

Abstract
White matter injury is a crucial component of human stroke, but it has often been neglected in preclinical studies. Most human stroke is associated with one or more comorbidities, including aging, hypertension, diabetes and metabolic syndrome including hyperlipidemia. The purpose of this review is to examine how age and hypertension impact stroke-induced white matter injury as well as white matter repair in both human stroke and preclinical models. It is essential that comorbidities be examined in preclinical trials as they may impact translatability to the clinic. In addition, understanding how comorbidities impact white matter injury and repair may provide new therapeutic opportunities for patients with those conditions.
Keywords: Aging, diffusion tensor imaging, hypertension, inflammation, myelin, stroke, white matter
1. Introduction
White matter (WM) constitutes half the human brain and plays an essential role in the transmission of electric signals within the functional neural network (Fields, 2010). Ischemic stroke causes prominent WM damage, thereby contributing to long-term sensorimotor and cognitive impairment. It has been estimated that ~64–86% of stroke patients have WM injury (Fu et al., 2005; Li et al., 2013).
Subcortical WM stroke represents 15–30% of all stroke subtypes in humans (Bamford et al., 1991). WM receives relatively low blood supply compared to grey matter, has relatively poor collateral circulation and WM components (e.g. oligodendrocytes and their progenitor cells) are sensitive to ischemia-induced oxidative stress and excitotoxicity, making WM highly vulnerable to ischemic injury (Husain and Juurlink, 1995; Iadecola et al., 2009; McDonald et al., 1998; Wang et al., 2016). On the other hand, cerebral ischemia rapidly elicits compensatory WM regeneration and repair, which benefits the structural and functional restoration of the brain (Moskowitz et al., 2010). WM repair consists of axonal regeneration and neural plasticity, as well as remyelination, where new myelin sheaths are formed around demyelinated axons. Remyelination restores nerve function and prevents further axonal degeneration, and it involves the recruitment and differentiation of oligodendrocyte progenitor cells (OPCs) into mature myelin-forming oligodendrocytes (Gensert and Goldman, 1997). Despite the importance of WM in brain function, the mechanisms underlying WM injury and plasticity after ischemic stroke, and its implication in developing therapeutic approaches, have been a neglected area of research until relatively recently.
Age is the most important non-modifiable risk factor for ischemic stroke and has significant impact on stroke occurrence and prognosis. After age 55, the risk of stroke nearly doubles every 10 years (Marini et al., 2001). In combination with other comorbid conditions, such as hypertension, hyperglycemia and hyperlipidemia, age predisposes the WM to structural and functional changes and impairs the regenerative capacity of WM, thereby predicting worse outcome after stroke (Chien et al., 2007; Hamner et al., 2011; Kurl et al., 2006). White matter hyperintensity (WMH), also called leukoaraiosis, is a radiologic finding detected by magnetic resonance imaging (MRI) in at least 50% of the aged population (de Leeuw et al., 2001; Longstreth et al., 1996; Munoz Maniega et al., 2017; Ryu et al., 2014). The presence and severity of WMH is associated with cognitive decline, increased risk of stroke, and poor clinical outcome after ischemic stroke (Ay et al., 2008; Debette and Markus, 2010; Kuller et al., 2004). Furthermore, myelin density declines with age in both humans and rodents, indicating that the capacity for oligodendrogenesis and WM regeneration may be compromised by WM senescence (Sim et al., 2002).
Even though stroke mainly afflicts the elderly and most stroke patients have one or more comorbid conditions, there are only limited studies concerning the effect of these comorbid conditions on WM injury and repair after stroke. Here, we review recent findings on the impact of aging and hypertension, two of the most important vascular comorbidities on WM disease. Understanding the mechanisms underlying the effects of each of these two conditions may facilitate the development of therapeutic strategies for WM stroke that are more translatable tothe clinic.
2. White matter structure
WM tracts are composed of both myelinated and non-myelinated axons that are essential for communication between different CNS areas. There is evidence both within the brain and in the peripheral nervous system that myelinated axons are more sensitive to ischemia or ischemia-like conditions than non-myelinated axons (Fujimura et al., 1991; Li et al., 2016a). This reflects the sensitivity of oligodendrocytes that produce myelin to ischemia (Fujimura et al., 1991; Li et al., 2016a), although it may also reflect the average greater diameter of myelinated axons with larger axons being more sensitive to ischemia (Fujimura et al., 1991). In addition, there is evidence that after ischemia, axonal sprouting occurs that results in increased non-myelinated axons (Sozmen et al., 2016).
Many studies on stroke-induced WM injury have focused on oligodendrocyte and axonal changes as the ultimate cause of altered neurological function and OPCs as a potential source of WM repair. However, as outlined below, other cell types within WM (Fig. 1) play an important role in stroke-induced WM injury and repair including microglial and endothelial cells. In grey matter, communication within the neurovascular unit (NVU; formed of endothelial cells, pericytes, astrocyte endfeet, smooth muscle cells, basement membranes and neuronal and microglial processes) plays an important role in regulating the cerebrovasculature function (e.g. blood flow and the BBB) (Iadecola, 2017; Zlokovic, 2008). NVU signaling is important in preventing ischemic brain injury, but the NVU is also an important site of injury after ischemia (e.g. BBB disruption and swollen astrocytic endfeet) (Jiang et al., 2018; Shi et al., 2016a). In WM, there is an analogous oligovascular signaling between oligodendrocytes/OPCs and other vascular cells (endothelial cells, pericytes, astrocyte endfeet, smooth muscle cells) (Arai and Lo, 2009c; Shindo et al., 2016). The pathways involved in such signaling are beginning to be elucidated (e.g. (Arai and Lo, 2009c; Rajani and Williams, 2017)). Understanding how those pathways are affected by stroke comorbidities may give insight into the mechanisms by which they impact WM injury.
Fig. 1. Key cellular components of white matter and the oligovascular signaling.
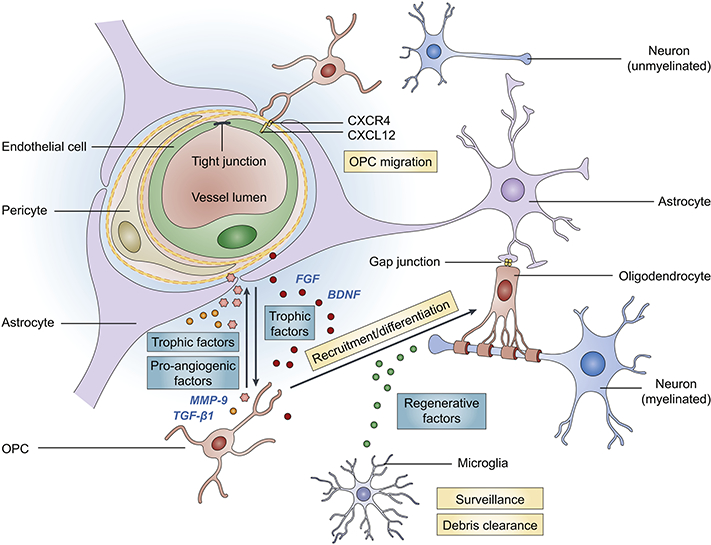
The ‘oligovascular unit’ in the white matter consists of oligodendrocyte progenitor cells (OPCs), myelinating oligodendrocytes, endothelial cells, astrocytes, pericytes and microglia. These key cellular components are functionally coupled and influence each other under physiological and pathological conditions. Astrocytes and oligodendrocytes are coupled by gap junctions (Orthmann-Murphy et al., 2007; Robinson et al., 1993). Endothelial cells promote the survival, proliferation and differentiation of OPCs through secretion of trophic factors, e.g., brain-derived neurotrophic factor (BDNF) and fibroblast growth factor (FGF) (Arai and Lo, 2009b). On the other hand, OPCs support the survival of endothelial cells and alter the permeability of BBB via various trophic factors and pro-angiogenic factors, such as transforming growth factor (TGF)-ßl and matrix metallopeptidase (MMP)-9 (Seo et al., 2014; Seo et al., 2013). The migration of OPCs along vasculature and OPC-endothelium interaction coordinated by CXCR4-CXCL12 signaling is essential for normal brain development (Tsai et al., 2016). Pericytes also critically regulate OPC differentiation/maturation and white matter homeostasis (Maki et al., 2018). Pericyte deficiency disrupts white matter microcirculation and facilitates the deposition of toxic blood-derived substances, leading to a loss of myelin, axons and oligodendrocytes (Montagne et al., 2018). Microglia, the resident immune cells in the brain, actively patrol the white matter and produce regenerative factors that support OPC differentiation. Microglia also play a pivotal role in clearing myelin debris and facilitating remyelination after brain injury.
3. Preclinical modeling
Preclinical models aid in elucidating the mechanisms by which stroke and associated comorbid conditions impact WM. However, the limitations in those models should be acknowledged. Such research has been performed in vitro on isolated cells (e.g. OPCs and oligodendrocytes) and cell lines, tissue preparations (e.g. the optic nerve), and using in vivo models (usually rodent). The advantages and disadvantages of such models have been described by Arai & Lo (Arai and Lo, 2009a). The in vitro experiments allow mechanistic examination of specific, or limited, cell types. Such experiments, however, do not fully replicate in vivo conditions, often lack the complex cellular interactions occurring in vivo (Arai and Lo, 2009a) and mostly use non-human cells. Current advances in human induced pluripotent stem cells, that can be used to produce multiple neural cell types including OPCs and oligodendrocytes (Goldman and Kuypers, 2015), is a major advance. The production of cerebral organoids (Lancaster et al., 2013) is a potential technique for examining the complex cellular interactions although there are ethical questions.
As described below, most stroke preclinical in vivo research is done on rodents that have greatly smaller brains and much less WM than humans. The effect of such scaling on WM injury is unclear and there is a need for more work on stroke-induced WM injury in larger gyrencephalic species with significant WM (Hainsworth et al., 2017), to confirm findings in smaller species if similar data cannot be directly obtained from patients.
A variety of rodent models have been used to study the effects of cerebral ischemia or hypoperfusion on WM. These include hypoperfusion caused by bilateral common carotid artery occlusion or stenosis, focal ischemia caused by middle cerebral artery occlusion (MCAO), and the spontaneous stroke that occurs in the stroke-prone spontaneously hypertensive rats (SHRSP). It should be noted that such models have grey as well as white matter ischemia adding complexity to determining underlying causes of injury. Recently, a model of WM ischemia has been developed involving stereotactic injection of an endothelial nitric oxide inhibitor, N5-(1- iminoethyl)-L-ornithine, into the corpus callosum in mice (Nunez et al., 2016). That should be useful for elucidating specific WM pathways.
A discussion of the limitations of animal models of aging and hypertension are beyond
4. Aging
4.1. Age-related WM structural alterations
Progressive brain atrophy and deterioration of WM ultrastructure during aging have been well documented. Lifespan studies based on postmortem investigations of human brains reveal a decline of the overall brain weight, beginning at about 45–50 years of age and reaching its lowest values (11% decrease from young adults) after 86 years of age (Dekaban, 1978). Such atrophy is accompanied by an elevation of brain water content, demyelination, and disruption of axonal structure (Meier-Ruge et al., 1992; Terao et al., 1994). Studies based on human and non-human primates implicate differential alterations of axons in the cortex and WM during normal aging (Peters, 2002). In cortex, no significant neuronal loss and very few loss of nerve fibers are observed during aging (Nielsen and Peters, 2000). Instead, degeneration of myelin sheaths of nerve fibers is prominent in the cortex and appears to account for normal aging-related cognitive decline (Peters et al., 2000; Peters and Sethares, 2002). This is in contrast to the WM where 27% the scope of this review. of the total length of nerve fibers are lost in old human brains (Tang et al., 1997). Age-related WM changes have further been examined by diffusion tensor imaging (DTI), a special type of diffusion-weighted MRI (DW-MRI) that is widely used to map WM tracts in the brain. Both the absolute volume of WM and the ratio of WM to total brain volume drop with age after about 40 years of age (Lebel et al., 2012). Age-related WM microstructural changes can also be assessed by various diffusivity parameters, the most commonly used being fractional anisotropy (FA) and mean diffusivity (MD). Several studies report a decrease of FA, an index of the degree of directionality of water diffusivity (Lebel et al., 2012; Sala et al., 2012; Sexton et al., 2014). In contrast, MD represents the average magnitude of water diffusion and is found to increase in WM tracts during aging (Lebel et al., 2012; Sala et al., 2012; Sexton et al., 2014). Furthermore, age-related changes of FA and MD are associated with increased radial diffusivity (RD; diffusivity perpendicular to the axonal fibers), a possible indicator of myelin injury (Sala et al., 2012; Song et al., 2003). In contrast, WM tracts display variable values of axial diffusivity (AD; diffusion parallel to the axon fibers) during aging, a possible indicator of axonal integrity (Sala et al., 2012; Song et al., 2003).
Age-associated axon and myelin alterations can also be detected histologically and underlying mechanisms examined. Ultrastructural examination using three-dimensional electron microscopy reveals larger axons and thicker myelin in the aged mouse optic nerve (Stahon et al., 2016). Furthermore, aged axons develop longer and thicker mitochondria, correlating to lower ATP levels and elevated production of nitric oxide, protein nitration, and lipid peroxidation (Stahon et al., 2016). Ca2+ homeostasis is disrupted in aged axons, as reflected by lower levels of calnexin and calreticulin, which results in deficient interactions between mitochondria and smooth endoplasmic reticulum (Stahon et al., 2016). On the other hand, one emerging theory on the mechanism of myelodegeneration is that the aberrant organization of axo-glial junctional complexes may underlie age-related myelin pathology (Shepherd et al., 2012). Normally, the anchor of myelin sheath to the axon is dependent on the integrity of paranodal axo-glial junctional complexes, and their disruption facilitates demyelination (Brophy, 2001; Marcus et al., 2002). The main components of such complexes are the so-called transverse bands, which are arrayed electron-dense structures bridging the peri-axonal space. Physiologically, paranodal complexes are dynamically reorganized, manifested by the loss of transverse bands in previously anchored loops followed by the lateral movement of newly juxtaposed loops to fill the gap (Shepherd et al., 2012). Transverse bands are reformed and turned over during this paranodal reorganization, thereby maintaining a stable paranodal length which is crucial for the organization of ion channels and function of the axon. In the aged CNS, the reformation of transverse bands is less efficient, leading to the loss of junctional components (Shepherd et al., 2012). These abnormal structural changes facilitate the degeneration of myelin and are likely to predispose the aged CNS to increased WM injury should a secondary insult occur. In line with this theory, disrupted axonal microdomain architecture resulting from abnormal axoglial signaling is frequently observed in the WM adjacent to lacunar and microinfarcts (Hinman et al., 2015). The disarrangement of node and paranode structure is associated with axonal injury and may lead to impaired nerve signal conduction of surviving axons after stroke (Hinman et al., 2015). Direct evidence on whether paranodal structural changes are important in age-related WM injury after stroke is, however, still lacking.
Chronic brain tissue hypoperfusion with aging is associated with WMHs (Bernbaum et al., 2015; Munoz Maniega et al., 2017; Promjunyakul et al., 2016; Shi et al., 2016b; van den Boom et al., 2003). However, whether low cerebral blood flow (CBF) is a cause or the result of WM injury is uncertain. A meta-analysis oflongitudinal studies suggested that low CBF may be the result of WMH rather than a cause, although more studies particularly focusing on WM blood flow changes with age are needed (Shi et al., 2016b).
4.2. Age-related injury mechanisms of the WM after ischemic stroke
Earlier studies employing rodent focal MCAO models to investigate the impact of aging on stroke outcome have generated controversial results. Some studies show an age-related enlargement of brain infarct (Sutherland et al., 1996), whereas others report smaller infarcts in aged subjects (Liu et al., 2009). This discrepancy may partly be accounted for by the confounding effects of high mortality rate in the aged cohort (44 versus 9% in young adult rats subjected to 60 minutes of MCAO) (Wang et al., 2003). More recently, researchers have used vasoconstrictor-induced WM stroke or permanent distal MCAO models, which have lower mortality and make it possible to evaluate long-term neurological functions after stroke. At 2 months after N5-(1-iminoethyl)-L-ornithine-induced WM stroke, aged mice (24-month old) developed more severe WM atrophy, disruption of corticostriatal connections, and forelimb motor deficits than young adult mice (Rosenzweig and Carmichael, 2013). Similarly, 18 months old aged mice demonstrated worse WM demyelination and long-term sensorimotor and cognitive deficits up to 35 days after distal MCAO (Suenaga et al., 2015).
4.2.1. Axonal mechanisms
An enhanced oxidative state of the brain and excitotoxicity are generally thought to underlie age-dependent exacerbation of WM injury after ischemic stroke (Fig. 2). Mitochondrial functions deteriorate during normal aging, leading to a decline of ATP production. Furthermore, the insufficient function of endogenous antioxidant systems (e.g. glutathione peroxidase) in the aged brain worsens oxidative stress (Espinoza et al., 2008). Increased oxidative damage is observed in both rodent WM stroke models (Li et al., 2011; Rosenzweig and Carmichael, 2013), and in post-mortem MRI-guided sampling from patients older than 65 years of age (Al-Mashhadi et al., 2015).
Fig. 2. Age-related injury and repair mechanisms of white matter after cerebral ischemia.
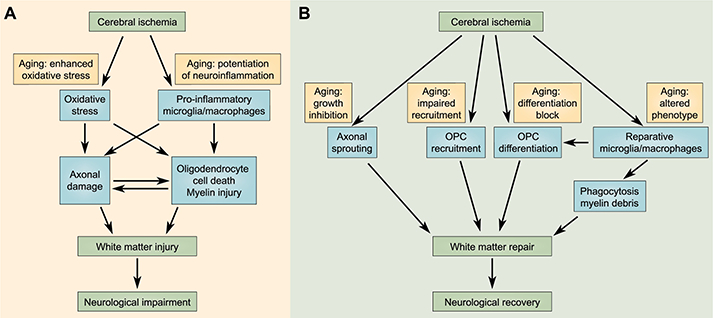
Aging profoundly influences white matter injury (A) and repair (B) after ischemic stroke. (A) Aging enhances oxidative stress and potentiates neuroinflammation after cerebral ischemia, thereby exacerbating axonal degeneration and oligodendrocyte cell death. The subsequent blockade of nerve signal transduction in the disrupted neural circuits contributes to post-stroke neurological impairments. (B) Various white matter repair mechanisms are compromised during aging. Aging directly inhibits axonal growth, oligodendrocyte progenitor cell (OPC) recruitment/differentiation, and remyelination after ischemia. Furthermore, aging shifts microglia/macrophages from a reparative inflammation-resolving phenotype toward a destructive pro-inflammatory phenotype, thereby indirectly blocking white matter repair after ischemic stroke.
Ischemia induces axonal injury through differential molecular mechanisms in young and aged subjects (Fig. 3). Studies using mouse optic nerve demonstrate that in young WM, removal of extracellular Ca2+ or blockade of reverse Na+/Ca2+ exchange (NCX) during ischemia robustly improves the functional recovery of axons, whereas blockade of NMDA receptors fails to promote axonal function (Baltan, 2009; Baltan, 2016). In contrast, the injury to aged WM after ischemia is primarily through Ca2+-independent mechanisms, as the same approach fails to save the oligodendrocytes and hinders axon function recovery (Baltan, 2009; Baltan, 2016). Ischemia rapidly triggers depletion of ATP and reversal of the NCX, leading to an elevation of intracellular Na+ and reversal of the Na+-dependent glutamate transporter (GLT1). The excessive glutamate may then activate AMPA/kainite receptors, eventually resulting in oligodendrocyte cell death and axonal injury in the WM (Baltan, 2014). In the aged brain, GLT1 is robustly upregulated, perhaps as a compensatory mechanism against the increased glutamate. However, ischemia-induced reversal of GLT1 results in earlier, more robust, and sustained glutamate release and severe excitotoxicity in the presence of higher GLT1 expression in aged WM (Baltan, 2009). Inhibiting the reversal of GLT1 or AMPA/kainate receptors protects aged axons against ischemic injury, an effect comparable to young axons. On the other hand, NMDA receptors are redistributed during normal aging such that in young WM, they are largely expressed in the cell bodies of oligodendrocytes, whereas in aged WM, they are also expressed by myelin processes (Baltan, 2016). This age-specific expression pattern may determine the interactions between oligodendrocytes and axons and their functional outcome after ischemia.
Fig. 3. Age-related molecular mechanisms of white matter injury after cerebral ischemia.
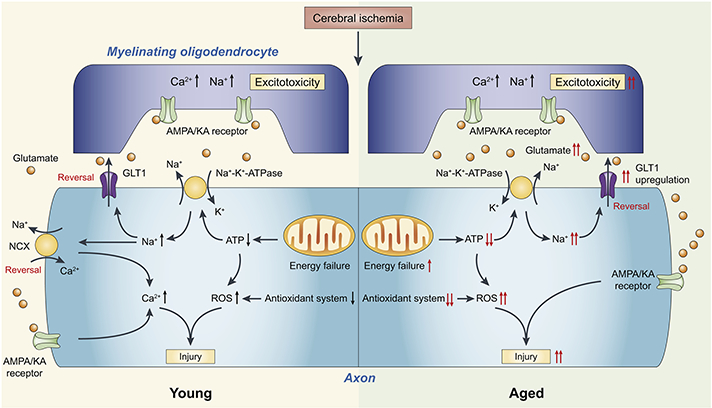
Cerebral ischemia leads to mitochondria dysfunction and ATP depletion. The resultant increase of intracellular Na+ and subsequent reversal of the Na+/Ca2+ exchanger (NCX) and Na+- dependent glutamate transporter (GLT1) lead to increased intracellular Ca2+ and an overload of glutamate (Baltan, 2009; Baltan, 2016). AMPA/kainite receptors are activated by the overloaded glutamate, leading to excitotoxic axonal and myelin injury. In aged WM, deteriorated mitochondria dysfunction and insufficient function of the endogenous antioxidant system both contribute to elevated production of ROS. Furthermore, GLT1 is upregulated in aged WM, resulting in more rapid and robust glutamate release and exacerbated excitotoxicity (Baltan, 2009; Baltan, 2016). Together, these changes contribute to increased vulnerability of aged WM to ischemic injury and worsen the outcome.
4.2.2. Vascular mechanisms
Age-dependent vascular alterations in the WM increase its susceptibility to hypofusion and ischemic injury. WM is intrinsically vulnerable to hypofusion, as it receives blood supply primarily through long arterioles from the border zone between the MCA and the anterior cerebral artery (Moody et al., 1990). Aging leads to a reduction of vascular density and a deficiency of cerebral vascular autoregulation, both exacerbating hypoperfusion in aged WM (Jin et al., 2017). Endothelial dysfunction associated with aging, such as enhanced vascular oxidative stress and impaired endothelium-dependent vasodilation in the cerebral circulation, is also likely to contribute to the chronic hypofusion of aged WM (Brown et al., 2007; Mayhan et al., 1990; Toth et al., 2017). Furthermore, tortuous arterioles are formed in aged WM and develop a coiled morphology (Brown et al., 2002), resulting in increased vessel length, declined kinetic energy, and reduced blood flow in the WM. The blood flow to the WM can further be deteriorated by excess collagen deposits in the walls of veins and venules in the deep WM of aged brains and subsequent constriction of the vessel lumen (Brown et al., 2002).
4.2.3. Inflammatory mechanisms
Aged brains display an enhanced pro-inflammatory state after ischemic stroke, with increased production of pro-inflammatory cytokines and greater microglial/macrophage activation (Rosenzweig and Carmichael, 2013). Microglia/macrophages can exert dual functions in brain injury and repair, dependent on their pro-inflammatory or anti-inflammatory functional phenotype (Hu et al., 2015; Jiang et al., 2016; Jin et al., 2017). Such phenotypic polarization of microglia/macrophages also has profound effects on WM injury and repair (Miron et al., 2013; Wang et al., 2015). After distal MC AO, 18 months old aged mice exhibited significantly fewer microglia/macrophages with an anti-inflammatory ‘M2-like’ phenotype compared to young adult mice (Suenaga et al., 2015). Importantly, this age-related shift of microglia/macrophages toward a pro-inflammatory phenotype is associated with exacerbated myelin degeneration and neurobehavioral deficits (Suenaga et al., 2015). In recent years, the use of unbiased whole- genome profiling has begun to identify the genomic changes occurring during the reprogramming of microglia/macrophages upon aging (Galatro et al., 2017). How such changes influence WM injury after stroke remains to be elucidated.
4.3. The effect of aging on WM repair after stroke
It is well established that the efficiency of WM repair declines with age, with deficits in both the regeneration of axons and the remyelination of injured axons after brain injury (Fig. 2B). Ischemic stroke elicits sprouting of spared axons in the peri-infarct region, which form new patterns of neuronal connections and are associated with recovery (Brown et al., 2009; Carmichael et al., 2001). Using whole-genome expression profiling of sprouting versus non-sprouting neurons after focal cortical ischemic stroke, Li et al. identified a “sprouting transeriptome” consisting of growth factor, cell adhesion, axonal guidance and cyto skeleta1 modifying molecules (Li et al., 2010). Importantly, this neuronal growth program differs between young adult and aged rodents, particularly in cytokine and chemokine, axonal guidance, bone morphogenic protein, cell adhesion and growth factor molecules. For example, EphA4 and Lingol, receptors for axonal growth inhibitory proteins (Ji et al., 2006; Wegmeyer et al., 2007), are upregulated in aged sprouting neurons (Li et al., 2010). These transcriptional profile changes indicate that reduced post-stroke recovery in aged subjects may be associated with blockade of axonal growth in the peri-infarct sprouting neurons.
Aging also impairs another key component of WM repair after stroke - remyelination of demyelinated axons. Studies of non-ischemic injury models have demonstrated clear impairment of remyelination in aged experimental animals. Using a focal demyelination model found that an age-associated decline in remyelination efficiency is attributed to an impairment of both the recruitment of OPCs and their subsequent differentiation into myelinating oligodendrocytes (Sim et al., 2002). OPC differentiation is critically determined by both environmental factors and cell- intrinsic mechanisms that change with age. Using a cuprizone-induced demyelination model, Shen et al. emphasize the importance of age-dependent epigenetic control of gene expression by histone deacetylases (HDACs) (Shen et al., 2008). In demyelinated young brains, oligodendrocyte differentiation inhibitors and neural stem cell markers are downregulated before the synthesis of new myelin. In contrast, in demyelinated old brains, the recruitment of HDACs and epigenetic modulation is inefficient, leading to the accumulation of transcriptional inhibitors and prevents the subsequent expression of myelin genes (Shen et al., 2008).
Such mechanisms are only beginning to be elucidated in stroke models. For example, a recent study by Sozmen et al. reveals a crucial role of Nogo receptor 1 (NgRl) signaling in blocking OPC maturation after WM stroke (Sozmen et al., 2016). In both aged and young adult mice, stroke induces NgR1 ligands and downregulates NgR1 inhibitors during the peak OPC maturation block. Blockade of Nogo signaling using an NgR1 antagonist reduced the transformation of OPCs into astrocytes and thereby enhanced oligodendrogenesis after stroke, which significantly improved post-stroke functional recovery in aged mice (Sozmen et al., 2016).
Aging may also alter the WM regeneration after stroke through indirect actions of other CNS cells on WM components, such as microglia/macrophages. Depending on their functional phenotype, microglia/macrophages differentially influence WM repair after brain injury. On the one hand, M2-like microglia/macrophages promote OPC differentiation and CNS remyelination after focal demyelination injury through production of regenerative factors (Miron et al., 2013). On the other hand, myelin debris are generated during injury-induced demyelination and contain inhibitors of OPC differentiation (Kotter et al., 2006). Efficient remyelination also depends on a supportive microenvironment where microglia/macrophages play a crucial rule in clearing cell and myelin debris. Aging alters a wide range of microglial functions (Koellhoffer et al., 2017) and, as reviewed above, the number of M2-like microglia/macrophages after ischemic stroke declines with age (Suenaga et al., 2015), exacerbating WM injury and hampering WM repair. Furthermore, the efficiency of myelin debris clearance declines with age, further delaying OPC differentiation and impeding remyelination (Ruckh et al., 2012). Microglia from aged mice have deficits in phagocytosis under baseline conditions and when activated with a functional shift towards a pro-inflammatory M1-like phenotype (Ritzel et al., 2015). Thus, during aging, aspects of microglial/macrophage functions become unfavorable for WM repair.
In recent years, studies focused on identifying molecular mechanisms that determine microglial/macrophage functions have provided insights into developing therapeutic strategies to improve WM repair after injury. For example, triggering receptor expressed on myeloid cells 2 (TREM2) sustains both microglial expansion during aging and the response to demyelination (Poliani et al., 2015). In response to cuprizone-induced demyelination, Trem2−/− microglia failed to amplify transcripts indicative of activation, phagocytosis, and lipid catabolism (Poliani et al., 2015). Consequently, Trem2 null mice exhibit impaired myelin debris clearance, axonal dystrophy, reduced oligodendrocytes, and persistent demyelination after prolonged cuprizone treatment (Poliani et al., 2015). Importantly, mutation of the TREM2 gene in humans causes Nasu-Hakola disease, for which WM degeneration is a prominent pathological feature (Kaneko et al., 2010). Considering these age-dependent alterations of microglial functions and their poor responses to demyelination in Trem2−/− mice, TREM2 functional deficiency may underlie the insufficient microglial clearance of myelin in the aging human.
Another example is the retinoid X receptor (RXR) pathway that is decreased with aging in both myelin-phagocytosing human monocytes and mouse macrophages (Natrajan et al., 2015). Young macrophages treated with an RXR antagonist mimic aging by reducing myelin debris uptake, whereas an RXR agonist partially restored myelin debris phagocytosis in aged macrophages (Natrajan et al., 2015). Young mice with macrophage-specific RXRa knockout had delayed clearance of myelin debris and slowed remyelination after experimental demyelination. The RXR agonist could also revise the gene expression profile in the monocytes of multiple sclerosis patients to a more ‘youthful’ state, enhancing myelin debris phagocytosis by patient cells (Natrajan et al., 2015). Whether similar strategies to modulate aged microglia/macrophages will benefit WM repair after ischemic stroke remains to be determined.
There are other agents that are being investigated for their ability to alter the phenotype of microglia/macrophages. For example, fingolimod, a sphingosine-1 -phosphate receptor modulator, shifts microglial/macrophage phenotype from Ml to M2 polarization, and it reduces WM injury in a bilateral common carotid artery stenosis (BCAS) model by reducing neuroinflammation and promoting oligodendrogenesis (Qin et al., 2017). Similarly, rosiglitazone, a peroxisome proliferator-activated receptor (PPAR)-γ agonist, shifts microglial/macrophage polarization from Ml to M2 after MCAO in mice, and it enhances OPC proliferation, increases the generation of mature oligodendrocytes and reduces WM injury (Han et al., 2015). PPAR-γ agonists do have pleiotropic effects that may reduce ischemic damage (Cai et al., 2018). Whether fingolimod and PPAR-γ agonists can impact stroke-induced WM injury in aged animals deserves investigation. It should also be noted that PPAR-γ agonists also impact other comorbidities for stroke that worsen WM injury - diabetes, hypertension and dyslipidemia.
5. Hypertension
5.1. Hypertension-related WM changes
Hypertension is the most important modifiable risk factor for ischemic stroke and an important risk factor for vascular cognitive impairment. Acute and chronic increases in blood pressure result in adaptive vascular remodeling throughout the body and subsequent impairment of multiple end-organs, including the brain (Scuteri et al., 2011). High blood pressure is also one of the most important risk factors for cerebral WM lesions in the aged population (de Leeuw et al., 2002; Xiong and Mok, 2011). Characterized by arteriolosclerosis, myelin loss and gliosis, WM lesions are frequently observed in cognitively intact and impaired elderly persons (Longstreth et al., 1996; van Swieten et al., 1991). An association between hypertension and WM lesions has been well documented in both cross-sectional studies (Dufouil et al., 2001; Shrestha et al., 2009) and longitudinal studies (Gottesman et al., 2010; Sachdev et al., 2007; Schmidt et al., 2003; White et al., 2011). Both systolic and diastolic blood pressures are significantly associated with annual WM lesion progression, and anti-hypertensive treatment can reduce WM lesion progression in the general population (Verhaaren et al., 2013). These WM lesions are closely associated with cognitive impairment and gait disorders in the aged population. Studies by Li et al. report decreased executive functions and attention in hypertensive versus normotensive patients, which is accounted for by altered connectivity in frontoparietal networks (Li et al., 2015). The integrity of WM tracts in the bilateral superior longitudinal fasciculus is impaired in the hypertensive subjects (Li et al., 2015). Thus, hypertension causes a specific pattern of cognitive decline, possibly due to deficits in the WM and functional connectivity in frontal and parietal lobes (Li et al., 2015; Li et al., 2016b).
Impaired cerebral autoregulation is believed to play a crucial role in mediating high blood pressure-induced WM injury. Autoregulation is an important compensatory mechanism to protect the brain from elevated perfusion pressure damage (Iadecola and Davisson, 2008). A constant and stable CBF prevents vascular leakage, WM damage and brain swelling (Iadecola and Davisson, 2008; Sam et al., 2016). A longitudinal study by Beason-Held et al. reported that the duration of hypertension contributes significantly to the patterns of CBF change over time. Compared to normal controls, hypertensive subjects had greater regional CBF decreases in prefrontal, anterior cingulate, and occipital cortex over time (Beason-Held et al., 2007). Furthermore, increased duration of hypertension was associated with decreased regional CBF in prefrontal and anterior cingulate cortex, areas that show functional decline with time in hypertensive subjects (Beason-Held et al., 2007).
5.2. Hypertension and WM injury in ischemic stroke
Age-dependent deterioration of stroke outcome associated with hypertension has been observed. Following focal cerebral ischemia, 3- and 12-month-old spontaneously hypertensive rats (SHRs) develop comparable infarct volume at both 3 and 14 days. However, recovery of behavioral deficits was significantly worse in the older animals (Liang et al., 2016). The influence of high blood pressure on the development of ischemic WM injury and cognitive dysfunction is not fully understood. Specifically, whether the exacerbated WM injury reflects a direct effect on WM components or secondary effects related to vascular changes remains to be elucidated. In a study using a chronic cerebral hypoperfusion model induced by bilateral common carotid artery occlusion (BCCAO), Choi et al. reported that SHRs develop earlier and more severe spatial memory deficits compared to normotensive rats (Choi et al., 2015). Despite a lack of gross structural changes in myelinated axons or oligodendrocyte numbers, BCCAO elicits subtle demyelination and paranodal structural alterations at the nodes of Ranvier, independent of hypertension. SHRSP develop WM disease, and this is associated with reduced tissue blood flow and abnormal blood vessels (Henning et al., 2010). Similarly, Weaver et al. found that SHR-SP fed a Japanese permissive diet and undergoing a unilateral carotid artery occlusion had marked reductions in WM tissue oxygenation that was associated with WM damage and microvessel hemorrhages (Weaver et al., 2014).
Another mechanism exacerbating WM injury after stroke is hypertension-induced brain microvascular endothelium dysfunction and BBB breakdown. Hypertension-induced BBB permeability facilitates extravasation of plasma proteins into brain, exacerbation of oxidative stress, inflammation and cerebral edema (Woywodt et al., 2003). Following BCCAO, BBB breakdown is predominantly observed in the WM of SHRs, suggesting a role of hypertension in BBB disruption in chronic cerebral hypoperfusion (Choi et al., 2015). In stroke-prone renal vascular hypertensive rats, tight junction proteins (occludin and ZO-1) are progressively lost from WM from 4 to 30 weeks after the onset of hypertension, and this occurred in concert with severe WM lesions (Fan et al., 2015). Stroke- and hypertension-induced WM tight junction changes are understudied, and it is still uncertain if such changes differ from grey matter or if the consequences of tight junction changes differ between the two types of tissue.
BBB disruption fosters neuroinflammation and may initiate a self-perpetuating cycle with increased matrix metalloproteinases (MMPs), BBB hyperpermeability and WM injury. Vascular dementia patients show expression of MMPs in regions with myelin loss (Rosenberg et al., 2001; Taheri et al., 2011; Topakian et al., 2010). In SHR-SP rats receiving 1% NaCl in their drinking water, apoptosis of mature oligodendrocytes is detected after MCAO, together with elevated MMP-3, MMP-9 and TNF-α, and BBB disruption (Jalal et al., 2012). In addition, OPCs can rapidly react to WM damage and produce MMP-9 that accelerates BBB breakdown (Rosenberg, 2009; Seo et al., 2013).
Lowering blood pressure in patients with chronic hypertension remains one of the most effective means of stroke prevention (Hermida et al., 2016). Importantly, however, there is evidence that some specific treatments of hypertension are more effective than others. For example, bilateral renal denervation can significantly reduce blood pressure in patients with treatment-resistant hypertension (Krum et al., 2009; Symplicity et al., 2010). In SHRSP, such denervation effectively prevents the onset of stroke and brain injury progression in hypertensive rats, and these beneficial effects are likely to involve attenuation of oxidative stress, reduced inflammation mediated by microglia/macrophages, and alleviation of BBB disruption (Nakagawa et al., 2013). However, treating animals with hydralazine, which caused a similar blood pressure lowering to renal denervation, did not significantly suppress the onset of stroke and brain injury in SHRSP.
Although aging and hypertension both have effects on WM injury after ischemia, there is still a relative paucity of studies examining the effects of hypertension in aged animals on such injury. As described above, both stroke comorbidities have axon/oligodendrocyte, cerebrovascular and inflammatory effects. The extent to which those effects are synergistic, enhancing WM injury, should be examined further. Such studies may help identify particularly important therapeutic targets for reducing WM injury in the elderly with hypertension.
5.3. Effect of aging on WM repair in hypertension
Hypertension is a negative regulator of endogenous brain regenerative processes after ischemic injury. Such inhibitory effects are exacerbated with aging. In a rat MCAO model, 12- month-old SHRs show significant less numbers of nestin+ neural stem/progenitor cells (NSPCs) in the peri-infarct area and subventricular zone than 3-month-old SHRs at day 3 after MCAO (Liang et al., 2016). Similarly, numbers of plate let-derived growth factor receptor alpha (PDGFRa)-positive OPCs in the corpus callosum are lower in 12-month-old SHRs at day 3 after MCAO (Liang et al., 2016). The reduced NSPC and OPC numbers appear to be associated with lower phosphorylation levels of cAMP response element-binding protein (CREB) (Liang et al., 2016). At day 14 after MCAO, NSPC and OPC numbers in 12-month-old SHRs recover to comparable levels as in 3-month-old SHRs, but proliferating NSPCs (Ki-67+nestin+ cells) and proliferating OPCs (Ki-67+PDGFRa+ cells) remain less in the aged brains (Liang et al., 2016). These findings suggest that aging may decrease post-stroke neurogenesis and oligodendrogene sis under hypertensive conditions. Another study by Wang et al. also supports age-related effects on white matter integrity after stroke in hypertensive subjects, using SHR-SP (Wang et al., 2011).
6. Perspectives
The last decade has seen a greater preclinical research focus on WM injury and repair after stroke, and that has been associated with advances in our understanding of the mechanisms involved in those processes. As yet, there are no post-stroke clinical therapies to limit WM injury or enhance WM repair. This review highlights one potential problem in translating preclinical data, mostly obtained in healthy young animals, to the clinic - the impact that comorbidities have on the WM response to stroke. The majority of stroke patients have one or more comorbidities and these may affect therapeutic efficacy. However, it should be noted that an understanding of how comorbidities impact WM injury/repair may help identify specific targets for patients with those conditions.
Another difficulty in translation is that some targets may have both beneficial and detrimental effects, where initial deleterious responses may actively participate in neurovascular restoration (Lo, 2008; Lo, 2010). In this regard, it is imperative to understand when, where, and how the transition from injury to repair occurs. For example, microglial activation after cerebral ischemia can enhance early brain injury but be involved in later brain repair (Guruswamy and ElAli, 2017; Hu et al., 2015). Comorbid conditions may affect not only the magnitude of such effects (e.g. aging reducing the ability of microglia to promote brain repair), but also their timing (e.g. delaying repair), complicating therapeutic interventions.
The presence of WMHs with age has been attributed to the relatively low blood flow of WM, poor collateral circulation and the potential selective vulnerability of oligodendrocytes and OPCs to oxidative stress and excitotoxicity (Wang et al., 2016). There may, however, be other contributory factors. For example, there has been an upsurge in interest in how alterations in fluid movement within the brain contribute to neurological conditions (Lundgaard et al., 2017; Plog and Nedergaard, 2018). Although the ‘glymphatic’ hypothesis posits fluid movement via astrocytes (Plog and Nedergaard, 2018), there is evidence that WM is a major route for edema movement within the brain (Ohata et al., 1990; Vorbrodt et al., 1985). Thus, conditions with BBB disruption and vasogenic edema may result in the preferential delivery of potentially neuro to xic/neuroinflammatory plasma constituents (e.g. prothrombin/thrombin, fibrinogen) to WM. Increased, low level, BBB disruption has been identified in aged patients and animals (Elahy et al., 2015; Erdo et al., 2017; Farrall and Wardlaw, 2009).
There have been significant advances in the tools available to study structural changes in WM after injury, including an array of MRI modalities such as DTI (Wang et al., 2016). Those structural assessments can be combined with resting state functional MRI to assess changes in connectivity in animals and in patients (Dijkhuizen et al., 2012; Gordon et al., 2018). Recent years, the development of new techniques is greatly facilitating preclinical research on WM structure and mechanisms of WM injury and repair. Clear Lipid-exchanged, Anatomically Rigid, Imaging/immunostaining compatible, Tissue hYdrogel (CLARITY) is a recently developed method that can transform intact brain tissues into optically transparent and macromolecule- permeable hydrogel-hybridized form, while preserving the fine-structure and crosslinked biomolecules (Chung et al., 2013). In combination with brain-wide immunohistochemistry and DTI, CLARITY reveals structure and connectivity of WM tracts in the brain and elucidates the relationship between water diffusion signal and its underlying biological basis (Chang et al., 2017). Additional technological advances which could be adapted to deeper structures of the CNS, such as two-photon imaging, can also be used to study WM structure and function in vivo.
Optogenetics, a technique to achieve temporally precise, noninvasive control of activity in well-defined neuronal populations (Boyden et al., 2005), is also being applied to WM research. Through precise control of neuronal activity, optogenetic approaches have begun to reveal the causal relationship between specific neuronal activity and WM repair mechanisms (e.g. oligodendrogenesis, remyelination) and improved functional outcome after brain injury (Gibson et al., 2014; Jin et al., 2015). On the other hand, optogenetic stimulation is being used as a therapy to improve WM connectivity and stroke recovery (Tennant et al., 2017).
There has also been much interest in the use of plasma and cerebrospinal fluid (CSF) biomarkers in brain injury diagnosis/prognosis. After ischemic stroke, Hjalmarsson et al. found that CSF concentrations of neuro filament light protein (an axonal component of myelinated tissue) correlated with WM injury (Hjalmarsson et al., 2014). CSF levels of that protein are also elevated in patients with vascular dementia compared to Alzheimer’s disease and healthycontrols (Vilar-Bergua et al., 2016).
7. Conclusions
WM injury is an extremely important component of stroke-induced brain injury in patients. A variety of stroke comorbidities including aging, hypertension and diabetes enhance WM injury and inhibit repair. It is essential that this be taken into account in both preclinical studies and clinical trials to improve stroke outcome. In addition, understanding how such comorbidities impact WM may provide new therapeutic opportunities.
ACKNOWLEDGMENTS
This work was supported by the American Heart Association grant 17SDG33630130 (to Y.S.), by the National Institutes of Health grant NS093399 (to R.F.K.) and by the Joyce & Don Massey Family Foundation (to R.F.K.). Y.S. is supported by the Competitive Medical Research Fund of the University of Pittsburgh Medical Center (UPMC) Health System, and the Pittsburgh Institute of Brain Disorders & Recovery startup funds. M.X. is supported by the China Scholarship Council award 201506100062. M.M.W. is supported by the VA Merit Awards BX003824 and BX003855. Y.G. is supported by the Ministry of Science and Technology of China Key R&D Plan 2017YFC1308403 and the National Natural Science Foundation of China grant 81571285. The funding sources had no involvement in the writing of this article and in the decision to submit the article for publication.
Footnotes
DECLARATIONS OF INTEREST
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Al-Mashhadi S, Simpson JE, Heath PR, Dickman M, Forster G, Matthews FE, Brayne C, Ince PG, Wharton SB, Medical Research Council Cognitive, F., Ageing S, 2015. Oxidative Glial Cell Damage Associated with White Matter Lesions in the Aging Human Brain. Brain Pathol. 25, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Lo EH, 2009a. Experimental models for analysis of oligodendrocyte pathophysiology in stroke. Exp Transl Stroke Med. 1, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Lo EH, 2009b. An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci. 29, 4351–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Lo EH, 2009c. Oligovascular signaling in white matter stroke. Biol Pharm Bull. 32, 1639–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ay H, Arsava EM, Rosand J, Furie KL, Singhal AB, Schaefer PW, Wu O, Gonzalez RG, Koroshetz WJ, Sorensen AG, 2008. Severity of leukoaraiosis and susceptibility to infarct growth in acute stroke. Stroke. 39, 1409–1413. [DOI] [PubMed] [Google Scholar]
- Baltan S, 2009. Ischemic injury to white matter: an age-dependent process. Neuroscientist. 15, 126–133. [DOI] [PubMed] [Google Scholar]
- Baltan S, 2014. Excitotoxicity and mitochondrial dysfunction underlie age-dependent ischemic white matter injury. Adv Neurobiol. 11, 151–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, 2016. Age-specific localization of NMDA receptors on oligodendrocytes dictates axon fonction recovery after ischemia. Neuropharmacology. 110, 626–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford J, Sandercock P, Dennis M, Burn J, Warlow C, 1991. Classification and natural history of clinically identifiable subtypes of cerebral infarction. Lancet. 337, 1521–1526. [DOI] [PubMed] [Google Scholar]
- Beason-Held LL, Moghekar A, Zonderman AB, Kraut MA, Resnick SM, 2007. Longitudinal changes in cerebral blood flow in the older hypertensive brain. Stroke. 38, 1766–1773. [DOI] [PubMed] [Google Scholar]
- Bernbaum M, Menon BK, Fick G, Smith EE, Goyal M, Frayne R, Coutts SB, 2015. Reduced blood flow in normal white matter predicts development of leukoaraiosis. J Cereb Blood Flow Metab. 35, 1610–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K, 2005. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 8, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Brophy PJ, 2001. Axoglial junctions: separate the channels or scramble the message. Curr Biol. 11, R555–557. [DOI] [PubMed] [Google Scholar]
- Brown CE, Aminoltejari K, Erb H, Winship IR, Murphy TH, 2009. In vivo voltage- sensitive dye imaging in adult mice reveals that somatosensory maps lost to stroke are replaced over weeks by new structural and functional circuits with prolonged modes of activation within both the peri-infarct zone and distant sites. J Neurosci. 29, 1719–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KA, Didion SP, Andresen JJ, Faraci FM, 2007. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler Thromb Vasc Biol. 27, 1941–1946. [DOI] [PubMed] [Google Scholar]
- Brown WR, Moody DM, Challa VR, Thore CR, Anstrom JA, 2002. Venous collagenosis and arteriolar tortuosity in leukoaraiosis. J Neurol Sci. 203–204, 159–163. [DOI] [PubMed] [Google Scholar]
- Cai W, Yang T, Liu H, Han L, Zhang K, Hu X, Zhang X, Yin KJ, Gao Y, Bennett MVL, Leak RK, Chen J, 2018. Peroxisome proliferator-activated receptor gamma (PPARgamma): A master gatekeeper in CNS injury and repair. Prog Neurobiol. 163–164, 27–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael ST, Wei L, Rovainen CM, Woolsey TA, 2001. New patterns of intracortical projections after focal cortical stroke. Neurobiol Dis. 8, 910–922. [DOI] [PubMed] [Google Scholar]
- Chang EH, Argyelan M, Aggarwal M, Chandon TS, Karlsgodt KH, Mori S, Malhotra AK, 2017. The role of myelination in measures of white matter integrity: Combination of diffusion tensor imaging and two-photon microscopy of CLARITY intact brains. Neuroimage. 147, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien KL, Hsu HC, Sung FC, Su TC, Chen MF, Lee YT, 2007. Metabolic syndrome as a risk factor for coronary heart disease and stroke: an 11-year prospective cohort in Taiwan community. Atherosclerosis. 194, 214–221. [DOI] [PubMed] [Google Scholar]
- Choi JY, Cui Y, Kim BG, 2015. Interaction between hypertension and cerebral hypoperfusion in the development of cognitive dysfunction and white matter pathology in rats. Neuroscience. 303, 115–125. [DOI] [PubMed] [Google Scholar]
- Chung K, Wallace J, Kim SY, Kalyanasundaram S, Andalman AS, Davidson TJ, et al. , 2013. Structural and molecular interrogation of intact biological systems. Nature. 497, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw FE, de Groot JC, Achten E, Oudkerk M, Ramos LM, Heijboer R, Hofman A, Jolles J, van Gijn J, Breteler MM, 2001. Prevalence of cerebral white matter lesions in elderly people: a population based magnetic resonance imaging study. The Rotterdam Scan Study. J Neurol Neurosurg Psychiatry. 70, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw FE, de Groot JC, Oudkerk M, Witteman JC, Hofman A, van Gijn J, Breteler MM, 2002. Hypertension and cerebral white matter lesions in a prospective cohort study. Brain. 125, 765–772. [DOI] [PubMed] [Google Scholar]
- Debette S, Markus HS, 2010. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ. 341, c3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekaban AS, 1978. Changes in brain weights during the span of human life: relation of brain weights to body heights and body weights. Ann Neurol. 4, 345–356. [DOI] [PubMed] [Google Scholar]
- Dijkhuizen RM, van der Marel K, Otte WM, Hoff EI, van der Zijden JP, van der Toorn A, van Meer MP, 2012. Functional MRI and diffusion tensor imaging of brain reorganization after experimental stroke. Transl Stroke Res. 3, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufouil C, de Kersaint-Gilly A, Besancon V, Levy C, Auffray E, Brunnereau L, Alperovitch A, Tzourio C, 2001. Longitudinal study of blood pressure and white matter hyperintensities: the EVA MRI Cohort. Neurology. 56, 921–926. [DOI] [PubMed] [Google Scholar]
- Elahy M, Jackaman C, Mamo JC, Lam V, Dhaliwal SS, Giles C, Nelson D, Takechi R, 2015. Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun Ageing. 12, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdo F, Denes L, de Lange E, 2017. Age-associated physiological and pathological changes at the blood-brain barrier: A review. J Cereb Blood Flow Metab. 37, 4–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza SE, Guo H, Fedarko N, DeZern A, Fried LP, Xue QL, Leng S, Beamer B, Walston JD, 2008. Glutathione peroxidase enzyme activity in aging. J Gerontol A Biol Sci Med Sci. 63, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Yang X, Tao Y, Lan L, Zheng L, Sun J, 2015. Tight junction disruption of blood- brain barrier in white matter lesions in chronic hypertensive rats. Neuroreport. 26, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Farrall AJ, Wardlaw JM, 2009. Blood-brain barrier: ageing and microvascular disease- systematic review and meta-analysis. Neurobiol Aging. 30, 337–352. [DOI] [PubMed] [Google Scholar]
- Fields RD, 2010. Neuroscience. Change in the brain’s white matter. Science. 330, 768–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu JH, Lu CZ, Hong Z, Dong Q, Luo Y, Wong KS, 2005. Extent of white matter lesions is related to acute subcortical infarcts and predicts further stroke risk in patients with first ever ischaemic stroke. J Neurol Neurosurg Psychiatry. 76, 793–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura H, Lacroix C, Said G, 1991. Vulnerability of nerve fibres to ischaemia. A quantitative light and electron microscope study. Brain. 114 ( Pt 4), 1929–1942. [DOI] [PubMed] [Google Scholar]
- Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, et al. , 2017. Transcriptomic analysis of purified human cortical microglia reveals age- associated changes. Nat Neurosci. 20, 1162–1171. [DOI] [PubMed] [Google Scholar]
- Gensert JM, Goldman JE, 1997. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron. 19, 197–203. [DOI] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, Barres BA, Woo PJ, Vogel H, Monje M,2014. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science. 344, 1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SA, Kuypers NJ, 2015. How to make an oligodendrocyte. Development. 142, 3983–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon EM, Scheibel RS, Zambrano-Vazquez L, Jia-Richards M, May GJ, Meyer E, Nelson SM, 2018. High-Fidelity Measures of Whole-Brain Functional Connectivity and White Matter Integrity Mediate Relationships between Traumatic Brain Injury and Post-Traumatic Stress Disorder Symptoms. J Neurotrauma. 35, 767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman RF, Coresh J, Catellier DJ, Sharrett AR, Rose KM, Coker LH, Shibata K, Knopman DS, Jack CR, Mosley TH Jr., 2010. Blood pressure and white- matter disease progression in a biethnic cohort: Atherosclerosis Risk in Communities (ARIC ) study. Stroke. 41, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruswamy R, ElAli A, 2017. Complex Roles of Microglial Cells in Ischemic Stroke Pathobiology: New Insights and Future Directions. Int J Mol Sci. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainsworth AH, Allan SM, Boltze J, Cunningham C, Farris C, Head E, et al. , 2017. Translational models for vascular cognitive impairment: a review including larger species. BMC Med. 15, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamner MA, Moller T, Ransom BR, 2011. Anaerobic function of CNS white matter declines with age. J Cereb Blood Flow Metab. 31, 996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Cai W, Mao L, Liu J, Li P, Leak RK, Xu Y, Hu X, Chen J, 2015. Rosiglitazone Promotes White Matter Integrity and Long-Term Functional Recovery After Focal Cerebral Ischemia. Stroke. 46, 2628–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning EC, Warach S, Spatz M, 2010. Hypertension-induced vascular remodeling contributes to reduced cerebral perfusion and the development of spontaneous stroke in aged SHRSP rats. J Cereb Blood Flow Metab. 30, 827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermida RC, Ayala DE, Smolensky MH, Fernandez JR, Mojon A, Portaluppi F, 2016. Chronotherapy with conventional blood pressure medications improves management of hypertension and reduces cardiovascular and stroke risks. Hypertens Res. 39, 277–292. [DOI] [PubMed] [Google Scholar]
- Hinman JD, Lee MD, Tung S, Vinters HV, Carmichael ST, 2015. Molecular disorganization of axons adjacent to human lacunar infarcts. Brain. 138, 736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjalmarsson C, Bjerke M, Andersson B, Blennow K, Zetterberg H, Aberg ND, Olsson B, Eckerstrom C, Bokemark L, Wallin A, 2014. Neuronal and glia-related biomarkers in cerebrospinal fluid of patients with acute ischemic stroke. J Cent Nerv Syst Dis. 6, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Chen J, 2015. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 11, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain J, Juurlink BH, 1995. Oligodendroglial precursor cell susceptibility to hypoxia is related to poor ability to cope with reactive oxygen species. Brain Res. 698, 86–94. [DOI] [PubMed] [Google Scholar]
- Iadecola C, 2017. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron. 96, 17–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Davisson RL, 2008. Hypertension and cerebrovascular dysfunction. Cell Metab. 7, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Park L, Capone C, 2009. Threats to the mind: aging, amyloid, and hypertension. Stroke. 40, S40–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalal FY, Yang Y, Thompson J, Lopez AC, Rosenberg GA, 2012. Myelin loss associated with neuroinflammation in hypertensive rats. Stroke. 43, 1115–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji B, Li M, Wu WT, Yick LW, Lee X, Shao Z, Wang J, So KF, McCoy JM, Pepinsky RB, Mi S, Relton JK, 2006. LINGO-1 antagonist promotes functional recovery and axonal sprouting after spinal cord injury. Mol Cell Neurosci. 33, 311–320. [DOI] [PubMed] [Google Scholar]
- Jiang X, Andjelkovic AV, Zhu L, Yang T, Bennett MVL, Chen J, Keep RF, Shi Y, 2018. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol. 163–164, 144–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Pu H, Hu X, Wei Z, Hong D, Zhang W, Gao Y, Chen J, Shi Y, 2016. A Post-stroke Therapeutic Regimen with Omega-3 Polyunsaturated Fatty Acids that Promotes White Matter Integrity and Beneficial Microglial Responses after Cerebral Ischemia. Transl Stroke Res. 7, 548–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D, Liu Y, Sun F, Wang X, Liu X, He Z, 2015. Restoration of skilled locomotion by sprouting corticospinal axons induced by co-deletion of PTEN and SOCS3. Nat Commun. 6, 8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin WN, Shi SX, Li Z, Li M, Wood K, Gonzales RJ, Liu Q, 2017. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab. 37, 2224–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Sano K, Nakayama J, Amano N, 2010. Nasu-Hakola disease: The first case reported by Nasu and review: The 50th Anniversary of Japanese Society of Neuropathology. Neuropathology. 30, 463–470. [DOI] [PubMed] [Google Scholar]
- Koellhoffer EC, McCullough LD, Ritzel RM, 2017. Old Maids: Aging and Its Impact on Microglia Function. Int J Mol Sci. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter MR, Li WW, Zhao C, Franklin RJ, 2006. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci. 26, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krum H, Schlaich M, Whitbourn R, Sobotka PA, Sadowski J, Bartus K, Kapelak B, Walton A, Sievert H, Thambar S, Abraham WT, Esler M, 2009. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof- of-principle cohort study. Lancet. 373, 1275–1281. [DOI] [PubMed] [Google Scholar]
- Kuller LH, Longstreth WT Jr., Arnold AM, Bernick C, Bryan RN, Beauchamp NJ Jr., Cardiovascular Health Study Collaborative Research, G., 2004. White matter hyperintensity on cranial magnetic resonance imaging: a predictor of stroke. Stroke. 35, 1821–1825. [DOI] [PubMed] [Google Scholar]
- Kurl S, Laukkanen JA, Niskanen L, Laaksonen D, Sivenius J, Nyyssonen K, Salonen JT, 2006. Metabolic syndrome and the risk of stroke in middle-aged men. Stroke. 37, 806–811. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA, 2013. Cerebral organoids model human brain development and microcephaly. Nature. 501, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel C, Gee M, Camicioli R, Wieler M, Martin W, Beaulieu C, 2012. Diffusion tensor imaging of white matter tract evolution over the lifespan. Neuroimage. 60, 340–352. [DOI] [PubMed] [Google Scholar]
- Li L, Simoni M, Kuker W, Schulz UG, Christie S, Wilcock GK, Rothwell PM, 2013. Population-based case-control study of white matter changes on brain imaging in transient ischemic attack and ischemic stroke. Stroke. 44, 3063–3070. [DOI] [PubMed] [Google Scholar]
- Li L, Velumian AA, Samoilova M, Fehlings MG, 2016a. A Novel Approach for Studying the Physiology and Pathophysiology of Myelinated and Non-Myelinated Axons in the CNS White Matter. PLoS One. 11, e0165637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Kong X, Ye R, Yang Q, Han J, Xiong L, 2011. Age-related differences in experimental stroke: possible involvement of mitochondrial dysfunction and oxidative damage. Rejuvenation Res. 14, 261–273. [DOI] [PubMed] [Google Scholar]
- Li S, Overman JJ, Katsman D, Kozlov SV, Donnelly CJ, Twiss JL, Giger RJ, Coppola G, Geschwind DH, Carmichael ST, 2010. An age-related sprouting transcriptome provides molecular control of axonal sprouting after stroke. Nat Neurosci. 13, 1496–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Liang Y, Chen Y, Zhang J, Wei D, Chen K, Shu N, Reiman EM, Zhang Z, 2015. Disrupted Frontoparietal Network Mediates White Matter Structure Dysfunction Associated with Cognitive Decline in Hypertension Patients. J Neurosci. 35, 10015–10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ma C, Sun X, Zhang J, Chen Y, Chen K, Zhang Z, 2016b. Disrupted white matter structure underlies cognitive deficit in hypertensive patients. Eur Radiol. 26, 2899–2907. [DOI] [PubMed] [Google Scholar]
- Liang AC, Mandeville ET, Maki T, Shindo A, Som AT, Egawa N, Itoh K, Chuang TT, McNeish JD, Holder JC, Lok J, Lo EH, Arai K, 2016. Effects of Aging on Neural Stem/Progenitor Cells and Oligodendrocyte Precursor Cells After Focal Cerebral Ischemia in Spontaneously Hypertensive Rats. Cell Transplant. 25, 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Yuan R, Benashski SE, McCullough LD, 2009. Changes in experimental stroke outcome across the life span. J Cereb Blood Flow Metab. 29, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH, 2008. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 14, 497–500. [DOI] [PubMed] [Google Scholar]
- Lo EH, 2010. Degeneration and repair in central nervous system disease. Nat Med. 16, 1205–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longstreth WT Jr., Manolio TA, Arnold A, Burke GL, Bryan N, Jungreis CA, Enright PL, O’Leary D, Fried L, 1996. Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. The Cardiovascular Health Study. Stroke. 27, 1274–1282. [DOI] [PubMed] [Google Scholar]
- Lundgaard I, Lu ML, Yang E, Peng W, Mestre H, Hitomi E, Deane R, Nedergaard M, 2017. Glymphatic clearance controls state-dependent changes in brain lactate concentration. J Cereb Blood Flow Metab. 37, 2112–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki T, Choi YK, Miyamoto N, Shindo A, Liang AC, Ahn BJ, et al. , 2018. A-Kinase Anchor Protein 12 Is Required for Oligodendrocyte Differentiation in Adult White Matter. Stem Cells. 36, 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus J, Dupree JL, Popko B, 2002. Myelin-associated glycoprotein and myelin galactolipids stabilize developing axo-glial interactions. J Cell Biol. 156, 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini C, Triggiani L, Cimini N, Ciancarelli I, De Santis F, Russo T, Baldassarre M, di Orio F, Carolei A, 2001. Proportion of older people in the community as a predictor of increasing stroke incidence. Neuroepidemiology. 20, 91–95. [DOI] [PubMed] [Google Scholar]
- Mayhan WG, Faraci FM, Baumbach GL, Heistad DD, 1990. Effects of aging on responses of cerebral arterioles. Am J Physiol. 258, H1138–1143. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP, 1998. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor- mediated excitotoxicity. Nat Med. 4, 291–297. [DOI] [PubMed] [Google Scholar]
- Meier-Ruge W, Ulrich J, Bruhlmann M, Meier E, 1992. Age-related white matter atrophy in the human brain. Ann N Y Acad Sci. 673, 260–269. [DOI] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJM, Ffrench-Constant C, 2013. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 16, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Nikolakopoulou AM, Zhao Z, Sagare AP, Si G, Lazic D, et al. , 2018. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat Med. 24, 326–337. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Moody DM, Bell MA, Challa VR, 1990. Features of the cerebral vascular pattern that predict vulnerability to perfusion or oxygenation deficiency: an anatomic study. AJNR Am J Neuroradiol. 11, 431–439. [PMC free article] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C, 2010. The science of stroke: mechanisms in search of treatments. Neuron. 67, 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz Maniega S, Chappell FM, Valdes Hernandez MC, Armitage PA, Makin SD, Heye AK, Thrippleton MJ, Sakka E, Shuler K, Dennis MS, Wardlaw JM, 2017. Integrity of normal-appearing white matter: Influence of age, visible lesion burden and hypertension in patients with small-vessel disease. J Cereb Blood Flow Metab. 37, 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Hasegawa Y, Uekawa K, Ma M, Katayama T, Sueta D, Toyama K, Kataoka K, Koibuchi N, Maeda M, Kuratsu J, Kim-Mitsuyama S, 2013. Renal denervation prevents stroke and brain injury via attenuation of oxidative stress in hypertensive rats. J Am Heart Assoc. 2, e000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natrajan MS, de la Fuente AG, Crawford AH, Linehan E, Nunez V, Johnson KR, Wu T, Fitzgerald DC, Ricote M, Bielekova B, Franklin RJ, 2015. Retinoid X receptor activation reverses age-related deficiencies in myelin debris phagocytosis and remyelination. Brain. 138, 3581–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen K, Peters A, 2000. The effects of aging on the frequency of nerve fibers in rhesus monkey striate cortex. Neurobiol Aging. 21, 621–628. [DOI] [PubMed] [Google Scholar]
- Nunez S, Doroudchi MM, Gleichman AJ, Ng KL, Llorente IL, Sozmen EG, Carmichael ST, Hinman JD, 2016. A Versatile Murine Model of Subcortical White Matter Stroke for the Study of Axonal Degeneration and White Matter Neurobiology. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata K, Marmarou A, Povlishock JT, 1990. An immunocytochemical study of protein clearance in brain infusion edema. Acta Neuropathol. 81, 162–177. [DOI] [PubMed] [Google Scholar]
- Orthmann-Murphy JL, Freidin M, Fischer E, Scherer SS, Abrams CK, 2007. Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J Neurosci. 27, 13949–13957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, 2002. Structural changes that occur during normal aging of primate cerebral hemispheres. Neurosci Biobehav Rev. 26, 733–741. [DOI] [PubMed] [Google Scholar]
- Peters A, Moss MB, Sethares C, 2000. Effects of aging on myelinated nerve fibers in monkey primary visual cortex. J Comp Neurol. 419, 364–376. [DOI] [PubMed] [Google Scholar]
- Peters A, Sethares C, 2002. Aging and the myelinated fibers in prefrontal cortex and corpus callosum of the monkey. J Comp Neurol. 442, 277–291. [DOI] [PubMed] [Google Scholar]
- Plog BA, Nedergaard M, 2018. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu Rev Pathol. 13, 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, Colonna M, 2015. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest. 125, 2161–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promjunyakul NO, Lahna DL, Kaye JA, Dodge HH, Erten-Lyons D, Rooney WD, Silbert LC, 2016. Comparison of cerebral blood flow and structural penumbras in relation to white matter hyperintensities: A multi-modal magnetic resonance imaging study. J Cereb Blood Flow Metab. 36, 1528–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C, Fan WH, Liu Q, Shang K, Murugan M, Wu LJ, Wang W, Tian DS, 2017. Fingolimod Protects Against Ischemic White Matter Damage by Modulating Microglia Toward M2 Polarization via STAT3 Pathway. Stroke. 48, 3336–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajani RM, Williams A, 2017. Endothelial cell-oligodendrocyte interactions in small vessel disease and aging. Clin Sci (Lond). 131, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzel RM, Patel AR, Pan S, Crapser J, Hammond M, Jellison E, McCullough LD, 2015. Age- and location-related changes in microglial function. Neurobiol Aging. 36, 2153–2163. [DOI] [PubMed] [Google Scholar]
- Robinson SR, Hampson EC, Munro MN, Vaney DI, 1993. Unidirectional coupling of gap junctions between neuroglia. Science. 262, 1072–1074. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, 2009. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 8, 205–216. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Sullivan N, Esiri MM, 2001. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke. 32, 1162–1168. [DOI] [PubMed] [Google Scholar]
- Rosenzweig S, Carmichael ST, 2013. Age-dependent exacerbation of white matter stroke outcomes: a role for oxidative damage and inflammatory mediators. Stroke. 44, 2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckh JM, Zhao JW, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ, Franklin RJ, 2012. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 10, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu WS, Woo SH, Schellingerhout D, Chung MK, Kim CK, Jang MU, et al. , 2014. Grading and interpretation of white matter hyperintensities using statistical maps. Stroke. 45, 3567–3575. [DOI] [PubMed] [Google Scholar]
- Sachdev P, Wen W, Chen X, Brodaty H, 2007. Progression of white matter hyperintensities in elderly individuals over 3 years. Neurology. 68, 214–222. [DOI] [PubMed] [Google Scholar]
- Sala S, Agosta F, Pagani E, Copetti M, Comi G, Filippi M, 2012. Microstructural changes and atrophy in brain white matter tracts with aging. Neurobiol Aging. 33, 488–498 e482. [DOI] [PubMed] [Google Scholar]
- Sam K, Peltenburg B, Conklin J, Sobczyk O, Poublanc J, Crawley AP, Mandell DM, Venkatraghavan L, Duffin J, Fisher JA, Black SE, Mikulis DJ, 2016. Cerebrovascular reactivity and white matter integrity. Neurology. 87, 2333–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R, Enzinger C, Ropele S, Schmidt H, Fazekas F, Austrian Stroke Prevention S, 2003. Progression of cerebral white matter lesions: 6-year results of the Austrian Stroke Prevention Study. Lancet. 361,2046–2048. [DOI] [PubMed] [Google Scholar]
- Scuteri A, Nilsson PM, Tzourio C, Redon J, Laurent S, 2011. Microvascular brain damage with aging and hypertension: pathophysiological consideration and clinical implications. J Hypertens. 29, 1469–1477. [DOI] [PubMed] [Google Scholar]
- Seo JH, Maki T, Maeda M, Miyamoto N, Liang AC, Hayakawa K, Pham LD, Suwa F, Taguchi A, Matsuyama T, Ihara M, Kim KW, Lo EH, Arai K, 2014. Oligodendrocyte precursor cells support blood-brain barrier integrity via TGF-beta signaling. PLoS One. 9, e103174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo JH, Miyamoto N, Hayakawa K, Pham LD, Maki T, Ayata C, Kim KW, Lo EH, Arai K, 2013. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J Clin Invest. 123, 782–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton CE, Walhovd KB, Storsve AB, Tamnes CK, Westlye LT, Johansen-Berg H, Fjell AM, 2014. Accelerated changes in white matter microstructure during aging: a longitudinal diffusion tensor imaging study. J Neurosci. 34, 15425–15436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJ, Casaccia-Bonnefil P, 2008. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci. 11, 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd MN, Pomicter AD, Velazco CS, Henderson SC, Dupree JL, 2012. Paranodal reorganization results in the depletion of transverse bands in the aged central nervous system. Neurobiol Aging. 33, 203 e213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Leak RK, Keep RF, Chen J, 2016a. Translational Stroke Research on Blood-Brain Barrier Damage: Challenges, Perspectives, and Goals. Transl Stroke Res. 7, 89–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Thrippleton MJ, Makin SD, Marshall I, Geerlings MI, de Craen AJ, van Buchem MA, Wardlaw JM, 2016b. Cerebral blood flow in small vessel disease: A systematic review and meta-analysis. J Cereb Blood Flow Metab. 36, 1653–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo A, Liang AC, Maki T, Miyamoto N, Tomimoto H, Lo EH, Arai K, 2016. Subcortical ischemic vascular disease: Roles of oligodendrocyte function in experimental models of subcortical white-matter injury. J Cereb Blood Flow Metab. 36, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha I, Takahashi T, Nomura E, Ohtsuki T, Ohshita T, Ueno H, Kohriyama T, Matsumoto M, 2009. Association between central systolic blood pressure, white matter lesions in cerebral MRI and carotid atherosclerosis. Hypertens Res. 32, 869–874. [DOI] [PubMed] [Google Scholar]
- Sim FJ, Zhao C, Penderis J, Franklin RJ, 2002. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci. 22, 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song SK, Sun SW, Ju WK, Lin SJ, Cross AH, Neufeld AH, 2003. Diffusion tensor imaging detects and differentiates axon and myelin degeneration in mouse optic nerve after retinal ischemia. Neuroimage. 20, 1714–1722. [DOI] [PubMed] [Google Scholar]
- Sozmen EG, Rosenzweig S, Llorente IL, DiTullio DJ, Machnicki M, Vinters HV, Havton LA, Giger RJ, Hinman JD, Carmichael ST, 2016. Nogo receptor blockade overcomes remyelination failure after white matter stroke and stimulates functional recovery in aged mice. Proc Natl Acad Sci U S A. 113, E8453–E8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahon KE, Bastian C, Griffith S, Kidd GJ, Brunet S, Baltan S, 2016. Age-Related Changes in Axonal and Mitochondrial Ultrastructure and Function in White Matter. J Neurosci. 36, 9990–10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga J, Hu X, Pu H, Shi Y, Hassan SH, Xu M, Leak RK, Stetler RA, Gao Y, Chen J, 2015. White matter injury and microglia/macrophage polarization are strongly linked with age-related long-term deficits in neurological function after stroke. Exp Neurol. 272, 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland GR, Dix GA, Auer RN, 1996. Effect of age in rodent models of focal and forebrain ischemia. Stroke. 27, 1663–1667; discussion 1668. [DOI] [PubMed] [Google Scholar]
- Symplicity HTNI, Esler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, Bohm M, 2010. Renal sympathetic denervation in patients with treatment-resistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet. 376, 1903–1909. [DOI] [PubMed] [Google Scholar]
- Taheri S, Gasparovic C, Huisa BN, Adair JC, Edmonds E, Prestopnik J, Grossetete M, Shah NJ, Wills J, Qualls C, Rosenberg GA, 2011. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke. 42, 2158–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Nyengaard JR, Pakkenberg B, Gundersen HJ, 1997. Age-induced white matter changes in the human brain: a stereological investigation. Neurobiol Aging. 18, 609–615. [DOI] [PubMed] [Google Scholar]
- Tennant KA, Taylor SL, White ER, Brown CE, 2017. Optogenetic rewiring of thalamocortical circuits to restore function in the stroke injured brain. Nat Commun. 8, 15879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao S, Sobue G, Hashizume Y, Shimada N, Mitsuma T, 1994. Age-related changes of the myelinated fibers in the human corticospinal tract: a quantitative analysis. Acta Neuropathol. 88, 137–142. [DOI] [PubMed] [Google Scholar]
- Topakian R, Barrick TR, Howe FA, Markus HS, 2010. Blood-brain barrier permeability is increased in normal-appearing white matter in patients with lacunar stroke and leucoaraiosis. J Neurol Neurosurg Psychiatry. 81, 192–197. [DOI] [PubMed] [Google Scholar]
- Toth P, Tarantini S, Csiszar A, Ungvari Z, 2017. Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol. 312, H1–H20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HH, Niu J, Munji R, Davalos D, Chang J, Zhang H, Tien AC, Kuo CJ, Chan JR, Daneman R, Fancy SP, 2016. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science. 351,379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom R, Lesnik Oberstein SA, Spilt A, Behloul F, Ferrari MD, Haan J, Westendorp RG, van Buchem MA, 2003. Cerebral hemodynamics and white matter hyperintensities in CADASIL. J Cereb Blood Flow Metab. 23, 599–604. [DOI] [PubMed] [Google Scholar]
- van Swieten JC, van den Hout JH, van Ketel BA, Hijdra A, Wokke JH, van Gijn J, 1991. Periventricular lesions in the white matter on magnetic resonance imaging in the elderly. A morphometric correlation with arteriolosclerosis and dilated perivascular spaces. Brain. 114 ( Pt 2), 761–774. [DOI] [PubMed] [Google Scholar]
- Verhaaren BF, Vernooij MW, de Boer R, Hofman A, Niessen WJ, van der Lugt A, Ikram MA, 2013. High blood pressure and cerebral white matter lesion progression in the general population. Hypertension. 61, 1354–1359. [DOI] [PubMed] [Google Scholar]
- Vilar-Bergua A, Riba-Llena I, Nafria C, Bustamante A, Llombart V, Delgado P, Montaner J, 2016. Blood and CSF biomarkers in brain subcortical ischemic vascular disease: Involved pathways and clinical applicability. J Cereb Blood Flow Metab. 36, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorbrodt AW, Lossinsky AS, Wisniewski HM, Suzuki R, Yamaguchi T, Masaoka H, Klatzo I, 1985. Ultrastructural observations on the transvascular route of protein removal in vasogenic brain edema. Acta Neuropathol. 66, 265–273. [DOI] [PubMed] [Google Scholar]
- Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y, Pu H, Li WW, Tang B, Wang Y, Gao Y, Zheng P, Bennett MV, Chen J, 2015. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci U S A. 112, 2853–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Tian WW, Song J, Guan YF, Miao CY, 2011. Deficiency of NG2+ cells contributes to the susceptibility of stroke-prone spontaneously hypertensive rats. CNS Neurosci Ther. 17, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RY, Wang PS, Yang YR, 2003. Effect of age in rats following middle cerebral artery occlusion. Gerontology. 49, 27–32. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu G, Hong D, Chen F, Ji X, Cao G, 2016. White matter injury in ischemic stroke. Prog Neurobiol. 141, 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver J, Jalal FY, Yang Y, Thompson J, Rosenberg GA, Liu KJ, 2014. Tissue oxygen is reduced in white matter of spontaneously hypertensive-stroke prone rats: a longitudinal study with electron paramagnetic resonance. J Cereb Blood Flow Metab. 34, 890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmeyer H, Egea J, Rabe N, Gezelius H, Filosa A, Enjin A, Varoqueaux F, Deininger K, Schnutgen F, Brose N, Klein R, Kullander K, Betz A, 2007. EphA4-dependent axon guidance is mediated by the RacGAP alpha2-chimaerin. Neuron. 55, 756–767. [DOI] [PubMed] [Google Scholar]
- White WB, Wolfson L, Wakefield DB, Hall CB, Campbell P, Moscufo N, Schmidt J, Kaplan RF, Pearlson G, Guttmann CR, 2011. Average daily blood pressure, not office blood pressure, is associated with progression of cerebrovascular disease and cognitive decline in older people. Circulation. 124, 2312–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woywodt A, Streiber F, de Groot K, Regelsberger H, Haller H, Haubitz M, 2003. Circulating endothelial cells as markers for ANCA-associated small-vessel vasculitis. Lancet. 361, 206–210. [DOI] [PubMed] [Google Scholar]
- Xiong YY, Mok V, 2011. Age-related white matter changes. J Aging Res. 2011, 617927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, 2008. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 57, 178–201. [DOI] [PubMed] [Google Scholar]