

Abstract
The A1762T and G1764A mutations in the basal core promoter (BCP) region and the G1896A mutation in the precore (PC) region of hepatitis B virus (HBV) genome are found commonly in HBeAg-negative patients. Experiments in vitro suggest that BCP and PC mutation reduce and abolish HBeAg expression, respectively. In the present study, the prevalence of the BCP and PC mutations were determined in 207 patients with HBeAg positive chronic hepatitis B from China and correlated with the titers of serum HBeAg. None of the patients received antiviral therapy. The HBV genotype was determined by direct sequencing of the HBsAg gene. The BCP and PC mutations were detected by the polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) and confirmed by DNA sequencing. The HBeAg titer was measured by the microparticle enzyme immunoassay. Fifty-one of the 207 patients (24.6%) were infected with genotype B and the remainder with genotype C. The BCP mutations were detected in 103 patients (50%) while the PC mutation was present in 43 (20.8%). Thirteen patients (6.3%) harbored both BCP and PC mutations. No significant difference in the titers of HBeAg was found between patients infected with the two HBV genotypes, but the presence of either the BCP or PC mutation was associated with reduced HBeAg titer (P < 0.05). The presence of both the BCP and PC mutations was accompanied by even lower HBeAg titer (P < 0.05). These findings confirm that in patients with HBeAg, the BCP and PC mutations reduced the expression of HBeAg.
Keywords: chronic hepatitis B, core promoter mutations, genotypes, HBeAg titer, precore mutation, restriction fragment length polymorphism
INTRODUCTION
Over 350 million people worldwide are infected chronically with hepatitis B virus (HBV), of whom 250 millions reside in Asia [Magnius and Norder, 1995; Kao and Chen, 2002]. HBV is classified into eight genotypes designated as A–H [Magnius and Norder, 1995; Chu et al., 2002a]. The most prevalent genotypes in Asia are hepatitis B virus genotype B and C. Infection with genotype C is associated with HBeAg seroconversion (loss of HBeAg and presence of the corresponding antibody, anti-HBe) a decade later than genotype B infection [Chu et al., 2002b; Yuen et al., 2003]. Such seroconversion leads frequently to the selection of mutations in the precore (PC) region [Carman et al., 1989; Okamoto et al., 1990; Tong et al., 1990] and/or basal core promoter (BCP) region of the HBV genome [Okamoto et al., 1994]. The most common PC mutation is a G to A transition at nucleotide (nt) 1896 (A1896) that creates a premature stop codon and abolishes HBeAg translation [Carman et al., 1989; Blum et al., 1991; Tong et al., 1991]. The most common BCP mutations are A to T at nt 1762 and G to A at nt 1764 (T1762/A1764) [Okamoto et al., 1994; Kidd-Ljunggren et al., 1997; Chan et al., 1999; Honda et al., 1999; Lindh et al., 1999]. The double mutation decreases the transcription of pre-C mRNA, the mRNA for HBeAg, and consequently reduces HBeAg expression [Buckwold et al., 1996; Moriyama et al., 1996; Parekh et al., 2003]. Previous studies in Japanese and South African patients revealed an association of BCP mutations in HBV genome with reduced titers of HBeAg [Takahashi et al., 1995; Kurosaki et al., 1996; Baptista et al., 1999]. In the present study, prevalence of the PC and BCP mutations was determined among HBeAg positive Chinese chronic hepatitis B patients and sought for their possible correlation with HBeAg titers.
PATIENTS AND METHODS
Patients
Between July and October 2003, 207 Chinese patients with HBeAg-positive chronic hepatitis B were recruited from six hospitals in Shanghai, Beijing, Guangzhou, and Changchun. All the patients met the following criteria: (1) 18–70 years of age; (2) HBsAg positive for at least 6 months before entry; (3) HBeAg positive and HBV DNA titer > 5 log10 copies/ml; (4) ALT levels within 2–10 times the upper limit of normal (ULN); (5) lack of evidence for auto-immune hepatitis or markers of infection with hepatitis C virus, hepatitis D virus, or HIV. Patients who had received any type of antiviral therapy were also excluded. The earliest available serum samples before Peg-interferon clinical trial were used for this study. The serum samples were stored at −20°C until use. The study was conducted in accordance with the ethics principles of the Declaration of Helsinki. Written informed consent was obtained from each participant.
Serological Assays
The presence of HBsAg, HBeAg, anti-HBe, anti-HCV, and anti-HDV antibodies was determined using commercial AxSYM MEI kits (Abbott Laboratories, North Chicago, IL). The HBeAg values were measured using the microparticle enzyme immunoassay and expressed as signal/cutoff. The HBV DNA level was quantified using a real-time fluorescence quantitative commercial kit (Shenzhen PG Biotech, Shenzhen, China), which has a detection limit of 500 HBV copies/ml. Assay calibration using the standard HBV DNA was supplied by the National Institute for the Control of Pharmaceutical and Biological Products, China.
HBV DNA Extraction, Amplification, and Genotype Detection
One hundred microliters of serum were mixed with equal volume of lysis buffer [25 mM Tris–HCl (pH 8.3), 10 mM EDTA, 1%SDS] and 500 µg/ml fresh proteinase K (TaKaRa Dalian, Dalian, China) and incubated at 65°C for 3 hr. DNA was extracted with phenol/chloroform, precipitated with ethanol, and resuspended in 20 µl distilled water. Genotyping was performed using a 1.3-kb fragment of the HBV genome covering the entire HBsAg gene (nt 2,816–886), which was amplified by the polymerase chain reaction (PCR). Primers S1 and AS (Table I), which occupied conserved regions, were designed according to Naito et al. [2001] and Kirschberg et al. [2004]. The PCR was performed in a volume of 50 ml containing 1 PCR buffer (10 mM Tris–HCl, pH 8.3, 50 mM KCl), 1.5 mM MgCl2, 0.2 mM each of dNTPs (TaKaRa Bio, Inc.), 1 µM of primers, 4.0 U of Taq DNA polymerase (TaKaRa Dalian) and 5 µl of template DNA. Thermal cycling conditions consisted of 95°C for 10 min followed by 30 cycles of 94°C for 30 sec, 55°C for 45 sec, and 72°C for 1 min, and a final extension step at 72°C for 5 min. Precautions were taken to avoid contamination. Serum samples from individuals without HBV infection were used as negative controls during PCR. The PCR products were subjected to direct sequencing. HBV genotype was determined according to phylogenetic analysis [Simmonds and Midgley, 2005] using the Vector NTI 9.0 TreeView software [Zhou et al., 2004]. Since sequences from each genotype were monophyletic, only the most recent common ancestor is shown for non-gibbon genotypes. The tree was constructed by neighbor-joining using Jukes-Cantor corrected distances in the MEGA2 package [Tamura et al., 2001], using 1000 bootstrap replicates.
TABLE I.
Primers Used in This Study and Their Locations on the HBV Genome
| Primer | Sequence (5′−3′) | Location | Polarity | Purpose |
|---|---|---|---|---|
| S1 | GTC ACC ATA TTC TTG GGA AC | 2,816 –2,835 | Forward | S gene PCR for genotyping |
| AS | CAT ATC CCA TGA AGT TAA GG | 886 –867 | Reverse | S gene PCR for genotyping |
| B1 | CAA GGT CTT GCA TAA GA GGA CT | 1,643 –1,664 | Forward | 1st PCR for BCP and PC mutations; 2nd PCR for BCP mutations |
| C1 | CCC CAC CTT ATG AGT CCA AG | 2,477 –2,458 | Reverse | 1st PCR for BCP and PC mutations |
| C-Xag | CCT CCT AGC TGT GCC TTG GCC TGC TTT | 1,869 –1,895 | Forward | 2nd PCR for PC mutation |
| C2 | TGA GAG CAG TAT GGT GAG GTG AAC AAT G | 2,066 –2,039 | Reverse | 2nd PCR for PC mutation |
| B-Bcl | CTA CAG CCT CCT AGT ACA ATG A | 1,786 –1,765 | Reverse | 2nd PCR for BCP mutations |
Boldface: mismatches in the C-Xag and B-Bcl primers to introduce the XagI and BclI sites for PC and BCP mutants, respectively.
Detection of Precore and Core Promoter Mutations by RFLP and DNA Sequencing
Nested and semi-nested PCR were used to detect the A1896 mutation in the precore region and T1762/A1764 double mutation in the core promoter region, respectively. First, a fragment spanning nt 1,643–2,477 was amplified using primers B1 and C1 (Table I) under the following conditions: 94°C for 5 min; 30 cycles of 94°C for 1 min, 56°C for 1 min, and 72°C for 1 min; and finally 72°C for 10 min. Next, two parallel PCR reactions were carried out to detect the BCP and PC mutations using restriction fragment length polymorphism (RFLP). Although the T1762/A1764 double mutation does not create a novel restriction site, introduction of artificial A1767T mutation into the antisense primer B-Bc1 produces a BclI cleavage site (TGATCA) in conjunction with the double mutation [Amini-Bavil-Olyaee et al., 2005]. Similarly, due to the artificial G1888C/T1889C/G1890T mutations in the sense primer C-Xag, an XagI cleavage site (CCTN5AGG) was created in the presence of the A1896 [Xing et al., 2000]. The first-round PCR product (0.1 µl) was reamplified using B1/B-Bc1 primer pair (for BCP mutations) or C-Xag/C2 primer pair (for PC mutation) under the following conditions: 94°C for 5 min; 32 cycles of 94°C for 50 sec, 56°C for 1 min, and 72°C for 55 sec; a final 72°C for 10 min. The sensitivity of this nested PCR method was 100 copies/ml. All necessary precautions were observed to prevent cross-contamination, and negative controls were included in each assay. All the tests were performed in duplicate to confirm the results.
PCR product of the B1/B-Bc1 primer pair was digested with BclI and separated on 3% agarose gel. The 144-bp (uncleaved) and 120-bp (cleaved) bands correspond to the wild-type sequence and T1762/A1764 double mutant, respectively. The presence of both 144-bp and 120-bp fragments suggests a mixture of wild-type virus and the BCP mutant. PCR product of the C-Xag/C2 primer pair was digested with XagI and also analyzed on 3% agarose gel. The 198-bp (uncleaved) and 176-bp (cleaved) bands correspond to wild-type virus and the A1896 mutant, respectively.
To confirm the presence of the BCP and PC mutations and the ratio between wild-type and mutant virus when a mixture was present, the first round PCR product of six samples was sequenced directly. The sequences were analyzed with software Vector NTI 9.0 and Chromas.
Statistical Analysis
Data were analyzed by: the Mann–Whitney rank sum test, the chi-square, Spearman correlation, Linear correlation and regression. A P value below 0.05 was considered statistically significant.
RESULTS
Prevalence of Genotypes B and C
A total of 207 patients were studied. Viral genotype was established by PCR and sequencing of the HBsAg gene (GenBank accession numbers: EU915485; EU 921790–EU921799; EU926420–EU926473; EU927150–EU927286), followed by phylogenetic analysis. Fifty-one isolates were classified into genotype B while the remaining 156 belonged to genotype C (Table II).
TABLE II.
Comparison of Clinical and Virological Features Between Genotype B and Genotype C Patients
| Factor | Total (n = 207) | Genotype B (n = 51) | Genotype C (n = 156) | P-value |
|---|---|---|---|---|
| Gender (M/F) | 173/34 ± (1/0.2) | 38/13 (1/0.34) | 135/21 (1/0.16) | 0.083 |
| Age (years)a | 32.2 ± 9.2 | 28.08 ± 7.77 | 33.43 ± 9.16 | 0.0002 |
| ALT (U/L)a | 176.6 ± 86.5 | 177 ± 100 | 177 ± 82 | 0.987 |
| HBeAg titer (S/CO)a | 195.2 ± 98.9 | 189 ± 99 | 197 ± 92 | 0.591 |
| HBV DNA titerb | 7.15 ± 0.89 | 7.08 ±1.15 | 7.17 ± 0.81 | 0.532 |
| T1762/A1764 | 103 | 14/51 (27.5%) | 89/135 (65.9%) | 0.001 |
| A1896 | 43 | 19/51 (37.2%) | 24/135 (17.8%) | 0.001 |
| T1762/A1764/A1896 | 13 | 2/51 (3.9%) | 11/135 (8.1%) | 0.527 |
Expressed as X ± SD.
log10 copies/ml.
Prevalence of PC and BCP Mutations
The presence of PC mutation was determined by digestion of the nested PCR product with XagI, which cleaves the A1896 mutant but not wild-type virus. As shown in the upper panel of Figure 1, samples contained a 198-bp band (wild-type), a 176-bp band (A1896 mutant), or both bands at various ratios. The wild-type was defined as dominant if the 198-bp band is stronger than the 176-bp band, and the A1896 mutant as dominant if the 176-bp band is stronger. In six samples, the presence of the wild-type, mutant, or mixed sequence was confirmed by direct sequencing of the first round PCR product (Fig. 1, lower panel and Fig. 3). Presence of the BCP mutation was established by digestion of the 144-bp semi-nested PCR product with BclI, which converts it into 120 bp if the T1762/A1764 double mutation is present. Again patterns of wild-type, mutant, and mixture at different ratios were observed (Fig. 2, top panel). A sample is considered as wild-type dominant if the 144-bp band is stronger than the 120-bp band. The result was confirmed by direct sequencing of the PCR product (Fig. 2, lower panel and Fig. 3).
Fig. 1.
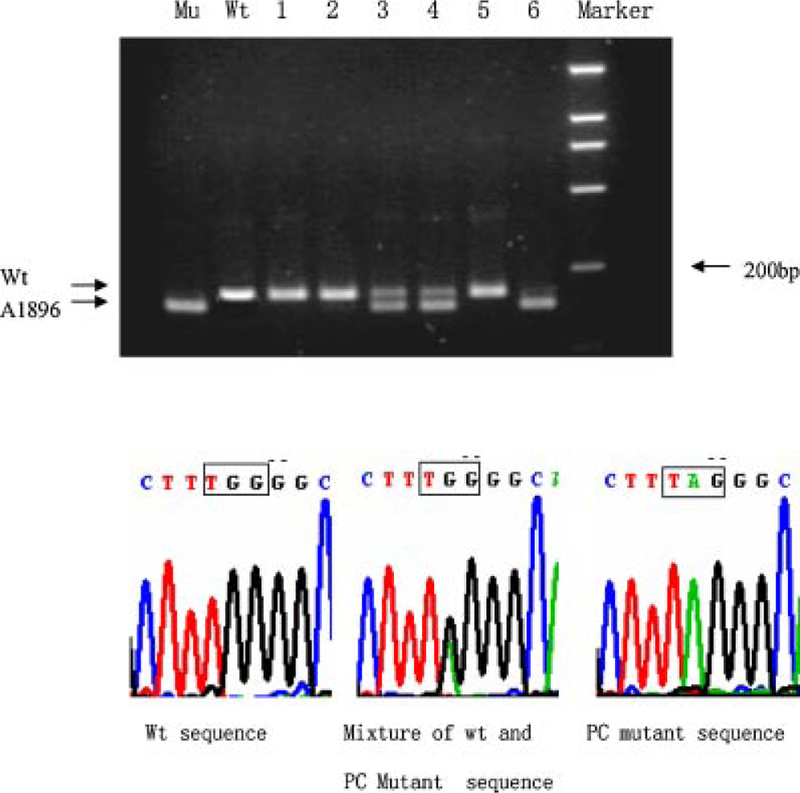
Detection of G1896A precore mutation by RFLP assay and direct sequencing. For the RFLP assay (top panel), the second round PCR product was digested with XagI and separated in 3% agarose gel. Positions of the wild-type band and A1896 band are indicated. Marker, 2,000-bp molecular size markers; Mu, control of PC mutant; Wt, control of PC wild-type; 1–6, digested DNA from six samples. For direct sequencing (lower panels), the first round PCR product was used. The three panels correspond to samples number 1 (left panel), 3 (middle panel), and 6 (right panel).
Fig. 3.

Sequence alignment of the six samples shown in Figures 1 and 2. The first round PCR product was sequenced directly. Shown here are nucleotides 1,687–1,946 covering both basal core promoter and precore regions.
Fig. 2.

Detection of T1762/G1764 BCP mutations by RFLP assay and direct sequencing. The six samples analyzed here are identical to those shown in Figure 1. For the RFLP assay (top panel), the second round PCR product was digested with BclI and separated in 3% agarose gel. Positions of the wild-type band and mutant band are indicated. Marker, 2,000-bp molecular size markers; Mu, control of BCP mutant; Wt, control of BCP wild-type; 1–6, digested DNA from the six samples. For direct sequencing (lower panels), the first round PCR product was used. The three panels correspond to samples number 2 (left panel),3 (middle panel), and 4 (right panel).
The PC mutant was detectable in 43 samples (20.8%), with 19 of them showing dominance of the mutant species (Table III). In the remaining 24 samples, the wild-type was dominant. The BCP mutant was present in 103 samples (50%), of which 77 showed dominance of the mutant (Table III). When both the PC and BCP mutations were considered (Table IV), the samples fall into four groups: wild-type sequence at both regions (74), presence of PC but no BCP mutant (30), presence of BCP but no PC mutant (90), presence of both PC and BCP mutants (13).
TABLE III.
HBeAg Titers in Relationship to Gender, Age, as Well as BCP and PC Mutations
| Factor | Subgroup | Number | HBeAg titer (S/CO)a | P-value | HBV DNAb | P-value |
|---|---|---|---|---|---|---|
| Sex | Male | 173 | 194.2 ± 94.5 | 7.20 ± 0.90 | ||
| Female | 34 | 200.1 ± 92.2 | 0.743 | 6.93 ± 0.89 | 0.125 | |
| Age (years) | <35 | 134 | 206.9 ± 87.5 | 7.14 ± 0.93 | ||
| ≥35 | 73 | 173.7 ± 101.9 | 0.015 | 7.18 ± 0.86 | 0.736 | |
| BCP mutations | Pure wild-type | 104 | 219.9 ± 85.2 | 7.16 ± 0.84 | ||
| With A1762T/G1764A mutants | 103 | 170.6 ± 96.1 | 0.0001 | 7.13 ± 0.94 | 0.85 | |
| Wild-type dominant | 26 | 209.1 ± 87 | 7.03 ± 0.87 | |||
| BCP mutant dominant | 77 | 159.6 ± 95.2 | 0.017 | 7.17 ± 0.97 | 0.64 | |
| PC mutation | Pure wild-type | 164 | 204.9 ± 87.9 | 7.20 ± 0.88 | ||
| With G1896A mutants | 43 | 157.9 ± 104.3 | 0.003 | 6.99 ± 0.89 | 0.095 | |
| Wild-type dominant | 24 | 183.0 ± 101.3 | 7.15 ± 0.84 | |||
| PC mutants dominant | 19 | 126.2 ± 108.8 | 0.084 | 6.70 ± 0.91 | 0.098 |
Expressed as X ± SD.
log10 copies/ml.
TABLE IV.
Impact of Combined BCP and PC Mutations on HBeAg Titers
| Group | Sequence | Number | HBeAg titer (S/CO)a | HBV DNAb |
|---|---|---|---|---|
| A | Wild-type | 74 | 228.5 ± 81.4 | 7.27 ± 0.84 |
| B | PC mutation alone | 30 | 192.2 ± 96.7 | 6.91 ± 0.83 |
| C | BCP mutations alone | 90 | 185.6 ± 88.7 | 7.16 ± 0.95 |
| D | BCP + PC mutations | 13 | 78.9 ± 89.2 | 7.03 ± 1.04 |
P values (HBeAg titers): A/B, 0.019; A/C, 0.0001; A/D, 0.0000; B/C, 0.552; B/D, 0.0008; C/D, 0.0003.
P values (HBV DNA): A/B, 0.132; A/C, 0.446; A/D, 0.363; B/C, 0.326; B/D, 0.915; C/D, 0.627.
Expressed as X ± SD.
log10 copies/ml
Comparison of HBeAg Titers and Mutation Patterns Between Genotypes B and C
Table II compares patients infected with genotypes B and C. There were no significant differences in serum ALT level and HBV DNA titer between the two genotypes. The mean HBeAg titer was slightly higher in genotype C than genotype B (197 vs. 189) but the difference was not statistically significant. Patients with genotype C isolates were older than those with genotype B isolates (mean age: 33.43 vs. 28.08; P = 0.0002) and had higher percentage of males (86%) than patients with genotype B (75%), although the difference did not reach statistical significance (P = 0.083). The genotype C isolates had a higher prevalence of BCP mutations but a lower prevalence of the PC mutation than genotype B (P = 0.001).
Correlation of HBeAg Titers With Patient Gender, Age, and Mutations in the BCP and PC Regions
Table III compares impact of factors other than viral genotype on HBeAg titer. The HBeAg titers did not differ between male and female patients, but were higher in younger patients (<35 years of age) in comparison to older patients (≥35 years) (206.9 ± 87.5 vs. 173.7 ± 101.9; P = 0.015). Since the prevalence of BCP and PC mutations differed in the younger and older patients, HBeAg titers were compared among patients infected with wild-type PC and BCP sequence using linear regression analysis. The regression coefficient between age and HBeAg titers was 1.651 (Fig. 4). Although the value did not reach statistical significance (P = 0.078), this is consistent with a negative correlation of age with HBeAg titers. No correlation was found for patients infected with the PC mutant/BCP WT sequence (Fig. 4). The HBeAg titers were lower in patients harboring the BCP mutant (whether alone or together with wild-type virus) than those harboring the pure wild-type sequence at this position (170.6 ± 96.1 vs. 219.9 ± 85.2; P = 0.001); of the former group, it was lower in those with dominance of the mutant than those with wild-type dominance (159.6 ± 95.2 vs. 209.1 ± 87; P = 0.017). The HBeAg titer was also lower in patients harboring the PC mutant than those with pure wild-type sequence at this position (170.6 ± 96.1 vs. 219.9 ± 85.2; P = 0.001). In those samples harboring the PC mutant, the mutant dominant group had lower HBeAg titer than the wild-type dominant group (126.2 ± 108.8 vs. 183 ± 101.3), although the difference did not reach statistical significance (P = 0.084).
Fig. 4.
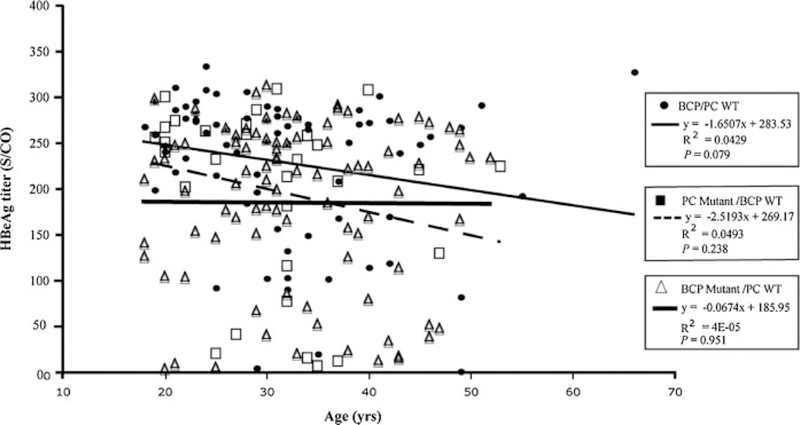
Correlation of HBeAg titers with age in patients infected with BCP/PC WT sequence (closed circle), PC mutant/BCP WT (solid square), and BCP mutant/PC WT (open triangle).
The impact of dual mutations in both the BCP and PC regions versus single region mutation on HBeAg titers were compared. As shown in Table IV, the HBeAg titer was 192.2 ± 96.7 in those harboring the PC mutation alone, 185.6 ± 88.7 in those harboring the BCP muta-tions alone (P = 0.552), but only 78.92 ± 89.2 in those with mutations in both regions (P < 0.001). Therefore, the presence of mutant populations at both the precore and core promoter regions led to lower HBeAg titer than the presence of mutation in either region alone.
Correlation of HBeAg Titers With HBV DNA
Figure 5 shows the relationship between HBeAg titers and viral load in patients infected with the PC WT/BCP WT sequence (P = 0.288), the PC mutant/BCP WT (P = 0.454), and the BCP mutant/PC WT sequence (P = 0.057). There was no statistically significant correlation although the value was nearly significant for the BCP mutant/PC WT group.
Fig. 5.
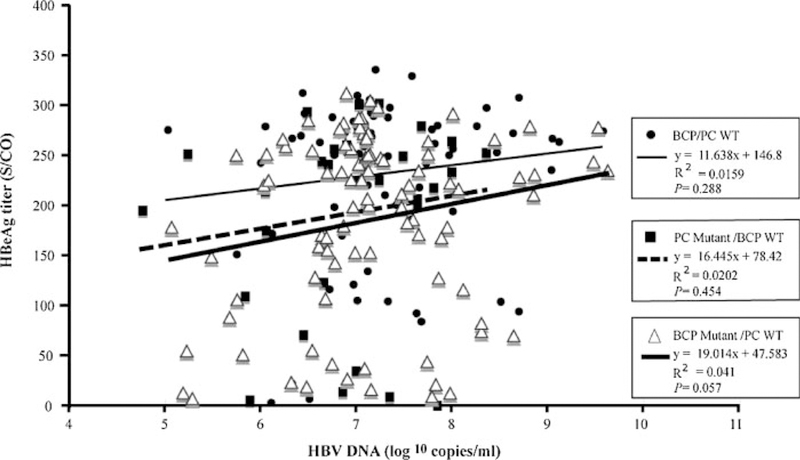
Correlation of the HBeAg titers with HBV DNA levels in patients infected with BCP/PC WT sequence (closed circle), PC mutant/BCP WT (solid square), and BCP mutant/PC WT (open triangle).
DISCUSSION
Genotypes B and C are the dominant HBV genotypes in East Asia. Their distribution shows a North-to-South Gradient, with genotype C predominating in the north and B in the south [Orito et al., 2001a; Zeng et al., 2005]. In the present study of HBeAg positive patients from six hospitals in China, it was found that the prevalence of genotype C (75.4%) was higher than genotype B (24.6%). Consistent with the earlier reports that genotype C patients clear HBeAg about a decade later than genotype B patients [Chu et al., 2002b; Yuen et al., 2003], this study also showed that patients with genotype C were 5 years older than patients with genotype B. These HBeAg positive patients of genotype C infection also had a higher prevalence of the A1762T/G1764A BCP mutations but lower prevalence of the G1896A PC mutation, as have been reported by many other investigators [Orito et al., 2001b; Chu et al., 2003; Sumi et al., 2003]. At present, the preferential development of the BCP mutations in genotype C as opposed to PC mutation in genotype B remains unknown. C1858, which is found in majority of genotype A isolates and some isolates of genotypes C and F, precludes the emergence of 1896 mutation because of the base pairing restrictions of the encapsidation signal [Li et al., 1993; Lok et al., 1994; Arauz-Ruiz et al., 1997; Alestig et al., 2001]. Since most samples in this study were analyzed by restriction fragment length polymorphism, the prevalence of C1858 variant in our genotype C isolates is unknown. On the other hand, the serum HBV DNA titer did not differ between patients infected with these two genotypes.
Despite the importance of the presence of HBeAg/anti-HBe in the pathogenesis, few studies have attempted to measure HBeAg quantitatively. In the present quantitative assay of HBeAg, no difference in HBeAg titers was found between patients infected with genotype B and C. However, the titers were higher in patients <35 years of age than those >35 years, which is consistent with age-dependent decline and loss of HBeAg expression. Importantly, presence of either precore or core promoter mutations was associated with reduced HBeAg titer. It is possible that this reduction of HBeAg titer is caused by immune clearance that reduces viral load and also provides a selection force for the precore and core promoter mutants. If this is true, HBV DNA replication should also decline in patients harboring such mutants. However, no difference was found in the titers of HBV DNA between patients with the wild-type or mutated precore or core promoter sequence. Thus, the reduced HBeAg titer is more likely a direct consequence of precore and core promoter mutations, which are known to reduce and abolish HBeAg expression, respectively [Carman et al., 1989; Blum et al., 1991; Tong et al., 1991; Buckwold et al., 1996; Moriyama et al., 1996; Parekh et al., 2003]. The reason why patients harboring the G1896A precore mutation continued to express HBeAg was the co-existence of wild-type virus (the mutant was never detected as a pure population in any of our patients). In fact patients with mutant precore sequence as the dominant species tended to have lower HBeAg titer than those with predominantly wild-type sequence, although the difference was not statistically significant probably due to small sample size (Table III). The coexistence of mutant and wild-type sequence also explains why presence of both precore and core promoter mutations led to a further reduction in HBeAg titer than presence of either mutation alone: these mutations probably were present in separate molecules and had additive effects. This issue could not be addressed in the present study because the second round of PCR did not amplify the two regions as a single DNA fragment.
In conclusion, the HBeAg titer correlated with the age of the patients and precore/core promoter mutations, but not with the viral genotype. Follow-up studies will establish whether patients with a lower HBeAg titer will seroconvert to anti-HBe sooner than those with a higher HBeAg titer. Indeed, a recent study demonstrated that patients with a lower HBeAg titer were more likely to lose HBeAg during interferon therapy [Fried et al., 2008].
Acknowledgments
Grant sponsor: Basic Research Fund of the Science & Technology Commission of Shanghai Municipality; Grant numbers: 07JC14009, 08410708600; Grant sponsor: Natural Science Foundation of China; Grant number: 30872242; Grant sponsor: Chinese High-tech Program; Grant number: 2006AA02A411.
Abbreviations used:
- ALT
alanine aminotransferase
- HBeAg
hepatitis B e antigen
- HBsAg
hepatitis B surface antigen
- BCP
basal core promoter
- HBV
hepatitis B virus
- PCR
polymerase chain reaction
- PC
precore
- RFLP
restriction fragment length polymorphism
- ULN
upper limit of normal
Footnotes
The authors have nothing to disclose regarding conflict of interest with respect to this manuscript.
REFERENCES
- Alestig E, Hannoun C, Horal P, Lindh M. 2001. Phylogenetic origin of hepatitis B virus strains with precore C-1858 variant. J Clin Microbiol 39:3200–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amini-Bavil-Olyaee S, Sarrami-Forooshani R, Adeli A, Mahboudi F, Sabahi F, Nafisi H, Zali MR, Azizi M. 2005. A novel accurate amplification created restriction site method for determination of the wild type and the precore mutant hepatitis B virus variants. J Virol Methods 127:19–23. [DOI] [PubMed] [Google Scholar]
- Arauz-Ruiz P, Norder H, Visona A, Magnius LO. 1997. Genotype F prevails in HBV infected patients of Hispanic origin in Central America and may carry the precore stop mutant. J Med Virol 51:305–312. [PubMed] [Google Scholar]
- Baptista M, Kramvis A, Kew MC. 1999. High prevalence of 1762(T) 1764(A) mutations in the basic core promoter of hepatitis B virus isolated from black Africans with hepatocellular carcinoma compared with asymptomatic carriers. Hepatology 29:946–953. [DOI] [PubMed] [Google Scholar]
- Blum H, Liang T, Galun E, Wands J. 1991. Persistence of hepatitis B viral DNA after serological recovery from hepatitis B virus infection. Hepatology 14:56–63. [DOI] [PubMed] [Google Scholar]
- Buckwold V, Xu Z, Chen M, Yen T, Ou J. 1996. Effects of a naturally occurring mutation in the hepatitis B virus basal core prooter on precore gene expression and viral replication. J Virol 70:5845–5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman WF, Hadziyannis S, Mcgarvey MJ, Jacyna MR, Karayiannis P, Makris A, Thomas C. 1989. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet 2:588–591. [DOI] [PubMed] [Google Scholar]
- Chan H, Hussain M, Lok A. 1999. Different hepatitis B virus genotypes are associated with different mutations in the core promoter and precore regions during hepatitis B e antigen seroconversion. Hepatology 29:976–984. [DOI] [PubMed] [Google Scholar]
- Chu C, Lok A. 2002a. Clinical significance of hepatitis B virus genotypes. Hepatology 35:1274–1276. [DOI] [PubMed] [Google Scholar]
- Chu C, Hussain M, Lok A. 2002b. Hepatitis B virus genotype B is associated with earlier HBeAg seroconversion compared with hepatitis B virus genotype C. Gastroenterology 122:1756–1762. [DOI] [PubMed] [Google Scholar]
- Chu C, Keeffe EB, Han SH, Perrillo RP, Min AD, Soldevila-Pico C, Carey W Jr., Luketic RB, Terrault VA, Lok NAS, and the U.S.HBV Epidemiology Study Group. 2003. Prevalence of HBV precore/core promoter variants in the United States. Hepatology 38:619–628. [DOI] [PubMed] [Google Scholar]
- Fried MW, Piratvisuth T, Lau GK, Maecellin P, Chow WC, Cookslev G, Luo KX, Paik SW, Liaw YF, Button P, Popescu M. 2008. HBeAg and hepatitis B virus DNA as outcome predictors during therapy with peginterferon Alfa-2a for HBeAg-positive chronic hepatitis B. Hepatology 47:428–434. [DOI] [PubMed] [Google Scholar]
- Honda A, Yokosuka O, Ehata T, Tagawa M, Imazeki F, Saisho H. 1999. Detection of mutations in the enhancer 2/core promoter region of hepatitis B virus in patients with chronic hepatitis B virus infection: Comparison with mutations in precore and core regions in relation to clinical status. J Med Virol 57:337–344. [PubMed] [Google Scholar]
- Kao JH, Chen DS. 2002. Global control of hepatitis B virus infection. Lancet Infect Dis 2:395–403. [DOI] [PubMed] [Google Scholar]
- Kidd-Ljunggren K, Oberg M, Kidd A. 1997. Hepatitis B virus X gene 1751 to 1764 mutations: Implications for HBeAg status and disease. J Gen Virol 78:1469–1478. [DOI] [PubMed] [Google Scholar]
- Kirschberg O, Schüttler C, Repp R, Schaefer S. 2004. A multiplex-PCR to identify hepatitis B virus genotypes A-F. J Clin Virol 29:39–43. [DOI] [PubMed] [Google Scholar]
- Kurosaki M, Enomoto N, Asahina Y, Sakuna I, Ikada T, Tozuka S, Izumi N, Marumo F, Sato C. 1996. Mutations in the core promoter region of hepatitis B virus in patients with chronic hepatitis B. J Med Virol 49:115–123. [DOI] [PubMed] [Google Scholar]
- Li J-S, Tong SP, Wen Y-M, Vitvitski L, Zhang Q, Trepo C. 1993. Hepatitis B virus genotype A rarely circulates as an HBeAg-minus mutant: Possible contribution of a single nucleotide in the precore region. J Virol 67:5402–5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindh M, Hannoun C, Dhillon AP, Norkrans G, Horal P. 1999. Core promoter mutations and genotypes in relation to viral replication and liver damage in East Asian hepatitis B virus carriers. J Infect Dis 179:775–782. [DOI] [PubMed] [Google Scholar]
- Lok AS, Akarca U, Greene S. 1994. Mutations in the pre-core region of hepatitis B virus serve to enhance the stability of the secondary structure of the pregenome encapsidation signal. Proc Natl Acad Sci USA 91:4077–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnius L, Norder H. 1995. Subtypes, genotypes and molecular epidemiology of the hepatitis B virus as reflected by sequence variability of the S-gene. Intervirology 38:24–34. [DOI] [PubMed] [Google Scholar]
- Moriyama K, Okamoto H, Tsuda F, Mayumi M. 1996. Reduced precore transcription and enhanced core-pregenome transcription of hepatitis B virus DNA after replacement of the precore-core promoter with sequences associated with e antigen-seronegative persistent infections. Virology 226:269–280. [DOI] [PubMed] [Google Scholar]
- Naito H, Hayashi S, Abe K. 2001. Rapid and specific genotyping system for hepatitis B virus corresponding to six major genotypes by PCR using type-specific primers. J Clin Microbiol 39:362–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Yotsumoto S, Akahane Y, Yamanaka T, Miyakawa Y, Sugai Y, Tsuda F, Tanaka T, Miyakawa Y, Mayumi M. 1990. Hepatitis B viruses with precore region defects prevail in persistently infected hosts along with seroconversion to the antibody against e antigen. J Virol 64:1298–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Yotsumoto S, Akahane Y, Sugai Y, Yoshiba M, Moriyama K, Tanaka T, Miyakawa Y, Mayumi M. 1994. Hepatitis B virus with mutations in the core promoter for an e antigen-negative phenotype in carriers with antibody to e antigen. J Virol 68:8102–8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orito E, Ichida T, Sakugawa H, Sata M, Horiike N, Hino K, Okita K, Okanoue T, Iino S, Tanaka E, Suzuki K, Watanabe H, Hige S, Mizokami M. 2001a. Geographic distribution of hepatitis B virus (HBV) genotype in patients with chronic HBV infection in Japan. Hepatology 34:590–594. [DOI] [PubMed] [Google Scholar]
- Orito E, Mizokami M, Sakugawa H, Michitaka K, Ishikawa K, Ichida T, Okanoue T, Yotsuvanaqi H, Iino S. 2001b. A case-control study for clinical and molecular biological differences between hepatitis B viruses of genotypes B and C. Hepatology 33:218–223. [DOI] [PubMed] [Google Scholar]
- Parekh S, Zoulim F, Ahn SH, Tsai A, Li JS, Kawai S, Khan N, Trépo C, Wands J, Tong SP. 2003. Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J Virol 77:6601–6612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P, Midgley S. 2005. Recombination in the genesis and evolution of hepatitis B virus genotypes. J Virol 79:15467–15476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi H, Yokosuka O, Seki N, Arai M, Imazeki F, Kurihara T, Kanda T, Fukai K, Kato M, Saisho H. 2003. Influence of hepatitis B virus genotypes on the progression of chronic type B liver disease. Hepatology 37:19–26. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Aoyama K, Ohno N, Iwata K, Akahane Y, Baba K, Yoshizawa H, Mishiro S. 1995. The precore/core promoter mutant (T1762A1764) of hepatitis B virus: Clinical significance and an easy method for detection. J Gen Virol 76:3159–3164. [DOI] [PubMed] [Google Scholar]
- Tamura K, Jakobsen I, Nei M. 2001. MEGA2: Molecular evolutionary genetics analysis software. Bioinformatics 17:1244–1245. [DOI] [PubMed] [Google Scholar]
- Tong SP, Li JS, Vitvitski L, Trépo C. 1990. Active hepatitis B virus replication in the presence of anti-HBe is associated with viral variants containing an inactive pre-C region. Virology 176:596–603. [DOI] [PubMed] [Google Scholar]
- Tong SP, Diot C, Gripon P, Li JS, Vitviski L, Trépo C, Guguen-Guillouzo C. 1991. In vitro replication competence of a cloned hepatitis B virus variant with a nonsense mutation in the distal pre-C region. Virology 181:733–737. [DOI] [PubMed] [Google Scholar]
- Xing L, Zhu C, Shi H, Han F. 2000. Detection of core promoter mutants in chronic hepatitis B virus. Chinese J Exp Clin Virol 2:163–165. [PubMed] [Google Scholar]
- Yuen M, Sablon E, Yuan H, Wong D, Hui C, Wong B, Chan A, Lai C. 2003. Significance of hepatitis B genotype in acute exacerbation, HBeAg seroconversion, cirrhosis-related complications, and hepa-tocellular carcinoma. Hepatology 37:562–567. [DOI] [PubMed] [Google Scholar]
- Zeng G, Wang Z, Wen S, Jiang J, Wang L, Cheng J, Tan D, Xiao F, Ma S, Li W, Luo K, Naoumov NV, Hou J. 2005. Geographic distribution, virologic and clinical characteristics of hepatitis B virus genotypes in China. J Viral Hepat 12:609–617. [DOI] [PubMed] [Google Scholar]
- Zhou J, Zeng X, Yin Y, Guo X, Zhang J. 2004. Sequence diversity analysis of CagA gene and corresponding protein in Helicobacter pylori. World Chin J Digestol 12:1307–1312. [Google Scholar]