

Abstract
Motivation
Metabolomics has shown great potential to improve the understanding of complex diseases, potentially leading to therapeutic target identification. However, no single analytical method allows monitoring all metabolites in a sample, resulting in incomplete metabolic fingerprints. This incompleteness constitutes a stumbling block to interpretation, raising the need for methods that can enrich those fingerprints. We propose MetaboRank, a new solution inspired by social network recommendation systems for the identification of metabolites potentially related to a metabolic fingerprint.
Results
MetaboRank method had been used to enrich metabolomics data obtained on cerebrospinal fluid samples from patients suffering from hepatic encephalopathy (HE). MetaboRank successfully recommended metabolites not present in the original fingerprint. The quality of recommendations was evaluated by using literature automatic search, in order to check that recommended metabolites could be related to the disease. Complementary mass spectrometry experiments and raw data analysis were performed to confirm these suggestions. In particular, MetaboRank recommended the overlooked α-ketoglutaramate as a metabolite which should be added to the metabolic fingerprint of HE, thus suggesting that metabolic fingerprints enhancement can provide new insight on complex diseases.
Availability and implementation
Method is implemented in the MetExplore server and is available at www.metexplore.fr. A tutorial is available at https://metexplore.toulouse.inra.fr/com/tutorials/MetaboRank/2017-MetaboRank.pdf.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Untargeted metabolomics studies allow monitoring a large range of small molecules (metabolome) in a tissue, an organism or a biofluid (Fiehn, 2002). When applied to human health research, a subset of this metabolome is considered as a metabolic fingerprint of a given pathology if it is statistically shared by a homogeneous group of patients in comparison to control subjects or another group of patients not affected by the pathology under study. These fingerprints constitute valuable supplementary knowledge that can be used for instance for patient stratification (Sreekumar et al., 2009). Metabolic fingerprints also provide important clues for drug discovery and precision medicine since they reflect the biochemical modulations of human metabolism induced by a pathology (Hocher and Adamski, 2017).
A commonly used approach to establish a link between metabolic fingerprints and endogenous metabolism consists in applying enrichment analysis (Chagoyen and Pazos, 2011; Persicke et al., 2012) on biochemical pathway collections provided by publicly available databases such as KEGG (Kanehisa et al., 2014). This technique aims at finding which metabolic pathways contain a significant number of metabolites that belong to the fingerprint. The notion of metabolic pathway is informative since it assigns functions to a set of reactions. Nevertheless, the definition of their functional boundaries (input and output compounds) often varies from one database to another (Altman et al., 2013) potentially leading to different interpretations when analyzing the same fingerprint in various databases. Fragmented view of metabolism offered by metabolic pathways is also a major limitation in global interpretation of fingerprints, especially when the systemic biochemical modulation associated with a disease is spanning several pathways.
To overcome these limits and take into account the full complexity of metabolism, metabolic fingerprints can be interpreted by considering them in the context of genome scale metabolic network which gathers all the biochemical reactions that can occur in a given organism (Mo and Palsson, 2009). Regarding human metabolism, networks reconstructed from genome annotation and manual curation had been made available to the community (Swainston et al., 2016; Thiele et al., 2013). However, the size of these networks (e.g. Recon2 human metabolic network contains 7440 reactions) makes the visual interpretation difficult and time-consuming. It thus requires the development of algorithms which reduce this complexity by finding the subset of reactions and metabolites (subnetwork) that are related to the metabolic fingerprint. Several of these methods have been proposed (Frainay and Jourdan, 2017) and applied to interpret metabolic fingerprints (Milreu et al., 2014). The results strongly support the use of network topology combined with graph-theory algorithms to give biological insight from a list of metabolites.
Most of these methods are based on path search between pairs of compounds in the fingerprint, based on the assumption that the sources and end products involved in the mechanism are known. However, in contrast to gene or protein studies, no single metabolomics technology allows monitoring every small molecule in a sample leading to potentially incomplete fingerprints. Moreover, due to technological or biological artifacts, the annotation of detected molecules is a challenging task which may discard some parts of the fingerprint before downstream interpretation (Creek et al., 2014; Neumann and Böcker, 2010). Fingerprint incompleteness can also be due to the fact that the matrix sampled to decipher metabolic modulations is not the tissue where the biochemical perturbations are occurring [e.g. metabolites are measured in cerebrospinal fluids (CSF) to study brain afflictions]. This gap may overshadow potential metabolic shifts and, consequently, leave aside important metabolites from the fingerprint, leading to misleading interpretations. Recent studies addressed the problem, inferring hidden paths in sparsely observed network (Lages et al., 2018; Massucci et al., 2016). However, none of them specifically assesses this problem in the context of metabolic networks, in particular regarding metabolomics results interpretation, where the unobserved part of the network can be difficult to accurately define. By taking into account the specificity of metabolism and its observation, the network approach presented in this article will on one hand help in biological interpretation, and on the other hand recommends candidate metabolites to enrich metabolic fingerprints.
Recommendation method development is an active research field in information retrieval community. The proposed algorithms have been intensively and successfully used for many applications, such as content recommendation in social networks like Twitter™ (Backstrom and Leskovec, 2011; Gupta et al., 2013; Liang et al., 2014; Liben-Nowell and Kleinberg, 2007). These algorithms typically suggest new people one might be interested in, based on its connections with people already present in its personal list of interest. We propose to extend this concept to metabolic networks, thus suggesting new metabolites of interest by taking into account how they are connected to metabolites already present in the metabolic fingerprint. Our approach, in contrast to the pioneering method borrowed from worldwide web analysis, does not assume that all the edges are equivalent, since relationships between metabolites are more complex to interpret than web page links. In fact, it is necessary to ensure the biological and chemical relevance of connections used to compute the recommendations.
We show how this approach, called MetaboRank, was successful in complementing the CSF metabolic fingerprint of the hepatic encephalopathy (HE) disease described in (Weiss et al., 2016). HE corresponds to the neurological or neuropsychological symptoms of acute or chronic liver failure and/or portosystemic shunt. The spectrum goes from mild neuropsychological symptoms to impaired level of consciousness, often leading to coma. Even if the physiopathology is still largely unrevealed, the major role of hyperammonemia in conjunction of inflammation is well established (Weiss et al., 2018). As a consequence, glutamine levels increase in the brain. However, the sole abundance of ammonemia does not scale with symptoms’ severity and it has been shown that associated inflammation, increased levels of TNF-alpha and IL-6, were much better correlated to symptoms’ severity.
In order to better decipher EH metabolic alterations, Weiss et al. used metabolomics to describe for the first time impaired metabolic pathways. Nevertheless, like many metabolomic experiments related to human health, the fingerprint was obtained from biofluid samples, overshadowing the importance of molecules that do not transit through the blood–brain barrier. Moreover, the heterogeneity of disease severity has led to high inter-individual variability at the metabolic level, making many abundance shifts weakly trustworthy. Those limitations emphasize the relevance of performing biological interpretation including knowledge outside the bounds of metabolomics scope.
In order to assess the relevance of hypotheses raised by our recommendation system, we applied automatic processing of literature data to associate concepts described in the literature to the recommended metabolites. We also reanalysed raw data and patient metadata to confirm some of the recommended metabolites.
MetaboRank is implemented in the freely accessible web server MetExplore (Cottret et al., 2010), allowing interactive analysis of the results.
2 System, methods and data
2.1 Centrality and recommendation systems
Many social network recommendation systems are based on the concept of network centrality, which aims at measuring the importance of a node. One of the best-known methods is the PageRank (PR) algorithm, which was used by Google to rank web pages in a search result according to their importance in the World Wide Web (Brin et al., 1998; Page et al., 1999). Since its first application, it has been successfully applied to many fields (Ma et al., 2008; Mihalcea et al., 2004), including biology (Allesina and Pascual, 2009; Iván and Grolmusz, 2011). PR defines the importance of a node as its probability to be encountered during a random walk in the network. In order to guarantee convergence of the random walk, stationary probability is obtained by adding to each node a probability to ‘jump’ to any other target node, restarting the walk from this target node. This ‘jump’ probability is specified in the algorithm through a parameter referred to as the damping factor. When those jumps are guided to favour some nodes according to a set of preferred ones, the term Personalized PageRank (PPR) is used (Haveliwala, 2002). PPR is well suited for fingerprint analysis since it has the ability to identify metabolites that are likely to be reached (produced) from molecules belonging to the fingerprint, by setting the jumps to target those metabolites of interest. We choose to define each metabolite of the fingerprint as equally important, thus with the same probability to be chosen following a jump. However, it is possible to set for each of them a different ‘restarting’ probability, based on prior assumption regarding their importance. Such personalization could be defined, for example, from fold changes or knowledge from the literature.
PPR can be considered as a downstream search that evaluates the scope of a list of metabolites. However, metabolites can also belong to a fingerprint because they are the outcomes of modulated metabolic processes. To enrich these results with upstream metabolites (i.e. potential precursors of fingerprint metabolites), we propose to compute the CheiRank (CR). CR and PR principles are similar except that for CR the links in the network are taken in reverse direction (Ermann and Shepelyansky, 2015). We adapted CR to create a Personalized CheiRank (PCR), which takes into account the list of metabolites in the fingerprint.
We propose to consider node centrality as a combination of these two measures. This two-dimension analysis combining PR and CR had been successfully used to analyse Wikipedia pages network (Zhirov et al., 2010) or world trade networks (Ermann and Shepelyansky, 2015), but has never been applied to metabolic networks.
Metabolic networks are usually highly centralized around few hubs, such as Coenzyme A or ATP (Jeong et al., 2000). It can thus be expected that these central nodes will always have a high PR or CR regardless of the content of the metabolic fingerprints. In order to limit this bias and emphasize metabolites whose centrality is higher compared with the general case, the scoring function is defined as the ratio between PPR (resp. PCR) and global PR (resp. CR).
In order to obtain relevant results, analysis of metabolic networks requires taking into account biochemical properties related to each reaction (Arita, 2004). To this end, MetaboRank is computed on a probability matrix encoding biochemical knowledge as described in the next section.
2.2 Adapting human genome scale metabolic network and defining transition probabilities
The last two decades have seen an exponential growth of metabolic network reconstructions, which are made available through public databases (Wimalaratne et al., 2014) or alongside articles (Hucka et al., 2003). However, for practical reasons, most methods designed to analyse metabolomic results in the context of those networks are database-dependent, restricting for example their use to KEGG (Kanehisa et al., 2014). In order to apply our analysis to networks coming from various sources as well as home-brewed networks, we propose a generic method that can be applied to any network described in the standard SBML format (Hucka et al., 2003) with sufficient information on metabolites.
We applied our method to Recon2 human genome scale metabolic network (Thiele et al., 2013). In this model, metabolites are assigned to cellular compartments (mitochondria, cytoplasm, etc.). Nevertheless, current global and untargeted metabolomics approaches do not provide information on cellular localization of metabolites. Hence, we created a modified version of Recon2 network by considering a metabolite belonging to several compartments as a single metabolite.
Metabolic networks can be turned into graph mathematical formalism by assigning network elements to nodes connected by edges. Several ways to turn a metabolic network into a graph exist (Lacroix et al., 2008). We chose to use the compound graph whereone metabolite is connected to another if they are respectively substrate and product of a reaction from the network. This formalism allows the integration of information about substrate-product transition on edges.
One of the main issues when analyzing metabolic graphs is the presence of side compounds, which are ubiquitous compounds involved in many biochemical reactions for annex purposes, such as energy carrier or proton donor. This leads to create edges between a ‘main’ substrate node and a side product (like water) node. When computing paths, these side compounds may cause an underestimation of distances by creating irrelevant shortcuts (Arita, 2004; Holme, 2009). One way to overcome this issue is to remove a set of side compounds based on expert knowledge or using degree threshold (Croes et al., 2005). However, several metabolites considered as side-compounds in most reactions may be implicated as ‘main’ compounds in other processes (typically their own biosynthesis pathway). Systemically removing those compounds will lead to the loss of relevant parts of the network. A more suitable approach consists of comparing molecular structure of substrates and products by using chemical similarity or atom mapping. This approach allows dissociating side compounds from main ones on a chemical basis (Blum and Kohlbacher, 2008; Rahman et al., 2005). In those cases, side compounds are not defined globally for the entire metabolic network, but in the context of each reaction they are involved in.
Hence, we applied a pre-processing step on the Recon2 uncompartmentalized metabolic network to avoid irrelevant transitions by computing atom-atom mapping using the Reaction Decoder Tool (Rahman et al., 2016). For a reaction, atom-atom mapping consists of establishing a one-to-one correspondence between the substrate and product atoms. This method requires structural description of the compounds which is encrypted using the SMILES format (Weininger, 1988). Since this information was not available for all metabolites in the Recon2 network, we automatically retrieved this knowledge form chemical databases using web services [PubChem, ChEBI (Davies et al., 2015), HMDB (Wishart et al., 2013)]. When only the InChI identifiers (Heller et al., 2015) were available, we converted them into SMILES using the Chemistry Development Toolkit (Steinbeck et al., 2003). Finally, all substrate-to-product transitions that do not involve the preservation of at least one carbon atom between the source and the target were removed.
The previously described steps allow building the graph on which PPR and PCR will be computed. The algorithm used to compute PPR considers every outgoing edges of a node to have the same probability to be traversed in the random walk. This model can induce a bias when applied to compound graphs. In fact, using equiprobable edges implies that a reaction with many products will be favoured against a reaction consuming the same compound but producing fewer metabolites. For instance, in Figure 1A, the network contains two reactions R1 and R2. If we consider a compound centric weighting policy, all corresponding edges in the compound graph will have the same probability of 1/3, as shown in Figure 1B. A more proper way would be to spread probabilities between reactions and then subdivide the probabilities of compound graph edges as it is shown in Figure 1C.
Fig. 1.

Effect of the number-of-product bias on compound graph transition probabilities. By overshadowing the reaction levels, seen on the bipartite graph (A), the use of compound graph will favour reactions that involve more products than other consuming reactions (B). The hybrid weighting policy (C) allows suppressing that bias
We define the probability policy for substrate-to-product transition as follows, with and as two connected nodes, the edge weight: 1 by default, proportion of atoms of m1 mapped on m2 in our study, the set of all products of reaction r, and the set of reactions consuming
By applying this probabilistic approach, random walks will go through edges as if computation was performed on the bipartite graph representation of the network. However, this policy still allows to benefiting from the compound graph capability, namely adding information about substrate-product chemical transitions in the probability computation, such as chemical similarity (Rahman et al., 2005), RPAIR tags scoring (Faust et al., 2009) or atom conservation (Blum and Kohlbacher, 2008), which has been chosen in this study, using atom mapping results. Each of those weighting strategies have been shown to produce meaningful results and can be chosen depending on the context of the study and data availability (see Frainay and Jourdan, 2017 for in depth comparison). This probability policy also allows weighting at reaction level, considering data obtained from transcriptomic or proteomic experiments for example, by changing the reaction factor in the first equation. The weight of an edge would be the probability of the substrate-to-product transition, multiplied by the probability of the reaction defined from data.
Finally, this policy can also be applied to multi-graphs where two metabolites can be connected by several edges when a chemical transition can be catalyzed by several enzymes.
The metabolic network adaptation of PPR (resp. PCR) will be called in the following MetaboRankout (resp. MetaboRankin). The combination of both ‘in’ and ‘out’ measures form the MetaboRank recommendation system.
2.3 Metabolic fingerprint of HE
Metabolomics experiments have been conducted on CSF samples from 14 patients suffering from HE against samples from 27 control patients without any proven neurological disease (Weiss et al., 2016). The CSF metabolome was analysed by LC/MS [Orbitrap-Exactive and Q-Exactive Plus: Positive and negative ESI Scanning from m/z 75 to m/z 1000. Mass resolution: 100 000 FWHM. The relevant LC/HRMS features were kept by applying analytical filters from XCMS data matrix as followed: correlation between dilution factor of diluted QC (Quality Control, mix of all sample) and their area corresponded and area. , Mean QC/mean Blank > 3, CV (QC) < 30%; Feature annotation using public databases, CAMERA R package and ESI-MS and HCD MSMS spectral in-house database] and the discriminating fingerprint was built using multivariate [SIMCA-P software (version 11.0, Umetrics, Umea, Sweden)] and univariate statistical analyses.
In order to focus on metabolites confidently identified and presenting the most trustworthy abundance changes, we extracted a core fingerprint from the one presented in (Weiss et al., 2016). We only kept metabolites with a relative abundance fold change between patient and control >2 times the standard deviation, a level 1 or 2 identification (Sumner et al., 2007) and considered as significant regarding Mann–Whitney test (P-value < 0.005) (see the full list in Supplementary Table S1).
2.4 Mining literature to corroborate and enrich suggestions
Social-network recommendation system efficiency is commonly evaluated by measuring the number of suggestions followed by a user during future web browsing. Unfortunately, this methodology cannot be applied to metabolic recommendation system assessment since recommendations in our case is not part of a decision process but is involved in data interpretation. More generally, assessing the quality of methods providing support to biological interpretation is still a key challenge in the field. In fact, since the disease mechanisms are still partially known, we do not have gold standard datasets (other validated biomarkers) to compare with our recommendation system suggestion.
In order to establish a link between metabolites of interest and HE, we used the Metab2MeSH tool (Sartor et al., 2012). MeSH (Medical Subject Headings) is a controlled vocabulary thesaurus hierarchically structured used to index scientific publications from the MedLine and PubMed databases. Metab2MeSH performs enrichment analysis to identify MeSH terms significantly associated with metabolite names, based on their occurrences in scientific publications. Compound names from the Recon2 model were converted to PubChem entry names using the Chemical Translation Service web service (Wohlgemuth et al., 2010) and used to retrieve associated MeSH terms using the Metab2MeSH web service.
3 Results
On the basis of HE core fingerprint, MetaboRankout and MetaboRankin were computed on all metabolites from the pre-processed metabolic graph. Isolated metabolites, including metabolites without structural information, where ignored from the final ranking, resulting in two ranked lists of 1450 metabolites (see Supplementary Table S2). In the following we will focus on compounds ranked in top 50th of MetaboRankout and MetaboRankin. The union of these two lists contains 72 metabolites and will be called in the following ‘suggestion list’.
To assess the quality of this suggestion list, we compared it to a list obtained by performing an automatic literature search. 38 compounds in recon2 were associated with the MeSH term ‘hepatic encephalopathy’ (MeSH id D006501) (see Fig. 2). Ten of them were found in the original metabolic fingerprint. Four others were found in the MetaboRankin 50th top ranked compounds, and eight were found in the MetaboRankout 50th top ranked compounds. Overall, the suggestion list allowed enriching the original fingerprint with 10 compounds known in the literature to be related to the disease. Fisher exact test has revealed that the suggestion list is significantly associated with the list of compounds found in the literature, with a P-value of 3.119e − 07. The receiver operating characteristic (ROC) curve presented Figure 3 suggests that the ranking makes solid performance at retrieving the metabolites associated with the disease, with an area under the curve (AUC, computed using trapezoidal approximation) of 0.768 for the MetaboRankout and 0.745 for the MetaboRankin. The best performance was achieved using the combination of the two ranking, where metabolites are ranked according to their best rank in any of the two other rankings, with an AUC of 0.812.
Fig. 2.
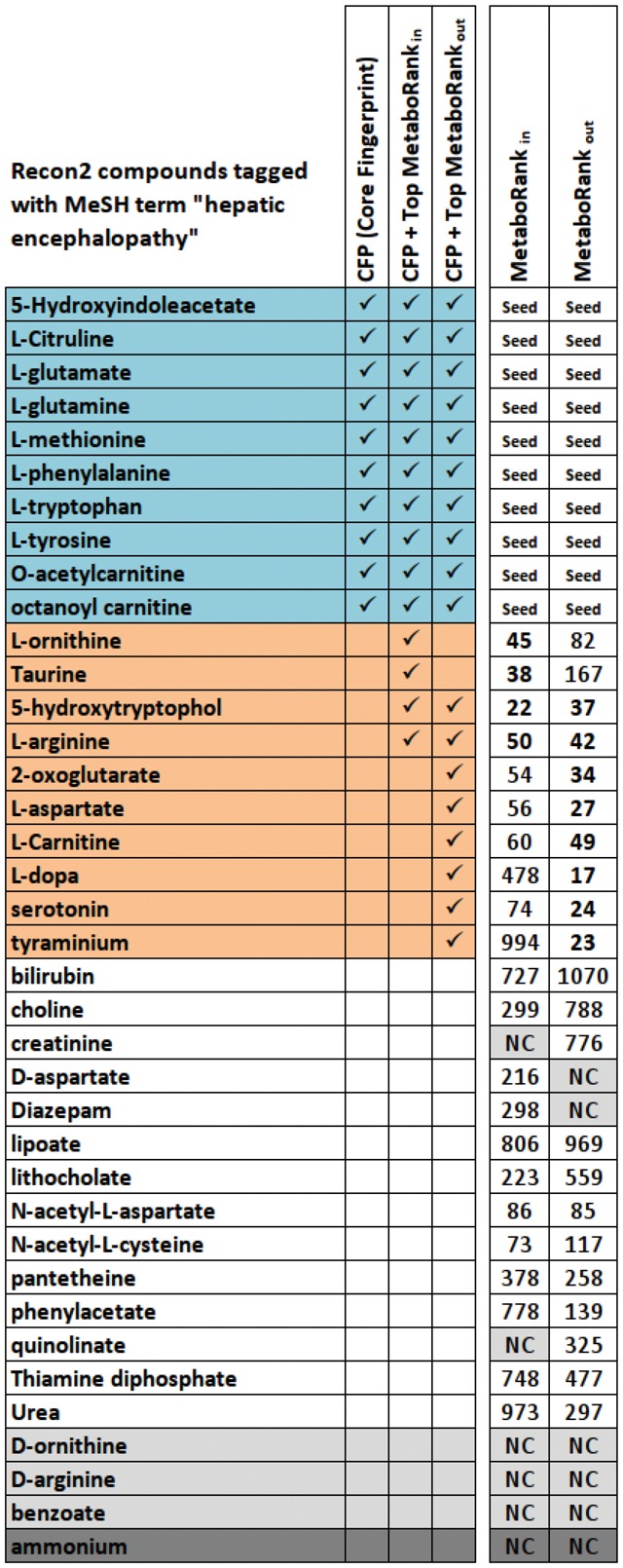
2D-rank of HE related compounds found in Recon2. HE related compounds were found using Metab2Mesh tool. Ten of them were present in the core fingerprint obtained from LCR metabolomic profile (blue cells), 10 others were present in the list of recommendations (union of top 50 MetaboRankout and MetaboRankin) (orange cells). The light grey cells contain compounds that are disconnected from the input list (NC), dark grey cells contain compounds that have been removed from the network (Color version of this figure is available at Bioinformatics online.)
Fig. 3.
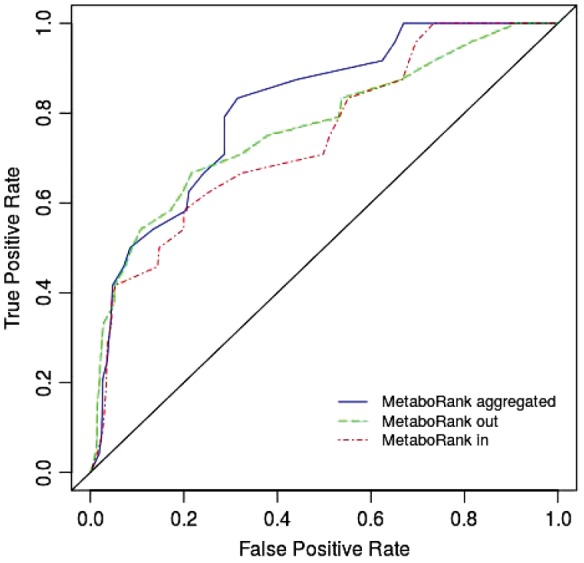
ROC curves for MetaboRanks. The True Positive Rate (TPR) corresponds to the proportion of metabolites associated with HE literature retrieved in the recommendation list. The False Positive Rate (FPR) corresponds to the proportion of metabolites non-significantly associated with the HE literature retrieved in the recommendation list. The curves represent the TPR and FPR of a recommendation list for different rank cutoffs. The black diagonal line corresponds to the expected value obtained from a random ranking (Color version of this figure is available at Bioinformatics online.)
Among the remaining 18 HE-related compounds found in the network, four were completely disconnected from the fingerprint, meaning that no single path can be found in the metabolic graph between them and any compound from the fingerprint (see grey lines in Fig. 2). The ammonium cannot be reached because we consider only substrate-to-product transitions that involve carbon atom conservation (see Section 2).
The presence of the D-forms of aspartate, ornithine and arginine in the literature based HE-related list might be due to erroneous compound annotations in literature, as they are very rare in nature. In fact, D and L-forms of those compounds match exactly the same HE-related publications in Pubmed, where the chirality is rarely specified.
The literature based HE-related list may also contain molecules associated with the disease which are not involved in the pathogenicity, but rather mentioned in literature for their intake effect on HE patients. Therefore, the endogenous form of the molecule might not be part of the modulated metabolic mechanism, and will not be related to the fingerprint. This could be the case for compounds like benzoate and Diazepam present in the Recon2 network and absent from our suggestion list. Sodium benzoate has been used for HE treatment (Misel et al., 2013) in order to activate an alternative pathway of waste nitrogen removal. Diazepam overdose has been shown to induce progressive encephalopathy (Rupasinghe and Jasinarachchi, 2011), and the administration of benzodiazepine medication to cirrhotic patient has been suggested contributing to neurological impairment (Perney et al., 1998). Diazepam has been detected in CSF samples, but has not been included in the HE metabolic fingerprint (Weiss et al., 2016). The input fingerprint was built from cirrhotic patient, mainly due to alcohol consumption. This drug-induced form of the disease would likely yield a different metabolic fingerprint and could explain why this compound is not suggested by the recommendation system. Finally, HE-related compounds might be missing from the suggestion list because of network incompleteness or too sparse fingerprint.
Some metabolites in the suggestion list may not yet be mentioned in literature focusing on HE, but they may be present in articles mentioning symptoms or diseases related to HE. To address this issue and enlarge the scope of our interpretation, we performed the literature analysis by starting from compounds in the suggestion list to decipher the ones that are not yet associated to HE in the literature, but which could be related to health impacts and symptoms strongly associated with HE. MeSH terms related to HE were extracted using a similarity metric that consider the number of co-occurrences between MeSH terms compared with an expected number of co-occurrences appearing ‘by chance’ (Smalheiser and Bonifield, 2016). Only MeSH terms with an odds ratio above 3 were considered. Figure 4 shows main MeSH terms from categories: diseases, signs and symptoms associated with HE. By overlaying the suggestion list onto this graph (size of nodes in Fig. 4) it appears that brain, liver and metabolic diseases are the main categories of diseases related to HE.
Fig. 4.

Suggested compounds mapped onto HE-related disease MeSH subnetwork. Nodes represent Mesh terms. Edges represent tie in the MeSH ontology. The strength of the association with HE is represented as the label font size. Node size represents the number of suggested compounds associated with the corresponding term and/or sub-term. For readability purpose the whole relationships of the MeSH ontology are not represented, only shortest path between each term is considered
Figure 5 shows in more details how the 53 metabolites of the suggestion list, annotated with at least one MeSH term (see Supplementary Material, Table S3), are related to liver and nervous system diseases and symptoms.
Fig. 5.

Association between brain and liver diseases MeSH terms and suggested compounds. Black and grey cells represent an unexpected number of co-occurrences between the compound name and the MeSH annotation in PubMed, defined accordingly to Smalheiser and Bonifield’s metric, with an odds ratio threshold of 3. Only suggested compounds that are found by Metab2Mesh tool are represented
The largest part of the suggestion list is associated with terms belonging to the ‘brain diseases’ category (31) and ‘liver diseases’ category (21) in which HE is classified. Twenty compounds were found associated with both liver and brain diseases. Fisher exact test reveals that the high-ranked list is significantly associated with brain and liver disease groups (significance level α = 0.01).
By looking to more detailed levels of the MeSH thesaurus in Figure 5, we can see that four compounds were associated with MeSH terms related to HE symptoms: coma, confusion and consciousness disorders. One is also associated with intracranial hypertension and brain oedema which often occurs in HE patients.
Few compounds were significantly associated with ‘liver failure’ tagged articles. However, many are overrepresented in corpus related to diseases causing the liver failure, and by extension causing HE: five were found associated with hepatitis, five with cirrhosis and other alcohol-related diseases.
Besides association with pathological status, the suggestion list is more generally associated with organs and cellular types (astrocytes, neurotransmitters, blood–brain barrier) that play a central role in the HE (additional MeSH terms from chemical and anatomy categories are provided in Supplementary Table S3).
Regarding association with chemicals related MeSH, 15 were associated with MeSH terms Glutamine, Glutamic acid or Ammonia, which are suggested to play a central role in the pathogenicity of the disease. Fifteen (six more) were associated with molecules used as treatment (Branched-chain amino-acids, Lactulose and sodium benzoate). One (plus one also associated with HE) is associated with bilirubin, which is a marker of liver failure, the main cause of the HE.
Some suggested metabolites are of particular interest in the context of HE. Kynurenic acid (glutamate receptor antagonist) synthesis is inhibited by hyperammonemia and it has been suggested to exacerbate neuro-excitatory effect of ammonia in HE(Albrecht and Jones, 1999). Kynurenic acid (kynurenate in the metabolic network) was not added to the original HE fingerprint because of mass spectrometry detection limit issues, and consequently a high variability of fold changes between patient and control group. However, a closer look to raw data shows a clear homogeneity in the control group and suggests deregulation specific to the HE patient group, corroborated by Mann–Whitney test (P-value < 0.0001) (Fig. 6). Moreover, N-Ω-Hydroxyarginine and N-Ω-l-Arginosuccinate are both involved in the pathway arginine-nitric oxide. It has also been suggested that the N-Ω-Hydroxyarginine inhibit the arginase that produces ornithine and urea from arginine. Finally, Serotonin (PR 24, CR 72) (Knell et al., 1974) and noradrenaline have been shown to increase extracellularly and could be related to the early neuropsychiatric symptom of HE (Shawcross and Jalan, 2005). However, none of these metabolites was detected using our LC/MS methods.
Fig. 6.
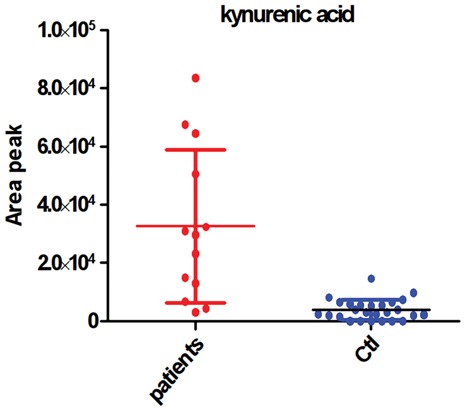
Kynurenic acid concentrations (arbitrary units) in CSF samples from HE and control patients. Data were obtained by LC/MS using a HILIC column and ESI mass spectrometry detection in the negative mode. Kynurenic acid identification level 1 according to (Sumner et al., 2007) (i.e. the same chromatographic retention time, accurate measured mass and MS/MS spectrum as those of the reference compound)
Some other suggested compounds appear to be of interest despite no significant association with relevant MeSH terms. For example, α-Ketoglutaramate, which has been found in CSF of patients with hepatic coma and has been suggested as a biomarker of HE (Halámková et al., 2012). However, very few studies investigated its mechanism, explaining the lack of association with disease MeSH term. This metabolite as well as the enzyme producing it have respectively been described as ‘overlooked’ and ‘underappreciated, but important’, regarding HE and other hyperammonemic diseases (Cooper and Kuhara, 2014).
4 Discussion
PR based methods have recently gained much interest for protein-protein interaction networks. For instance, the Protein Rank (Freschi, 2007) is designed for protein functional annotation, using PPR favouring protein with a selected function as random walk seeds. Another example is the SubNet approach, providing a sub-network extraction from interaction network based on PR scoring (Zhang and Zhang, 2013). It uses a ‘global’ PR implementation where random walks can start from any compounds, but favour starts from nodes of interest by adding a constant parameter to bias the damping. Previously, Iván and Grolmusz also proposed to use PPR for protein-protein interaction networks (Iván and Grolmusz, 2011) and successfully retrieved cancer related proteins from proteomics data of melanoma patients. A similar approach, known as GeneRank (Morrison et al., 2005), has been proposed for gene candidate priorization, using gene correlation networks.
In contrary to protein or gene association networks, centrality-based method has been far less applied to metabolic networks. Faust and colleagues also proposed a random walks based approach [available through the NeAT web server (Brohée et al., 2008)] to extract relevant sub-networks from metabolic networks (Faust et al., 2010), using reactant-pair information to avoid side compounds [RPAIR (Kotera et al., 2004)]. However, this method is mainly focused on KEGG networks and assumes that the list of input metabolites is complete (as it considers only walks linking them) and therefore serves a different purpose than the method proposed here.
The closest implementation was introduced by Bánky et al. (2013) who also used PPR. However, the computation is done on reaction graph and is dedicated to protein target identification. They avoid overscoring hubs by dividing the PPR by the degree of the node. We chose to divide by the global PR, because the carbon conservation rule drastically changes the topology of the network (water is no longer a hub for example) and makes the degree less straightforward to interpret. Finally, they do not use the PCR thus potentially missing upstream metabolites which could be of interest for the interpretation.
Our method is based on a PPR implementation (also known as PageRank with prior) combined with a PCR for precursor suggestion. To the best of our knowledge, this is the first method that allows identifying potential precursors since most of previous studies were limited to PPR. Since our method is focused on metabolic networks, we also added a network pre-processing and a custom transition probability matrix to avoid metabolic network pitfalls, namely side compounds shortcuts and reactions number of product bias. To the best of our knowledge, MetaboRank is the first recommendation system for interpretation of list of metabolites and the first use of the two-dimensional PPR-PCR computation applied to metabolic networks.
A recent study, also took advantage of the PR/CR for providing biological insights (Lages et al., 2018). By analysing the rewiring of signalling networks from cancer cell line and healthy cell line, using PR/CR variations, the authors were able to successfully recover cancer related proteins and suggest meaningful hidden relationships. Though the shared use of 2D analysis using PR and CR, the two methods serve different aims and scopes. While the work of Lages et al. uses a reduced version of the Google matrix, the method proposed in this article uses a personalized version of the two metrics, with several adjustments specific to the analysis of metabolic networks. Other approaches not related to PageRank nor path search could serve similar purpose. The recent work of Massucci et al. (2016), despite not specifically aiming at suggesting nodes of interest, proposes a general Bayesian framework for the inference of perturbation propagation in a network with unobserved nodes, which could allow ranking metabolites according to their probability of being affected by a perturbation. Further development would still be required to extend this general method to metabolic networks, by taking into account reaction directions and side compounds. Furthermore, the use of information regarding observed unaltered nodes to lower the perturbation probability of its unobserved neighbours can be problematic in the context of a metabolomics experiments. In fact, the alteration of a metabolite can only be assessed in the particular biological compartment sampled, at a specific time, making the interpretation of a non-altered metabolite far from trivial.
The damping factor parameter used during the computation of PR and CR is usually chosen empirically, and most applications follow the suggestion of 0.85 from the seminal paper by Brin and Page (Page et al., 1999). Some studies, designed to reveal the impact of the damping factor choice on the ranking of web pages, suggest that the algorithm is not excessively sensitive to the variation of the damping factor (at least on web graphs) and that the value of 0.85 seems appropriate when avoiding false negative constitute a priority (Boldi et al., 2005). Unfortunately, it has never been assessed on metabolic networks and there is no clear recommendation for this type of network. Intuitively, we can see that choosing a low damping factor will decrease the likelihood of encountering long walks. The lack of consensus for an appropriate length of a metabolic pathway complicates the definition of a criterion for choosing the most appropriate damping factor. However, we have shown that the default value proposed in the original paper was still sufficient to obtain meaningful suggestions well related to HE.
One criticism against topological methods applied to metabolic networks is the incompleteness and erroneousness of those networks. Metabolic network content is likely to change over time given that reactions are continually edited, removed or added during manual curation loops (Thiele and Palsson, 2010). The PageRank seems to be relevant for dealing with this instability since it has been claimed to be more robust to small changes in the network topology (Ng et al., 2001) thanks to the damping process that obfuscates less relevant parts of the network (far from the nodes of interest).
5 Conclusion
MetaboRank is a new method to interpret metabolic fingerprints obtained from metabolomics experiments, in the form of a recommendation system. Several adjustments to the original PageRank approach had been made to ensure the biological relevance of obtained results. MetaboRank suggested metabolites that could be related to the disease, from which several were confirmed by the literature. In particular, MetaboRank recommended the overlooked α-Ketoglutaramate as a metabolite that should be added to the fingerprint of HE, thus suggesting that strengthening metabolic fingerprints can provide new insight on complex diseases.
Notably, obtained results show great value for the interpretation of metabolites that were on the edge of significance due to high inter-individual variability. In fact, beside the different level of disease severity between patients, high inter-individual variability may come from pathogenic metabolites involved in highly dynamic processes. This variability makes it difficult to distinguish them from unrelated metabolites, leading to discard them during mechanistic interpretation while still tightly connected to other molecules from the fingerprint. Highly dynamic processes are therefore a key challenge in metabolomics. The recommendation system was able to emphasize two metabolites falling in that case, Taurine and Carnitine, that also appear to play a critical role in the disease according to the literature.
MetaboRank can be applied to metabolomics results from a large range of organisms as it can take any network from a SBML file as input. Furthermore, the proposed mathematical model allows integrating various data at the compound, reaction and reactant pair level. We believe that this method has the potential to facilitate metabolic network exploration by focusing on most relevant metabolites and could help the elucidation of perturbed metabolic mechanisms and the identification of new drug targets. It can also be combined with mechanistic interpretation methods such as pathway enrichment or sub-network extraction. Computed scores can be used as a weighting scheme before subnetwork extraction, such as paths or Steiner Tree computation, which would prior high-scored compounds (Frainay and Jourdan, 2017).
MetaboRank also shows a great potential in the metabolite tedious identification process. In the HE application, some suggested metabolites, like kynurenic acid, had been a posteriori added to the metabolic fingerprint by going back to raw data. Iterative loops between the manual identification from raw data and the suggestion algorithm thus allow refining the metabolic fingerprint and increasing confidence in the mechanistic interpretations inferred from the suggestion list. Cross validation of the algorithm in the missing data prediction context can be found in Supplementary Material.
This work could be extended by integrating various data at the compound level, reaction level and reactant pair level, using custom transition probabilities based on other omics data or by modifying the topology of the network. The prior vector can also be set to favour some starting nodes among others during the damping phase, based for example on the fold changes obtained from metabolomics results.
Supplementary Material
Acknowledgements
‘PageRank’ is a registered trademark of Google. Inc.
C.F would like to thank Dr Dima Shepelyansky (Paul Sabatier University, Toulouse, France) and Dr Leonardo Ermann (CNEA, Argentina) for their insightful courses about Google Matrix and fruitful discussions at the Luchon Summer School.
The authors also thank Dr Sara Fontanella for proof-reading the manuscript.
Funding
This work was supported by the French Ministry of Research and National Research Agency as part of the French MetaboHUB; the national metabolomics and fluxomics infrastructure [Grant ANR-INBS-0010]; and by PhenoMeNal project, European Commission’s Horizon 2020 programme [Grant 654241].
Conflict of Interest: none declared.
References
- Albrecht J., Jones E.A. (1999) Hepatic encephalopathy: molecular mechanisms underlying the clinical syndrome. J. Neurol. Sci, 170, 138–146. [DOI] [PubMed] [Google Scholar]
- Allesina S., Pascual M. (2009) Googling food webs: can an eigenvector measure species’ importance for coextinctions? PLoS Comput. Biol., 5, e1000494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman T., et al. (2013) A systematic comparison of the MetaCyc and KEGG pathway databases. BMC Bioinformatics, 14, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita M. (2004) The metabolic world of Escherichia coli is not small. Proc. Natl. Acad. Sci. USA, 101, 1543–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backstrom L., Leskovec J. (2011) Supervised random walks. In: Proceedings of the Fourth ACM International Conference on Web Search and Data Mining – WSDM ‘11, p. 635. ACM Press, New York, NY, USA. [Google Scholar]
- Bánky D., et al. (2013) Equal opportunity for low-degree network nodes: a pagerank-based method for protein target identification in metabolic graphs. PLoS One, 8, e54204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum T., Kohlbacher O. (2008) Using atom mapping rules for an improved detection of relevant routes in weighted metabolic networks. J. Comput. Biol., 15, 565–576. [DOI] [PubMed] [Google Scholar]
- Boldi P., et al. (2005) PageRank as a function of the damping factor. In: Proceedings of the 14th International Conference on World Wide Web – WWW ‘05, p. 557. ACM Press, New York, NY, USA. [Google Scholar]
- Brin S., et al. (1998) The anatomy of a large-scale hypertextual Web search engine. Comput. Netw. ISDN Syst., 30, 107–117. [Google Scholar]
- Brohée S., et al. (2008) NeAT: a toolbox for the analysis of biological networks, clusters, classes and pathways. Nucleic Acids Res., 36, W444–W451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagoyen M., Pazos F. (2011) MBRole: enrichment analysis of metabolomic data. Bioinformatics, 27, 730–731. [DOI] [PubMed] [Google Scholar]
- Cooper A.J.L., Kuhara T. (2014) α-Ketoglutaramate: an overlooked metabolite of glutamine and a biomarker for hepatic encephalopathy and inborn errors of the urea cycle. Metab. Brain Dis., 29, 991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottret L., et al. (2010) MetExplore: a web server to link metabolomic experiments and genome-scale metabolic networks. Nucleic Acids Res., 38, W132–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creek D.J., et al. (2014) Metabolite identification: are you sure? And how do your peers gauge your confidence? Metabolomics, 10, 350–353. [Google Scholar]
- Croes D., et al. (2005) Metabolic PathFinding: inferring relevant pathways in biochemical networks. Nucleic Acids Res., 33, W326–W330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M., et al. (2015) ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res., 43, W612–W620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermann L., Shepelyansky D.L. (2015) Google matrix analysis of the multiproduct world trade network. Eur. Phys. J. B, 88, 84. [Google Scholar]
- Faust K., et al. (2009) Metabolic pathfinding using RPAIR annotation. J. Mol. Biol., 388, 390–414. [DOI] [PubMed] [Google Scholar]
- Faust K., et al. (2010) Pathway discovery in metabolic networks by subgraph extraction. Bioinformatics, 26, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O. (2002) Metabolomics – The Link between Genotypes and Phenotypes. Springer, Dordrecht, pp. 155–171. [PubMed] [Google Scholar]
- Frainay C., Jourdan F. (2017) Computational methods to identify metabolic sub-networks based on metabolomic profiles. Brief. Bioinform., 18, 43–56. [DOI] [PubMed] [Google Scholar]
- Freschi V. (2007) Protein function prediction from interaction networks using a random walk ranking algorithm. In: 2007 IEEE 7th International Symposium on BioInformatics and BioEngineering, pp. 42–48. IEEE. [Google Scholar]
- Gupta P., et al. (2013) WTF, the who to follow service at Twitter. In: Proceedings of the 22nd International Conference on World Wide Web – WWW ‘13, pp. 505–514. ACM Press, New York, NY, USA. [Google Scholar]
- Halámková L., et al. (2012) Enzymatic analysis of α-ketoglutaramate—a biomarker for hyperammonemia. Talanta, 100, 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haveliwala T.H. (2002) Topic-sensitive PageRank. In: Proceedings of the Eleventh International Conference on World Wide Web – WWW ‘02, p. 517. ACM Press, New York, NY, USA. [Google Scholar]
- Heller S.R., et al. (2015) InChI, the IUPAC international chemical identifier. J. Cheminform., 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocher B., Adamski J. (2017) Metabolomics for clinical use and research in chronic kidney disease. Nat. Rev. Nephrol., 13, 269–284. [DOI] [PubMed] [Google Scholar]
- Holme P. (2009) Model validation of simple-graph representations of metabolism. J. R. Soc. Interface, 6, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucka M., et al. (2003) The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics, 19, 524–531. [DOI] [PubMed] [Google Scholar]
- Iván G., Grolmusz V. (2011) When the web meets the cell: using personalized PageRank for analyzing protein interaction networks. Bioinformatics, 27, 405–407. [DOI] [PubMed] [Google Scholar]
- Jeong H., et al. (2000) The large-scale organization of metabolic networks. Nature, 407, 651–654. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., et al. (2014) Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res., 42, D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knell A.J., et al. (1974) Dopamine and serotonin metabolism in hepatic encephalopathy. Br. Med. J., 1, 549–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotera M., et al. (2004) RPAIR: a reactant-pair database representing chemical changes in enzymatic reactions. Genome Informatics, 15, 62. [Google Scholar]
- Lacroix V., et al. (2008) An introduction to metabolic networks and their structural analysis. IEEE/ACM Trans. Comput. Biol. Bioinform., 5, 594–617. [DOI] [PubMed] [Google Scholar]
- Lages J., et al. (2018) Inferring hidden causal relations between pathway members using reduced Google matrix of directed biological networks. PLoS One, 13, e0190812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang B., et al. (2014) Searching for people to follow in social networks. Expert Syst. Appl., 41, 7455–7465. [Google Scholar]
- Liben-Nowell D., Kleinberg J. (2007) The link-prediction problem for social networks. J. Am. Soc. Inf. Sci. Technol., 58, 1019–1031. [Google Scholar]
- Ma N., et al. (2008) Bringing PageRank to the citation analysis. Inf. Process. Manag., 44, 800–810. [Google Scholar]
- Massucci F.A., et al. (2016) Inferring propagation paths for sparsely observed perturbations on complex networks. Sci. Adv., 2, e1501638–e1501638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalcea R., et al. (2004) PageRank on semantic networks, with application to word sense disambiguation. In: Proceedings of the 20th International Conference on Computational Linguistics – COLING ‘04. Association for Computational Linguistics, p. 1126–es. Morristown, NJ, USA. [Google Scholar]
- Milreu P.V., et al. (2014) Telling metabolic stories to explore metabolomics data: a case study on the yeast response to cadmium exposure. Bioinformatics, 30, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misel M.L., et al. (2013) Sodium benzoate for treatment of hepatic encephalopathy. Gastroenterol. Hepatol. (N.Y.), 9, 219–227. [PMC free article] [PubMed] [Google Scholar]
- Mo M.L., Palsson B.Ø. (2009) Understanding human metabolic physiology: a genome-to-systems approach. Trends Biotechnol., 27, 37–44. [DOI] [PubMed] [Google Scholar]
- Morrison J.L., et al. (2005) GeneRank: using search engine technology for the analysis of microarray experiments. BMC Bioinformatics, 6, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S., Böcker S. (2010) Computational mass spectrometry for metabolomics: identification of metabolites and small molecules. Anal. Bioanal. Chem., 398, 2779–2788. [DOI] [PubMed] [Google Scholar]
- Ng A.Y., et al. (2001) Stable algorithms for link analysis. In: Proceedings of the 24th Annual International ACM SIGIR Conference on Research and Development in Information Retrieval – SIGIR ‘01, pp. 258–266. ACM Press, New York, NY, USA. [Google Scholar]
- Page L., et al. (1999) The PageRank Citation Ranking: Bringing Order to the Web . Stanford InfoLab, Stanford, CA, USA. [Google Scholar]
- Perney P., et al. (1998) Plasma and CSF benzodiazepine receptor ligand concentrations in cirrhotic patients with hepatic encephalopathy: relationship to severity of encephalopathy and to pharmaceutical benzodiazepine intake. Metab. Brain Dis., 13, 201–210. [DOI] [PubMed] [Google Scholar]
- Persicke M., et al. (2012) MSEA: metabolite set enrichment analysis in the MeltDB metabolomics software platform: metabolic profiling of Corynebacterium glutamicum as an example. Metabolomics, 8, 310–322. [Google Scholar]
- Rahman S.A., et al. (2005) Metabolic pathway analysis web service (Pathway Hunter Tool at CUBIC). Bioinformatics, 21, 1189–1193. [DOI] [PubMed] [Google Scholar]
- Rahman S.A., et al. (2016) Reaction Decoder Tool (RDT): extracting features from chemical reactions. Bioinformatics, 32, 2065–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupasinghe J., Jasinarachchi M. (2011) Progressive encephalopathy with cerebral oedema and infarctions associated with valproate and diazepam overdose. J. Clin. Neurosci., 18, 710–711. [DOI] [PubMed] [Google Scholar]
- Sartor M.A., et al. (2012) Metab2MeSH: annotating compounds with medical subject headings. Bioinformatics, 28, 1408–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawcross D., Jalan R. (2005) The pathophysiologic basis of hepatic encephalopathy: central role for ammonia and inflammation. Cell. Mol. Life Sci., 62, 2295–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalheiser N., Bonifield G. (2016) Two similarity metrics for medical subject headings (MeSH): an aid to biomedical text mining and author name disambiguation. J. Biomed. Discov. Collab, 7, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreekumar A., et al. (2009) Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature, 457, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Steinbeck C., et al. (2003) The Chemistry Development Kit (CDK): an open-source Java library for Chemo- and Bioinformatics. J. Chem. Inf. Comput. Sci., 43, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner L.W., et al. (2007) Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics, 3, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swainston N., et al. (2016) Recon 2.2: from reconstruction to model of human metabolism. Metabolomics, 12, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I., et al. (2013) A community-driven global reconstruction of human metabolism. Nat. Biotechnol., 31, 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I., Palsson B.Ø. (2010) A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc., 5, 93–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weininger D. (1988) SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model., 28, 31–36. [Google Scholar]
- Weiss N., et al. (2016) Cerebrospinal fluid metabolomics highlights dysregulation of energy metabolism in overt hepatic encephalopathy. J. Hepatol., 65, 1120–1130. [DOI] [PubMed] [Google Scholar]
- Weiss N., et al. (2018) Understanding hepatic encephalopathy. Intensive Care Med, 44, 231–234. [DOI] [PubMed] [Google Scholar]
- Wimalaratne S.M., et al. (2014) BioModels linked dataset. BMC Syst. Biol., 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S., et al. (2013) HMDB 3.0 – the human metabolome database in 2013. Nucleic Acids Res., 41, D801–D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlgemuth G., et al. (2010) The Chemical Translation Service – a web-based tool to improve standardization of metabolomic reports. Bioinformatics, 26, 2647–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Zhang Z.D. (2013) SubNet: a Java application for subnetwork extraction. Bioinformatics, 29, 2509–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhirov A.O., et al. (2010) Two-dimensional ranking of Wikipedia articles. Eur. Phys. J. B, 77, 523–531. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.