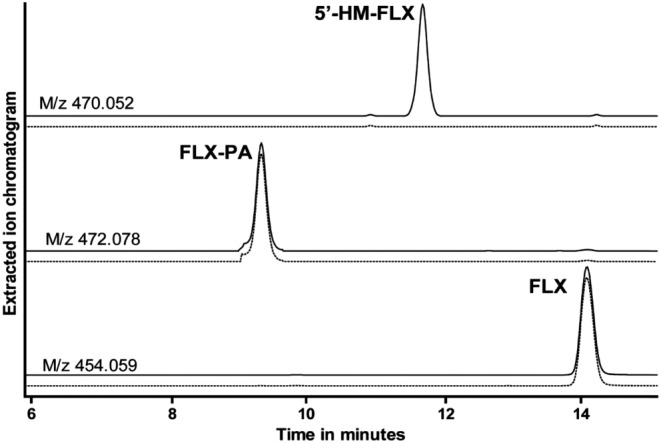
LC–MS analysis of an incubation of 100 μM flucloxacillin incubated for 60 min with 0.5 mg·mL−1 pooled human liver microsomes with (solid line) or without (dotted line) NADPH‐regenerating system. (A) Extracted ion chromatogram of m/z 470.052 corresponding to the [M + H]+ of 5′‐HM‐FLX; (B) extracted ion chromatogram of m/z 472.078 corresponding to the [M + H]+ of the penicilloic acid of 5′‐HM‐FLX (FLX‐PA); (C) extracted ion chromatogram of m/z 454.059 corresponding to the [M + H]+ of flucloxacillin (FLX).
Figure 3.
Concentration dependency of the rate of flucloxacillin hydroxylation as catalysed by 0.5 mg·mL−1 pooled HLM (A), 100 nM CYP3A4 Supersomes (B), 100 nM CYP3A7 Supersomes (C) and 100 nM CYP2C9 Supersomes (D). Each condition was incubated in duplicate, and each data point represents as mean whereas error bars represent the range of duplicate measurements. Enzymes kinetic parameters obtained by non‐linear regression according to the Michaelis–Menten equation were (A) HLM: K M 284 ± 38 μM and Vmax 30 ± 2 pmol·min−1·mg−1 protein; (B) CYP3A4 Supersomes: K M 124 ± 15 μM and Vmax 197 ± 9 pmol·min−1·nmol−1 P450, (C) CYP3A7 Supersomes: K M of 65 ± 8 μM and Vmax 193 ± 7 pmol·min−1·nmol−1 P450 and (D) CYP2C9 Supersomes: K M of 508 ± 82 μM and Vmax 67 ± 7 pmol·min−1·nmol−1 P450.
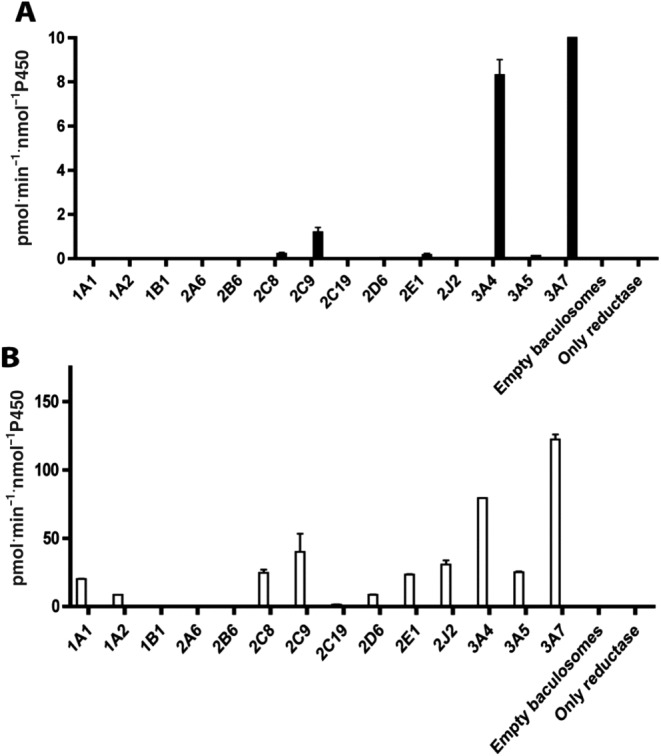
To characterize which human CYP isoenzymes catalyse the formation of 5′‐HM‐FLX, eleven different recombinant CYP Supersomes were incubated with 10 and 100 μM FLX. These concentrations were chosen to discriminate between active isoforms at the average Cmax in patients taking 1000 mg orally (100 μM) and high affinity enzymes active at lower substrate concentration (10 μM). At a flucloxacillin concentration of 10 μM, recombinant CYP3A4, CYP3A7 and to a lesser extent CYP2C9 showed the highest activity, in terms of the formation of 5′‐HM‐FLX (Figure 4A). At 100 μM flucloxacillin, CYP3A7 was the most active CYP followed by CYP3A4. Low activities of 5′‐HM‐FLX formation were observed, in decreasing order, in incubations with CYP2C9, CYP2J2, CYP2C8, CYP2E1, CYP3A5, CYP1A1, CYP2D6 and CYP1A2 (Figure 4B). As CYP3A4, CYP3A7 and CYP2C9 Supersomes showed highest activity at both concentrations, the enzyme kinetics of 5′‐HM‐FLX formation was also determined for these recombinant CYPs (Figure 3B–D). Similar to the incubations with pooled HLM (Figure 3A), Michaelis–Menten kinetics were observed. Using the Michaelis–Menten model, the following enzyme kinetic parameters were determined: CYP3A4, K M 124 ± 15 μM, Vmax 197 ± 9 pmol·min−1·nmol−1 P450; CYP3A7: K M 65 ± 8 μM, Vmax 193 ± 7 pmol·min−1·nmol−1 P450; and CYP2C9: K M of 508 ± 82 μM, Vmax 67 ± 7 pmol·min−1·nmol−1 P450.
Figure 4.
Specific activities of formation of 5′‐HM‐FLX in incubations of flucloxacillin with CYP‐containing Supersomes (100 nM). (A) Incubations in the presence of 10 μM flucloxacillin (FLX); (B) incubations in the presence of 100 μM flucloxacillin . Data represent mean of duplicate experiments; the error bars represent the range of duplicate measurements.
Effect of isoenzyme‐selective CYP inhibitors on flucloxacillin metabolism by HLM
Figure 5 shows the effects of isoenzyme‐selective CYP inhibitors on the oxidative metabolism of flucloxacillin by pooled HLM. The three inhibitors of CYP3A isoforms, KCZ, miconazole and troleandomycin, showed a relatively modest and differential inhibitory effect on 5′‐HM‐FLX formation. Even at the highest concentration of ketoconazole, which inhibits the CYP3A family, still only 50% inhibition was observed at both concentrations of flucloxacillin. The other CYP3A4 inhibitors showed only 20 to 40% inhibition at 10 μM flucloxacillin concentration. Although recombinant CYP2C9 showed much lower activity when compared to CYP3A4 (Figures 3 and 4), an almost 90% inhibition of 5′‐HM‐FLX formation was observed in the presence of sulfaphenazole, which is considered to be a selective CYP2C9 inhibitor. However, fluvoxamine, the other CYP2C9 inhibitor used, did not show significant inhibition of 5′‐HM‐FLX formation.
Figure 5.
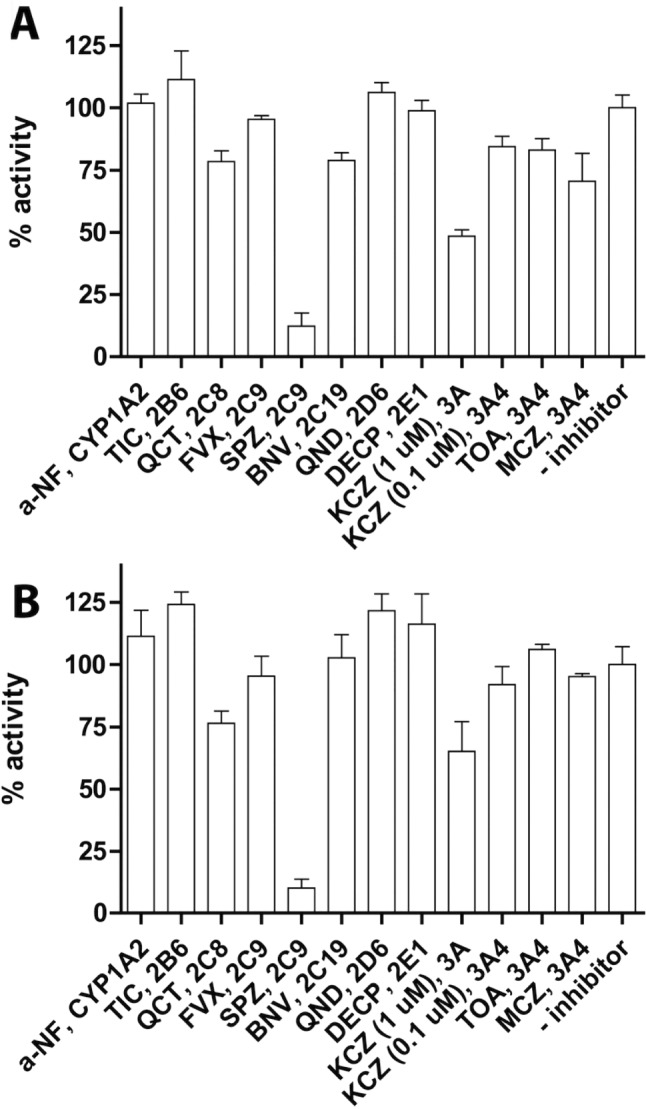
The inhibition of 5′‐HM‐FLX formation by pooled HLM (0.5 mg·mL−1) by isoform‐specific inhibitors of CYPs. (A) Incubations performed in the presence of 10 μM flucloxacillin (B) incubations performed in the presence of 100 μM flucloxacillin. Activities are relative to the non‐inhibited reactions which showed specific activities of 0.52 pmol·min−1·mg−1 protein (A) and 7.4 pmol·min−1·mg−1 protein (B) respectively; 100% was defined as the mean of enzyme activities in the absence of inhibitor, and 0% was defined as no enzyme activity. Data represent the averages of the three independent experiments in which each condition was performed in duplicate; error bars represent the SD of the six individual measurements. The inhibitors used were: α‐naphtoflavone (a‐NF), ticlopidine (TIC), quercetin (QCT), fluvoxamine (FVX), sulfaphenazole (SPZ), (+)‐N‐3‐benzyl‐nirvanol (BNV), quinidine (QND), diethylthiocarbamate (DECP), ketoconazole (KCZ), troleandomycin (TOA) and miconazole (MCZ); isoform specificity is also indicated and concentrations are shown in Methods.
To study whether sulfaphenazole has the ability to inhibit CYP3A‐mediated hydroxylation of flucloxacillin, the inhibitor was added at different concentrations to incubations of CYP3A4 and CYP3A7, with flucloxacillin and testosterone as substrates. As shown in Figure 6A, B, sulfaphenazole appeared to be a potent inhibitor of flucloxacillin hydroxylation by recombinant CYP3A4 and CYP3A7 with an IC50 of 4.1 ± 0.4 μM and 6.1 ± 0.5 μM respectively. At these sulfaphenazole concentrations, no inhibition of CYP3A4‐catalysed 6β‐hydroxylation of testosterone was observed. In HLM, sulfaphenazole inhibited both diclofenac 4′‐hydroxylation and flucloxacillin hydroxylation but not testosterone hydroxylation, with an IC50 of 2.2 ± 1.1 μM for diclofenac and an IC50 of 13 ± 1.3 μM for flucloxacillin (Figure 6C).
Figure 6.
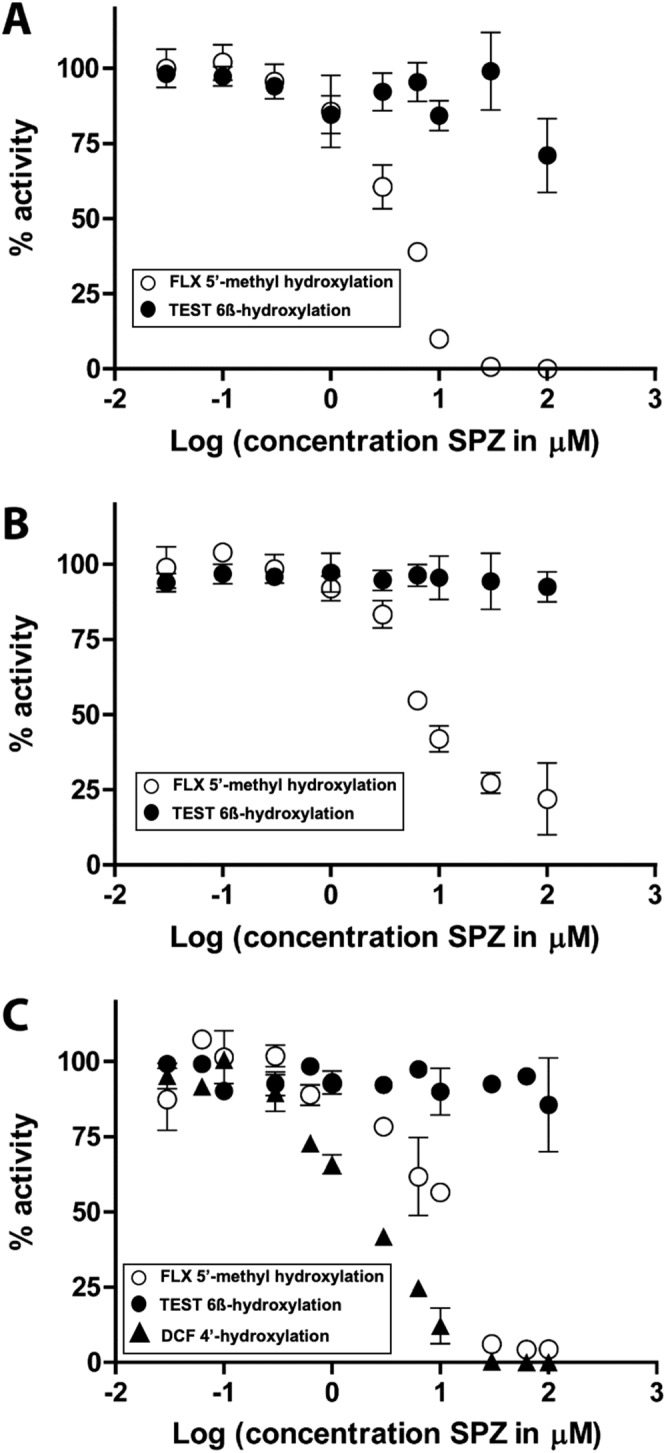
Concentration‐dependent inhibition by sulfaphenazole (SPZ) of 5′‐hydroxylation of flucloxacillin, 6β‐hydroxylation of testosterone and 4′‐hydroxylation of diclofenac as catalysed by CYP3A4 Supersomes (A), CYP3A7 Supersomes (B) and HLM (C); 100% was defined as the mean of enzyme activities in the absence of inhibitor, and 0% was defined as no enzyme activity (e.g. full inhibition). Each condition was incubated in duplicate, and results are shown as mean with the range of duplicate measurements as error bars.
Variability of oxidative metabolism of flucloxacillin by individual HLM fractions and correlation analysis
Figure 7 shows the variability of oxidative flucloxacillin metabolism which was observed in incubations of flucloxacillin with HLM fractions from 16 donors. When incubating with 10 μM flucloxacillin, a 6.5‐fold difference was found between the lowest (S1342) and the highest (S1441) metabolizing donor. When using 100 μM flucloxacillin as substrate concentration, the difference between the lowest (S1342) and the highest (S1343) metabolizing donor was 3.3‐fold. As shown in Figure 8 and Table 1, the activities of 5′‐HM‐FLX formation showed very poor correlations with activities of most CYP‐specific reactions. At both concentrations of flucloxacillin, the highest correlation coefficients were found with CYP2C9‐catalysed diclofenac 4′‐hydroxylation, with Pearson correlation coefficients of 0.78 and 0.58, respectively, and CYP3A‐catalysed 6β‐hydroxylation, with correlation coefficients of 0.69 and 0.58 respectively. Visual inspection of the data points of the correlation between CYP2C9 activity and flucloxacillin hydroxylation shows a trend that the line does not cross the X‐axis at the origin but at 280 pmol·min−1·mg−1 protein . As none of the correlation coefficients approached unity may suggest that more than one CYP iosform contributes to flucloxacillin hydroxylation.
Figure 7.
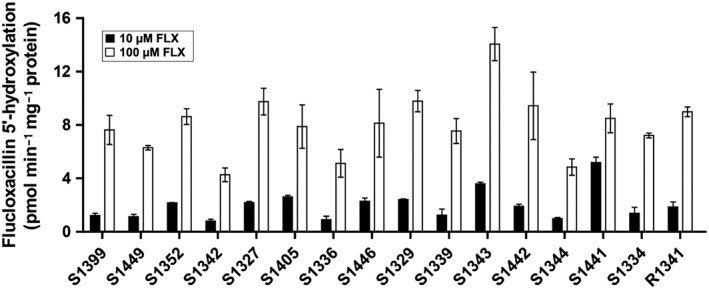
Variability in specific activity of 5′‐hydroxylation of flucloxacillin as catalysed by HLM from 16 individual donors. Specific activities obtained in the presence of 10 μM or 100 μM flucloxacillin (FLX), are shown. Each data point was measured in duplicate and is presented as the mean with the range of duplicate measurements.
Figure 8.
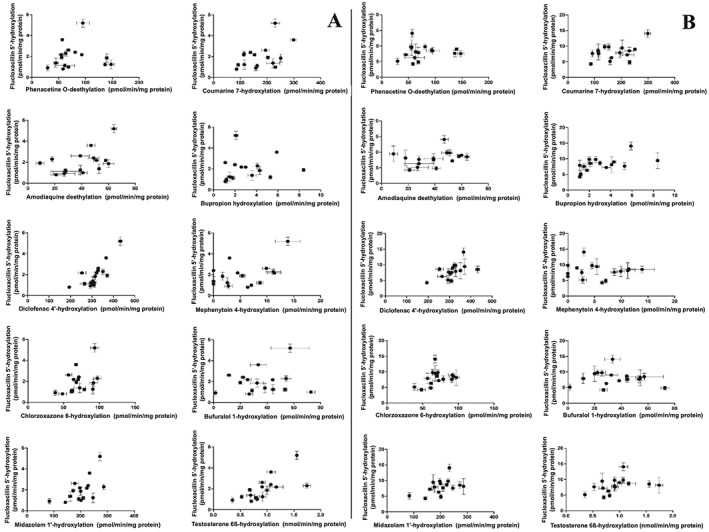
Correlation analysis of CYP‐specific reactions and flucloxacillin (FLX) hydroxylation at 10 μM flucloxacillin (A) or 100 μM flucloxacillin (B) using HLM of 16 different donors. Each data point was measured in duplicate and shown as mean with the range of duplicate measurements. Pearson correlation coefficients and corresponding P‐values are tabulated in Table 1.
Table 1.
Statistical analysis of the correlation between activities of flucloxacillin (FLX) hydroxylation and isoenzyme specific reactions catalysed by HLM from 16 different donors
| Isoenzyme‐specific reaction | CYP | 10 μM FLX | 100 μM FLX | ||
|---|---|---|---|---|---|
| Pearson's r | P‐value | Pearson's r | P‐value | ||
| Phenacetin O‐deethylation | CYP1A2 | 0.04 | 0.87 | 0.10 | 0.70 |
| Coumarin 7‐hydroxylation | CYP2A6 | 0.45 | 0.078 | 0.45 | 0.093 |
| Buproprion hydroxylation | CYP2B6 | 0.16 | 0.54 | 0.49 | 0.16 |
| Amodiaquine N‐deethylation | CYP2C8 | 0.50 | 0.051 | 0.044 | 0.87 |
| Diclofenac 4′‐hydroxylation | CYP2C9 | 0.78 | 0.0004 | 0.58 | 0.019 |
| Mephenytoin 4′‐hydroxylation | CYP2C19 | 0.45 | 0.079 | −0.059 | 0.83 |
| Bufuralol 1‐hydroxylation | CYP2D6 | 0.15 | 0.59 | −0.069 | 0.80 |
| Chlorzoxazone 6‐hydroxylation | CYP2E1 | 0.42 | 0.10 | 0.36 | 0.17 |
| Midazolam 1′‐hydroxylation | CYP3A4 | 0.58 | 0.012 | 0.48 | 0.061 |
| Testosterone 6β‐hydroxylation | CYP3A4 | 0.69 | 0.003 | 0.39 | 0.14 |
Discussion
In earlier work, 5′‐HM‐FLX appeared to be highly toxic to biliary epithelial cells and variability in the internal exposure to 5′‐HM‐FLX should be considered as an important factor determining susceptibility to liver injury (Lakehal et al., 2001). Therefore, the aim of the present study was to further characterize the CYPs involved in hydroxylation of flucloxacillin by using recombinant CYPs, specific inhibitors and correlation analysis. Furthermore, the enzyme kinetic parameters of flucloxacillin hydroxylation were quantified using biosynthesized 5′‐HM‐FLX, as the reference compound.
The results of the present study confirm that, upon incubation of flucloxacillin with HLM or recombinant CYPs, 5′‐HM‐FLX is the only oxidative metabolite formed (Figure 2), consistent with previous studies (Lakehal et al., 2001). By using recombinant CYPs, at both substrate concentrations used, CYP3A4 and CYP3A7 appeared to be the most active enzymes. At 10 μM flucloxacillin, CYP2C9 showed a 8‐ to 10‐fold lower activity (Figure 4A). At 100 μM flucloxacillin, low activities were also observed with CYP1A1, CYP1A2, CYP2C8, CYP2E1, CYP2J2 and CYP3A5. The fact that Lakehal et al. (2001) did not detect 5′‐HM‐FLX in incubations with recombinant CYP1A2, CYP2C9 and CYP3A5 might be attributed to the less sensitive analytical method used (HPLC–UV detection vs. LC–MS) and/or the use of yeast‐expressed CYPs with lower specific activity than the Supersomes used in the present study. Also, compared to the present study, incubations with recombinant CYPs were performed at lower temperature (28°C vs. 37°C) and lower enzyme concentration (50 nM vs. 100 nM CYP).
The present study confirms the previous study of Lakehal et al. (2001) that CYP3A4 is likely to play a major role in the formation of 5′‐HM‐FLX, as it is, on average, the most abundant CYP in human liver (mean 93 pmol·mg−1 protein) (Achour et al., 2014). However, the meta‐analysis of Achour et al. demonstrated that in the 713 human livers analysed, CYP3A4 levels varied markedly, ranging from 0 to 601 pmol·mg−1 protein (Achour et al., 2014). This large variability can contribute to the interindividual variability in plasma concentrations of 5′‐HM‐FLX (Maier‐Salamon et al., 2017). As mentioned, next to CYP3A4, another CYP, CYP3A7, showed high activity in flucloxacillin hydroxylation. CYP3A7 was originally considered to be a CYP only expressed in fetal tissues (Schuetz et al., 1994). However, more recent studies have indicated that in approximately 10% of adult livers, CYP3A7 is expressed to a significant extent, up to 90 pmol·mg−1 protein, which equals the mean level of CYP3A4 (Sim et al., 2005; Ohtsuki et al., 2012). This markedly increased CYP3A7 expression appears to be strongly associated with the CYP3A7*1C allele which has arisen through the conversion of 60 bp of the promotor of CYP3A4 into the corresponding region of CYP3A7 (Burk et al., 2002). To what extent the variability in CYP3A7 expression contributes to the variability of flucloxacillin hydroxylation remains to be established and is complicated by the present lack of specific inhibitors for the CYP3A isoforms. The relatively poor correlation between the rate of flucloxacillin hydroxylation with the CYP‐specific reactions (Figure 8 and Table 1) may be explained by the involvement of more than one CYP enzyme.
Involvement of CYP3A isoforms in drug metabolism is well known to result in very diverse types of enzyme kinetics and poorly predictable drug–drug interactions. Dependent on the substrate used, for all CYP3A isoforms, enzyme kinetics can appear as normal Michaelis–Menten kinetics or atypical kinetics such as autoactivation and substrate inhibition (Williams et al., 2002; Atkins, 2005). As possible explanations for this complex behaviour, multiple binding sites in the active site allowing accommodation of multiple substrate molecules or the presence of an effector binding site have been proposed (Ekins et al., 2003; Houston and Galetin, 2005). Because of the multiple binding sites in CYP3A enzymes, also the type of drug–drug interactions appears complex and strongly substrate dependent (Stresser et al., 2000; Ekins et al., 2003). Therefore, to predict the ability to cause CYP3A4 inhibition, the use of multiple CYP3A4 substrates has been recommended (Stresser et al., 2000). The inhibition experiments in the present study seem to confirm the complicated inhibition pattern of CYP3A reactions. The established CYP3A4 inhibitors ketoconazole, troleandomycin and miconazole showed relatively low degree of inhibition at the concentrations used. Remarkably, sulfaphenazole, which is considered to be a highly selective inhibitor of CYP2C9 (Khojasteh et al., 2011), showed clear inhibition of flucloxacillin hydroxylation by pooled HLM, CYP3A4 and CYP3A7, whereas the 6β‐hydroxylation of testosterone was not affected. These results suggest that flucloxacillin has a unique binding mode to the active site to CYP3A4, explaining the relatively weak inhibition by 1 μM ketaconazole and the unexpectedly strong inhibition by sulfaphenazole. A similar situation has been described recently in a study where sulfaphenazole significantly inhibited bioactivation of hydroxylapatinib by HLM, although recombinant CYP2C9 showed only very low activity (Towles et al., 2016). Therefore, the CYP3A family in rare occurrences can be inhibited by sulfaphenazole in a substrate‐dependent manner. The results of the present study contrast with the results of Lakehal et al. which reported no inhibition of HLM‐catalysed hydroxylation of flucloxacillin by 25 μM sulfaphenazole (Lakehal et al., 2001). The reason for the discrepancy between these results remains to be elucidated. A possible explanation might be the different sources of enzymes used, because the ratio between CYP and reductase as well as the membrane composition can affect the specific activity of CYPs (Kim et al., 2003).
In conclusion, the results of this present study confirm the involvement of CYP3A4 in the hydroxylation of flucloxacillin, whereas CYP3A7 and CYP2C9 may also contribute significantly in individuals expressing high hepatic levels. The large variability of both hepatic CYP3A and CYP2C9 levels may contribute to the variability of plasma levels of 5′‐HM‐FLX, which is suspected to play a role in flucloxacillin induced DILI by causing cytotoxicity to biliary epithelial cells. The strong inhibition of CYP3A‐catalysed hydroxylation of flucloxacillin by the commonly used CYP2C9 inhibitor sulfaphenazole suggest that unanticipated drug–drug interactions can occur with co‐administered drugs.
Author contributions
S.J.D. and F.D. performed the experiments; S.J.D. and J.N.M.C. contributed to the writing; S.J.D., N.P.E.V. and J.N.M.C. contributed to the study design.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by the European Community under the Innovative Medicines Initiative (IMI) programme through grant agreement number 115336.
Dekker, S. J. , Dohmen, F. , Vermeulen, N. P. E. , and Commandeur, J. N. M. (2019) Characterization of kinetics of human cytochrome P450s involved in bioactivation of flucloxacillin: inhibition of CYP3A‐catalysed hydroxylation by sulfaphenazole. British Journal of Pharmacology, 176: 466–477. 10.1111/bph.14548.
References
- Achour B, Barber J, Rostami‐Hodjegan A (2014). Expression of hepatic drug‐metabolizing cytochrome P450 enzymes and their intercorrelations: a meta‐analysis. Drug Metab Dispos 42: 1349–1356. [DOI] [PubMed] [Google Scholar]
- Adam D, Koeppe P, Heilmann HD (1983). Pharmacokinetics of amoxicillin and flucloxacillin following the simultaneous intravenous administration of 4 g and 1 g, respectively. Infection 11: 150–154. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews E, Daly AK (2008). Flucloxacillin‐induced liver injury. Toxicology 254: 158–163. [DOI] [PubMed] [Google Scholar]
- Atkins WM (2005). Non‐Michaelis‐Menten kinetics in cytochrome P450‐catalyzed reactions. Annu Rev Pharmacol Toxicol 45: 291–310. [DOI] [PubMed] [Google Scholar]
- Burk O, Tegude H, Koch I, Hustert E, Wolbold R, Glaeser H et al (2002). Molecular mechanisms of polymorphic CYP3A7 expression in adult human liver and intestine. J Biol Chem 277: 24280–24288. [DOI] [PubMed] [Google Scholar]
- Chaudhuri A, Wade SL (2018). Flucloxacillin‐warfarin interaction: an under‐appreciated phenomenon. Intern Med J 48: 860–863. [DOI] [PubMed] [Google Scholar]
- Comuth WJ, Comuth JA, Hauer RNW, Malingré MM (2012). Interaction of flucloxacillin and quinidine. Eur J Clin Pharmacol 68: 891–893. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Brit J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cynke E, Binet I, Haefeli WE, Thiel G (1999). Flucloxacillin & cyclosporine A: an unrecognized but relevant interaction in renal transplant recipients. Kidney Int 55: 1156–1157. [Google Scholar]
- Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Pe'er I, Floratos A et al (2009). DILIGEN Study; International SAE Consortium. HLA‐B*5701 genotype is a major determinant of drug‐induced liver injury due to flucloxacillin. Nat Genet 41: 816–819. [DOI] [PubMed] [Google Scholar]
- De Abajo FJ, Montero D, Madurga M, Garcia Rodriguez LA (2004). Acute and clinically relevant drug‐induced liver injury: a population based case‐control study. Br J Clin Pharmacol 58: 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Braver‐Sewradj SP, den Braver MW, van Dijk M, Zhang Y, Dekker SJ, Wijaya L (2018). Inter‐individual variability in activity of the major drug metabolizing enzymes in liver homogenates of 20 individuals. Curr Drug Metab 19: 370–381. [DOI] [PubMed] [Google Scholar]
- Derby LE, Jick H, Henry DA, Dean AD (1993). Cholestatic hepatitis associated with flucloxacillin. Med J Aust 1581: 596–600. [DOI] [PubMed] [Google Scholar]
- Dijkmans AC, den Hartigh J, van Dissel JT, Burggraaf J (2012). A simplified oral flucloxacillin absorption test for patients requiring long‐term treatment. Therap Drug Monit 34: 356–358. [DOI] [PubMed] [Google Scholar]
- Eckstein RP, Dowsett JF, Lunzer MR (1993). Flucloxacillin induced liver disease: histopathological findings at biopsy and autopsy. Pathology 25: 223–228. [DOI] [PubMed] [Google Scholar]
- Ekins S, Stresser DM, Williams JA (2003). In vitro and pharmacophore insights into CYP3A enzymes. TIPS 24: 161–166. [DOI] [PubMed] [Google Scholar]
- Fairley CK, McNeil JJ, Desmond P, Smallwood R, Young H, Forbes A et al (1993). Risk factors for development of flucloxacillin associated jaundice. Br Med J 306: 233–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gath J, Charles B, Sampson J, Smithurst B (1995). Pharmacokinetics and bioavailability of flucloxacillin in elderly hospitalized patients. J Clin Pharmacol 35: 31–36. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS guide to pharmacology in 2018: updates and expansion to encompass the new guide to immunopharmacology. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston JB, Kenworthy KE (2000). In vitro‐in vivo scaling of CYP kinetic data not consistent with the classical Michaelis‐Menten model. Drug Metab Dispos 28: 246–254. [PubMed] [Google Scholar]
- Houston JB, Galetin A (2005). Modelling atypical CYP3A4 kinetics: principles and pragmatism. Arch Biochem Biophys 433: 351–360. [DOI] [PubMed] [Google Scholar]
- Huwyler J, Wright M, Gutmann H, Drewe J (2006). Induction of cytochrome P450 3A4 and P‐glycoprotein by the isoxazolyl‐penicillin antibiotic flucloxacillin. Curr Drug Metab 7: 119–126. [DOI] [PubMed] [Google Scholar]
- Jenkins RE, Meng X, Elliott VL, Kitteringham NR, Pirmohamed M, Park BK (2009). Characterisation of flucloxacillin and 5‐hydroxymethyl flucloxacillin haptenated HSA in vitro and in vivo . Proteomics Clin Appl 3: 720–729. [DOI] [PubMed] [Google Scholar]
- Khojasteh SC, Prabhu S, Kenny JR, Halladay JS, Lu AYH (2011). Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: a re‐evaluation of P450 isoform selectivity. Eur J Drug Metab Pharmacokinet 36: 1–16. [DOI] [PubMed] [Google Scholar]
- Kim K, Ahn T, Yun C (2003). Membrane properties induced by anionic phospholipids and phosphatidylethanolamine are critical for the membrane binding and catalytic activity of human cytochrome P450 3A4. Biochemistry 42: 15377–15387. [DOI] [PubMed] [Google Scholar]
- Koek GH, Stricker BHC, Blok APR, Schalm SW, Desmet VJ (1994). Flucloxacillin‐associated hepatic injury. Liver 14: 225–229. [DOI] [PubMed] [Google Scholar]
- Kumar V, Wahlstrom JL, Rock DA, Warren CJ, Gorman LA, Tracy TS (2006). CYP2C9 inhibition: impact of probe selection and pharmacogenetics on in vitro inhibition profiles. Drug Metab Dispos 34: 1966–1975. [DOI] [PubMed] [Google Scholar]
- Lakehal F, Dansette PM, Becquemont L, Lasnier E, Delelo R, Balladur P et al (2001). Indirect cytotoxicity of flucloxacillin toward human biliary epithelium via metabolite formation in hepatocytes. Chem Res Toxicol 14: 694–701. [DOI] [PubMed] [Google Scholar]
- Landersdorfer CB, Kirkpatrick CMJ, Kinzig‐Schippers M, Bulitta JB, Holzgrabe U, Drusano GL et al (2007). Population pharmacokinetics at two dose levels and pharmacodynamic profiling of flucloxacillin. Antimicrob Agents Chemother 51: 3290–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder K, Turnidge JD, Korman TM, Grayson ML (1999). The clinical efficacy of continuous‐infusion flucloxacillin in serious staphylococcal sepsis. J Antimicrob Chemother 43: 113–118. [DOI] [PubMed] [Google Scholar]
- Luirink RA, Dekker SJ, Capoferri L, Janssen LFH, Kuiper CL, Ari ME et al (2018). A combined computational and experimental study on selective flucloxacillin hydroxylation by cytochrome P450 BM3 variants. J Inorg Biochem 184: 115–122. [DOI] [PubMed] [Google Scholar]
- Maier‐Salamon A, Elgendy SA, Meyer B, Vossen M, Thalhammer T, Thalhammer F et al (2017). Pharmacokinetics of flucloxacillin and its metabolites in patients with renal failure: impact on liver toxicity. Int J Clin Pharmacol Ther 55: 701–711. [DOI] [PubMed] [Google Scholar]
- Monshi MM, Faulkner L, Gibson A, Jenkins RE, Farrell J, Earnshaw CJ et al (2013). Human leukocyte antigen (HLA)‐B*57:01‐restricted activation of drug‐specic T cells provides the immunological basis for flucloxacillin‐induced liver injury. Hepatology 57: 727–739. [DOI] [PubMed] [Google Scholar]
- Muilwijk EW, Dekkers BGJ, Henriet SSV, Verweij PE, Witjes B, Lashof AMLO et al (2017). Flucloxacillin results in suboptimal plasma voriconazole concentrations. Antimicrob Agents Chemother 6: e00915–e00917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa T, Shiraga T, Takagi A (2009). Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull 28: 1805–1808. [DOI] [PubMed] [Google Scholar]
- Ohtsuki S, Schaefer O, Kawakami H, Inoue T, Liehner S, Saito A et al (2012). Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP‐glucuronosyltransferases as a novel approach for the characterization of individual human liver: comparison with mRNA levels and activities. Drug Metab Dispos 40: 83–92. [DOI] [PubMed] [Google Scholar]
- Røder BL, Frimodt‐Møller N, Espersen F, Rasmussen SN (1995). Dicloxacillin and flucloxacillin: pharmacokinetics, protein binding and serum bactericidal titers in healthy subjects after oral administration. Infection 23: 107–112. [DOI] [PubMed] [Google Scholar]
- Russmann S, Kaye JA, Jick SS, Jick H (2005). Risk of cholestatic liver disease associated with flucloxacillin and flucloxacillin prescribing habits in the UK: cohort study using data from the UK General Practice Research Database. Br J Clin Pharmacol 60: 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz JD, Beach DL, Guzelian PS (1994). Selective expression of cytochrome P450 3A mRNAs in embryonic and adult human liver. Pharmacogenetics 4: 11–20. [DOI] [PubMed] [Google Scholar]
- Sim SC, Edwards RJ, Boobis AR, Ingelman‐Sundberg M (2005). CYP3A7 protein expression is high in a fraction of adult human livers and partially associated with the CYP3A7*1C allele. Pharmacogenet Genomics 15: 625–631. [DOI] [PubMed] [Google Scholar]
- Stresser DM, Blanchard AP, Turner SD, Erve JCL, Dandeneau AA, Miller VP et al (2000). Substrate‐dependent modulation of CYP3A4 catalytic activity: analysis of 27 test compounds with four fluorimetric substrates. Drug Metab Dispos 28: 1440–1448. [PubMed] [Google Scholar]
- Sutherland R, Croydon EAP, Rolinson GN (1970). Flucloxacillin, a new isoxazolyl penicillin, compared with oxacillin, cloxacillin, and dicloxacillin. Br Med J 4: 455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailor A, Faulkner L, Naisbitt DJ, Park BK (2015). The chemical, genetic and immunological basis of idiosyncratic drug‐induced liver injury. Hum Exp Toxicol 34: 1310–1317. [DOI] [PubMed] [Google Scholar]
- Thijssen HHW, Wolters J (1982). The metabolic disposition of flucloxacillin in patients with impaired kidney function. Eur J Clin Pharmacol 22: 429–434. [DOI] [PubMed] [Google Scholar]
- Towles JK, Clark RN, Wahlin MD, Uttamsingh V, Rettie AE, Jackson KD (2016). Cytochrome P450 3A4 and CYP3A5‐catalyzed bioactivation of lapatinib. Drug Metab Dispos 44: 1584–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht J (2007). Idiosyncratic drug reactions: current understanding. Annu Rev Pharmacol Toxicol 47: 513–539. [DOI] [PubMed] [Google Scholar]
- Williams JA, Ring BJ, Cantrell VE, Jones DR, Eckstein J, Ruterbories K et al (2002). Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab Dispos 30: 883–891. [DOI] [PubMed] [Google Scholar]
- Wuillemin N, Adam J, Fontana S, Krähenbühl S, Pichler WJ, Yerly D (2013). HLA haplotype determines hapten or p‐i T cell reactivity to flucloxacillin. J Immunol 190: 4956–4964. [DOI] [PubMed] [Google Scholar]
- Wuillemin N, Terracciano L, Beltraminelli H, Schlapbach C, Fontana S, Krahenbuhl S et al (2014). T cells infiltrate the liver and kill hepatocytes in HLA‐B(*)57:01‐associated oxacillin‐induced liver injury. Am J Pathol 184: 1677–1682. [DOI] [PubMed] [Google Scholar]