

Abstract
Heterochromatin, characterized by histone H3 lysine 9 (H3K9) methylation, assembles on repetitive regions including centromeres. Although centromeric heterochromatin is important for correct segregation of chromosomes, its exact role in maintaining centromere integrity remains elusive. Here, we found in fission yeast that heterochromatin suppresses gross chromosomal rearrangements (GCRs) at centromeres. Mutations in Clr4/Suv39 methyltransferase increased the formation of isochromosomes, whose breakpoints were located in centromere repeats. H3K9A and H3K9R mutations also increased GCRs, suggesting that Clr4 suppresses centromeric GCRs via H3K9 methylation. HP1 homologs Swi6 and Chp2 and the RNAi component Chp1 were the chromodomain proteins essential for full suppression of GCRs. Remarkably, mutations in RNA polymerase II (RNAPII) or Tfs1/TFIIS, the transcription factor that facilitates restart of RNAPII after backtracking, specifically bypassed the requirement of Clr4 for suppressing GCRs. These results demonstrate that heterochromatin suppresses GCRs by repressing Tfs1-dependent transcription of centromere repeats.
Akiko Okita et al. demonstrate that heterochromatin suppresses gross chromosomal rearrangements by repressing Tfs1/TFIIS-dependent transcription of repetitive sequences. This study underscores the role of heterochromatin for maintaining chromosome stability.
Introduction
Repetitive DNA elements such as centromere repeats and transposable elements are prevalent in eukaryotic genomes and occupy at least 50% of the human genome1. The presence of repetitive elements is a threat to genome stability. Recombination events such as crossover and break-induced replication (BIR) between repetitive elements give rise to gross chromosomal rearrangements (GCRs), which cause cell death and genetic diseases including cancer2,3. Most of the repetitive elements, including centromere repeats, are present in heterochromatin domains and transcriptionally silenced4. Transcriptional de-repression of repetitive elements (also called satellite DNA) has been observed in a variety of cancers5,6, suggesting a link between GCRs and transcription of repetitive elements.
Heterochromatin is marked by histone H3 lysine 9 (H3K9) methylation that is catalyzed by specific methyltransferases such as fission yeast Clr4 and mammalian Suv397. A clr4 deletion increases RNA polymerase II (RNAPII) localization and de-represses transcription at centromere repeats8, demonstrating that H3K9 methylation causes transcriptional silencing. The H3K9 methylation mark is recognized by chromodomain proteins such as Heterochromatin Protein 1 (HP1)9,10, which creates phase-separated compartments in the nucleus11. RNA interference (RNAi) that utilizes small RNAs mediates heterochromatin assembly12,13. In fission yeast, the RNA-induced transcriptional silencing (RITS) complex, which consists of small RNAs, Ago1, Chp1, and Tas3, localizes to the centromeres through the Chp1 chromodomain protein and Ago1 that captures small RNAs8,14–18. The RITS complex recruits the Clr4-Rik1-Cul4 (CLRC) complex and facilitates H3K9 methylation at the centromeres. In addition to RNAi, the exosome-dependent RNA degradation also contributes to transcriptional silencing. Cid14 is an essential component of the Trf4/Air2/Mtr4 polyadenylation (TRAMP) complex that promotes exosome-dependent degradation of RNAs including centromere transcripts19. Mlo3 RNA-binding protein, the homolog of budding yeast Yra1 and mammalian Aly/REF, is required for the export of poly(A)+ RNA from the nucleus20–22. Yra1 directly binds to the C-terminal domain of RNAPII23, facilitating the transcription-coupled loading of RNA export factors. Like RNAPII, Mlo3 localizes to the gene body of the euchromatin, and it binds to centromere repeats in the absence of Clr424. Mlo3 also interacts with Cid14 and facilitates the exosome-dependent RNA degradation24. Loss of either Mlo3 or Cid14 restores H3K9 methylation in ago1∆ cells25, probably via the recruitment of the CLRC complex to non-degraded nascent transcripts at the centromeres.
Centromeres play an essential role in the correct segregation of chromosomes. Centromeres comprise species-specific centromere repeats in many eukaryotes and are one of the fragile sites of the chromosomes. Chromosome breakages frequently occur at centromeres during tumorigenesis, and the centromere sequence and position change rapidly during the process of evolution26,27. Robertsonian translocation that occurs around centromeres of acrocentric chromosomes is the most common type of chromosomal abnormality observed in humans (1 per 1000 individuals)28. The formation of isochromosomes, whose arms are mirror images of each other, is mediated by inverted repeats at the centromeres in Schizosaccharomyces pombe and Candida albicans29,30. Heterochromatin ensures sister chromatid cohesion at the centromeres31 and prevents incorrect attachment of spindle microtubules to kinetochores32. However, heterochromatin is not always formed at the centromeres: heterochromatin assembly is sometimes lacking at the centromeres that are devoid of repetitive elements33–35. Heterochromatin may have an important role especially when the centromeres consist of repetitive elements.
Heterochromatin plays an important role in the maintenance of genome integrity. Suv39 knockout mice exhibit chromosome aneuploidy and predisposition to develop cancer36. Loss of H3K9 methylation in Caenorhabditis elegans increases instability of repetitive elements probably through the formation of RNA:DNA hybrids37. In fission yeast, heterochromatin appears to prevent replication fork collapse and DNA recombination at the centromeres38,39. Heterochromatin prevents DNA double-strand break formation at the centromeres in meiosis40. However, how heterochromatin affects GCRs between centromere repeats remains elusive.
Here, we found that heterochromatin suppresses GCRs at the centromeres of fission yeast. Deletion of Clr4 increased the formation of isochromosomes, whose breakpoints were located in centromere repeats. Amino acid substitutions in H3K9 (i.e., H3K9A and H3K9R) also increased GCR rates, suggesting that Clr4 suppresses centromeric GCRs through H3K9 methylation. Mutations in the HP1 homologs, Swi6 and Chp2, and the RNAi component Chp1 synergistically increased the GCR rate, showing that both HP1 and RNAi machinery are required to suppress GCRs. Mutations in the C-terminal domain (CTD) of RNAPII impaired chromatin binding of RNAPII and reduced GCRs in clr4∆ cells. Tfs1/TFIIS is the transcription factor that facilitates restart of transcription elongation when RNAPII is paused and backtracked on template DNA41,42. Strikingly, tfs1∆ specifically bypassed the requirement of Clr4 for GCR suppression, without changing chromatin binding levels of RNAPII. These data demonstrate that heterochromatin suppresses GCRs by repressing Tfs1/TFIIS-dependent transcription of repetitive sequences.
Results
Clr4 suppresses GCRs through H3K9 methylation
Clr4 is essential for H3K9 methylation in fission yeast. To understand the role of heterochromatin in genome stability, we disrupted the clr4 gene and determined the rate of spontaneous GCRs29. We detected otherwise lethal GCRs in haploid cells, using an extra-chromosome ChL derived from chromosome 3 (chr3)29,43 (Fig. 1a). Cells harboring ChL (Leu+ Ura+ Ade+) were grown in the minimum medium supplemented with uracil and adenine (EMM + UA), and then plated onto YNB + UA and YNB supplemented with 5-fluoroorotic acid and adenine (5FOA + A) to count Leu+ and Leu+ Ura– colonies, respectively. The clr4∆ strain produced slightly fewer Leu+ colonies than wild type (Fig. 1b), probably due to high incidence of chromosome loss. However, clr4∆ formed more Leu+ Ura– colonies than wild type (Fig. 1b). Leu+ Ura– colonies were replicated onto EMM + U plates to test whether they are Ade+ or Ade– (see Methods). Almost all Leu+ Ura– colonies were Leu+ Ura– Ade–. Using the numbers of Leu+ and Leu+ Ura– Ade– cells (see Methods), we determined the GCR rate by means of a fluctuation analysis44 and found that it was strongly increased by clr4∆ (Fig. 1c, gray dots). Because clr4∆ de-represses the silent mating-type locus mat2P-mat3M and occasionally forms diploid cells45, clr4∆ might increase GCRs by potentiating expression of the meiotic genes including Rec12/Spo11, which creates DNA double-strand breaks40,46. However, clr4∆ increased GCR rate even in the absence of mat2P-mat3M (Fig. 1c, blue dots) and rec12 (Fig. 1c, orange dots). These results show that Clr4 suppresses spontaneous GCRs in mitotic cells. Nevertheless, mat2-3∆ strains were used hereafter to exclude any possible effects of the silent mating-type locus de-repression.
Fig. 1.

Clr4 methyltransferase suppresses gross chromosomal rearrangements (GCRs) through H3K9 methylation. a Illustration of an extra-chromosome ChL. Positions of LEU2, ura4+, ade6+, and centromere 3 (cen3) are indicated. When GCRs associated with the loss of ura4+ and ade6+ take place, Leu+ Ura+ Ade+ cells become Leu+ Ura– Ade– cells. b Wild-type and clr4∆ strains (TNF5676 and 5702, respectively) grown in EMM + UA were plated onto YNB + UA (2 × 102 cells) and 5FOA + A (2 × 104 cells) media to count Leu+ and Leu+ Ura– colonies, respectively. Plates were incubated at 30 °C for 6–9 days. wt, wild type. c GCR rates of wild-type, clr4∆, mat2-3∆, mat2-3∆ clr4∆, mat2-3∆ rec12∆, and mat2-3∆ rec12∆ clr4∆ strains (TNF3896, 5440, 5676, 5702, 5701, and 5766, respectively). Each dot represents the GCR rate determined using a single colony formed on EMM + UA plates in scatter plots. Lines represent the median. The GCR rate relative to that of the wild-type clr4+ strain is indicated on the top of each column. Statistical significance of differences between pairs of strains was determined using the two-tailed Mann–Whitney test. ****P < 0.0001. d GCR rates of wild-type, clr4∆, rik1∆, clr4-set, mlo3KA, mlo3KR, H3K9, H3K9A, and H3K9R strains in the mat2-3∆ background (TNF5676, 5702, 6121, 6958, 6155, 6157, 5738, 6223, and 5802, respectively). The GCR rate relative to that of wild type is indicated on the top of each column. In the cases of H3K9, H3K9A, and H3K9R strains, the GCR rate relative to that of the wild-type H3K9 strain is also shown in parentheses. Statistical significance of differences relative to wild type (the top of each column), and of differences between pairs of strains was determined using the two-tailed Mann–Whitney test
Next, we sought to elucidate how Clr4 suppresses GCRs. Rik1, a component of the CLRC complex is required for the localization of Clr4 to heterochromatin regions8,47. Like clr4∆, rik1∆ increased GCR rate (Fig. 1d, blue dots), suggesting that chromatin localization of Clr4 is required to suppress GCRs. The R/HφφNH (φ = hydrophobic residues) motif in the SET domain constitutes the binding site of S-adenosyl-L-methionine (SAM), which is essential for the methyl transfer7,48,49. It has been shown that single amino acid substitutions in the SET domain impair methyltransferase activity of recombinant Clr4 in vitro, but the mutant strains show residual levels of H3K9 methylation at centromeres in vivo9. To examine if the methyltransferase activity of Clr4 is required to suppress GCRs, we introduced alanine substitutions at the three evolutionally conserved residues, R406, N409, and H410, in the R/HφφNH motif of the SET domain (Supplementary Fig. 1a). We prepared extracts from the yeast that expressed wild-type Flag-Clr4 or mutant Flag-Clr4-set protein from the native promoter, performed Western blotting using anti-Flag antibodies, and found that the clr4-set mutation only slightly reduces the protein level (Supplementary Fig. 1b). Chromatin immunoprecipitation (ChIP) showed that clr4-set completely abolished di-methylation and tri-methylation of H3K9 (H3K9me2 and H3K9me3, respectively) at the centromeres (Supplementary Fig. 1c). Like clr4∆, clr4-set increased GCR rate (Fig. 1d, blue dots). Clr4 methyltransferase has other targets including Mlo324,50, in addition to histone H3. Neither alanine (mlo3KA) nor arginine (mlo3KR) substitution in Mlo3 methylation sites significantly changed GCR rate (P = 0.93 and 0.73, respectively) (Fig. 1d, blue dots). We examined the effect of H3K9 mutations in the H3-H4×1 strain background where two out of three H3-H4 genes in the genome have been eliminated51. Reducing the copy number of H3-H4 genes by itself slightly increased GCR rate (Fig. 1d, magenta dots). Either alanine (H3K9A) or arginine (H3K9R) substitution further increased GCR rate, showing the importance of H3K9 in GCR suppression. These results suggest that Clr4 suppresses GCRs through H3K9 methylation.
Clr4 and Rik1 suppress isochromosome formation at centromeres
Kinetochore chromatin, characterized by the centromere-specific H3 variant CENP-A, is formed on the central sequence (cnt), whereas heterochromatin assembles on the flanking inverted repeats (imr, dg, dh, and irc) (Fig. 2a)52. Loss of ura4+ and ade6+ from ChL results either from translocation, truncation, or isochromosome formation (Fig. 2b)29,53,54. Isochromosomes are produced by recombination between inverted repeats at the centromeres. To determine whether heterochromatin affects GCRs at the centromeres, chromosomal DNAs of parental and independent GCR clones of wild-type, clr4∆, and rik1∆ strains were prepared in agarose plugs, separated by broad-range pulse field gel electrophoresis (PFGE), and stained with ethidium bromide (EtBr) (Fig. 2c and Supplementary Fig. 2a). In wild type, among the 32 GCR products examined, there were two translocations larger than the parental ChL (Fig. 2c, wt #3; Supplementary Fig. 2a, wt #27). Other GCR products were smaller than the parental ChL. The parental ChL was detected, but the small GCR products were not detected by Southern blotting using probe A that hybridizes to the right side of cen3 (Fig. 2b, c and Supplementary Fig. 2a), suggesting that they have completely lost the right arm of the parental ChL. The size of the truncated chromosome that have lost the entire region of the right arm would be ~220 kb (Fig. 2b). Short-range PFGE showed that small GCR products were in the range of 300–400 kb but not ~220 kb (Fig. 2b, d, and Supplementary Fig. 2b), indicating that they were isochromosomes but not truncations. Variable sizes of individual isochromosomes may be explained by the difference in the copy number of dg and dh repeats29. The total length of cen3 becomes longer in isochromosomes when recombination between a pair of inverted repeats (imr3, dg, or dh) occurs and the right side of cen3 gains an increased number of dg and dh repeats29. Around 6% of GCR products were translocations in this study, where the minimal medium was utilized (Fig. 2e). In contrast, in our previous study, where we used rich medium, ~50% of GCR products were translocations53. The difference may be due to severe growth disadvantage of the cells containing a translocation in the minimal medium. Similar to the case with wild type (30 out of 32), most of the GCR products formed in clr4∆ (30 out of 30) and rik1∆ (30 out of 32) strains were isochromosomes (Fig. 2e, P > 0.4, the two-tailed Fisher’s exact test). Given the high rates of GCRs in clr4∆ and rik1∆ strains (Fig. 1d), these data show that Clr4 and Rik1 suppress GCRs especially the isochromosome formation.
Fig. 2.

Clr4 and Rik1 suppress the formation of isochromosomes whose breakpoints are located in centromere repeats. a Repetitive sequences in cen3 of ChL are shown. Units of centromere repeats are indicated as arrows. b Illustration of the gross chromosomal rearrangement (GCR) products that have lost ura4+ and ade6+ from ChL: translocation, truncation, and isochromosome. The position of probe A used in Southern hybridization is indicated as filled box. c Chromosomal DNAs of wild-type, clr4∆, and rik1∆ strains (TNF5676, 5702, and 6121, respectively) were separated by broad-range pulse field gel electrophoresis (PFGE) and stained with ethidium bromide (EtBr). Positions of chr1, chr2, chr3, and ChL (5.7, 4.6, ~3.5, and 0.5 Mb, respectively) in the parental strain are indicated on the left of the panel. DNAs were transferred onto a nylon membrane and hybridized with probe A. P, Parental. d Chromosomal DNAs were separated by short-range PFGE and stained with EtBr. Sizes of the λ DNA ladder are indicated on the left of the panel. e Pie charts depict proportions of different types of GCRs. f Breakpoints were determined by PCR reactions using GCR products recovered from agarose gel. Both sides of cnt3–imr3 junctions were amplified in the reaction containing im1, cn1, and cn2 primers. irc3L and irc3R were amplified using rc1 and rc2 primers, and the PCR products were digested by ApoI and separated by agarose gel electrophoresis. A, ApoI. Uncropped images of depicted gels and blots are shown in Supplementary Figs. 10 and 11
To see whether breakpoints are located in centromere repeats, GCR products were recovered from agarose gel and analyzed by PCR. In all samples examined, both sides of cnt3–imr3 junctions were amplified (Fig. 2f and Supplementary Fig. 2c, cnt3–imr3). However, ApoI restriction fragments (136 and 138 bp) of the irc3 PCR product that are indicative of the right side of irc3 (irc3R) were not detected in all isochromosomes (Fig. 2f and Supplementary Fig. 2c, irc3 (ApoI digest)). We further confirmed that the boundary between cen3 and arm regions was specifically missing on the right side in all isochromosomes (Supplementary Fig. 2d). Together with the absence of the probe A region in isochromosomes, these results show that Clr4 and Rik1 suppress the formation of isochromosomes whose breakpoints are located in centromere repeats.
Chromodomain proteins are required for GCR suppression
Clr4, Swi6, Chp2, and Chp1 bind to H3K9me2 and H3K9me3 through the chromodomain9,10,55–57 (Fig. 3). H3K9me2 and H3K9me3 are present at similar levels in chromatin-bound histones58. RNAi-dependent transcriptional gene silencing occurs on H3K9me2 chromatin. Transition from H3K9me2 to H3K9me3 depends on Clr4 chromodomain and is required for stable binding of Swi6 to nucleosomes58. To identify chromodomain proteins important for GCR suppression, we determined GCR rates of the mutant strains of chromodomain proteins (Fig. 3). It has been shown that clr4-W31G in Clr4 chromodomain impairs its centromere localization and reduces H3K9me3 but not H3K9me2 levels9,57,58 (Supplementary Fig. 1a). clr4-W31G only slightly increased GCR rate compared to clr4∆, indicating that H3K9me3 plays a minor role in GCR suppression. Neither swi6∆ nor chp2∆ significantly affected GCR rate (P = 0.08 and 0.76, respectively). A previous study has also shown that swi6∆ does not significantly increase GCRs38. However, the swi6∆ chp2∆ double mutation increased GCR rate, showing that Swi6 and Chp2 redundantly suppress GCRs. Note that GCR rate of clr4∆ is 16-fold higher than that of chp2∆ swi6∆ (P < 0.0001), indicating that H3K9 methylation suppresses GCRs only partly through HP1 homologs. Deletion of Chp1, the chromodomain subunit of the RITS complex, increased GCR rate, suggesting that RNAi machinery is required for GCR suppression. swi6∆ chp2∆ and chp1∆ synergistically increased GCR rate to the level similar to that of clr4∆. Collectively, these results demonstrate that both HP1 homologs and RNAi component Chp1 are the chromodomain proteins that are essential for full suppression of GCRs.
Fig. 3.
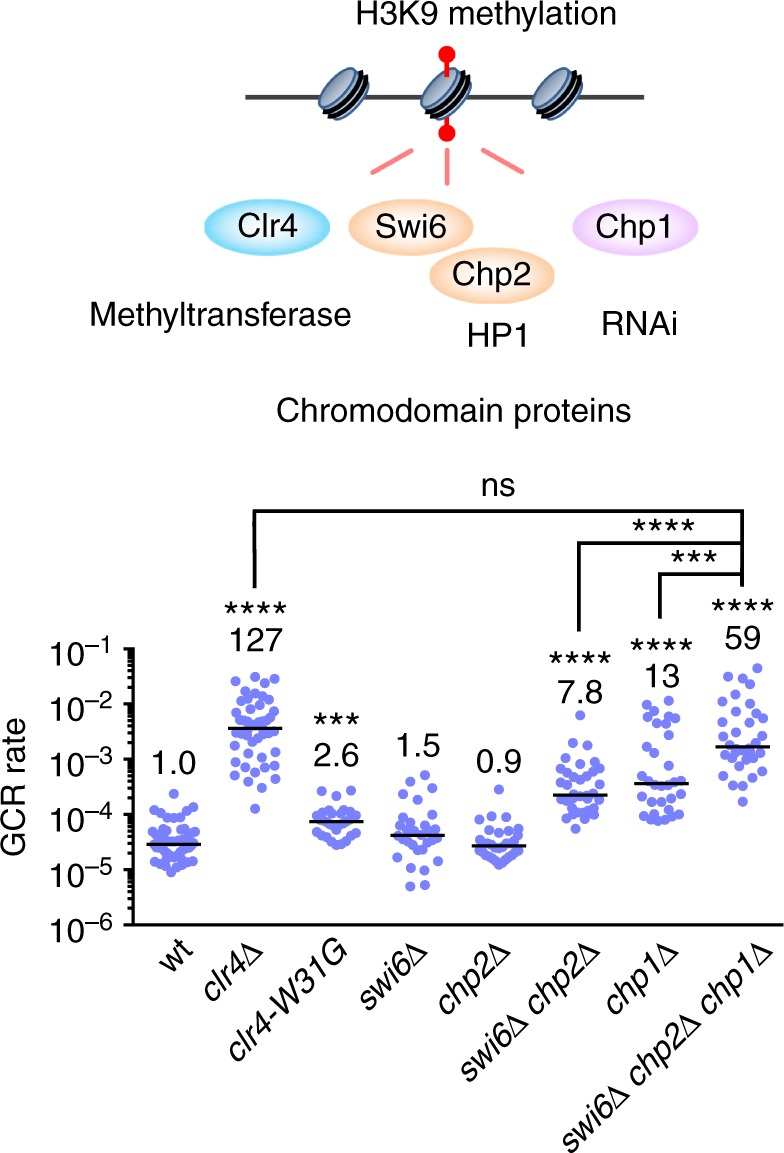
Both HP1 homologs, Swi6 and Chp2, and the RNAi component Chp1 are the chromodomain proteins essential for full suppression of gross chromosomal rearrangements (GCRs). The chromodomain proteins Clr4, Swi6, Chp2, and Chp1 that bind to H3K9 methylation marks are illustrated. GCR rates of wild-type, clr4∆, clr4-W31G, swi6∆, chp2∆, swi6∆ chp2∆, chp1∆, and swi6∆ chp2∆ chp1∆ strains (TNF5676, 5702, 6012, 5706, 5685, 5900, 5708, and 6151, respectively) are shown. The two-tailed Mann-Whitney test. ***P < 0.001, ****P < 0.0001; ns, not significant
RNAi machinery is required for GCR suppression at centromeres
The RNA-directed RNA polymerase Rdp1 creates double-stranded RNAs from noncoding RNAs transcribed from centromere repeats59. Dcr1 cleaves double-stranded RNAs to produce small RNAs. Loading of small RNAs onto Ago1 occurs in the Argonaute small interfering RNA chaperon (ARC) complex that contains Ago1, Arb1, and Arb260. Then, Ago1 bound to small RNAs forms the RITS complex with Chp1 and Tas3, and localizes to the centromeres through Chp1 and through base pairing between small RNAs and nascent transcripts at the centromeres14,16 (Fig. 4). To establish whether RNAi machinery is required to suppress GCRs at the centromeres, we disrupted these RNAi factors and determined their GCR rates (Fig. 4). Interestingly, ago1∆ increased GCR rate even greater than clr4∆, suggesting that Ago1 not only facilitates H3K9 methylation but also plays some other role to suppress GCRs. GCR rate of ago1∆ was higher than those of chp1∆, tas3∆, arb1∆, and arb2∆ (P ≤ 0.0002), suggesting that Ago1 suppresses GCRs partly through the formation of ARC and RITS complexes. GCR rate of ago1∆ was also higher than those of rdp1∆ and dcr1∆ (P < 0.0001), probably due to a Dcr1-independent pathway of small RNA production that uses the exosome61. Analysis of GCR products formed in ago1∆ cells showed that most of them (15 out of 16) were isochromosomes whose breakpoints were located in centromere repeats (Supplementary Fig. 3). These results show that RNAi machinery plays an essential role in GCR suppression at the centromeres.
Fig. 4.
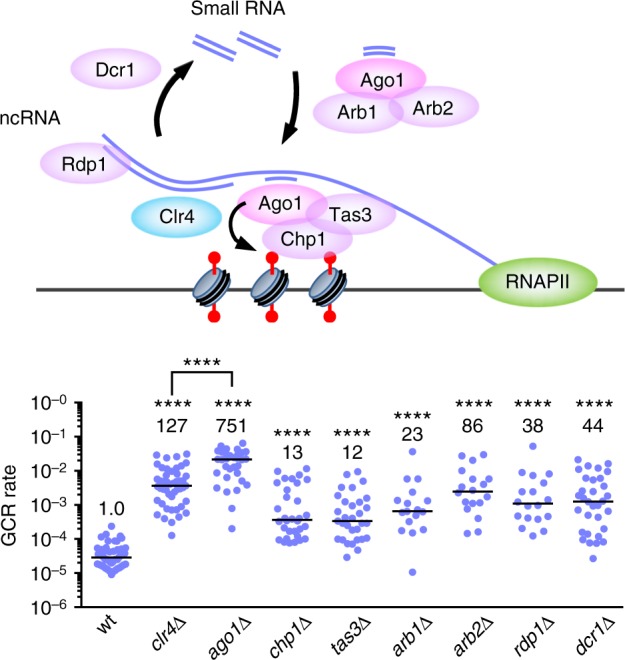
RNAi machinery plays an essential role to suppress gross chromosomal rearrangements (GCRs) at the centromeres. Illustrated is the RNAi system that utilizes small RNAs and facilitates H3K9 methylation at the centromeres. GCR rates of wild-type, clr4∆, ago1∆, chp1∆, tas3∆, arb1∆, arb2∆, rdp1∆, and dcr1∆ strains (TNF5676, 5702, 5688, 5708, 7335, 7337, 7331, 7333, and 5687, respectively) are shown. The two-tailed Mann–Whitney test. ****P < 0.0001. ncRNA, noncoding RNA. arb2∆ caused a higher rate of GCRs than arb1∆ (P < 0.05), suggesting that Arb2 has an Arb1-independent function to suppress GCRs
Ago1 represses RNAPII chromatin binding and GCRs at centromeres
To examine whether Ago1 suppresses GCRs only via H3K9 methylation or not, we took advantage of cid14∆ and mlo3∆ mutations that restore H3K9me2 levels in ago1∆ cells24,25. cid14∆ did not significantly change GCR rate in ago1∆ cells (Fig. 5a, magenta dots, P = 0.14). Most of the GCR products formed in cid14∆ ago1∆ cells were isochromosomes whose breakpoints were located in centromere repeats (14 out of 16 samples) (Supplementary Fig. 4). These results show that the restoration of H3K9me2 levels by cid14∆ is not sufficient to suppress centromeric GCRs in ago1∆ cells (also see below). In contrast to cid14∆, mlo3∆ reduced GCR rate in ago1∆ cells (Fig. 5a, magenta dots).
Fig. 5.
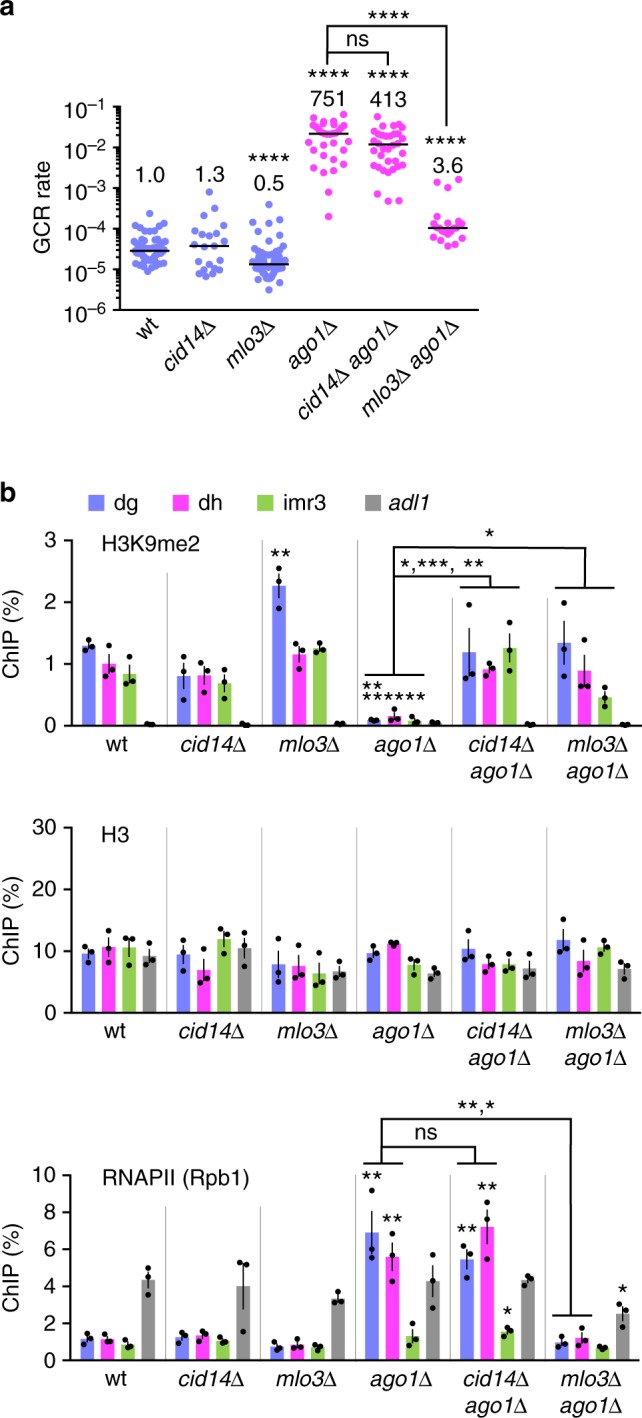
mlo3∆ but not cid14∆ reduces RNAPII chromatin binding and suppresses gross chromosomal rearrangements (GCRs) at the centromeres in ago1∆ cells. a GCR rates of wild-type, cid14∆, mlo3∆, ago1∆, cid14∆ ago1∆, and mlo3∆ ago1∆ strains (TNF5676, 6153, 5764, 5688, 6411, and 6188, respectively). The two-tailed Mann–Whitney test. ****P < 0.0001; ns, not significant. b Chromatin immunoprecipitation (ChIP) analysis was performed to determine H3K9me2, H3 and RNAPII (Rpb1) levels at centromere repeats (dg, dh, and imr3) and at a non-centromeric region of chr2 (adl1) in wild-type, cid14∆, mlo3∆, ago1∆, cid14∆ ago1∆, and mlo3∆ ago1∆ strains (TNF5921, 6276, 5923, 5922, 6550, and 6210, respectively). DNA levels were quantified by real-time PCR, and percentages of input DNA were obtained. Data are presented as the mean ± s.e.m. from three biologically independent experiments. Dots represent individual measurements from distinct samples. Statistical significance of differences relative to wild type (top of bars), and of differences between pairs of mutant strains was determined using the two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001
To find the difference between cid14∆ and mlo3∆, we performed ChIP experiments and determined H3K9me2 and H3 levels at centromere repeats (dg, dh, and imr3) and at a non-centromere region (adl1). In wild type, H3K9me2 was specifically detected at dg, dh, and imr3, but not at adl1 (Fig. 5b, H3K9me2). As expected, ago1∆ reduced H3K9me2 level at centromere repeats, and both cid14∆ and mlo3∆ restored it24,25. Similar levels of H3 were observed in all strains examined (Fig. 5b, H3), showing that the mutations affect histone modification rather than nucleosome occupancy. Because Mlo3 is involved in transcription, as well as RNA export and degradation23,24,63, we further determined RNAPII chromatin binding levels. In wild type, the localization of Rpb1, the catalytic subunit of RNAPII, was limited at the centromeres as compared to its level at adl1 (Fig. 5b, RNAPII (Rpb1)). ago1∆ increased RNAPII levels at dg and dh to the level comparable to that at adl1. Note that ago1∆ did not increase RNAPII levels at imr3 significantly (P = 0.29), suggesting that intrinsic transcriptional activity of imr3 is low64. cid14∆ did not significantly change RNAPII levels in ago1∆ cells (P ≥ 0.24), suggesting that Ago1 acts downstream of H3K9me2 to reduce RNAPII localization at the centromeres. In contrast to cid14∆, mlo3∆ reduced RNAPII levels at dg and dh, consistent with previous reports25. Epe1 is a putative H3K9 demethylase that antagonizes heterochromatin assembly62,65. Loss of Epe1 restored H3K9me2 and reduced RNAPII chromatin occupancy and GCR rate in ago1∆ cells (Supplementary Fig. 5). Whereas cid14∆, mlo3∆, and epe1∆ restored H3K9me2, only mlo3∆ and epe1∆ reduced RNAPII occupancy and GCRs in ago1∆ cells. These results show that repression of RNAPII might be required for GCR suppression.
Loss of Mlo3 reduces GCRs in the absence of H3K9 methylation
To examine whether repression of RNAPII suppresses GCRs even in the absence of H3K9 methylation, we deleted mlo3 in the clr4∆ mutant and found that mlo3∆ greatly reduced GCR rate in clr4∆ cells (Fig. 6a, magenta dots). It has been shown that mlo3∆ restores chromatin binding of Rik1 in ago1∆ cells25. However, mlo3∆ suppressed GCRs independently of Rik1, as mlo3∆ also reduced GCRs of rik1∆ cells. The homologous recombination factor Rad51 is required to suppress isochromosome formation, but it is not essential for transcription silencing at centromeres29,53. Contrary to clr4∆ cells, mlo3∆ did not reduce GCR rate in rad51∆ cells (Fig. 6a, gray dots; Supplementary Fig. 6), showing that mlo3∆ specifically affects GCRs that occur in heterochromatin-deficient cells. ChIP experiments showed that clr4∆, like ago1∆, increased RNAPII but not H3 levels at dg and dh, and that mlo3∆ reduced RNAPII levels in clr4∆ cells at dg, dh, and adl1 sites (Fig. 6b). As expected, mlo3∆ did not restore H3K9me2 and H3K9me3 in clr4∆ cells (Fig. 6c). Repression of histone acetylation is another feature of heterochromatin51. Sir2, Clr3, and Clr6 catalyze deacetylation of histones at different sites, including H3K9 and H3K14, and are involved in transcriptional silencing at the centromeres66–68. We found that they are also required for GCR suppression (Supplementary Fig. 7). However, like H3K9 methylation, mlo3∆ did not significantly change H3K9 and H3K14 acetylation levels (Fig. 6c, P ≥ 0.24 and ≥ 0.33, respectively, for H3K9ac and H3K14ac), suggesting that Mlo3 directly affects chromatin binding of RNAPII. These results suggest that the CLRC complex suppresses centromeric GCRs by repressing RNAPII.
Fig. 6.

Repression of RNAPII suppresses centromeric gross chromosomal rearrangements (GCRs) in the absence of H3K9 methylation. a GCR rates of wild-type, mlo3∆, clr4∆, mlo3∆ clr4∆, rik1∆, mlo3∆ rik1∆, rad51∆, and mlo3∆ rad51∆ strains (TNF5676, 5764, 5702, 5824, 6121, 6378, 6244, and 6383, respectively). The two-tailed Mann-Whitney test. ***P < 0.001, ****P < 0.0001. b Chromatin immunoprecipitation (ChIP) analysis of RNAPII (Rpb1) and H3 in wild-type, mlo3∆, clr4∆, and mlo3∆ clr4∆ strains (TNF5921, 5923, 5948, and 5925, respectively). The two-tailed Student’s t-test. *P < 0.05, **P < 0.01. c ChIP analysis of H3K9me2, H3K9me3, H3K9ac, and H3K14ac in wild-type, mlo3∆, clr4∆, and mlo3∆ clr4∆ strains. mlo3∆ reduced the level of H3K9me3 but not that of H3K9me2, suggesting that Mlo3 is required for the transition from H3K9me2 to H3K9me3 state
RNAPII and Tfs1/TFIIS cause GCRs in the absence of Clr4
Rpb1 CTD consists of YSPTSPS heptapeptide repeats69. Ser7 of the CTD is required for transcription of noncoding small nuclear RNAs in human cells70. Changing all the Ser7 to Ala, rpb1-S7A, reduces chromatin-bound RNAs and H3K9me2 levels at fission yeast centromeres71,72. To obtain the direct evidence that RNAPII is involved in centromeric GCRs, we created the rpb1-S7A strain that harbored ChL. Consistent with low levels of H3K9me272 (Supplementary Fig. 8a), rpb1-S7A slightly increased GCR rate in otherwise wild-type background (Fig. 7a, blue dots). However, rpb1-S7A greatly reduced the GCR rate in clr4∆ cells (Fig. 7a, magenta dots), showing that RNAPII is involved in centromeric GCRs that occur in clr4∆ cells. Whereas rpb1-S7A did not restore H3K9 methylation in clr4∆ cells (Supplementary Fig. 8a), rpb1-S7A reduced Rpb1 localization at centromere repeats and at adl1 and act1 genes either in the presence or absence of Clr4 (Fig. 7b and Supplementary Fig. 8b). rpb1-S7A also reduced chromatin binding of Rpb3 another subunit of RNAPII73 (Supplementary Fig. 8c), suggesting that RNAPII CTD Ser7 is required for chromatin binding of the RNAPII complex. Because levels of RNAPII chromatin binding do not always reflect levels of transcription, we detected noncoding RNAs transcribed from centromere repeats. Northern blotting using total RNAs prepared from yeast cells showed that clr4∆ increased the amounts of dg, dh, and (less prominently) imr3 RNAs (Fig. 7c and Supplementary Fig. 9a). rpb1-S7A slightly increased dg and dh RNAs in otherwise wild-type background, as expected72. However, in clr4∆ cells, rpb1-S7A reduced the levels of centromeric noncoding RNAs most prominently at imr3 where transcription levels are low. These results show that RNAPII CTD Ser7 is required for a subset of transcription events in clr4∆ cells. We detected adl1 RNAs of ~2.5 and ~5 kb: the long RNAs were the readthrough transcripts that encompassed the downstream converging gene spbc713.07c74. Interestingly, rpb1-S7A specifically reduced the long RNAs of adl1. Re-hybridization of the membrane showed that rpb1-S7A did not affect the transcription of the act1 gene that has no converging genes nearby (Supplementary Fig. 9a). These results suggest that RNAPII CTD Ser7 is required for a specific type of transcription that causes centromeric GCRs in the clr4∆ mutant.
Fig. 7.

Clr4 suppresses centromeric gross chromosomal rearrangements (GCRs) by repressing transcription that is dependent on RNAPII CTD Ser7 and Tfs1/TFIIS. a GCR rates of wild-type, rpb1-S7A, tfs1∆, ell1∆, leo1∆, spt4∆, clr4∆, rpb1-S7A clr4∆, tfs1∆ clr4∆, ell1∆ clr4∆, leo1∆ clr4∆, and spt4∆ clr4∆ strains (TNF5676, 6848, 6688, 7042, 7130, 7055, 5702, 6850, 6726, 7063, 7154, and 7057, respectively). The two-tailed Mann-Whitney test. **P < 0.01, ****P < 0.0001; ns, not significant. b Chromatin immunoprecipitation (ChIP) analysis of RNAPII (Rpb1) and H3 in wild-type, rpb1-S7A, tfs1∆, clr4∆, rpb1-S7A clr4∆, and tfs1∆ clr4∆ strains (TNF5921, 6862, 6722, 5948, 6864, and 6799, respectively). The two-tailed Student’s t-test. *P < 0.05, ***P < 0.001. c Northern blotting using total RNAs prepared from log phase cultures of rpb1-S7A, wild-type, tfs1∆, rpb1-S7A clr4∆, clr4∆, and tfs1∆ clr4∆ strains. Illustrated are the positions of DNA probes used in Northern blotting (magenta bars) and the readthrough transcript of adl1 (a green arrow). RNAs were separated by 1.0% agarose gel under denatured condition, stained with ethidium bromide (EtBr) (the bottom panel), transferred onto a nylon membrane, and hybridized with specific probes (the top panel). Uncropped images of depicted gels and blots are shown in Supplementary Fig. 12. d A model that explains how heterochromatin suppresses GCRs at centromeres. With the aid of the RNAi system, Clr4 catalyzes H3K9 methylation at centromeres. H3K9 methylation marks are recognized by the chromodomain proteins including Clr4, Swi6, Chp2, and Chp1. Both HP1 homologs, Swi6 and Chp2, and an RNAi component Chp1 are required for full suppression of GCRs. In addition to the Clr4 recruitment, RNAi machinery may prevent transcription of noncoding RNAs from centromere repeats to suppress GCRs. RNAPII transcription that depends on CTD Ser7, Mlo3, and Tfs1/TFIIS causes centromeric GCRs possibly by removing DNA binding proteins, such as replication factors, from DNA. e Tfs1/TFIIS-dependent transcription might remove the roadblock that binds to DNA and produce R-loops, which facilitate interaction between centromere repeats at non-allelic positions and cause crossover and/or break-induced replication (BIR) that leads to GCRs
To gain insights into how transcription causes GCRs in the absence of Clr4, we disrupted the genes encoding transcription factors Tfs1, Ell1, Leo1, and Spt475. Among them, Tfs1/TFIIS is the only factor that has been shown to facilitate restart of transcription elongation by trimming 3′-ends of nascent RNAs when RNAPII is paused and backtracked on template DNA41,42. In clr4+ cells, spt4∆ increased GCR rate (Fig. 7a, blue dots), probably because spt4∆ impairs transcriptional silencing76. In clr4∆ cells, only tfs1∆ greatly reduced GCR rate (Fig. 7a, magenta dots). In contrast to rpb1-S7A, tfs1∆ did not significantly change RNAPII levels (Fig. 7b, P ≥ 0.22), indicating that RNAPII chromatin binding per se does not cause GCR events. However, tfs1∆ slightly reduced the levels of centromeric noncoding RNAs in clr4∆ cells (Fig. 7c and Supplementary Fig. 9a). Interestingly, tfs1∆ also reduced adl1 readthrough transcripts, suggesting that Tfs1 facilitates transcription passing through termination sites. tfs1∆ did not reduce GCRs in rad51∆ cells (Supplementary Fig. 9b), showing that tfs1∆ specifically suppresses GCRs that occur in heterochromatin-deficient cells. These results suggest that a specific type of transcription elongation that depends on Tfs1/TFIIS causes GCRs at the centromeres.
Discussion
Here, we found that heterochromatin suppresses GCRs mediated by centromere repeats. Deletion of Clr4 or Rik1 strongly increased spontaneous formation of isochromosomes whose breakpoints were located in centromere repeats. Mutations in either the SET domain of Clr4 or H3K9 increased GCR rate, suggesting that Clr4 suppresses centromeric GCRs through H3K9 methylation. HP1 homologs Swi6 and Chp2 and the RNAi component Chp1 were the chromodomain proteins essential for full suppression of GCRs. mlo3∆ and rpb1-S7A impaired chromatin binding of RNAPII and reduced GCRs in the clr4∆ mutant, showing that Clr4-dependent H3K9 methylation suppresses GCRs by repressing RNAPII. Strikingly, deletion of Tfs1/TFIIS, which facilitates restart of paused and backtracked RNAPII, greatly reduced GCRs in the clr4∆ mutant without changing RNAPII chromatin binding levels. These results suggest that heterochromatin suppresses the Tfs1-dependent transcription that leads to GCRs between centromere repeats.
Chromodomain proteins are the readers of the H3K9 methylation mark. Among them, Swi6 is required for stable binding of cohesin complexes at the centromeres, and it also facilitates early replication of the centromeres by recruiting Dbf4/Dfp1-dependent kinase (DDK)31,77,78. However, swi6∆ did not increase GCR rate, indicating that neither cohesin enrichment nor replication timing control at the centromeres is essential to suppress GCRs. In contrast to each single mutation, the swi6∆ chp2∆ double mutation increased GCR rate, indicating that the redundant function of Swi6 and Chp2 such as inhibition of RNAPII localization at the centromeres10,56 is important for GCR suppression. A previous report has also shown that swi6∆ does not alter GCR rate but increases the proportion of isochromosomes among GCR products38. Similar to the case with GCRs, meiotic recombination at the centromeres is increased only when both Swi6 and Chp2 are eliminated40. Nonetheless, HP1 homologs are not the only chromodomain proteins required to suppress GCRs, as GCR rate of swi6∆ chp2∆ was not as high as that of clr4∆ cells. swi6∆ chp2∆ and chp1∆ synergistically increased GCR rate to the level similar to that of clr4∆, suggesting that both HP1 and the RNAi machinery are required for the full suppression of GCRs (Fig. 7d).
The RITS complex recruits the CLRC complex and facilitates H3K9 methylation at the centromeres. Loss of Clr4 eliminates H3K9 di-methylation and tri-methylation and increases RNAPII localization at centromere repeats8,58. Like clr4∆, ago1∆ reduced H3K9 methylation and increased RNAPII localization, resulting in high incidence of isochromosome formation. However, GCR rate of ago1∆ was higher than that of clr4∆, showing that Ago1 has a Clr4-independent function in GCR suppression. A discrepancy between RNAi mutants (ago1∆, dcr1∆, and rdp1∆) and clr4∆ phenotypes has been observed in the formation of uniparental disomy (UPD), a pair of homologous chromosomes originating from only one parent79. However, in contrast to the case of UPD, Ago1 may have a unique function to suppress GCRs, which is independent of other RNAi factors including Dcr1, Rdp1, Chp1, Tas3, Arb1, and Arb2. Human Ago1 directly binds to RNAPII80, and Drosophila Ago2 interacts with the negative elongation factor NELF and represses heat-shock genes under normal conditions81. Thus, Ago1 might directly affect transcription and suppress GCRs. This study provides the evidence that transcriptional repression is important to suppress GCRs between centromere repeats. Mlo3 as well as RNAPII localizes to the centromeres in clr4∆ cells8,24, and Yra1/Mlo3 directly binds to the CTD of Rpb1 in budding yeast23. mlo3∆ reduced chromatin binding of RNAPII and bypassed the requirement of Clr4 methyltransferase to suppress GCRs at the centromeres. In addition, mutations in the largest subunit of RNAPII, rpb1-S7A, also reduced RNAPII chromatin binding and bypassed the requirement of Clr4 for GCR suppression. Transcriptional repression by heterochromatin seems to be important to suppress homology-mediated GCRs not only at the centromeres but also at other chromosomal loci, e.g., subtelomeres82.
How does Tfs1/TFIIS-dependent transcription cause GCRs that are mediated by centromere repeats? During transcription, DNA supercoils are formed in front of and behind RNAPII, hybridization of RNA to the template DNA creates R-loops, nucleosomes are disassembled and reassembled, and proteins are detached from template DNA. RNAPII is paused and backtracked when it encounters DNA-binding proteins83,84. After backtracking, Tfs1/TFIIS cleaves nascent RNAs by enhancing the intrinsic nuclease activity of RNAPII and facilitates the restart of RNAPII41,42. Here, we found that tfs1∆ greatly reduced GCRs in clr4∆ cells. In contrast to GCRs, tfs1∆ did not significantly change the level of RNAPII chromatin binding and only slightly reduced the amount of centromeric noncoding RNAs, showing that neither RNAPII binding nor transcription per se induces GCRs. Interestingly, the effect of tfs1∆ on transcription levels was most prominent at imr3 repeats where the intrinsic transcription activity is low. Tfs1 facilitates passing through the transcription termination site and produces readthrough transcripts of the adl1 gene. We propose a model in which, with the aid of Tfs1, RNAPII competes with DNA-binding proteins that block transcription elongation (Fig. 7e). Interestingly, clr4∆ impairs the centromere localization of the Smc5-Smc6 complex that promotes a conservative way of homologous recombination85. Thus, the removal of DNA-binding proteins such as Smc5-Smc6 can cause illegitimate recombination. Tfs1-dependent transcription may also block the progression of replication forks, as the replication fork protection machinery is important to suppress recombination at centromeres in the heterochromatin mutant38. Rad52 recombinase binds to the centromeres during S phase in an RNAi mutant, and Rad51 recombinase is essential in RNAi mutants39. Drosophila lacking Suv39 histone methyltransferase accumulates spontaneous DNA damage in heterochromatin86. Dnmt3a and Dnmt3b that catalyze DNA methylation, another epigenetic mark of heterochromatin in mammalian cells, suppress recombination at the centromeres87. However, our previous study has shown that clr4∆ increases recombination between inverted repeats in the centromeres by ~2-fold54, arguing against the notion that clr4∆ merely increases DNA lesions that cause GCRs. Clr4 may affect the choice of recombination pathways at the centromeres. Either crossover or break-induced replication (BIR) between centromere inverted repeats results in isochromosome formation53,88. Rad51 suppresses isochromosome formation by promoting non-crossover recombination at the centromeres53. In the absence of Rad51, GCRs occur in a manner-dependent on the crossover-specific endonuclease Mus8153, demonstrating that crossover is the mechanism of GCRs in rad51∆ cells. Tfs1-dependent transcription passing through pausing or termination sites may extend the length of RNA:DNA hybrids and produce R-loops (Fig. 7e). In mammals, R-loops are sometimes formed at transcription termination sites, and BRCA1 recombinase binds to DNA damage derived from R-loops89. A recent paper showed that R-loops induce BIR in budding yeast90. R-loops that contain single-stranded DNA may facilitate the pairing between homologous sequences and initiate BIR (Fig. 7e). Further studies are required to address how Tfs1-dependent transcription causes GCRs between centromere repeats. We also found that tfs1∆, mlo3∆, and rpb1-S7A reduced chromosome loss in the clr4∆ background (Supplementary Fig. 9c), suggesting that transcriptional repression is important not only for GCR suppression but also for correct segregation of chromosomes. Unlike rpb1-S7A and mlo3∆, tfs1∆ reduced hypersensitivity to the microtubule-destabilizing drug, thiabendazole (TBZ) of clr4∆ cells (Supplementary Fig. 9d), making Tfs1/TFIIS-dependent transcription a critical target of heterochromatin to maintain the integrity and the function of the centromeres.
It is believed that heterochromatin assembles on the centromeres to ensure faithful segregation of chromosomes31,32,91. However, heterochromatin is not always formed at the centromeres. In fission yeast strain CBS2777 and pathogenic fungus Candida lusitaniae, no heterochromatin or transcriptional silencing was observed at the centromeres that were devoid of repeat sequences33,34. In chicken DT40 cells, heterochromatin is assembled at the repetitive centromeres but not at the non-repetitive centromeres35. Together with these links between heterochromatin and DNA repeats, our studies suggest that one of the important roles of centromeric heterochromatin is to suppress GCRs that are mediated by centromere repeats. Interestingly, de-repression of repetitive sequences including centromeric satellite DNA is observed in some kinds of cancer cells6. During the process of aging, heterochromatin is globally lost and frequencies of genome alterations increase92–94. We propose that heterochromatin represses transcription of noncoding repeats in the genome to prevent GCRs between the repetitive sequences.
Methods
Strains and media
Fission yeast strains used in this study are listed in Supplementary Table 1. Yeast cells were grown at 30 °C in YE, EMM, YNB, and 5FOA media supplemented with appropriate amino acids at a final concentration of 225 mg L−1 as described previously54. YNB medium contained 1.7 g L−1 of yeast nitrogen base (BD Biosciences, Difco 233520), 5 g L−1 of ammonium sulfate (Nacalai Tesque, 02619-15), and 2% glucose. YNB medium was supplemented with 1 g L−1 of 5-fluoroorotic acid (Apollo Scientific, PC4054) and 56 mg L−1 of uracil to make 5FOA medium. Solid media contained 1.5% agarose (Nacalai Tesque, 01028-85). Yeast transformation was performed by the lithium acetate method. The transformants that contain the kanamycin, hygromycin, or nourseothricin resistance gene were selected on the media supplemented with G418 (Nacalai Tesque, 09380-86), hygromycin B (Nacalai Tesque, 09287-84), or clonNAT (Werner BioAgents, 96736-11-7) at a final concentration of 100 µg mL−1. clr4-R406A,N409A,H410A (clr4-set), mlo3K165A,K167A (mlo3KA), and mlo3K165R,K167R (mlo3KR) mutant strains were created by the pop-in/pop-out gene replacement95: pTN1220 plasmid containing the wild-type ura4+ and mutant clr4-set genes was digested with NgoMIV and introduced into ura4-D18 mutant cells. pTN1179 containing ura4+ and mlo3KA and pTN1178 containing ura4+ and mlo3KR were digested with HpaI and introduced into ura4-D18 cells. Ura+ transformants were selected on EMM plates, and then, Ura– progenies resulting from ura4+ pop-out were selected on 5FOA plates. We performed PCR and DNA sequencing to confirm correct integration of the clr4 and mlo3 mutations.
Plasmids
A 1.8 kb HindIII–SspI fragment containing the ura4+ gene was introduced between HindIII–EcoRV sites of pBluescript II KS+ (Stratagene) to make pTN782. clr4-set, mlo3KA, and mlo3KR mutant genes were constructed by a two-step PCR method. From yeast genomic DNA, a 0.7 kb PCR fragment was produced using clr4-1 and clr4-NHR-F primers, and a 1.0 kb fragment using clr4-NHR-R and clr4-2 primers, independently. These partially overlapping PCR fragments were mixed and used for the 2nd PCR in the presence of clr4-1 and clr4-2 primers. A 1.4 kb SpeI–PvuII restriction fragment prepared from the 2nd PCR product was introduced between SpeI–NaeI sites of pTN782 to make pTN1220. A 2.0 kb genomic region that contains the mlo3+ gene was amplified using mlo3-1 and mlo3-5, and digested with XbaI at one site. A 1.9 kb restriction fragment with XbaI–blunt ends was introduced between XbaI–NaeI sites of pTN782 to make pTN1169. From yeast genomic DNA, a 1.0 kb PCR fragment was produced using mlo3-1 and mlo3-KA-R primers, and a 0.7 kb fragment using mlo3-KA-F and mlo3-4 primers. These partially overlapping PCR fragments were mixed and used for the 2nd PCR in the presence of mlo3-1 and mlo3-4 primers. A 1.0 kb SacI–XbaI restriction fragment of the 2nd PCR product that contains the mlo3KA mutation was introduced between SacI–XbaI sites of pTN1169 to make pTN1179. mlo3-KR-R and mlo3-KR-F primers were used in place of mlo3-KA-R and mlo3-KA-F to make pTN1178 that contains the mlo3KR mutation. A 9.6 kb XbaI–EcoRI fragment containing cen1 sequence from pRS14096 was introduced between XbaI–EcoRI sites of pUC19 to make pTN834. From yeast genomic DNA, a 1.7 kb region that contains a portion of dh was amplified using dh-1 and dh-2 primers. A 1.5 kb NheI–ClaI restriction fragment of the PCR product was introduced between SpeI–ClaI sites of pBluescript II KS+ to make pTN770. A 2.3 kb region that contains a portion of imr3 was amplified using otr3-2 and imr3-XhoI-R primers. A 1.7 kb PvuII–MfeI restriction fragment of the PCR product was introduced between HincII–EcoRI sites of pBluescript II KS+ to make pTN1226. A 0.9 kb region that contains a portion of the adl1 gene was amplified using adl1-F and adl1-R primers. A 0.9 kb XbaI–ApaI restriction fragment of the PCR product was introduced between XbaI–ApaI sites of pBluescript II KS+ to make pTN1227. A 2.1 kb region that contains a portion of the act1 gene was amplified using act1-F and act1-R primers. A 1.9 kb XhoI–EcoRV restriction fragment of the PCR product was introduced between XhoI–EcoRV sites of pBluescript II KS+ to make pTN1225. DNA sequencing confirmed that no mutations were introduced during PCR amplification.
Gross chromosomal rearrangement (GCR) assay
Rates of spontaneous GCR were determined by the fluctuation analysis. Yeast cells were incubated for 6–9 days on EMM + UA plates, and 10 mL of EMM + UA medium was inoculated with a single colony formed on the EMM + UA plates. After 2-days incubation, cells were plated onto YNB + UA and 5FOA + A media. After incubation for 6–12 days, the number of colonies formed on YNB + UA and 5FOA + A plates were counted to determine the number of Leu+ and that of Leu+ Ura– cells, respectively. Leu+ Ura– colonies formed on 5FOA + A plates were incubated on EMM + UA plates and then replicated onto EMM + A and EMM + U plates to confirm Ura– and to inspect Ade+/–, respectively. The number of Leu+ Ura– Ade– cells indicative of GCR was obtained by subtracting the number of Leu+ Ura– Ade+ cells from that of Leu+ Ura– cells. Using the number of Leu+ cells and that of Leu+ Ura– Ade– cells in 10 mL of EMM + UA culture, we determined GCR rate by means of the fluctuation analysis44.
Pulse field gel electrophoresis (PFGE)
From parental and GCR (Leu+ Ura– Ade–) clones obtained from biologically independent experiments, chromosomal DNAs were prepared in 1.6% low melting agarose plugs (Nacalai Tesque, 01161-12) as described previously54. Chromosomal DNAs were separated in 0.55% Certified Megabase agarose gel (Bio-Rad, 161-3109) using CHEF-DRII system (Bio-Rad) under the following conditions. Broad-range PFGE: 1500 s pulse time at 2 V cm−1 for 42 h and then, 180 s pulse time at 2.4 V cm−1 for 4 h, at 4 °C in 1× TAE buffer (40 mM Tris-acetate, 1 mM EDTA). Short-range PFGE: from 40 to 70 s pulse time at 4.2 V cm−1 for 24 h, at 4 °C in 0.5× TBE buffer (89 mM Tris-borate, 2 mM EDTA). After electrophoresis, DNAs were stained with 0.2 µg mL−1 of EtBr (Nacalai Tesque, 14631-94) and detected using a Typhoon FLA9000 (GE Healthcare).
Southern blotting
After EtBr staining, agarose gel was irradiated with 300 mJ ultraviolet (UV) light using GS Gene Linker (Bio-Rad) for DNA fragmentation, and then soaked into 800 mL of alkaline buffer (1.2 M NaCl, 0.4 M NaOH) for 40 min with gentle shaking to denature DNA. DNA was transferred to ClearTrans nylon membrane 0.45 µm (Wako, 039-22673) by capillary action in 25 mM sodium phosphate buffer (pH 6.5) and covalently attached to the membrane by 150 mJ UV irradiation. A 0.6 kb EcoRI–EcoRI fragment prepared from pTN75529, α-32P-dCTP (PerkinElmer Life Sciences, NEG013H), and Random primer labeling kit ver. 2 (Takara, 6045) were used to prepare radioactive probe A. Radioactive signals were detected using BAS2500 phosphorimager (Fuji Film).
PCR analysis of GCR products
After separation of chromosomal DNA by PFGE, GCR products were recovered from agarose gel using a FastGene Gel/PCR Extraction kit (Nippon Genetics, FG-91302). KOD FX Neo polymerase (Toyobo, KFX-201) was utilized to amplify cnt3–imr3 junctions, whereas Q5 polymerase (New England Biolabs, M0491) was used to amplify irc3. PCR products were separated by 1.7% Seakem GTG agarose gel (Lonza, 50070) electrophoresis in 1× TBE buffer, stained with 0.2 µg mL−1 of EtBr, and detected using a Typhoon FLA9000. PCR primers used in this assay are listed in Supplementary Table 2.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described previously54. 1.5 × 108 cells from log-phase cultures in YE media supplemented with leucine, uracil, adenine, and histidine (YE4S) were collected by centrifugation, and suspended in 60 mL of EMM. After the addition of formaldehyde (Sigma-Aldrich, F8775) to a final concentration of 1%, the cell suspension was vigorously mixed for 15 min at room temperature. The cell suspension was further mixed for 5 min, after the addition of 3 mL of 2.5 M glycine to neutralize the crosslinker. Mouse antibodies against H3K9me297, H3K9me397, H3K9ac98, H3K14ac98, Rpb1 (Millipore, CTD4H8, 05-623), and FLAG (Sigma-Aldrich, F1804), and rabbit antibodies against histone H3 (Abcam, ab1791) were used. Mouse and rabbit antibodies were attached to Dynabeads M-280 sheep anti-Mouse IgG (Invitrogen, 11202D) and Dynabeads M-280 sheep anti-Rabbit IgG (Invitrogen, 11204D), respectively. DNAs in whole-cell extracts and immunoprecipitates were quantified by real-time PCR using SYBR FAST (Thermo Fisher, 4385614) in a StepOnePlus real-time PCR system (Applied Biosystems). The primers used in ChIP are listed in Supplementary Table 3.
Northern blotting
Northern blotting was carried out as described previously53. From 1 × 109 log-phase cells grown in YE4S media, RNA was extracted by heating and freezing cells in the presence of phenol and SDS. 10 µg of total RNAs was suspended in 8.5 µL of MOPS buffer (20 mM MOPS pH 7.0, 2 mM NaAc, 1 mM EDTA) supplemented with 8% formaldehyde, 50% deionized formamide, and 10 µg mL−1 EtBr, and heat denatured by incubation at 55 °C for 60 min. RNAs were separated by 1.0% PrimeGel agarose LE (TaKaRa, 5801 A) gel/2.2 M formaldehyde electrophoresis in MOPS buffer. RNAs stained with EtBr were detected using a Typhoon FLA9000. After soaking the gel in 50 mM NaOH (Nacalai Tesque, 31511-05) for 20 min, RNAs were transferred to a ClearTrans nylon membrane 0.45 µm by capillary action in alkaline transfer buffer (10 mM NaOH, 3 M NaCl) and covalently attached to the membrane by 150 mJ UV irradiation. A 2.0 kb KpnI–KpnI fragment prepared from pTN834, a 0.9 kb NsiI–XbaI fragment from pTN770, a 1.7 kb PstI–XhoI fragment from pTN1226, a 0.9 kb XbaI–ApaI fragment from pTN1227, and a 1.9 kb XhoI–EcoRV fragment from pTN1225, were used to prepare radioactive DNA probes for the detection of dg, dh, imr3, adl1, and act1 RNAs, respectively. Radioactive signals were detected using a BAS2500 phosphorimager.
Statistical analysis
The two-tailed Mann–Whitney test and the two-tailed Fisher’s exact test were performed using GraphPad Prism version 6.0 g for Mac (GraphPad Software). The two-tailed Student’s t-test was performed using Excel (Microsoft).
Supplementary information
Acknowledgements
We thank Damien Hermand (University of Namur, Belgium), Robin C. Allshire (University of Edinburgh, UK), Shiv I.S. Grewal (National Institute of Health), and Makoto Kimura (RIKEN, Japan) for rpb1-S7A, H3K9A, H3K9R, clr4-W31G, and Flag-rpb3 strains, respectively. This work was supported by JSPS KAKENHI grants (JP26114711 and JP18K06060 to T.N.; JP17H01417 and JP18H05527 to H.K.).
Author contributions
T.N. supervised this study. A.K.O. and T.N. conceived the study and designed the experiments. A.K.O., D.W., J.S., F.Z., and T.N. carried out experiments. Y.M., H.K., and T.K. provided reagents. A.K.O. and T.N. wrote the manuscript. H.M., Y.M., H.K., T.S.T., and T.K. critically discussed the findings. All the authors reviewed and approved the final version of the manuscript.
Data availability
The datasets generated during the current study are available in the Dryad repository99. The plasmids created in the study can be obtained from National Bio Resource Project (NBRP) in Japan.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s42003-018-0251-z.
References
- 1.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Carvalho CM, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016;17:224–238. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weischenfeldt J, Symmons O, Spitz F, Korbel JO. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 2013;14:125–138. doi: 10.1038/nrg3373. [DOI] [PubMed] [Google Scholar]
- 4.Pardue ML, Gall JG. Chromosomal localization of mouse satellite DNA. Science. 1970;168:1356–1358. doi: 10.1126/science.168.3937.1356. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Q, et al. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011;477:179–184. doi: 10.1038/nature10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ting DT, et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011;331:593–596. doi: 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rea S, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 8.Chen ES, et al. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature. 2008;451:734–737. doi: 10.1038/nature06561. [DOI] [PubMed] [Google Scholar]
- 9.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- 10.Fischer T, et al. Diverse roles of HP1 proteins in heterochromatin assembly and functions in fission yeast. Proc. Natl Acad. Sci. USA. 2009;106:8998–9003. doi: 10.1073/pnas.0813063106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tatarakis A, Behrouzi R, Moazed D. Evolving models of heterochromatin: from foci to liquid droplets. Mol. Cell. 2017;67:725–727. doi: 10.1016/j.molcel.2017.08.022. [DOI] [PubMed] [Google Scholar]
- 12.Volpe TA, et al. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- 13.Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015;16:71–84. doi: 10.1038/nrg3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verdel A, et al. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303:672–676. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bayne EH, et al. Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell. 2010;140:666–677. doi: 10.1016/j.cell.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bühler M, Verdel A, Moazed D. Tethering RITS to a nascent transcript initiates RNAi- and heterochromatin-dependent gene silencing. Cell. 2006;125:873–886. doi: 10.1016/j.cell.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 17.Kato H, et al. RNA polymerase II is required for RNAi-dependent heterochromatin assembly. Science. 2005;309:467–469. doi: 10.1126/science.1114955. [DOI] [PubMed] [Google Scholar]
- 18.Djupedal I, et al. RNA Pol II subunit Rpb7 promotes centromeric transcription and RNAi-directed chromatin silencing. Genes Dev. 2005;19:2301–2306. doi: 10.1101/gad.344205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bühler M, Haas W, Gygi SP, Moazed D. RNAi-dependent and -independent RNA turnover mechanisms contribute to heterochromatic gene silencing. Cell. 2007;129:707–721. doi: 10.1016/j.cell.2007.03.038. [DOI] [PubMed] [Google Scholar]
- 20.Thakurta AG, Gopal G, Yoon JH, Kozak L, Dhar R. Homolog of BRCA2-interacting Dss1p and Uap56p link Mlo3p and Rae1p for mRNA export in fission yeast. EMBO J. 2005;24:2512–2523. doi: 10.1038/sj.emboj.7600713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strässer K, Hurt E. Yra1p, a conserved nuclear RNA-binding protein, interacts directly with Mex67p and is required for mRNA export. EMBO J. 2000;19:410–420. doi: 10.1093/emboj/19.3.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Z, et al. The protein Aly links pre-messenger-RNA splicing to nuclear export in metazoans. Nature. 2000;407:401–405. doi: 10.1038/35030160. [DOI] [PubMed] [Google Scholar]
- 23.MacKellar AL, Greenleaf AL. Cotranscriptional association of mRNA export factor Yra1 with C-terminal domain of RNA polymerase II. J. Biol. Chem. 2011;286:36385–36395. doi: 10.1074/jbc.M111.268144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang K, et al. Clr4/Suv39 and RNA quality control factors cooperate to trigger RNAi and suppress antisense RNA. Science. 2011;331:1624–1627. doi: 10.1126/science.1198712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reyes-Turcu FE, Zhang K, Zofall M, Chen E, Grewal SI. Defects in RNA quality control factors reveal RNAi-independent nucleation of heterochromatin. Nat. Struct. Mol. Biol. 2011;18:1132–1138. doi: 10.1038/nsmb.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez AC, van Wely KH. Centromere fission, not telomere erosion, triggers chromosomal instability in human carcinomas. Carcinogenesis. 2011;32:796–803. doi: 10.1093/carcin/bgr069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosin LF, Mellone BG. Centromeres drive a hard bargain. Trends Genet. 2017;33:101–117. doi: 10.1016/j.tig.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Therman E, Susman B, Denniston C. The nonrandom participation of human acrocentric chromosomes in Robertsonian translocations. Ann. Hum. Genet. 1989;53:49–65. doi: 10.1111/j.1469-1809.1989.tb01121.x. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura K, et al. Rad51 suppresses gross chromosomal rearrangement at centromere in Schizosaccharomyces pombe. EMBO J. 2008;27:3036–3046. doi: 10.1038/emboj.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selmecki A, Forche A, Berman J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science. 2006;313:367–370. doi: 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernard P, et al. Requirement of heterochromatin for cohesion at centromeres. Science. 2001;294:2539–2542. doi: 10.1126/science.1064027. [DOI] [PubMed] [Google Scholar]
- 32.Abe Y, et al. HP1-assisted Aurora B kinase activity prevents chromosome segregation errors. Dev. Cell. 2016;36:487–497. doi: 10.1016/j.devcel.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown WR, et al. Kinetochore assembly and heterochromatin formation occur autonomously in Schizosaccharomyces pombe. Proc. Natl Acad. Sci. USA. 2014;111:1903–1908. doi: 10.1073/pnas.1216934111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kapoor S, Zhu L, Froyd C, Liu T, Rusche LN. Regional centromeres in the yeast Candida lusitaniae lack pericentromeric heterochromatin. Proc. Natl Acad. Sci. USA. 2015;112:12139–12144. doi: 10.1073/pnas.1508749112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shang WH, et al. Chromosome engineering allows the efficient isolation of vertebrate neocentromeres. Dev. Cell. 2013;24:635–648. doi: 10.1016/j.devcel.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters AH, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–337. doi: 10.1016/S0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 37.Zeller P, et al. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 2016;48:1385–1395. doi: 10.1038/ng.3672. [DOI] [PubMed] [Google Scholar]
- 38.Li PC, et al. Replication fork stability is essential for the maintenance of centromere integrity in the absence of heterochromatin. Cell Rep. 2013;3:638–645. doi: 10.1016/j.celrep.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaratiegui M, et al. RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature. 2011;479:135–138. doi: 10.1038/nature10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ellermeier C, et al. RNAi and heterochromatin repress centromeric meiotic recombination. Proc. Natl Acad. Sci. USA. 2010;107:8701–8705. doi: 10.1073/pnas.0914160107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Izban MG, Luse DS. The RNA polymerase II ternary complex cleaves the nascent transcript in a 3’ → 5’ direction in the presence of elongation factor SII. Genes Dev. 1992;6:1342–1356. doi: 10.1101/gad.6.7.1342. [DOI] [PubMed] [Google Scholar]
- 42.Kettenberger H, Armache KJ, Cramer P. Architecture of the RNA polymerase II-TFIIS complex and implications for mRNA cleavage. Cell. 2003;114:347–357. doi: 10.1016/S0092-8674(03)00598-1. [DOI] [PubMed] [Google Scholar]
- 43.Niwa O, Matsumoto T, Yanagida M. Construction of a mini-chromosome by deletion and its mitotic and meiotic behaviour in fission yeast. Mol. Gen. Genet. 1986;203:397–405. doi: 10.1007/BF00422063. [DOI] [Google Scholar]
- 44.Lin M, Chang CJ, Green NS. A new method for estimating high mutation rates in cultured cells. Mutat. Res. 1996;351:105–116. doi: 10.1016/0027-5107(95)00209-X. [DOI] [PubMed] [Google Scholar]
- 45.Ekwall K, Ruusala T. Mutations in rik1, clr2, clr3 and clr4 genes asymmetrically derepress the silent mating-type loci in fission yeast. Genetics. 1994;136:53–64. doi: 10.1093/genetics/136.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keeney S, Giroux CN, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell. 1997;88:375–384. doi: 10.1016/S0092-8674(00)81876-0. [DOI] [PubMed] [Google Scholar]
- 47.Jia S, Kobayashi R, Grewal SI. Ubiquitin ligase component Cul4 associates with Clr4 histone methyltransferase to assemble heterochromatin. Nat. Cell Biol. 2005;7:1007–1013. doi: 10.1038/ncb1300. [DOI] [PubMed] [Google Scholar]
- 48.Min J, Zhang X, Cheng X, Grewal SI, Xu RM. Structure of the SET domain histone lysine methyltransferase Clr4. Nat. Struct. Biol. 2002;9:828–832. doi: 10.1038/nsb860. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, et al. Structure of the Neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell. 2002;111:117–127. doi: 10.1016/S0092-8674(02)00999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kusevic D, Kudithipudi S, Iglesias N, Moazed D, Jeltsch A. Clr4 specificity and catalytic activity beyond H3K9 methylation. Biochimie. 2017;135:83–88. doi: 10.1016/j.biochi.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 51.Mellone BG, et al. Centromere silencing and function in fission yeast is governed by the amino terminus of histone H3. Curr. Biol. 2003;13:1748–1757. doi: 10.1016/j.cub.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 52.Allshire RC, Ekwall K. Epigenetic regulation of chromatin states in Schizosaccharomyces pombe. Cold Spring Harb. Perspect. Biol. 2015;7:a018770. doi: 10.1101/cshperspect.a018770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Onaka AT, et al. Rad51 and Rad54 promote noncrossover recombination between centromere repeats on the same chromatid to prevent isochromosome formation. Nucleic Acids Res. 2016;44:10744–10757. doi: 10.1093/nar/gkw874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zafar F, et al. Regulation of mitotic recombination between DNA repeats in centromeres. Nucleic Acids Res. 2017;45:11222–11235. doi: 10.1093/nar/gkx763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bannister AJ, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 56.Sadaie M, et al. Balance between distinct HP1 family proteins controls heterochromatin assembly in fission yeast. Mol. Cell. Biol. 2008;28:6973–6988. doi: 10.1128/MCB.00791-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang K, Mosch K, Fischle W, Grewal SI. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat. Struct. Mol. Biol. 2008;15:381–388. doi: 10.1038/nsmb.1406. [DOI] [PubMed] [Google Scholar]
- 58.Jih G, et al. Unique roles for histone H3K9me states in RNAi and heritable silencing of transcription. Nature. 2017;547:463–467. doi: 10.1038/nature23267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Motamedi MR, et al. Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding centromeric RNAs. Cell. 2004;119:789–802. doi: 10.1016/j.cell.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 60.Holoch D, Moazed D. Small-RNA loading licenses Argonaute for assembly into a transcriptional silencing complex. Nat. Struct. Mol. Biol. 2015;22:328–335. doi: 10.1038/nsmb.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Halic M, Moazed D. Dicer-independent primal RNAs trigger RNAi and heterochromatin formation. Cell. 2010;140:504–516. doi: 10.1016/j.cell.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Strässer K, et al. TREX is a conserved complex coupling transcription with messenger RNA export. Nature. 2002;417:304–308. doi: 10.1038/nature746. [DOI] [PubMed] [Google Scholar]
- 63.Cam HP, et al. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat. Genet. 2005;37:809–819. doi: 10.1038/ng1602. [DOI] [PubMed] [Google Scholar]
- 64.Iglesias N, et al. Automethylation-induced conformational switch in Clr4 (Suv39h) maintains epigenetic stability. Nature. 2018;560:504–508. doi: 10.1038/s41586-018-0398-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aygün O, Mehta S, Grewal SI. HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat. Struct. Mol. Biol. 2013;20:547–554. doi: 10.1038/nsmb.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alper BJ, et al. Sir2 is required for Clr4 to initiate centromeric heterochromatin assembly in fission yeast. EMBO J. 2013;32:2321–2335. doi: 10.1038/emboj.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buscaino A, et al. Distinct roles for Sir2 and RNAi in centromeric heterochromatin nucleation, spreading and maintenance. EMBO J. 2013;32:1250–1264. doi: 10.1038/emboj.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grewal SI, Bonaduce MJ, Klar AJ. Histone deacetylase homologs regulate epigenetic inheritance of transcriptional silencing and chromosome segregation in fission yeast. Genetics. 1998;150:563–576. doi: 10.1093/genetics/150.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harlen KM, Churchman LS. The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat. Rev. Mol. Cell Biol. 2017;18:263–273. doi: 10.1038/nrm.2017.10. [DOI] [PubMed] [Google Scholar]
- 70.Egloff S, et al. Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science. 2007;318:1777–1779. doi: 10.1126/science.1145989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cassart C, Drogat J, Migeot V, Hermand D. Distinct requirement of RNA polymerase II CTD phosphorylations in budding and fission yeast. Transcription. 2012;3:231–234. doi: 10.4161/trns.21066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kajitani T, et al. Ser7 of RNAPII-CTD facilitates heterochromatin formation by linking ncRNA to RNAi. Proc. Natl Acad. Sci. USA. 2017;114:E11208–E11217. doi: 10.1073/pnas.1714579115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kimura M, Suzuki H, Ishihama A. Formation of a carboxy-terminal domain phosphatase (Fcp1)/TFIIF/RNA polymerase II (pol II) complex in Schizosaccharomyces pombe involves direct interaction between Fcp1 and the Rpb4 subunit of pol II. Mol. Cell. Biol. 2002;22:1577–1588. doi: 10.1128/MCB.22.5.1577-1588.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McDowall MD, et al. PomBase 2015: updates to the fission yeast database. Nucleic Acids Res. 2015;43:D656–D661. doi: 10.1093/nar/gku1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou Q, Li T, Price DH. RNA polymerase II elongation control. Annu. Rev. Biochem. 2012;81:119–143. doi: 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crotti LB, Basrai MA. Functional roles for evolutionarily conserved Spt4p at centromeres and heterochromatin in Saccharomyces cerevisiae. EMBO J. 2004;23:1804–1814. doi: 10.1038/sj.emboj.7600161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hayashi MT, Takahashi TS, Nakagawa T, Nakayama J, Masukata H. The heterochromatin protein Swi6/HP1 activates replication origins at the pericentromeric region and silent mating-type locus. Nat. Cell Biol. 2009;11:357–362. doi: 10.1038/ncb1845. [DOI] [PubMed] [Google Scholar]
- 78.Bailis JM, Bernard P, Antonelli R, Allshire RC, Forsburg SL. Hsk1-Dfp1 is required for heterochromatin-mediated cohesion at centromeres. Nat. Cell Biol. 2003;5:1111–1116. doi: 10.1038/ncb1069. [DOI] [PubMed] [Google Scholar]
- 79.Folco HD, et al. Untimely expression of gametogenic genes in vegetative cells causes uniparental disomy. Nature. 2017;543:126–130. doi: 10.1038/nature21372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim DH, Villeneuve LM, Morris KV, Rossi JJ. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat. Struct. Mol. Biol. 2006;13:793–797. doi: 10.1038/nsmb1142. [DOI] [PubMed] [Google Scholar]
- 81.Cernilogar FM, et al. Chromatin-associated RNA interference components contribute to transcriptional regulation in Drosophila. Nature. 2011;480:391–395. doi: 10.1038/nature10492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bisht KK, Arora S, Ahmed S, Singh J. Role of heterochromatin in suppressing subtelomeric recombination in fission yeast. Yeast. 2008;25:537–548. doi: 10.1002/yea.1603. [DOI] [PubMed] [Google Scholar]
- 83.Garcia-Muse T, Aguilera A. Transcription-replication conflicts: how they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016;17:553–563. doi: 10.1038/nrm.2016.88. [DOI] [PubMed] [Google Scholar]
- 84.Kireeva ML, et al. Nature of the nucleosomal barrier to RNA polymerase II. Mol. Cell. 2005;18:97–108. doi: 10.1016/j.molcel.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 85.Pebernard S, Schaffer L, Campbell D, Head SR, Boddy MN. Localization of Smc5/6 to centromeres and telomeres requires heterochromatin and SUMO, respectively. EMBO J. 2008;27:3011–3023. doi: 10.1038/emboj.2008.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Peng JC, Karpen GH. Heterochromatic genome stability requires regulators of histone H3 K9 methylation. PLoS. Genet. 2009;5:e1000435. doi: 10.1371/journal.pgen.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jaco I, Canela A, Vera E, Blasco MA. Centromere mitotic recombination in mammalian cells. J. Cell Biol. 2008;181:885–892. doi: 10.1083/jcb.200803042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tinline-Purvis H, et al. Failed gene conversion leads to extensive end processing and chromosomal rearrangements in fission yeast. EMBO J. 2009;28:3400–3412. doi: 10.1038/emboj.2009.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hatchi E, et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell. 2015;57:636–647. doi: 10.1016/j.molcel.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Amon, J. D. & Koshland, D. RNase H enables efficient repair of R-loop induced DNA damage. eLife5, e20533 (2016). [DOI] [PMC free article] [PubMed]
- 91.Volpe T, et al. RNA interference is required for normal centromere function in fission yeast. Chromosome Res. 2003;11:137–146. doi: 10.1023/A:1022815931524. [DOI] [PubMed] [Google Scholar]
- 92.Wang J, Jia ST, Jia S. New insights into the regulation of heterochromatin. Trends Genet. 2016;32:284–294. doi: 10.1016/j.tig.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Risques RA, Kennedy SR. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS. Genet. 2018;14:e1007108. doi: 10.1371/journal.pgen.1007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vijg J, Suh Y. Genome instability and aging. Annu. Rev. Physiol. 2013;75:645–668. doi: 10.1146/annurev-physiol-030212-183715. [DOI] [PubMed] [Google Scholar]
- 95.Gao J, et al. Rapid, efficient and precise allele replacement in the fission yeast Schizosaccharomyces pombe. Curr. Genet. 2014;60:109–119. doi: 10.1007/s00294-013-0406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chikashige Y, et al. Composite motifs and repeat symmetry in S. pombe centromeres: direct analysis by integration of NotI restriction sites. Cell. 1989;57:739–751. doi: 10.1016/0092-8674(89)90789-7. [DOI] [PubMed] [Google Scholar]
- 97.Hayashi-Takanaka Y, et al. Tracking epigenetic histone modifications in single cells using Fab-based live endogenous modification labeling. Nucleic Acids Res. 2011;39:6475–6488. doi: 10.1093/nar/gkr343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Karmodiya K, Krebs AR, Oulad-Abdelghani M, Kimura H, Tora L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genom. 2012;13:424. doi: 10.1186/1471-2164-13-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Okita, A. K. et al. Data from: heterochromatin suppresses gross chromosomal rearrangements at centromeres by repressing Tfs1/TFIIS-dependent transcription. Dryad Digital Repository. 10.5061/dryad.c8n0748. (2019). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available in the Dryad repository99. The plasmids created in the study can be obtained from National Bio Resource Project (NBRP) in Japan.