

Abstract
The selection of effective genes that accurately predict chemotherapy responses might improve cancer outcomes. We compare optimized gene signatures for cisplatin, carboplatin, and oxaliplatin responses in the same cell lines and validate each signature using data from patients with cancer. Supervised support vector machine learning is used to derive gene sets whose expression is related to the cell line GI50 values by backwards feature selection with cross-validation. Specific genes and functional pathways distinguishing sensitive from resistant cell lines are identified by contrasting signatures obtained at extreme and median GI50 thresholds. Ensembles of gene signatures at different thresholds are combined to reduce the dependence on specific GI50 values for predicting drug responses. The most accurate gene signatures for each platin are: cisplatin: BARD1, BCL2, BCL2L1, CDKN2C, FAAP24, FEN1, MAP3K1, MAPK13, MAPK3, NFKB1, NFKB2, SLC22A5, SLC31A2, TLR4, and TWIST1; carboplatin: AKT1, EIF3K, ERCC1, GNGT1, GSR, MTHFR, NEDD4L, NLRP1, NRAS, RAF1, SGK1, TIGD1, TP53, VEGFB, and VEGFC; and oxaliplatin: BRAF, FCGR2A, IGF1, MSH2, NAGK, NFE2L2, NQO1, PANK3, SLC47A1, SLCO1B1, and UGT1A1. Data from The Cancer Genome Atlas (TCGA) patients with bladder, ovarian, and colorectal cancer were used to test the cisplatin, carboplatin, and oxaliplatin signatures, resulting in 71.0%, 60.2%, and 54.5% accuracies in predicting disease recurrence and 59%, 61%, and 72% accuracies in predicting remission, respectively. One cisplatin signature predicted 100% of recurrence in non-smoking patients with bladder cancer (57% disease-free; N = 19), and 79% recurrence in smokers (62% disease-free; N = 35). This approach should be adaptable to other studies of chemotherapy responses, regardless of the drug or cancer types.
Subject terms: Predictive medicine, Predictive markers
Oncology: Computers decode cancer drug responses
Machine learning has identified genetic signatures that predict how patients will respond to three of the most widely used cancer drugs. Chemotherapy regimens are usually based on how groups of people with similar cancers respond to them, but genetic differences can render the drugs more or less effective in individual patients. Machine learning provides a way of sifting through large amounts of data to identify patterns—in this case, in gene signatures associated with cancer recurrence and remission. The authors investigated cellular responses to cisplatin, carboplatin, and oxaliplatin and identified signatures in 11–15 genes which were the most predictive for each drug. The compositions of these signatures are also tailored to how well these therapies prevent growth of cancer cells. Accuracy varied, but one cisplatin signature was able to predict all instances of disease recurrence in non-smokers with bladder cancer.
Introduction
Chemotherapy regimens are selected based on overall outcomes for specific types and subtypes of cancer pathology, progression to metastasis, other high-risk indications, and prognosis,1,2 and variability in tumor resistance has led to the use of tiered, sequential strategies for the selection of agents based on their overall efficacy.3 Our group and other researchers have developed machine learning (ML)-based gene signatures (i.e., predictive models) aimed at predicting responses to specific chemotherapeutic agents and minimizing chemoresistance based on the inhibition of growth or drug targets (GI50 or IC50, respectively).4–6 In this study, we present integrated ML models of platin drug responses (cis-, carbo-, and oxaliplatin), and evaluate them using clinical outcome data that were not used to construct the signatures. Previous studies have reviewed the genes,7 gene products,8 and specific individual pathways that are activated and repressed by drugs,9 but comprehensive models of the global cellular response to drugs are lacking. We use integrated ML-based signatures based on the expression of multiple genes to predict key responses to each of these platin agents, for the first time, at different resistance levels.
Cisplatin, carboplatin, and oxaliplatin are each widely prescribed compounds with antineoplastic effects. While each drug contains platinum and forms adducts with tumor DNA, their effectiveness differs for specific types of cancers, such as bladder (cisplatin), ovarian (cisplatin and carboplatin), and colorectal cancer (oxaliplatin). Carboplatin differs in structure from cisplatin, exchanging the dichloride ligands in the latter with a cyclobutane dicarboxylic acid (CBDCA) group, while oxaliplatin is paired with both a diaminocyclohexane (DACH) ligand and a bidentate oxalate group. These chelating ligands have greater stability and solubility in aqueous solutions, which lead to differences in drug toxicity compared to cisplatin.10 Oxaliplatin is up to two times more cytotoxic than cisplatin, but it forms fewer DNA adducts.11 The large hydrophobic DACH ligand that overlaps with the major groove is thought to prevent the binding of certain DNA repair enzymes, such as the POL polymerases, and may contribute to the low cross-resistance between oxaliplatin and the other two platin drugs.10 While all three drugs enter the cell via copper transporters, organic cation transporters are oxaliplatin-specific and likely play a role in its efficacy in colorectal cancer (CRC) cells where these transporters are commonly overexpressed.7 Oxaliplatin specifically interferes with both DNA and RNA synthesis, unlike cisplatin, which only infers with DNA.12 These intrinsic properties of the platinum drugs lead to differences in their activity and resistance profiles, despite their similar modes of action.
We derived gene signatures to predict drug responses at different sensitivity and resistance levels for each of these agents. Our group and other researchers have used supervised learning algorithms, including random forest models;13 support vector machine (SVM) models;6 neural networks;14 and linear regression models5 for these predictions. Pathway and network analyses of gene expression (GE) have been used to identify hundreds of genes that are potentially up- and down-regulated upon cisplatin treatment.15 Cisplatin-specific gene signatures have been developed with integrative approaches such as elastic net regression using the inferred pathway activity obtained from data from bladder cancer cell lines.16 These methods have implicated genes that have not been described previously. Supervised ML with biochemically relevant genes has also been useful for predicting drug response.6 A concern with each of these ML approaches is that an insufficient number of samples coupled with a large number of features, i.e., GE changes, in each sample may result in overfitting of the model, affecting its generalizability with other sources of data.17 We therefore reduce the number of dimensions by selecting genes that are biologically relevant to the drugs under observation.6,17 In this study, genes included in the final signatures have well-defined roles in responses to their corresponding drugs (Supplementary References, Section A). Additional selection criteria are necessary when the number of genes implicated in peer-reviewed reports is still prohibitively large compared to the sample size.
Biochemically-inspired gene signatures have shown good performance in predicting treatment responses. A paclitaxel ML signature based on tumor GE had a higher success at predicting the pathological complete response rate (pCR18) for sensitive patients (84% of patients with no/minimal residual disease) than gene signatures based on a differential GE analysis.6 For gemcitabine, a signature derived from both expression and copy number (CN) data from breast cancer cell lines was derived and subsequently applied to the analysis of nucleic acids from archived patient samples. Multiple other outcome measures used to validate gene signatures include prognosis,5 Miller–Payne response,19 and disease recurrence. Binary SVM classifiers based on discrete time thresholds have been used to classify continuous outcome measures such as prognosis and recurrence. In contrast, pCR is simpler to interpret with binary SVM models. Nevertheless, differences in clinical recurrence have been noted between patients with known pCR and those who do not exhibit disease pathology.18 This source of variability in defining patient responses can confound the transferability of SVM models between different datasets.
We apply biochemically-inspired ML to predict and compare the cellular and patient responses to cisplatin, carboplatin, and oxaliplatin. We train models and perform model selection for the classification of platin resistance using data from cancer cell lines, and validate the results using patient GE and clinical outcome data. Our previous gene signatures derived from cell lines were based on median GI50 values for each drug.6 Models (i.e., gene signatures) learned and selected using the cell line data have not been re-trained prior to application to the patient data since GI50 values are not available for patient samples. This approach has been a necessary compromise; however, in the present study, we derive different signatures at the highest and the lowest levels of drug resistance. A series of candidate gene signatures are derived by shifting the GI50 thresholds that distinguish sensitivity from resistance. The frequency of genes selected at median and extreme thresholds highlights pathways that most likely define these responses among different patient subsets.
Results
Selection of platin drug-related genes
We documented genes in the peer-reviewed literature associated with drug effectiveness or responses (Supplementary References, Section B). For cisplatin, carboplatin, and oxaliplatin, 179, 90, and 288 genes were implicated, respectively (Supplementary Table S1). Multiple factor analysis (MFA) was used to determine which genes correlated with the GI50 in breast cancer cell lines through either GE and/or CN,13 significantly reducing the sizes of the gene sets for cisplatin (N = 39), carboplatin (N = 28), and oxaliplatin (N = 55). Genes with significant relationships to GI50 and the directions of correlations (positive or inverse) are indicated in Figs. 1–3. The diverse functions of these genes included apoptosis, DNA repair, transcription, cell growth, metabolism, immune system, signal transduction, and membrane transport. Analyses of IC50 and GE levels for cisplatin-treated bladder cancer cell lines confirmed these relationships based on the GI50 values of different breast cancer lines. IC50 values were related to GE for CFLAR, FEN1, MAPK3, MSH2, NFKB1, PNKP, PRKAA2, and PRKCA.20 Similarly, IC50 values obtained from separate bladder cell lines included in the Genomics of Drug Sensitivity in Cancer project (CancerRxGene; http://www.cancerrxgene.org; N = 17)21 correlated with GE for CFLAR, FEN1, and NFKB1, as well as ATP7B, BARD1, MAP3K1, NFKB2, SLC31A2, and SNAI1.
Fig. 2.

Schematic of platinum drug sensitivity and resistance genes that showed MFA correlation with the GI50 values for carboplatin. Refer to the legend of Fig. 1 for details
Fig. 1.

Schematic of platinum drug sensitivity and resistance genes that showed MFA correlation with the GI50 values for cisplatin. The gene products corresponding to those used to derive the SVM are indicated within boxes in the context of their cellular mechanisms of action and regulation of drug response. GE and CN correlations with inhibitory drug concentrations are based on the MFA of breast (GI50) and bladder (IC50) cancer cell line data. Gene products within the best-performing gene signature are embedded within color-coded ovals; whereas the other correlated gene products are embedded within rectangles
Fig. 3.

Schematic of platinum drug sensitivity and resistance genes that showed MFA correlation with the GI50 values for oxaliplatin. Refer to the legend of Fig. 1 for details
We performed an MFA of the GI50 values for cisplatin, carboplatin, and oxaliplatin, without considering either GE or CN. Responses to cis- and carboplatin were directly correlated (a 6.2° separation between vectors), but neither was related to the oxaliplatin response (Fig. 4). Cisplatin-resistant cell lines are generally sensitive to oxaliplatin.22–24
Fig. 4.
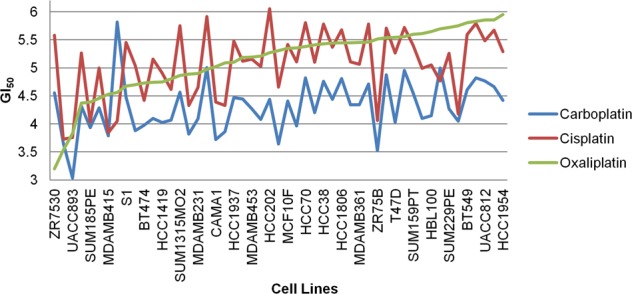
GI50 values for cell lines treated with the three platin drugs were plotted in order of ascending oxaliplatin GI50. For most cell lines, a trend between the GI50 values for cisplatin and carboplatin was observed, reflecting the correlation between the two drugs detected using MFA. Despite this correlation, carboplatin shows a much smaller variance (0.22) than cisplatin (0.37; the oxaliplatin variance is 0.34)
SVM-based signatures were initially derived for each platin drug using breast cancer cell line GE data. A 13-gene signature for cisplatin that predicts whether observed growth inhibition is above or below the median GI50 threshold (5.2% cross-validation misclassification rate) consisted of BARD1, BCL2L1, FAAP24, CFLAR, MAP3K1, MAPK3, NFKB1, POLQ, PRKAA2, SLC22A5, SLC31A2, TLR4, and TWIST1. A similarly derived carboplatin signature included AKT1, ATP7B, EGF, EIF3I, ERCC1, GNGT1, HRAS, MTR, NRAS, OPRM1, RAD50, RAF1, SCN10A, SGK1, TIGD1, TP53, and VEGFB (10.4% cross-validation misclassification). For oxaliplatin, the final SVM gene signature consisted of AGXT, APOBEC2, BRAF, CLCN6, FCGR2A, IGF1, MPO, MSH2, NAGK, NAT2, NFE2L2, NOTCH1, PANK3, PRSS1, and UGT1A1 (2.1% cross-validation misclassification). A cisplatin SVM generated from 17 bladder cancer cell lines in CancerRxGene resulted in 2 equally accurate signatures (with 11.8% cross-validation misclassification) consisting of either PNKP and PRKCA, or ATP7B, CFLAR, FEN1, MAPK3, NFKB1, and SLC22A11. These gene signatures were not useful for predicting patient outcomes due to the limited size of the training set.
GI50 threshold-independent modeling
In our previous studies, we set the median GI50 value as the threshold to distinguish drug resistance and sensitivity.5,6 An important question is whether the genes contributing to drug responses are consistent among different cell lines, each with their own unique GI50 values. Different ML gene signatures were obtained by shifting the GI50 threshold, which changed the labels of resistant and sensitive cell lines. After feature selection, the compositions of the corresponding gene signatures for each threshold were compared. Finally, ensemble averaging of all of these optimized SVMs with Gaussian kernels derived for different GI50 thresholds was used to create a single aggregated, threshold-independent, ML-based predictive model comprised of all genes that were selected in any of the threshold-specific models (i.e., a composite gene signature).
Kinase (MAPK3 and MAP3K1) genes and apoptotic family members (BCL2 and BCL2L1) were the most common genes in the cisplatin signatures at different GI50 thresholds, with consistent representation of error-prone and base-excision DNA repair genes as well (Fig. 5a and Supplementary Table S2A). The kinases were more concentrated in signatures with lower drug sensitivity thresholds, whereas BCL2 and BCL2L1 were more ubiquitous at all levels. The error-prone polymerases POLD1 and POLQ were more frequently detected in gene signatures with lower sensitivity thresholds, while the flap endonuclease FEN1 tended to be present at high levels of resistance. Thresholded gene signatures for carboplatin-related genes commonly contained the apoptotic family member AKT1, transcription regulation genes ETS2 and TP53, as well as cell growth factors VEGFB and VEGFC, although the latter were less common at lower sensitivity thresholds (Fig. 5b). Common oxaliplatin-related genes included the transporters SLCO1B1 and GRTP1 (but not SLC47A1), transcription-related genes NFE2L2, PARP15, and CLCN6, and multiple metabolism-related genes (Fig. 5c).
Fig. 5.

Variation in the composition of the gene signatures obtained using misclassification-based SVMs at different GI50 thresholds for a cisplatin, b carboplatin, and c oxaliplatin. GI50 intervals are indicated on the left, with the number of cell lines with GI50 values within the indicated intervals shown in brackets. Each box represents the density of genes appearing in optimized Gaussian SVM gene signatures in those functional categories, with darker gray indicating frequent genes in the indicated GI50 threshold intervals and lighter gray indicating less commonly selected genes. The number of thresholded gene signatures used to derive the density plot within each interval is equal to (or greater than, in the case of multiple equally performing gene signatures) the number of cell lines within that GI50 interval
GI50-thresholded ML models were also derived using the log-loss function to evaluate whether an alternative loss function (for classification) would differ significantly from the misclassification-based gene signatures (by both the distribution of selected genes and by model accuracy to patient data). The log-loss function penalizes false classifications, whose value ranges from zero (or completely accurate) to 1 (or completely inaccurate; Supplementary Table S3). The overall distribution of genes across GI50 thresholds exhibited many distinct similarities to the gene signatures derived by misclassification. For both sets of cisplatin gene signatures, BCL2, BCL2L1, and FEN1 were common in low-to-moderate GI50 thresholds, while NFKB1 was enriched at high thresholds (Fig. 5a and Supplementary Figure S1A). For carboplatin, AKT1, VEGFB, and VEGFC were similarly distributed across GI50 thresholds with both methods, although VEGFB was less frequently represented in log-loss-based gene signatures at low GI50 values (Fig. 5b and Supplementary Figure S1B). In both sets of oxaliplatin gene signatures, SIAE and SLC47A1 were present at high frequencies across all GI50 thresholds, whereas ABCG2 was present less frequently (<50% inclusion; Fig. 5c and Supplementary Figure S1C). Differences between signatures selected by minimizing log-loss and misclassification rates were observed. EGF and ERCC1 were selected at a greater frequency at a moderate carboplatin GI50 using the log-loss function, rather than misclassification. Similarly, in oxaliplatin signature genes, APOBEC2, HLA-B, LTA, and MPO, were selected considerably more often using the log-loss function. Therefore, while the misclassification- and log-loss-based gene signatures are not interchangeable, overall, they are quite similar to one another.
Log-loss gene signatures were initially constructed either using (a) a modified version of the misclassification-based method, or (b) the backwards feature selection (BFS) software described by Zhao et al.25 Multiple signatures with low log-loss values can have different compositions, consistent with the possibility that various diverse gene combinations may give rise to signatures with satisfactory performance. However, these signatures often contain a larger number of gene features than the misclassification-based signatures, raising the possibility that they might be more prone to overfitting. This concern was addressed by generating gene signatures by minimizing log-loss using both methods. The median GI50-thresholded cisplatin gene signature generated using the log-loss modified software [ATP7B, BCL2L1, CDKN2C, CFLAR, ERCC2, ERCC6, FAAP24, FOS, GSTO1, GSTP1, MAP3K1, MAPK13, MAPK3, MSH2, MT2A, PNKP, POLD1, POLQ, PRKAA2, PRKCA, PRKCB, SLC22A5, SLC31A2, SNAI1, TLR4, and TP63] shares 15/19 genes with the signature generated using the BFS software25 [ATP7B, BARD1, BCL2, BCL2L1, ERCC2, FAAP24, FEN1, FOS, MAP3K1, MAPK13, MAPK3, MSH2, MT2A, NFKB1, PNKP, POLQ, PRKCB, SLC22A5, and SNAI1].
Impacts of features in gene signatures
Each gene was independently excluded and model accuracy was reassessed within every SVM signature to determine the contributions of individual genes to the overall cross-validation accuracy of a gene signature (Supplementary Table S2A, S2B, and S2C contain cis-, carbo-, and oxaliplatin gene signatures, respectively). The elimination of ERCC2, POLD1, BARD1, BCL2, PRKCA, and PRKCB consistently significantly increased the misclassification error (average > 16% increase) in moderate threshold cisplatin SVMs (GI50 thresholds: 5.1–5.5). ERCC2 and POLD1 perform critical functions in nucleotide and base excision repair, respectively. PRKCA and PRKCB are paralogs with significant roles in signal transduction. BARD1 has been shown to reduce the expression of the apoptotic BCL2 gene in the mitochondria,26 and has a key role in genomic stability through its association with BRCA1. The genes NFKB1, NFKB2, TWIST1, TP63, PRKAA2, and MSH2 showed a high variance in increased misclassification between different gene signatures. The variance of these genes may be due to epistatic interactions with other biological components, including the other genes in the SVM. For example, NFKB1 and NFKB2 are jointly included in 7 SVMs generated at a moderate GI50 threshold. Possible epistasis was observed, as the removal of either of these genes, but not necessarily both, exerted a substantial impact on model misclassification rates (≥18.0% increase). The misclassification variance of NFKB1 with NFKB2 was significantly lower than in SVM gene signatures lacking NFKB2.
Derivation of gene signatures from data obtained from patients with bladder carcinoma
Gene signatures derived from cell line data were validated using data from patients with cancer. We also developed SVMs using the cisplatin and/or carboplatin-treated TCGA (The Cancer Genome Atlas) data from patients with bladder urothelial carcinoma using post-treatment time to relapse as a surrogate criterion for different GI50 resistance thresholds to explore the similarities in the gene signatures in the data obtained from these patients (as performed in Mucaki et al.;27 Supplementary Table S4). Similar trends to cell line SVMs were apparent: POLQ was frequently included in gene signatures with a recurrence threshold of a longer duration, while FEN1 was a marker of resistance when the time to relapse was shorter. However, BCL2, which is present in a majority of breast cancer cell lines SVMs, was present in only one gene signature derived from TCGA data. Similarly, MSH2 was rarely selected using cell lines, yet appeared in nearly all patient-derived SVMs with >1-year recurrence. However, independently-derived patient SVMs were not able to be used for any other analyses.
Validation of cell line-based models using data from patients with cancer
GI50-thresholded models for each platin drug, which were generated with the breast cancer cell line data, produced 70 cisplatin, 83 carboplatin, and 83 oxaliplatin SVM gene signatures. Each of the thresholded gene signatures was applied to available platin-treated patient datasets to understand how the choice of GI50 threshold for training on cell line data impacted the predictive accuracy when the resulting gene signatures were assessed according to patient outcomes.28–32 In this study, cisplatin gene signatures were validated using data from patients with bladder cancer, carboplatin signatures were validated using data from patients with ovarian cancer, and oxaliplatin signatures were validated using data from patients with CRC. While the available data contained the necessary GE information, the clinical response metadata differed between studies. The responses of patients with bladder cancer to cisplatin were described as post-treatment survival by Als et al.,31 whereas patients with CRC treated with oxaliplatin were categorized as responders and non-responders by Tsuji et al.32 TCGA provided two different measures that we used to assess the predictive accuracy of our gene signatures—clinical response to chemotherapy and disease-free survival. Signature accuracy was similar using either measure (Supplementary Table S5A); however, recurrence and disease-free survival were used as the primary measures of responses, as these outcomes were more consistently recorded among the TCGA datasets tested. Patients in the study by Als et al.31 with a ≥5-year post-treatment survival were labeled as sensitive to treatment. The differences between these metadata may partially contribute to differences in the prediction accuracy of the thresholded SVM gene signatures.
At higher resistance thresholds for any platin drug (low GI50), where more cell lines were labeled sensitive, the positive class (disease-free survival) was correctly classified, while the negative class (recurrence) was highly misclassified (Supplementary Figures S2 and S3). The reverse was true for gene signatures derived using lower resistance thresholds (high GI50). For these reasons, SVMs generated at these extreme thresholds were not very useful at predicting patient outcomes. When used to predict recurrence in the TCGA datasets, sensitivity and specificity appeared to be maximized in gene signatures where the GI50 threshold for resistance was set near (but not necessarily at) the median (Supplementary Figure S2 and Supplementary Table S5A to S5C). While this pattern held true for the data reported by Tsuji et al.,32 oxaliplatin gene signatures, where GI50 thresholds were set above the median, better separated patients with primary and metastatic CRC (best signature predicting 92.6% of metastases and 60.7% of primary cancers; Supplementary Table S5C). Although less consistent, cisplatin gene signatures generated with thresholds above the median GI50 performed better when evaluating the patient dataset reported by Als et al. (Supplementary Figure S3).31
Gene signatures were individually evaluated for their accuracy in TCGA patients using various recurrence times post-treatment to classify resistant and sensitive patients (0.5–5 years; Supplementary Table S6A-C). The best-performing cisplatin signature (hereby identified as Cis1; Table 1) accurately predicted 71.0% of the recurrence of bladder cancer in patients who experienced recurrence after 18 months (N = 31; 58.5% accurate for disease-free patients [N = 41]). The best-performing carboplatin gene signature (designated Car1 [Table 1]) predicted the recurrence of ovarian cancer after 4 years at an accuracy of 60.2% (N = 302; 61.0% accurate for disease-free patients [N = 108]). For oxaliplatin, the best-performing gene signature (designated Oxa1 [Table 1]) accurately predicted 71.6% of the disease-free TCGA patients with CRC after 1 year (N = 88; 54.5% accuracy in predicting recurrence [N = 11]). These gene signatures (based on GE measured with the Affymetrix Gene Chip Human Exon 1.0 ST arrays), TCGA sample expression data, and SVMs based on bladder cell line data (based on expression measured using the Affymetrix U133A microarray) were added to the online web‐based SVM calculator (http://chemotherapy.cytognomix.com; introduced in Dorman et al.6) to predict platin responses.
Table 1.
Gene signatures that best predicted the responses of TCGA patients
| Gene signature ID | Cancer type tested | GI50 threshold | Signature (C; σ) |
|---|---|---|---|
| Cis1 (Cisplatin) | Bladder | 5.11 | BARD1, BCL2, BCL2L1, CDKN2C, FAAP24, FEN1, MAP3K1, MAPK13, MAPK3, NFKB1, NFKB2, SLC22A5, SLC31A2, TLR4, TWIST1 (100,000; 100) |
| Cis2 (Cisplatin) | Bladder | 5.12 | BARD1, BCL2L1, CFLAR, FAAP24, MAP3K1, MAPK3, NFKB1, POLQ, PRKAA2, SLC22A5, SLC31A2, TLR4, TWIST1 (10,000; 100) |
| Cis3 (Cisplatin) | Bladder | 5.60 | BCL2, CFLAR, ERCC2, ERCC6, FAAP24, FEN1, MAP3K1, NFKB1, NFKB2, PNKP, POLQ, PRKCB, SLC22A5, SNAI1, TLR4 (100,000; 100) |
| Cis12 (Cisplatin) | Bladder | 5.40 | ATP7B, BCL2, BCL2L1, CDKN2C, ERCC2, FAAP24, GSTO1, MAP3K1, MAPK3, MT2A, NFKB1, NFKB2, POLD1, POLQ, PRKCB, SNAI1, TLR4, TP63 (10,000; 100) |
| Cis14 (Cisplatin) | Bladder | 5.16 | BARD1, BCL2, BCL2L1, CDKN2C, FAAP24, FEN1, FOS, GSTP1, MAP3K1, MAPK13, MAPK3, MSH2, NFKB1, POLD1, POLQ, PRKAA2, PRKCB, SLC22A5, SLC31A2, SNAI1, TWIST1 (10,000; 100) |
| Cis17 (Cisplatin) | Bladder | 5.10 | ATP7B, BCL2, BCL2L1, FEN1, GSTP1, MAP3K1, MAPK3, MT2A, NFKB1, PNKP, POLQ, PRKAA2, PRKCB, SLC31A2, TLR4, TP63 (100,000; 100) |
| Car1 (Carboplatin) | Ovarian | 4.22 | AKT1, EIF3K, ERCC1, GNGT1, GSR, MTHFR, NEDD4L, NLRP1, NRAS, RAF1, SGK1, TIGD1, TP53, VEGFB, VEGFC (100,000; 100) |
| Car9 (Carboplatin) | Ovarian | 4.32 | AKT1, ATP7B, EIF3I, ETS2, GNGT1, HRAS, KRAS, LIG3, MTHFR, MTR, NRAS, RAD50, SCN10A, TIGD1, TP53, VEGFB (10,000; 100) |
| Car51 (Carboplatin) | Ovarian | 4.34 | AKT1, EGF, EIF3I, ERCC1, ETS2, GNGT1, KRAS, MTHFR, MTR, NEDD4L, NLRP1, NRAS, RAD50, RAF1, SGK1, TIGD1, TP53, VEGFB, VEGFC (10,000; 100) |
| Car73 (Carboplatin) | Ovarian | 4.09 | AKT1, ATP7B, ETS2, GNGT1, HRAS, NLRP1, SCN10A, VEGFB (100,000; 1000) |
| Oxa1 (Oxaliplatin) | Colorectal | 5.10 | BRAF, FCGR2A, IGF1, MSH2, NAGK, NFE2L2, NQO1, PANK3, SLC47A1, SLCO1B1, UGT1A1 (10; 10) |
| Oxa21 (Oxaliplatin) | Colorectal | 5.10 | BRAF, IGF1, IGF1R, KLF3, MSH2, NAT2, NFE2L2, NQO1, PANK3, PRSS1, SIAE, SLC47A1, SLCO1B1, UGT1A1 (1000; 100) |
C—the box-constraint; σ—the kernel-scale (sigma)
Bolded gene signatures are those that exhibited the best overall performance in discriminating among TCGA patient outcomes
The TCGA bladder cancer dataset contained 19 patients treated with carboplatin (but not cisplatin), which enabled an evaluation of the specificity of cisplatin models relative to patients who were not treated with this drug. The cisplatin model that best predicted outcomes of carboplatin-treated patients with bladder cancer in TCGA was not Cis1 (the best-performing cisplatin model) but rather Cis12 at 2 years post-treatment (80% accurate for responding patients [N = 5]; 93% for recurrent patients [N = 14]). Cis12 contains 9 genes that are not present in Cis1, including ATP7B, a gene present in many of our carboplatin models. The presence of this gene may have a significant impact on the overall accuracy of Cis12 in determining the outcomes of the carboplatin-treated patients with bladder cancer. We also evaluated these 19 patients to determine the carboplatin-specific gene signatures, and the signature that best predicted the response of these patients (Car73) was 84% accurate for patients after 1 year of treatment (100% for responding patients [N = 11]; 62.5% accuracy for recurrent [N = 8]). Interestingly, Car73 shares the same ATP7B gene with Cis12. Two additional carboplatin gene signatures were tied for overall accuracy (84%; Car9 and Car51), but more successfully predicted non-responsive patients (87.5%; 82% accuracy for responding patients). AKT1, ETS2, GNGT1, and VEGFB were shared among these carboplatin gene signatures.
Distances from the hyperplane for all SVMs generated were determined for patients with a short recurrence time to evaluate the consistency of the predicted responses of TCGA patients with bladder cancer who were treated with cisplatin (<6 months, N = 10; Supplementary Figure S4). Despite showing similar levels of resistance to treatment, distances differed between patients. While these patients were expected to be indicated as highly cisplatin-resistant (hyperplane distance < 0), two patients (TCGA-XF-A9SU and TCGA-FJ-A871) were predicted to be sensitive by nearly all SVM gene signatures. Similar variations were also observed in patients with either a long recurrence time (>4 years) or no recurrence after 6 years (Supplementary Figure S5).
An aggregate, threshold-independent model was generated for each individual platin drug at different GI50 thresholds using ensemble ML, which involves the averaging of hyperplane distances for each model to generate a composite score for each TCGA patient tested (i.e., a composite gene signature). Hyperplane distances across all 70 cisplatin gene signatures were similar, with a mean score of −0.22 and a standard deviation of 3.5 hyperplane units (hu) across the set of patient data. The ensemble model classified disease-free patients diagnosed with bladder cancer with 59% accuracy and those with recurrent disease with 47% accuracy. Limiting ensemble averaging to only cisplatin gene signatures generated at a moderate GI50 threshold (ranging from 5.10 to 5.50) did not significantly improve accuracy (44% for disease-free patients and 66% for recurrent patients; Supplementary Table S7A). For carboplatin, ensemble ML did not produce significantly better predictions than random, regardless of the GI50 threshold interval selected (Supplementary Table S7B) or the similar mean hyperplane distances (−0.11 ± 3.9 hu). For oxaliplatin, the ensemble ML model (mean = −0.12 ± 2.7 hu) was most accurate after 1 year (60% accuracy for disease-free patients and 73% for recurrent patients; Supplementary Table S7C). Similar to cisplatin, limiting this analysis to oxaliplatin SVM gene signatures with moderate GI50 thresholds did not significantly increase accuracy.
K-fold cross-validation
The misclassification-based cisplatin, carboplatin, and oxaliplatin gene signatures were also evaluated with k-fold cross-validation of TCGA data from patients with bladder, ovarian, and colorectal cancer, respectively. This cross-validation approach was independent of the cell line data; namely, the genes and hyper-parameters of signatures were used, but the GE data used were exclusively derived from patients. Patients were evenly distributed in 5 groups with an equal (or near-equal) ratio of disease-free and recurrent patients. The majority of the cisplatin gene signatures showed an overall accuracy >50%. The cisplatin gene signature that performed best under the k-fold analysis (6-resistance level; BARD1, BCL2, BCL2L1, PRKAA2, PRKCA, PRKCB, and TWIST1) showed an overall accuracy of 71.2% (84.4% accurate for sensitive patients and 53.9% accurate for resistant patients). The accuracy of the carboplatin and oxaliplatin gene signatures did not exceed 60%. In general, treating the patient data as a held-out test set yielded higher performance estimates than training and evaluating the models on the patient data using k-fold cross-validation.
Predicting cisplatin responses in patients based on smoking history
Tobacco smoking is known as the risk factor with the greatest contribution to the development of bladder cancer.33 Patients with head and neck cancer who smoke while undergoing cisplatin and radiotherapy treatment have been shown to have a shorter overall survival rate.34 We therefore subdivided the patients based on their smoking history and tested the thresholded gene signatures (Supplementary Tables S8 and S9). When testing patients who were lifelong non-smokers, the prediction accuracy of Cis1 predicted all non-smoking patients who were recurrent after 18 months as cisplatin-resistant (N = 5). Prediction accuracy for disease-free patients was 57.1% (N = 14). Another gene signature (Cis18; Supplementary Table S8) performed equally well for non-smokers, and these two gene signatures shared the genes BCL2, BCL2L1, FAAP24, MAP3K1, MAPK13, MAPK3, and SLC31A2. The threshold-independent analysis predicted the disease-free status equally well, but recurrence was less accurate (66.7%). Notably, non-smokers comprised a small subset of the patients tested (N = 19). The threshold-independent prediction of recurrence in patients with a smoking history was 46% accurate (N = 13), while disease-free patients were correctly predicted at a rate of 58% (N = 19). Recurrence in these patients was best predicted by a gene signature built at the median GI50 threshold (Cis2). Accuracy improved for both disease-free (57.7–61.9%) and recurrent patients (76.0–78.6%) when excluding patients who quit smoking more than 15 years before the diagnosis. This SVM included the CFLAR and PRKAA2 genes, which were not present in the two gene signatures that performed well for non-smokers.
We gradually altered the expression of each signature gene until the misclassification was corrected to determine which genes in these gene signatures led to the discordant predictions of patient outcomes. Alterations in the expression of MAP3K1, MAPK3, SLC22A5, and SLC31A2 corrected discordant predictions of patient outcome. Alterations in BCL2L1 expression were more likely to correct the discordant predictions of Cis1 (4 of 5) than Cis2 (2 of 4). If the change exceeded ≥3 times the highest or lowest expression of that gene and the prediction remained unchanged between different patients, then the impact of that gene on the signature was considered to be limited. By these criteria, the expression of PRKAA2, NFKB1, NFKB2, and TWIST1 was not able to be altered to correct a discordant prediction.
Cytosine methylation levels of genes in cisplatin models
Tobacco smoking has a significant impact on cytosine methylation levels in the genome.35 CpG island methylation is associated with smoking in pack years in a subset of the TCGA patients with bladder urothelial carcinoma.28 We suspected that the level of methylation measured in the SVMs that performed best for smoking and non-smoking patients might differ and exert possible concomitant effects on GE. When ranking each gene from Cis1 by the highest methylation level and GE, 88 of 1080 patient–gene combinations showed the expected inverse correlation between methylation levels and GE (i.e., high methylation and low GE). Methylation and GE levels were more frequently inversely than directly correlated (i.e., high methylation and high GE; N = 17). However, the direct correlation was more common in patients with a recent smoking history (70.5%). This pattern was also observed for Cis2, which best predicted recurrence in smokers. In cases where methylation and GE were directly correlated, we propose that smoking may alter expression through other effects, e.g., mutagenic effects, rather than solely by epigenetic inactivation through methylation.
Discussion
Using GE signatures, we derived both GI50 threshold-dependent and -independent ML models that predict the chemotherapy responses to cisplatin, carboplatin, and oxaliplatin, respectively. The cisplatin gene signature Cis1 (Supplementary Table S6A) most accurately predicted the response of patients with bladder cancer after 18 months, and Car1 (Supplementary Table S6B) best predicted the response of patients with ovarian cancer after 4 years. Oxa1 (Supplementary Table S6C) more accurately predicted disease-free patients than patients with recurrent disease at the 1-year treatment threshold. The thresholds that best represented the time-to-recurrence differed between the platin drugs in patients with each cancer type. Cisplatin gene signatures exhibited noticeably improved performance when smoking history was taken into account.
The three platin drugs produced distinctly different gene signatures. The composition of the initial gene sets exhibited some overlap between platin drugs (N = 67 between any two platins), but the expression of only ATP7B, BCL2, and MSH2 was correlated with the GI50 values of more than one platin drug. The expression of BCL2L1, GSTP1, MAP3K1, MAPK3, MT1A, and MT2 was correlated with cisplatin GI50 values, but not with carboplatin and/or oxaliplatin GI50. Similarly, the carboplatin GI50 was correlated with AKT1, EGF, ERCC1, KRAS, LIG3, MTHFR, MTR, RAD50, and TP53, while oxaliplatin GI50 was correlated with ATM, BCL2, CLCN6, ERCC2, ERCC6, and UGT1A1. Despite the close similarity between cisplatin and carboplatin GI50 responses (see Fig. 4), MFA only related the expression of one gene (ATP7B) to GI50 levels of both drugs. BCL2 and MSH2 correlated with both the cisplatin and oxaliplatin GI50 values (BCL2 levels did not correlate with carboplatin GI50). The increase in misclassification caused by the elimination of MSH2 from any gene signature in which it was present was significant; for example, misclassification of Cis14 and Oxa21 (Table 1) was increased by 28.2% and 19.1%, respectively (Supplementary Table S2A and S2C). These differences may reflect the spectrum of activity, sensitivity, and toxicity of these signature genes.22–24,36,37
Our previous validation using patient expression and CN data for other chemotherapy drugs on other datasets6,27 exhibited better performance than what is reported in this study. We investigated the possibility that disease and molecular heterogeneity in platin-treated patients may have affected the accuracy of our results. Model predictions were re-evaluated after stratifying clinical features, such as time-to-disease recurrence, cancer stage, and metastatic lymph node count. Patients with advanced stage breast cancer (stage III and IV) were analyzed separately from patients with earlier stage diagnoses (stage I and II). The cisplatin gene signature Cis1 performed best on stage IV patients (overall accuracy of 72.4% at a 2-year recurrence threshold), while Oxa1 similarly performed best in predicting late stage cancers (74.5% accurate for stage III and 71.4% accurate for stage IV at a 2-year recurrence threshold). Cis5 was also more accurate for patients with later stage cancers (72.4% overall accuracy at 18 months). The accuracies of gene signatures were similar across all stages (e.g., Car1 ranged from 58 to 74%). Cisplatin-treated patients with bladder cancer and oxaliplatin-treated patients with CRC in TCGA were also stratified by lymph node status (N0, N1, and N2 [the dataset of patients with bladder cancer only included two N3 patients, which were combined with N2 patients in the analysis; N3 was not presented in patients with CRC]). In TCGA patients with bladder cancer, Cis1 exhibited ~60% accuracy across all categories; however, it performed better in sensitive N0 and N1 patients than N2. Cis2 was less accurate for N2 patients than for N0 and N1 patients. Sensitive N2 patients were more likely to be misclassified (<40%) than relapsed N2 patients. In TCGA patients with CRC, Oxa1 was 88% accurate in identifying N2 patients (95% accurate for sensitive N2 patients [n = 19], and 67% accurate for relapsed N2 patients [n = 6]). Oxaliplatin gene signatures were less accurate for N1 patients than N0 and N2 patients. Thus, heterogeneity in the disease stage and metastatic phenotypes adversely confounds the overall accuracies of our predictions.
Gene signature models derived from cell lines and tested on patients differed in their respective outcome measures. The exact GI50 cell line threshold that best predicts patient outcomes is not known, and different groups use different methods to establish thresholds for GI50 values.38,39 Therefore, we developed ML models for platin drugs that predict drug responses without relying on arbitrarily set GI50 thresholds. For cisplatin, SVM ensemble averaging generated at different resistance thresholds showed a small increase in accuracy compared with most gene signatures, better representing the sensitive, disease-free class (59% accuracy). Interestingly, ensemble averaging of only the gene signatures built using a moderate GI50 threshold yielded results that better represented the resistance class. This result more closely matched the accuracy of Cis1, and may be due to the greater overall impact of Cis1 on the ensemble prediction. When limiting ensemble averaging to only those gene signatures with the highest area under the curve (AUC) at each resistance threshold, differences in predictions were negligible. Ensemble ML potentially avoids problems with poor performance and overfitting by combining gene signatures that individually perform slightly better than chance.40
Reconciliation of gene signatures without features known to be related to chemoresistance with tumor biology is challenging. Our thresholding approach may reveal potentially important genes and pathways associated with platin resistance. A preferable method would be to explore pathways related to signature genes to improve accuracy, identify potential targets for further study of chemoresistance, and expand the model parameters by considering alternate states other than those captured in the original signature.41 Signatures for resistance may be useful for developing targeted interventions to re-sensitize tumors. For example, the mismatch repair (MMR) gene MSH2 is commonly present in gene signatures at high resistance levels for oxaliplatin, which is of interest because MMR deficiency has been shown to predict oxaliplatin resistance.37 Indeed, MLH1-, MSH2-, and MSH6-deficient cells are more susceptible to oxaliplatin, although an MMR deficiency is associated with cisplatin resistance.36 The autoimmune disease-associated gene SIAE, which exhibits a strong negative correlation with the oxaliplatin response in patients with advanced CRC,42 was selected in the majority of thresholded oxaliplatin gene signatures (Supplementary Table S2C). The gene BCL2, which was commonly selected for cisplatin (Fig. 5a), was rarely selected for oxaliplatin (Fig. 5c). At the highest levels of resistance to cisplatin, gene signatures were enriched for genes belonging to DNA repair, anti-oxidant response, and apoptotic pathways, as well as drug transporters (Fig. 5a). These gene pathways are known to be involved in cisplatin resistance43,44 and these specific genes may be explored in subsequent studies designed to identify their contributions to the chemotherapy response in a biochemical context.
Log-loss evaluates the accuracy of a classifier by penalizing erroneous classifications and is relevant in cases where data are imbalanced and/or have an unequally distributed error cost. We assessed whether ML gene signatures based on log-loss minimization improved the accuracy of predicting patient responses (Supplementary Table S3) and compared them to gene signatures generated by minimizing cell line misclassification. When gene signatures generated from both methods were highly similar (generated at the same GI50 threshold, consisting of a similar number of genes and ≥80% shared genes), the prediction accuracy of outcomes of TCGA patients with cancer was nearly indistinguishable, as accuracy can vary over different relapse thresholds. When significant differences in predictions were observed, the misclassification-based gene signatures were generally more accurate (Cis1, Cis17, and the “12-Resistant” carboplatin gene signature were +8.3%, +5.6%, and +3.9% more accurate than the log-loss gene signature, respectively). Oxaliplatin gene signatures were dissimilar across all GI50 thresholds, as the log-loss minimized ML gene signatures often contained greater numbers of genes than the misclassification-based gene signatures. Many of these larger gene signatures were less accurate in predicting patient outcomes than gene signatures that minimized misclassification rates, consistent with the observation that this evaluation and model selection method is more prone to overfitting. This pattern was also noted for gene signatures generated at extreme GI50 thresholds for all three platin drugs, in which the response was, by definition, somewhat imbalanced.
The prediction of responses to combination chemotherapy with the gene signatures described here may be feasible. Not included in the present analysis were signatures for methotrexate, vinblastine, and doxorubicin, which comprise the MVAC cocktail used to treat bladder cancer. This lack of analysis was primarily due to the small number of patients treated with this drug combination in the TCGA bladder cancer dataset (N = 11). Individual signatures for several of these drugs have been derived and analyzed using the patient data from Molecular Taxonomy of Breast Cancer International Consortium (METABRIC).27 A reasonable approach to predicting responses to combination chemotherapy would be to first determine the probability of sensitivity or resistance to individual drugs, after accounting for the misclassification rate for each (defined as d1, …, dk). The ML classifiers output these probabilities, analogous to their misclassification rates in a set of patients treated identically. If the model predicts that the patient is sensitive to drug d1 with 90% probability, sensitive to drug d2 with 5% probability, and the errors are independent, then the probability of sensitivity to the combination is 1 − (1 − 0.9) * (1 − 0.05) = 90.5%, and the probability of resistance is 9.5%, assuming no synergistic effects between drugs. If interactions or dependence among errors are suspected, the combined probability of resistance to the pair d12 could be estimated based on the features that are shared by the signatures of both drugs. The probability of sensitivity to the combination would then be: 1 − (1 − d12) * (1 − d3) *…* (1 − dk).
The predictive accuracy for the same gene signature might be able to provide good differentiation between the two datasets. Cis3 (Supplementary Table S6A) had an AUC of 0.64 when validated against TCGA patients with bladder cancer. However, the AUC was lower when applied to the dataset reported by Als et al.31 (AUC = 0.18). Patient metadata in the latter study only indicated patient survival times, while we based the expected TCGA patient outcomes on time to disease recurrence. As the basis of our expected outcomes differed between datasets, these differences may serve as a confounding factor when determining the accuracy of gene signatures. The datasets also differed in how expression was measured (microarray vs. RNA-seq). The relevance of gene signatures based on training and testing data from different platforms can affect the accuracy of validation, which might not be improved by data normalization. In the present study, datasets were subjected to z-score normalization. Other techniques to correct for some of these effects have been described and could be applied in subsequent studies.45
In summary, we describe GI50 or IC50 threshold-independent ML gene signatures that predict the chemotherapy responses of patients with cancer to platin agents. Ensemble ML produced combined signatures that were more accurate than most individual gene signatures generated with different thresholds. Genes associated with cisplatin response included genes that exacerbate resistance in patients with a history of smoking. The methodology described here should be adaptable to other drugs and cancer types. With a range of gene signatures for multiple drugs, the efficacy of treatment might be improved by tailoring treatment to a patient’s specific tumor biology and reduce treatment duration by limiting the number of different therapeutic regimens prescribed before achieving a successful response.46
Materials and methods
Data and preprocessing
Cell-line datasets
Microarray GE and data obtained from breast cancer cell lines were used to train ML-based gene signatures of drug responses based on respective growth or target inhibition data (GI50 or IC50). Cell lines were treated with either cisplatin (N = 39), carboplatin (N = 46), or oxaliplatin (N = 47).13 Bladder cancer cell line GE and IC50 measurements for cisplatin were obtained from CancerRxGene (N = 17).46 However, all models (gene signatures) used to evaluate patient data were trained on breast cancer cell line data because the number of bladder cancer cell lines was insufficient to produce accurate signatures.
Datasets from patients with cancer
RNA-seq GE and survival measurements were downloaded from TCGA for patients with bladder urothelial carcinoma (N = 72 patients treated with cisplatin),28 ovarian epithelial tumors (N = 410 treated with carboplatin),29 and colorectal adenocarcinoma (N = 99 treated with oxaliplatin).30 GE values for cisplatin-treated patients with cell carcinoma of the urothelium (N = 30)31 and oxaliplatin-treated patients with CRC (N = 83)32 were obtained from the Gene Expression Omnibus. Clinical metadata and GE for TCGA patients were obtained from Genomic Data Commons (https://gdc.cancer.gov/), while methylation HM450 (Illumina) data for these patients were downloaded from cBioPortal.47
Development and pre-processing of biochemically-inspired gene sets
Initial gene sets used to develop signatures for each drug were identified from previous publications (see Supplementary References, Section B) and databases, such as PharmGKB and DrugBank.48,49 The evidence supporting each gene contained in the final signatures is independent scientific evidence that the genes selected are not the result of spurious associations. The final gene sets were chosen using MFA with the breast cancer cell line data to analyze interactions between GE, CN, and GI50 data for the drug of interest.50 Genes whose GE and/or CN showed a direct or inverse correlation with GI50 were selected for SVM training. Because the number of genes related to the GI50 for oxaliplatin exceeded the number of cell lines available for training, we limited the input for the oxaliplatin ML model to those genes whose GE were related to the GI50. Similarly, the number of correlated genes in cisplatin-treated cells exceeded the number of cell lines. For cisplatin, genes whose expression correlated with the GI50 were eliminated if they showed no or little expression in TCGA patients with bladder cancer (i.e., RNA-seq counts by Expectation Maximization [RSEM] were <5.0 for the majority of individuals). This approach reduced the overall number of genes for the SVM analysis, and thus helped to avoid a data to size sample imbalance. For cisplatin, the MFA was repeated using IC50 values for 17 bladder cancer cell lines; however, the available CN data for these genes generally showed a lack of variation in the cell lines. Instead, the available IC50 values for three other cancer drugs (doxorubicin, methotrexate, and vinblastine) were compared with the IC50 of cisplatin using MFA.
The direct application of an SVM model to patient data without a normalization approach is imprecise when training and testing data are not obtained using similar methodology (i.e., different microarray platforms). Expression levels were normalized by conversion to z-scores using MATLAB to compare the cell line GE microarray data and the patient RNA-seq GE datasets.51 Although log2 intensity values from microarray data were not available for TCGA samples, RNA-seq-based GE data and log2 intensities from microarray data are highly correlated.52
Machine learning
SVMs were trained with breast cancer cell line GE datasets13 with the Statistics Toolbox in MATLAB51 using a method similar to the procedure reported by Dorman et al.6 Rather than employing a linear kernel, we used a Gaussian kernel function (fitcsvm) and then tested the data with leave-one-out cross-validation (using the options “crossval” and “leaveout”). A greedy BFS algorithm was used to improve the classification accuracy.53 BFS leaves out individual genes from the initial MFA-qualified gene set and then trains a cross-validated Gaussian kernel SVM on the training samples, removing the gene with the highest misclassification rate. The procedure is repeated until all genes have been evaluated. The gene subset with the lowest misclassification rate6 or log-loss statistic25 based on cross-validation is selected as the gene signature for subsequent testing with patient GE and clinical data. K-fold cross-validation of the misclassification-based gene signatures was performed using MATLAB software, as described in Zhao et al.25
SVMs minimized using the log-loss classification function were also generated with both the software described in Zhao et al. (uses a multiclass compatible “fitcecoc” function)25 and with a modified version of the software described above (using “fitSVMPosterior” to compute posterior probabilities). Computed probabilities differed between “fitSVMPosterior” and “fitcecoc” (range: 0.02–0.04); thus, the resulting gene signatures will differ between the two programs. When given unbalanced data (e.g., lower resistance thresholds), “fitSVMPosterior” will warn that some classes are not represented, and thus those folds will not predict the labels for those missing classes. The log-loss gene signatures described in this manuscript were generated with the multiclass compatible “fitcecoc” function software.25
Derivation of gene signatures for different drug resistance thresholds
We have previously set a conventional GI50 threshold that distinguishes sensitivity from resistance at the median of the range of drug concentrations that inhibited cell growth by 50%.6 We hypothesized that different gene signatures would be derived for different levels of drug resistance by varying this threshold. ML experiments for classifying resistance or sensitivity at GI50 values generated a series of optimized Gaussian SVM gene signatures whose performances were assessed with patient expression data for each signature. A heat map illustrating the frequencies of genes appearing in these gene signatures was created with the R language hist2d function.
A composite gene signature was created by ensemble averaging of all gene signatures generated at each resistance threshold. Ensemble averaging combines signatures by averaging the weighted accuracy of a set of related models.40 The decision function for the ensemble classifier is the mean of the decision function scores of the component classifiers, weighted by the AUC.
Significance of cell line-derived gene signatures
The significance of the derived SVMs (regardless of whether the observed performance of the gene signatures could have arisen by chance) was first assessed using a permutation analysis with randomized cell line labels and with random sets of genes, as described previously.6 Using the median cisplatin GI50 as the resistance threshold, 10,000 gene signatures based on random gene selection (15 genes) had higher rates of misclassification than the best median SVM gene signatures (2 signatures with 7.7% misclassification). Cisplatin, carboplatin, and oxaliplatin GE data for random cell line label combinations (n = 10,000) generated only 8, 1, and 1 signatures, respectively, with lower error rates than the best biochemically-inspired signatures. When minimizing for log-loss (rather than misclassification), the random gene analysis (10,000 iterations; median cisplatin GI50 threshold) only resulted in gene signatures with a higher log-loss than the signature generated with the initial cisplatin gene set. The log-loss-based random label analysis (n = 2000 combinations) resulted in 3.4% of random label gene signatures with a lower log-loss than the cisplatin signature at the same GI50 threshold (5.27). This finding was not entirely surprising since it depends on the GI50 threshold used for labeling. The differences between GI50 values for cell lines close to the median GI50 used in this analysis were almost negligible (e.g., 5.11 vs. 5.12) and likely within the measurement error for these values.
Regarding the specificity of the cisplatin gene signatures, the best-performing cisplatin gene signatures (Cis1 and Cis2) were used to evaluate participants who were treated with other drugs (using an 18-month post-treatment threshold). Among these patients, 36.5% of patients who were disease-free were predicted accurately with the Cis1 signature (N = 178; 22% less accurate than platin-treated patients), and 62.9% of patients with recurrent disease were predicted accurately (N = 70; 8.1% less accurate). Cis2 was 43.8% accurate at predicting disease-free non-platin-treated patients (N = 178; 12.3% lower accuracy) and 60.0% accurate at predicting patients who relapsed (N = 70; 2.9% less accurate). GE changes in patients treated with platin drugs were better modeled by cancer cell-line-based predictors than in patients receiving other drug treatments.
Supplementary information
Acknowledgements
Katherina Baranova contributed to the initial cisplatin gene signatures and Dimo Angelov developed automated feature selection. The authors thank Murray Junop for commenting on the manuscript. P.K.R. is supported by NSERC (RGPIN-2015-06290), Canadian Foundation for Innovation, Canada Research Chairs, and CytoGnomix. Compute Canada and Shared Hierarchical Academic Research Computing Network (SHARCNET) provided high-performance computing and storage facilities.
Author contributions
P.K.R. and D.J.L. designed the methodology. E.J.M. and J.Z.L.Z. performed the analyses. E.J.M. and P.K.R. wrote the manuscript.
Competing interests
P.K.R. cofounded CytoGnomix, Inc., which hosts the interactive resource described in this study for prediction of responses to chemotherapy agents. The other authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41392-018-0034-5) contains supplementary material, which is available to authorized users.
References
- 1.Cardoso F, et al. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012;23:vii11–vii19. doi: 10.1093/annonc/mds232. [DOI] [PubMed] [Google Scholar]
- 2.Oostendorp LJ, Stalmeier PF, Donders ART, van der Graaf WT, Ottevanger PB. Efficacy and safety of palliative chemotherapy for patients with advanced breast cancer pretreated with anthracyclines and taxanes: a systematic review. Lancet Oncol. 2011;12:1053–1061. doi: 10.1016/S1470-2045(11)70045-6. [DOI] [PubMed] [Google Scholar]
- 3.Alfarouk KO, et al. Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell Int. 2015;15:71. doi: 10.1186/s12935-015-0221-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gąsowska-Bodnar A, et al. Survivin expression as a prognostic factor in patients with epithelial ovarian cancer or primary peritoneal cancer treated with neoadjuvant chemotherapy. Int. J. Gynecol. Cancer. 2014;24:687–696. doi: 10.1097/IGC.0000000000000108. [DOI] [PubMed] [Google Scholar]
- 5.Hatzis C, et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA. 2011;305:1873–1881. doi: 10.1001/jama.2011.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorman SN, et al. Genomic signatures for paclitaxel and gemcitabine resistance in breast cancer derived by machine learning. Mol. Oncol. 2016;10:85–100. doi: 10.1016/j.molonc.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang S, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66:8847–8857. doi: 10.1158/0008-5472.CAN-06-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poisson, L. M. et al. A metabolomic approach to identifying platinum resistance in ovarian cancer. J. Ovarian Res. 8, 13 (2015). [DOI] [PMC free article] [PubMed]
- 9.Cadoná FC, et al. Guaraná a caffeine-rich food increases oxaliplatin sensitivity of colorectal HT-29 cells by apoptosis pathway modulation. Anticancer Agents Med. Chem. 2016;16:1055–1065. doi: 10.2174/1871520616666151217121138. [DOI] [PubMed] [Google Scholar]
- 10.Kasparkova J, Vojtiskova M, Natile G, Brabec V. Unique properties of DNA interstrand cross-links of antitumor oxaliplatin and the effect of chirality of the carrier ligand. Chem. – Eur. J. 2008;14:1330–1341. doi: 10.1002/chem.200701352. [DOI] [PubMed] [Google Scholar]
- 11.Woynarowski JM, et al. Oxaliplatin-induced damage of cellular DNA. Mol. Pharmacol. 2000;58:920–927. doi: 10.1124/mol.58.5.920. [DOI] [PubMed] [Google Scholar]
- 12.Tashiro T, Kawada Y, Sakurai Y, Kidani Y. Antitumor activity of a new platinum complex, oxalato (trans-l-1,2-diaminocyclohexane)platinum (II): new experimental data. Biomed. Pharmacother. 1989;43:251–260. doi: 10.1016/0753-3322(89)90004-8. [DOI] [PubMed] [Google Scholar]
- 13.Daemen A, et al. Modeling precision treatment of breast cancer. Genome Biol. 2013;14:R110. doi: 10.1186/gb-2013-14-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuan Y, et al. Identification of the biomarkers for the prediction of efficacy in first-line chemotherapy of metastatic colorectal cancer patients using SELDI-TOF-MS and artificial neural networks. Hepatogastroenterology. 2012;59:2461–2465. doi: 10.5754/hge12127. [DOI] [PubMed] [Google Scholar]
- 15.L’Espérance S, Bachvarova M, Tetu B, Mes-Masson AM, Bachvarov D. Global gene expression analysis of early response to chemotherapy treatment in ovarian cancer spheroids. BMC Genomics. 2008;9:99. doi: 10.1186/1471-2164-9-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nickerson, M. L. et al. Molecular analysis of urothelial cancer cell lines for modeling tumor biology and drug response. Oncogene10.1038/onc.2016.172 (2016). [DOI] [PMC free article] [PubMed]
- 17.Yuryev A. Gene expression profiling for targeted cancer treatment. Expert Opin. Drug Discov. 2015;10:91–99. doi: 10.1517/17460441.2015.971007. [DOI] [PubMed] [Google Scholar]
- 18.Sataloff DM, et al. Pathologic response to induction chemotherapy in locally advanced carcinoma of the breast: a determinant of outcome. J. Am. Coll. Surg. 1995;180:297–306. [PubMed] [Google Scholar]
- 19.Ogston KN, et al. A new histological grading system to assess response of breast cancers to primary chemotherapy: prognostic significance and survival. Breast. 2003;12:320–327. doi: 10.1016/S0960-9776(03)00106-1. [DOI] [PubMed] [Google Scholar]
- 20.Earl J, et al. The UBC-40 Urothelial Bladder Cancer cell line index: a genomic resource for functional studies. BMC Genomics. 2015;16:403. doi: 10.1186/s12864-015-1450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang W, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955–D961. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rixe O, et al. Oxaliplatin, tetraplatin, cisplatin, and carboplatin: spectrum of activity in drug-resistant cell lines and in the cell lines of the national cancer institute’s anticancer drug screen panel. Biochem. Pharmacol. 1996;52:1855–1865. doi: 10.1016/S0006-2952(97)81490-6. [DOI] [PubMed] [Google Scholar]
- 23.Mehmood RK. Review of cisplatin and oxaliplatin in current immunogenic and monoclonal antibody treatments. Oncol. Rev. 2014;8:256. doi: 10.4081/oncol.2014.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kweekel DM, Gelderblom H, Guchelaar HJ. Pharmacology of oxaliplatin and the use of pharmacogenomics to individualize therapy. Cancer Treat. Rev. 2005;31:90–105. doi: 10.1016/j.ctrv.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 25.Zhao JZL, Mucaki EJ, Rogan PK. Predicting ionizing radiation exposure using biochemically-inspired genomic machine learning. F1000Research. 2018;7:233. doi: 10.12688/f1000research.14048.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tembe V, et al. The BARD1 BRCT domain contributes to p53 binding, cytoplasmic and mitochondrial localization, and apoptotic function. Cell Signal. 2015;27:1763–1771. doi: 10.1016/j.cellsig.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 27.Mucaki EJ, et al. Predicting outcomes of hormone and chemotherapy in the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) study by biochemically-inspired machine learning. F1000Research. 2017;5:2124. doi: 10.12688/f1000research.9417.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robertson AG, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017;171:540–556. doi: 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Als AB, et al. Emmprin and survivin predict response and survival following cisplatin-containing chemotherapy in patients with advanced bladder cancer. Clin. Cancer Res. 2007;13:4407–4414. doi: 10.1158/1078-0432.CCR-07-0109. [DOI] [PubMed] [Google Scholar]
- 32.Tsuji S, et al. Potential responders to FOLFOX therapy for colorectal cancer by Random Forests analysis. Br. J. Cancer. 2012;106:126–132. doi: 10.1038/bjc.2011.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freedman ND, Silverman DT, Hollenbeck AR, Schatzkin A, Abnet CC. Association between smoking and risk of bladder cancer among men and women. JAMA. 2011;306:737–745. doi: 10.1001/jama.2011.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fortin A, Wang CS, Vigneault E. Influence of smoking and alcohol drinking behaviors on treatment outcomes of patients with squamous cell carcinomas of the head and neck. Int. J. Radiat. Oncol. Biol. Phys. 2009;74:1062–1069. doi: 10.1016/j.ijrobp.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 35.Joehanes, R. et al. Epigenetic signatures of cigarette smoking. Circ. Cardiovasc. Genet. 10.1161/CIRCGENETICS.116.001506 (2016). [DOI] [PMC free article] [PubMed]
- 36.Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E. Cellular and molecular pharmacology of Oxaliplatin1. Mol. Cancer Ther. 2002;1:227–235. [PubMed] [Google Scholar]
- 37.Alex, A. K. et al. Response to chemotherapy and prognosis in metastatic colorectal cancer with DNA deficient mismatch repair. Clin. Colorectal Cancer10.1016/j.clcc.2016.11.001 (2016). [DOI] [PubMed]
- 38.Sos ML, et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J. Clin. Invest. 2009;119:1727–1740. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laderas, T. G., Heiser, L. M. & Sönmez, K. A network-based model of oncogenic collaboration for prediction of drug sensitivity. Front. Genet. 6, 341 (2015). [DOI] [PMC free article] [PubMed]
- 40.Clemen RT. Combining forecasts: a review and annotated bibliography. Int. J. Forecast. 1989;5:559–583. doi: 10.1016/0169-2070(89)90012-5. [DOI] [Google Scholar]
- 41.Airley, R. Cancer Chemotherapy (Wiley-Blackwell, Hoboken, New Jersey 2009).
- 42.Li XX, et al. RNA-seq identifies determinants of oxaliplatin sensitivity in colorectal cancer cell lines. Int. J. Clin. Exp. Pathol. 2014;7:3763–3770. [PMC free article] [PubMed] [Google Scholar]
- 43.Borst P, Rottenberg S, Jonkers J. How do real tumors become resistant to cisplatin? Cell Cycle. 2008;7:1353–1359. doi: 10.4161/cc.7.10.5930. [DOI] [PubMed] [Google Scholar]
- 44.Wernyj R, Morin P. Molecular mechanisms of platinum resistance: still searching for the Achilles' heel. Drug Resist. Updat. 2004;7:227–232. doi: 10.1016/j.drup.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 45.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 46.Akamatsu N, Nakajima H, Ono M, Miura Y. Increase in acetyl CoA synthetase activity after phenobarbital treatment. Biochem. Pharmacol. 1975;24:1725–1727. doi: 10.1016/0006-2952(75)90013-1. [DOI] [PubMed] [Google Scholar]
- 47.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whirl-Carrillo M, et al. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012;92:414–417. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Law V, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abdi H, Williams LJ. Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat. 2010;2:433–459. doi: 10.1002/wics.101. [DOI] [Google Scholar]
- 51.MATLAB and Statistics Toolbox Release 2012b (The MathWorks, Inc., Natick, Massachusetts, United States).
- 52.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bermingham ML, et al. Application of high-dimensional feature selection: evaluation for genomic prediction in man. Sci. Rep. 2015;5:10312. doi: 10.1038/srep10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.