

One Sentence Summary:
Combined human and animal model studies conclusively implicate microbiota-triggered oral mucosal Th17 cells as drivers of local immunopathology and therapeutic targets in periodontitis.
Abstract
Periodontitis is one of the most common human inflammatory diseases, yet the mechanisms that drive immunopathology and could be therapeutically targeted are not well defined. Here, we demonstrate an expansion of resident memory Th17 cells in human periodontitis. Phenocopying humans, Th17 cells expanded in murine experimental periodontitis through local proliferation. Unlike homeostatic oral Th17 cells, which accumulate in a commensal-independent and IL-6-dependent manner, periodontitis-associated expansion of Th17 cells was dependent upon the local dysbiotic microbiome and required both IL-6 and IL-23. Importantly, Th17 cells and associated neutrophil accumulation were necessary for inflammatory tissue destruction in experimental periodontitis. Genetic or pharmacological inhibition of Th17 cell differentiation conferred protection from immunopathology. Studies in a unique patient population with a genetic defect in Th17 cell differentiation established human relevance for our murine experimental studies. Indeed, in the oral cavity, human Th17 cell defects were associated with diminished periodontal inflammation and bone loss, despite increased prevalence of recurrent oral fungal infections. Our study highlights distinct functions of Th17 cells in oral immunity and inflammation and paves the way to a new targeted therapeutic approach for the treatment of periodontitis.
Introduction
Periodontitis is one of the most prevalent human inflammatory diseases and poses a significant economic and public health burden (1). In this condition, exaggerated inflammatory responses in the oral mucosal tissues surrounding the dentition (gingiva) lead to immunopathology and destruction of supporting bone (2). To date, the pathogenic drivers of exaggerated destructive inflammation are incompletely understood and treatment largely aims at reduction of microbial stimulation rather than targeting of specific immune pathways through host-modulation therapies (2). In the lesions of human chronic periodontitis, expression of the cytokine interleukin (IL-) 17 (3–6) and an abundance of T helper (Th) 17 cells have been reported, but not causally linked, to periodontal disease pathogenesis (3, 5–7). Here, we test the hypothesis that Th17 cells are drivers of pathogenic mucosal inflammation in periodontitis.
In various mucosal surfaces, Th17 cells are critical regulators of barrier immunity. Yet, amplification and dysregulation of IL-17-secreting cells in the setting of disease has been linked to immunopathology (8–10). Th17 cells have been implicated in the development of psoriasis in the skin and colitis at the lower gastrointestinal (GI) tract (8–10). The physiologic immune-protective role of Th17 cells is particularly evident at the oral mucosal barrier. Defects in Th17 cells and in IL-17 cytokine signaling underlie susceptibility to oral fungal infection (candidiasis) in both human and murine models (11). Specifically, patients with primary immunodeficiencies affecting either Th17 cell differentiation or function (12, 13) all have susceptibility to oral candidiasis. Whereas the homeostatic role of Th17 cells at the oral mucosa is well delineated, the role of Th17 cells in the development of oral immunopathology, particularly in humans, has not been conclusively defined.
Importantly, periodontitis provides an attractive setting to study Th17-associated chronic inflammation in humans (14). This disease is particularly common and readily accessible for obtaining mucosal biopsy samples and corresponding microbiome samples directly from specific mucosal microenvironments, allowing for direct evaluation of the host-microbiome interplay at a specific site. Furthermore, Th17 cells in the oral mucosa have a well-established role in oral antifungal immunity (11), but are also associated with local inflammatory lesions in periodontitis. Therefore, oral tissue is a unique setting in which to dissect distinct regulation and function of Th17 cells in tissue immunity and inflammation.
Previous work in our laboratory has highlighted unique requirements for homeostatic development of Th17 cells at the oral mucosa (15). Unlike other barrier sites, such as the skin and lower GI tract, Th17 cells may arise at the oral mucosa independently of commensal colonization. Indeed, ongoing damage through mastication is a unique tissue-specific trigger for the development of homeostatic Th17 cells in oral tissues in health (15). Yet, the mechanisms implicated in the amplification and dysregulation of Th17 cells in chronic periodontitis are poorly understood. We aimed to characterize the phenotypic and functional characteristics of human periodontitis-associated Th17 cells and investigated Th17 cell induction and functionality in periodontitis through complementary studies in experimental models and human systems.
Here, we document expansion of memory tissue-resident Th17 cells that secrete cytokines linked to pathogenicity in human periodontitis. Mirroring human findings, in experimental periodontitis Th17 cells preferentially expanded (compared to other IL-17+ cells) through local proliferation. Whereas health-associated Th17 cells accumulated in a commensal-independent and IL-6-dependent manner (15), in periodontitis, the disease-associated microbiota triggered the expansion of local Th17 cells in a manner dependent on both IL-6 and IL-23, revealing divergent regulation of oral Th17 cells in health versus disease. Importantly, Th17 cells were required for inflammatory periodontal tissue destruction. Selective inhibition of Th17 cell differentiation, using genetic or pharmacological approaches, inhibited periodontitis, suggesting Th17 cells as a therapeutic target in periodontitis. Moreover, in humans, a genetic defect in Th17 cell differentiation was associated with reduced periodontal inflammation and bone loss. Collectively, our use of concurrent and consistent evidence from both experimental and clinical models reveal microbially-driven Th17 cells as pathogenic drivers in periodontitis.
Results
Th17 cell signature in the oral inflammatory disease periodontitis in humans
Human lesions of periodontitis were characterized by significant infiltration of immune cells with high representation of T cells within the infiltrates (6, 16) (Fig. 1A-C). Amongst known T helper associated factors, IL-17 had the highest expression, suggesting a bias towards Th17 cell differentiation in lesions of periodontitis (Fig. 1D) (16, 17). Consistent with increased IL-17A expression, the number and proportion of CD45+IL-17+ cells was significantly increased in periodontitis compared to health (p<0.05, Fig. 1E-F, Fig. S1A). Characterization of the subtypes of IL-17+ cells, revealed that the overwhelming majority (~ 80%) are CD4+IL-17+ T cells (Th17 cells) with minimal CD8+T IL-17+, TCRγδ+IL-17+ T cells and innate lymphocytes (CD45+/CD3-/CD19-/CD20-/CD1a-/CD11c-/CD14-/FcɛR1α-/CD16-/CD34-IL-17+) (Fig. 1G). Indeed, Th17 cells were significantly increased in periodontitis lesions compared to health (p<0.05, Fig. 1H-I, Fig. S1B) and, importantly, their proportion correlated with disease severity as reflected by tissue destruction-bone loss in millimiters (Fig. 1J). Th17 cells in periodontitis were almost exclusively memory cells (CD45RO+CD45RA-~99%) and predominantly tissue resident cells, primarily resident effector memory (rTem~60%) and secondarily resident central memory T cells (rTcm~20%) (Fig. 1K-M). Periodontitis-associated Th17 cells co-produced cytokines linked to pathogenicity (10) such as Granulocyte-Macrophage-Colony-Stimulating-Factor (GM-CSF~ 30%) and Intereferonγ (IFNγ~ 15%), whereas a subset of Th17 cells co-produced IL-22 (~ 15%) (Fig. 1N-O).
Fig. 1. Th17 cells in human periodontitis.
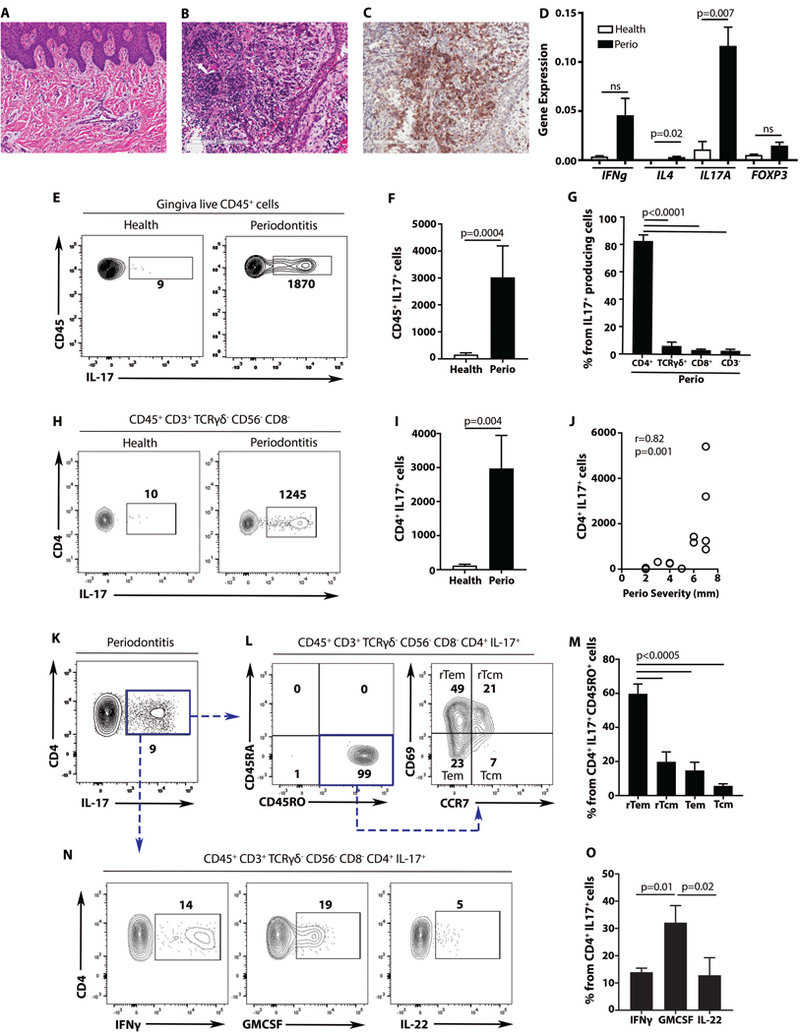
H&E of healthy (A) and periodontitis (B) gingiva. (C) CD3/T cell immunohistochemical staining in periodontitis (original magnification 15x). (D) mRNA expression for IFNg, IL4, IL17A and FOXP3 in health and periodontitis (n=3/6, health/periodontitis, unpaired t test, mean±SEM). (E-G) IL-17+CD45+ in health and periodontitis. FACS plot (E) and graph (F) showing numbers of CD45+IL-17+ cells per standardized biopsy (n=11/7, health/periodontitis, Mann-Whitney test, mean±SEM). (G) IL-17+ cellular sources in periodontitis (one-way ANOVA, Holm-Sidak’s multiple comparisons test, mean±SEM). (H-J) IL-17+CD4+ in health and periodontitis. FACS plot (H) and graph (I) showing numbers of CD4+IL-17+ cells per standardized biopsy (n=11/7, health/periodontitis, unpaired t test, mean±SEM). (J) Spearman correlation of CD4+IL-17+ cells with bone loss in mm (n=18 patients). (K-O) Th17 cells in periodontitis. FACS plot of CD4+IL-17+ (K), CD4+IL-17+ cells expressing CD45RO, CD45A, CCR7, CD69 (L). Frequencies of CD4+IL-17+ resident effector (rTem), resident central (rTcm), effector (Tem) and central (Tcm) memory T cells (M). CD4+IL-17+ cells co-producing IFNγ, GMCSF or IL-22, FACS Plot (N) and graph (O) (n=4–9, ANOVA and Holm-Sidak’s (M) or Tukey’s (O) multiple comparisons tests, mean±SEM). All p values are indicated in graphs.
Selective expansion of Th17 cells in experimental periodontitis
To mechanistically dissect the role of Th17 cells in periodontal inflammation, we utilized an established murine model of experimental periodontitis, ligature-induced periodontitis (LIP). In LIP, atraumatic placement of a silk suture (ligature) around the second molar tooth leads to local accumulation of bacteria and local gingival inflammation followed by destruction of tooth-supporting bone (18) (Fig. S2A-D). Within the inflammatory lesions of experimental periodontitis, there was a profound increase in Il17a gene expression (30- to 80-fold increase over baseline) (Fig. 2A), while other T helper-associated cytokines and inflammatory mediators were essentially unaltered (Fig. S2E).
Fig. 2. Preferential expansion of Th17 cells in experimental periodontitis.
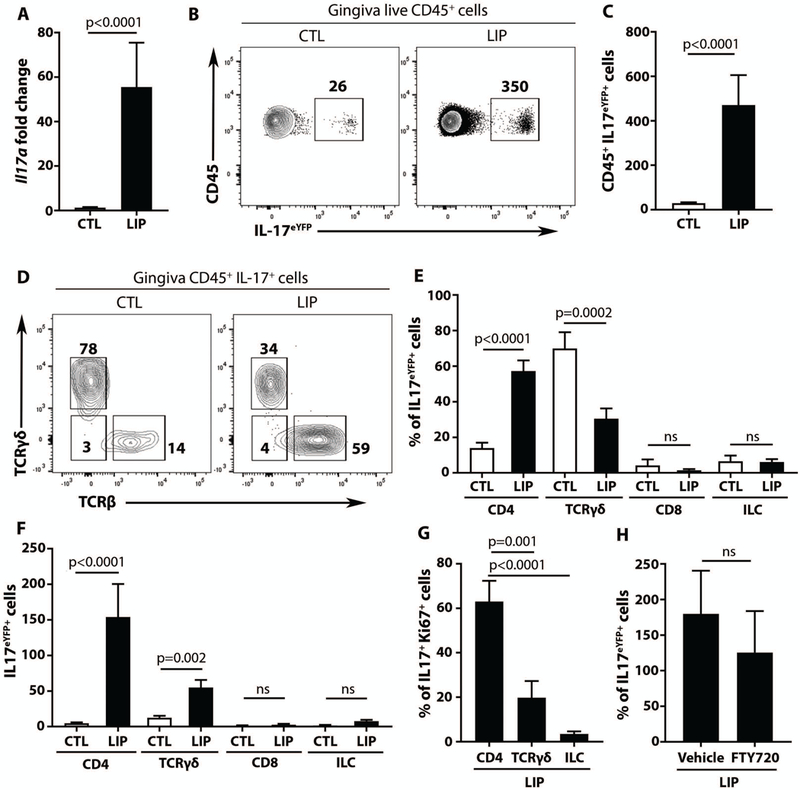
(A) IL-17a mRNA expression in gingival tissues at baseline (CTL) and after ligature induced periodontitis (LIP) (n=10 mice per group, 2 separate experiments, Mann Whitney test, mean±SEM). (B-C) IL-17+CD45+ cells at baseline and LIP, in IL-17acreR26ReYFP mice. FACS plot (B) and graph (C) indicating numbers of CD45eYFP+ cells per standardized tissue (n=12 per group, 3 separate experiments, unpaired t test, mean±SEM). (D-F) Proportions and numbers of IL-17+ cells at baseline and LIP. FACS plots (D) and graph (E) showing percentage of eYFP+ cells. (F) Graph showing numbers of eYFP+, per standardized gingival tissue (n=10, 3 separate experiments). (G) Ki67+ staining in IL-17+ cells in LIP (n=6, 2 separate experiments, one-way ANOVA and Tukey’s multiple comparisons test, mean±SEM for E-G). (H) Numbers of eYFP+ cells per gingival tissue in LIP with/without FTY720 (n=5, 2 separate experiments, Mann-Whitney test, mean±SEM). All p values are indicated in graphs.
To investigate the cellular sources of IL-17, a reporter mouse designed to map the fate of cells that have activated IL-17 expression (IL-17acreR26ReYFP) was employed. This IL-17 reporter mouse was subjected to experimental periodontitis and cellular sources of IL-17 at baseline and after 5 days of LIP were evaluated. Consistent with an increase in IL-17a gene expression during LIP, and similar to the human findings (Fig. 1E-G), CD45+IL-17+ cell number and (Fig. 2B, C) CD4+IL-17+ cell proportion and number (Fig. 2D, E, F, Fig. S2F-H) significantly increased in disease lesions (all, p<0.05). These data confirm this as a highly relevant animal model to dissect Th17 cell triggering and functionality in periodontal inflammation.
Interestingly, distinct cellular sources of IL-17 were involved in periodontal homeostasis versus inflammation. At baseline, TCRγδ+ T cells were the major IL-17 source and represented 60–70% of total IL-17+ cells. TCRαβ+CD4+IL-17+ (Th17) cells constituted approximately 10% of total CD45+IL-17+ cells followed by ILC-IL-17+ cells (TCRβ-, TCRγδ-, Ly6C-, Ly6G- B220-, CD11b-, CD11c-, Thy-1.2, CD90.2+) and a minimal proportion of CD8+IL-17+ cells at steady state (Fig. 2D, E). In contrast, in the lesions of experimental periodontitis, the dominant IL-17+ cell population were Th17 cells (Fig 2D, E). We attributed the accumulation of Th17 cells to increased local proliferation. Indeed, proliferating (Ki67+) IL-17+ cells in the lesions of periodontitis were primarily Th17 cells, followed by γδT IL-17+ cells (Fig. 2G). Moreover, consistent with local expansion of Th17 cells, numbers of Th17 cells in LIP were essentially unchanged when mice were treated with the lymph node egress inhibitor FTY720 (Fig. 2H). Interestingly, CD4+ T cells expanding in LIP were predominantly Nur77eGFP+. These findings suggest that CD4+ T cells in LIP have active TCR engagement (Fig. S3A-C). Collectively, our data document preferential local expansion of Th17 cells in experimental periodontitis.
Expansion of Th17 cells in periodontitis is dependent on IL-6 and IL-23
We next examined cytokine requirements for accumulation of Th17 cells during LIP. IL-6 has been shown to be required for homeostatic Th17 cell accumulation in gingiva (15). Indeed, Il6-deficient mice had significantly reduced (almost undetectable) Th17 cells at baseline (CTL) and a significant reduction of Th17 cell accumulation during LIP (Fig. 3A-B, both p<0.05). IL-1 and IL-23 were also examined, as both have been linked to Th17 differentiation and/or expansion in other settings (9). Interestingly, IL-1 was dispensable for gingival Th17 cell accumulation in LIP (Fig. 3C-D) whereas IL-23 was required (Fig. 3E-F). Indeed, Il1r1-deficient mice had comparable Th17 cell numbers with wildtype (WT) controls, while Il23a (p19)-deficient mice displayed a significant reduction in Th17 cell accumulation during LIP (p<0.05). Taken together, these data highlight divergent regulation of Th17 cells in gingiva in health and disease (LIP), with IL-6 being the necessary trigger for homeostatic accumulation of Th17 cells, whereas both IL-6 and IL-23 (but not IL-1) are required for Th17 cell expansion in disease.
Fig. 3. Cytokine requirements for Th17 cell accumulation during experimental periodontitis.
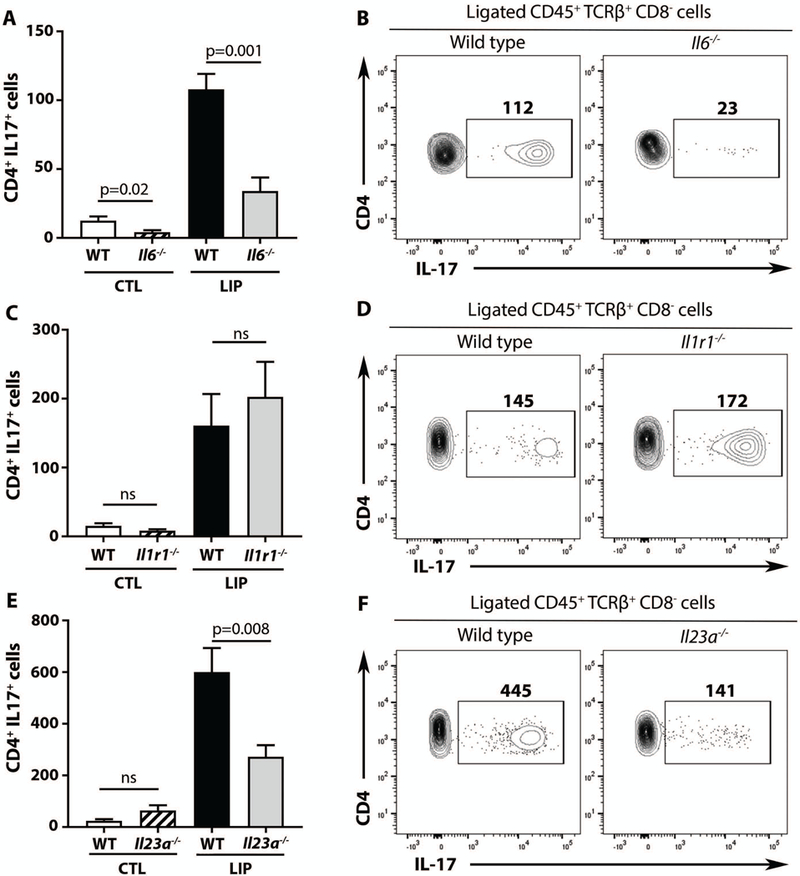
(A-F) IL-17 production by CD4+ cells at baseline (CTL) and LIP. Bar graphs and FACS plots show numbers of Th17 cells at CTL and after LIP in Il6−/− and Il6+/+ mice (A-B), Il1r1−/− and Il1r1+/+ mice (C, D), Il23a−/− and Il23a+/+ mice (E-F). All data are representative of 2–3 separate experiments, n=6–8 for each group, p values determined by Mann-Whitney test, mean±SEM.
Expansion of Th17 cells in periodontitis is triggered by the microbiome
In comparison to health, LIP featured local microbial communities with shifts in the relative abundance of commensal species and significant alterations in community composition and structure (Fig. 4A-C, p<0.05). Therefore, LIP is associated with alterations to local microbial communities consistent with dysbiosis, which might trigger local gingival Th17 cell amplification in periodontitis. To assess the contribution of the microbiome to Th17 cell accumulation, mice were placed on a broad-spectrum systemic antibiotic cocktail (Doripenem-Vancomycin-Neomycin) for 2 weeks and subsequently were subjected to LIP experiments in the presence of continuous antibiotic treatment. In the presence of broad spectrum antibiotics (ATB), the numbers of CD45+IL-17+ cells remained unchanged despite LIP (Fig. 4D). Although antibiotic treatment significantly impacted Th17 cell proportions (Fig. S4A, p<0.05) and cell numbers, it did not affect other IL-17 cellular sources (Fig. 4E). Indeed, Th17 cell numbers were significantly lower during LIP in the ATB group (p<0.05), while the numbers of TCRγδ+IL-17+ and ILCIL-17+ cells were not affected (Fig. 4E), suggesting that (i) the microbiota specifically triggers Th17 cell accumulation in the gingival mucosa and (ii) that Th17 and not other IL-17+ cells are likely to drive periodontitis. Reduction of Th17 cell accumulation in the presence of antibiotics correlated with nearly absent proliferation (as assessed by Ki67 staining) of Th17 cells (Fig 4F).
Fig.4. Expansion of Th17 cells in periodontitis disease-associated bacteria.
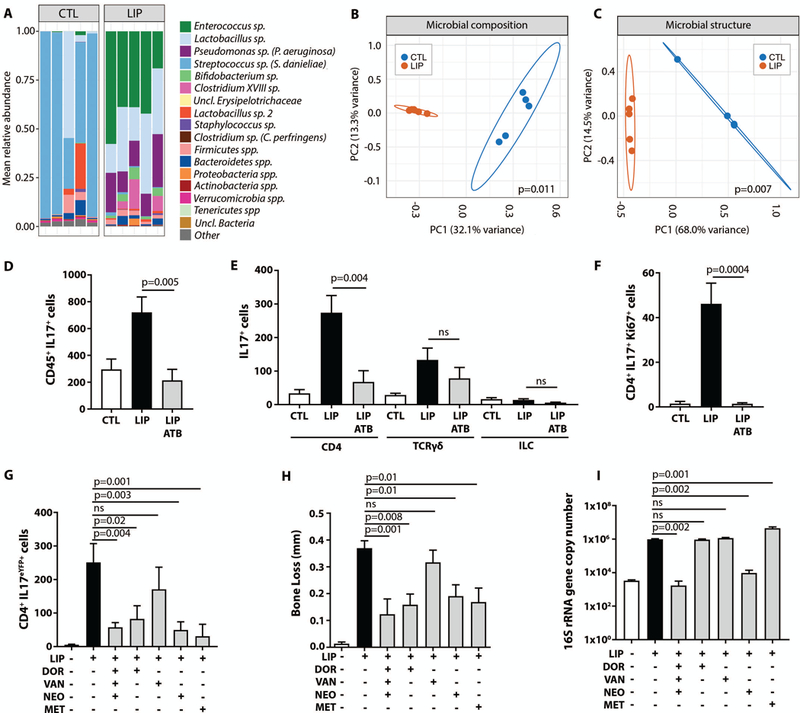
(A) Microbiome composition at the OTU level. Most abundant OTUs are classified at the species-level (10) and less dominant OTUs are shown combined at phylum level. (B) Principal Coordinates Analysis (PCoA) plot of global microbial community composition and (C) community structure at baseline (CTL) and LIP, p values were determined using AMOVA and 95% confidence ellipse were depicted. (D) Numbers of CD45+IL-17+ cells at CTL and after LIP without/with broad-spectrum antibiotic cocktail (ATB = Doripenem-Vancomycin-Neomycin) (n=7 per group, 2 separate experiments, one-way ANOVA and Tukey’s multiple comparisons test, mean±SEM). (E) Numbers of IL-17+(CD4, TCRγδ and ILC) at CTL and after LIP without/with antibiotics (ATB) (n=7 per group, 2 separate experiments). (F) Numbers of CD4+IL-17+Ki67+ cells CTL and LIP with/without antibiotics (ATB) (n=7, 2 separate experiments, Kruskal-Wallis test and Dunn’s multiple comparisons test, mean±SEM for E-F). (G) CD4+IL-17+ cell numbers , (H) Bone loss in millimeters and (I) Total oral microbial biomass at CTL and after LIP with/without antibiotic treatment (DOR=Doripenem; VAN=Vancomycin; NEO=Neomycin; MET=Metronidazole) (n=6–9 per group, 2 separate experiments, one-way ANOVA and Tukey’s multiple comparisons test, mean± SEM). All p values are indicated in graphs.
Dysbiotic changes in the microbiome trigger Th17 cell accumulation in Periodontitis
To assess the spectrum of bacteria that mediate Th17 cell accumulation, animals were placed on different single regimen antibiotics during LIP. Treatment with the broad-spectrum antibiotic Doripenem (which targets Gram-positive, Gram-negative and anaerobic bacteria) was effective in inhibiting Th17 cell expansion (Fig. 4G). Vancomycin and ampicillin, which typically target Gram-positive bacteria did not affect Th17 cells (Fig. 4G, Fig. S4B). Neomycin and metronidazole both significantly inhibited Th17 cell accumulation, when used as single agents (Fig. 4G, p<0.05). Neomycin is considered effective against Gram-negative and some Gram-positive bacteria and metronidazole targets mostly anaerobic bacteria. Importantly, antibiotics that inhibited Th17 cells led to a concurrent significant inhibition of periodontal bone loss (p<0.05), while vancomycin, which did not inhibit Th17 cells, also failed to inhibit periodontal bone loss (Fig. 4H). These findings strongly suggest a causative link between periodontitis-associated microbiota, Th17 cell accumulation and periodontal bone loss.
Next, we inquired whether Th17 cell accumulation was dependent on an increase in bacterial biomass. We reasoned that if increased biomass was necessary for Th17 accumulation, then antibiotics which inhibit Th17 cells would also lead to a decrease in bacterial biomass. Whereas combination antibiotics and neomycin alone were effective in both reduction of microbial load and Th17 cells, doripenem and metronidazole were efficient in inhibiting Th17 cell accumulation without reduction of microbial biomass (Fig. 4I). These data indicate that, rather than global reduction in biomass, specific alterations in microbial communities are likely to be responsible for microbial-Th17 triggering in periodontitis.
Finally, the specific shifts in microbial communities which are linked to Th17 cell triggering were characterized. 16SrRNA gene based sequencing of LIP-associated communities in the presence or absence of different antibiotic regimens revealed effects of each antibiotic on the oral microbiome. (Fig. S4C). Combination of broad spectrum antibiotics as well as neomycin targeted all bacteria associated with LIP, through significant reduction in bacterial biomass. Unlike neomycin, doripenem and metronidazole (which also inhibit Th17 cell accumulation) did not target major constituents of LIP-associated communities (Cluster 1: Enterococcus sp., Lactobacillus sp. and Pseudomonas sp.) (Fig. S4C). Interestingly, doripenem and metronidazole did inhibit other LIP- associated bacteria (Cluster 2: Porphyromonadaceae spp., Lachnospiraceae spp., Erysipelotrichaceae spp., among other taxa); however, these constituents were also targeted by vancomycin, which did not inhibit Th17 cell accumulation (Fig. S4C). Different antibiotic regimens also resulted in an increase in differential abundance of select species.Therefore, our analysis did not reveal single bacterial candidates that may trigger Th17 expansion but demonstrated unique shifts of bacterial depletion and overgrowth which occurred with each antibiotic regimen. These data suggest that changes in the balance of the community may underlie dysbiosis and pathogenicity.
Inhibition of Th17 cell differentiation confers protection from inflammatory bone loss
To definitively implicate Th17 cells as potential drivers of periodontal inflammatory bone loss, a murine model with a genetic defect in Th17 cell differentiation was employed. In this model, the Th17 cell transcription factor Stat3 was deleted within the CD4 compartment (Cd4creStat3fl/fl mice). Cd4creStat3fl/fl mice did not have detectable Th17 cells within their gingival mucosa, neither at steady state nor after induction of experimental periodontitis (Fig. 5A, Fig. S5A). In contrast, other cellular sources of IL-17 (TCRγδ cells and ILC) remained essentially intact at both steady state and after experimental periodontitis (Fig. 5C-F), thus validating this model for targeting of Th17 cells. Lack of Th17 cells in Cd4creStat3fl/fl resulted in a significant decrease in total CD45+IL-17+ cells (p<0.05) only upon experimental periodontitis (i.e. no difference in total IL-17+ cells was detectable at steady state) (Fig. 5G, H, Fig S5B). Therefore, Th17 cells have minimal contribution to the IL-17 pool at steady state but a major contribution to IL-17+ cell expansion during disease.
Fig. 5. Genetic inhibition of Th17 cells protects from periodontal bone loss.

IL-17+ cells in Cd4CreStat3 fl/fl mice and littermate controls before and after LIP. Representative FACS plots from LIP and graphs showing number of IL-17+ cells from CTL and LIP, for (A-B) TCRβ+CD4+IL-17+, (C-D) TCRγδ+IL-17+, (E-F) ILC(Lin-CD90.2+),(lineage=TCRβ, TCRγδ, Ly6C, Ly6G, B220, CD11b, CD11c,), (G-H) CD45+IL-17+. (n=7 per group, 3 separate experiments). (I) Bone loss (in mm) after LIP in Cd4CreStat3 fl/fl mice and littermate controls (n=9, 3 separate experiments). All p values were determined by unpaired t test and graphs showing mean±SEM. All p values are indicated in graphs.
Importantly, Cd4creStat3fl/fll mice displayed significantly reduced bone loss compared to littermate controls (Fig. 5I, p<0.05). Finally, whereas inhibition of Stat3 in CD4 T cells has been linked to expansion of the Foxp3+CD4+ T regulatory cells (Tregs) (19, 20) in other disease models, in LIP we did not detect changes in the proportion or absolute numbers of Tregs in Cd4creStat3fl/fll mice (Fig. S5C-E), while RORγt and RORγt+Foxp3+ cells were significantly reduced in Cd4creStat3fl/fll mice (Fig. S5F-K, p<0.05). Therefore, the decreased disease severity observed in Cd4creStat3fl/fll mice may be attributed directly to the lack of Th17 cells rather than to a compensatory increase in Tregs (21).
To confirm these results using an independent approach, we employed mice with a specific deletion of the master regulator of Th17 cells, Rorc, within the T cell compartment (LckcreRorcfl/fl mice). Similar to Cd4creStat3fl/fl, LckcreRorcfl/fl mice displayed significantly reduced numbers of Th17 cells at steady state and during periodontitis (p<0.05) without alterations in the γδT or ILC IL-17+ populations and had reduced periodontal bone loss compared to their littermate controls (Fig. 6A) and (Fig. S6A-D).
Fig. 6. RORγt targeting protects from inflammatory bone loss and reveals mechanisms of Th17 cell-driven periodontal inflammation.
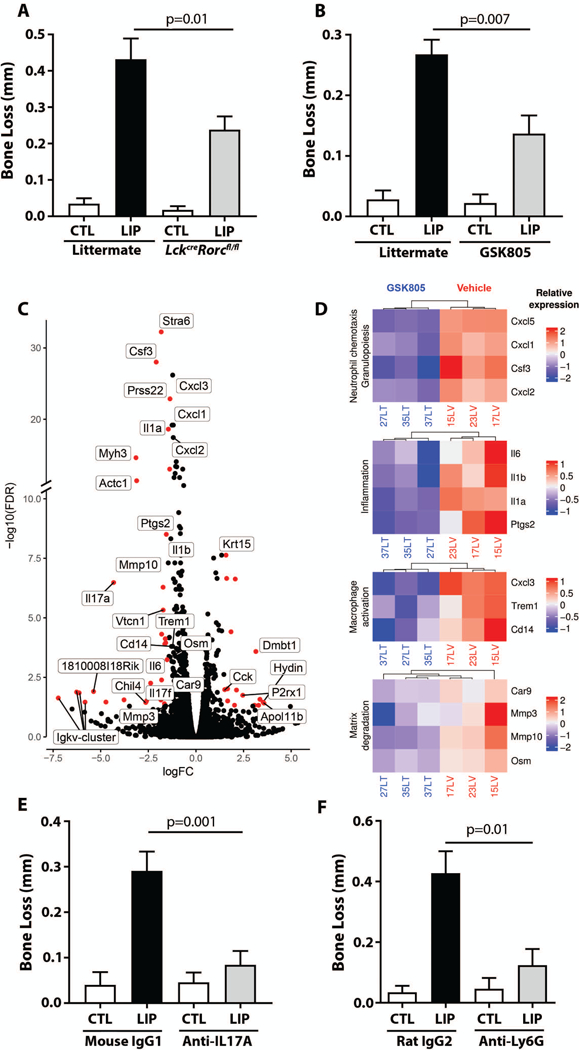
(A) Bone loss (in mm) after LIP in LckCreRorcfl/fl mice and littermate controls (n=5 per group, 2 separate experiments). (B) Bone loss (in mm) after LIP in the presence/absence of RORγt inhibitor (GSK805) (n=6, 2 separate experiments). (C) Volcano plot of genes differentially expressed during LIP in the presence/absence of RORγt inhibitor (GSK805). Genes in red are p<0.05 and fold-change>1.5. (D) Heatmap depicts genes of interest from the top 30 downregulated genes with RORγt inhibitor (GSK805) in LIP, FDR ≤ 0.05. (E) Bone loss (in mm) with LIP in mice treated with anti-IL-17A or isotype control (n=8 per group) (F) Bone loss (in mm) with LIP in mice treated with anti-Ly6G or isotype control (n=5 per group, Mann-Whitney test). All other p values determined by unpaired t test and graphs depict mean±SEM unless otherwise stated.
Collectively, these data demonstrate that genetic inhibition of Th17 cell differentiation blocks the overall expansion of CD45+IL-17+ cells during experimental periodontitis and confers significant protection from periodontal inflammatory bone loss. Therefore, Th17 cells are directly implicated as a major pathogenic cell subtype in this inflammatory disease, which supports focus on Th17 cell-targeted therapeutic approaches.
Pharmacologic targeting of RORγt reduces inflammatory bone loss suggesting an IL-17 and neutrophil mediated immunopathology in periodontitis
Next, the effect of GSK805 –a small-molecule inhibitor of RORγt– mediated transcription, Th17 cell development and function (22), was evaluated in experimental periodontitis. To this end, mice were subjected to LIP in the presence or absence of oral GSK805. GSK805-treated mice displayed significantly reduced disease severity (measured as bone loss, p<0.05) compared to control vehicle-treated mice (Fig. 6B). Consistent with previous reports (22), GSK805 treatment preferentially targeted the expansion of Th17 cells during experimental periodontitis and did not affect steady-state sources of IL-17 (Fig. S7 A-D), thus confirming GSK805 as a selective Th17 inhibitor in the setting of periodontitis.
These experiments additionally provided a platform to evaluate mechanisms by which RORγt /Th17 cell inhibition suppresses periodontal bone loss. For this, RNA sequencing of gingival tissues was employed in the presence/absence of RORγt/Th17 inhibition. The global transcriptomic consequences of RORγt inhibition during experimental periodontitis were evaluated. RORγt targeting inhibited IL-17A/F (Fig. 6C-D, Fig. S8) and downstream targets, such as factors mediating granulopoiesis and neutrophil recruitment (Csf3, Cxcl1, Cxcl2, Cxcl5) (Fig 6C, D 1st top panel). Pro-inflammatory genes (Il1β, Il1α, Il6, Ptgs2) were also inhibited (Fig 6C-D 2nd panel), in line with a role of IL-17 as an amplifier of inflammatory responses. Expression of genes associated with myeloid cell activation (Cd14, Trem-1, Cxcl3) was also significantly reduced (Fig. 6D, 3rd panel). Finally, expression of molecules linked to tissue destruction and bone loss (Mmp3, Mmp10, Pprss22, Osm, Car9) (Fig. 6D, 4th bottom panel) was downregulated. In sum, our transcriptome analysis following RORγt/Th17 targeted treatment provided detailed insight into top regulated genes and pathways which collectively suggest inhibition of IL-17 signaling primarily affecting the function of neutrophils and other myeloid cells.
We next investigated whether IL-17 and related neutrophil immunopathology is implicated in periodontal bone loss. In line with IL-17 playing a primary role in Th17 cell-mediated immune immunopathology, antibody-mediated inhibition of IL-17 led to significant reduction of periodontal bone loss (Fig. 6E, p<0.05). Although neutrophils are suspected to mediate tissue destruction in periodontitis, no direct cause-and-effect relationship has been reported thus far. To this end, reduction of neutrophil numbers (Fig. S8B) by means of anti-Ly6G treatment led to significant inhibition of periodontal bone loss (Fig. 6F, p<0.05). The involvement of neutrophils in periodontal tissue destruction is consistent with neutrophil accumulation in lesions of periodontitis in mice and humans (Fig. S8C-E). Collectively, our data suggest that Th17 cell-derived IL-17 and neutrophils mediate pathology in periodontitis.
Humans with defects in Th17 differentiation present with reduced periodontal inflammation and bone loss
If Th17 cells are important in human periodontitis as our preclinical data suggests, one would expect protection against periodontal inflammation and bone loss when Th17 cells are reduced or inhibited. To evaluate the clinical consequences of Th17 cell inhibition in humans, we studied patients with the Mendelian disorder Autosomal Dominant Hyper-IgE Syndrome (AD-HIES), a unique real-life setting to decipher the patho-physiologic role of Th17 cells in humans. AD-HIES patients bear an autosomal dominant, loss-of-function mutation in the STAT3 gene (12), which leads to reduced STAT3 signaling and consequent defective differentiation of Th17 cells (23). Patients do not have circulating Th17 cells and their T cells display significantly reduced rates of in vitro differentiation into Th17 (23). Importantly, amongst the described Th17 cell human defects, AD-HIES is the most common, thereby allowing for evaluation of a fairly large Mendelian cohort rather than select patient cases.
Consistent with blunted Th17 cell differentiation, AD-HIES patients exhibited significantly reduced numbers of Th17 cells within gingival tissues as compared to age- and gender-matched healthy volunteers and periodontitis patients (Fig. 7A-B). In line with the functional consequences of blunted tissue-Th17 cell responses and previous reports (24), we documented a selective susceptibility to oral candidiasis in this population with 85% reporting recurrent oral thrush (Fig. 7C). Therefore, these patients constitute an appropriate cohort for investigating the consequences of local oral mucosal Th17 cell deficiency in periodontal immunity and inflammation.
Fig. 7. Patients with genetic defects in Th17 cell differentiation present with reduced susceptibility to periodontal inflammation (A-B).
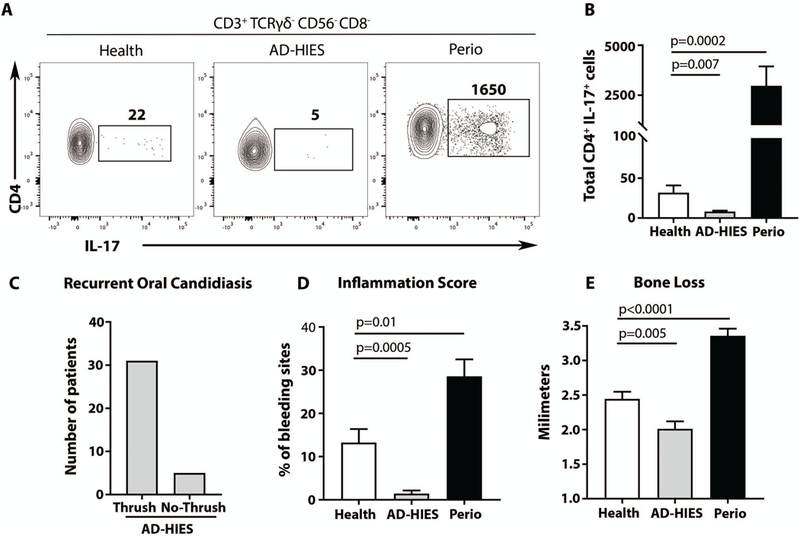
IL-17 production by CD4+ (CD45+CD3+TRCγδ-CD56-CD8-) human gingival cells in health, AD-HIES and periodontitis patients. Representative FACS plots (A) and graph (B) indicating numbers of CD4+IL-17+ cells per standardized gingival biopsy (n=7–10). (C) Susceptibility to recurrent oral candidiasis in AD-HIES. Bar graph shows the number of patients with history of recurrent oral candidiasis (Thrush, n=31) or no history of candida infections (No-Thrush, n=5). (D-E). Periodontitis susceptibility in health, AD-HIES and periodontitis patients (Perio). (D) Periodontal inflammation in health, AD-HIES and periodontitis (n=29/25/27 patients). Bar graph shows frequency of bleeding sites (Inflammation score, per patient). (E) Periodontal bone loss in health, AD-HIES and Periodontitis (n=29/25/27 patients). Bar graph shows clinical attachment (level of bone loss). All p values in this figure were determined by one-way ANOVA and Holm-Sidak’s test, mean±SEM.
Our large cohort of adult AD-HIES patients (n=35) with confirmed STAT3 mutations was clinically evaluated for presence and history of periodontal inflammation and tissue destruction. In stark contrast to their susceptibility to oral fungal infection, AD-HIES patients did not demonstrate susceptibility to periodontal disease. In fact, detailed clinical evaluation revealed significantly reduced periodontal inflammation and attenuated bone loss (reduced clinical attachment loss, p<0.05) in AD-HIES patients compared to age/gender matched healthy controls and periodontitis patients (Fig. 7D-E). These findings suggest that blunted Th17 cell responses confer protection from periodontal inflammatory disease in humans. Therefore, our clinical data from AD-HIES patients provides the human relevance of our murine mechanistic experiments and reinforce the distinct roles of oral Th17 cells in immunoprotection and immunopathology.
Discussion
At mucosal barrier sites, the local immune system is tasked with the delicate act of maintaining a dynamic balance between host and environmental stimuli (25). While much work in recent years has focused on the regulation of barrier immunity at the GI tract and skin, little is understood regarding the critical elements of oral barrier immunity, despite its unique features. The oral mucosa is constantly exposed to myriad stimuli (15), including a rich and diverse commensal microbial community. It is the initial portal of encounter for microbes, food and airborne antigens as they enter the GI tract, all in the context of mechanical stress (mastication) (26). Hence, it is of great significance to understand how immunity is regulated at this critical barrier.
In our current work, we focused on the Th17 cell subset, which is pivotal for oral anti-fungal immunity (11, 24) but has also been documented to expand in the oral inflammatory disease, periodontitis (6, 27, 28). Our studies, using complementary human and murine datasets, demonstrate divergent regulation of oral Th17 cells in health and disease and implicate Th17 cells as drivers of disease pathogenesis and plausible therapeutic targets.
We investigated mechanisms by which Th17 cells are triggered to expand in the setting of periodontitis and potentially participate in disease pathogenesis. In human lesions of periodontitis we document an expansion of Th17 cells that secrete cytokines associated with pathogenic activity (10, 29) and are predominantly resident memory (particularly resident effector memory rTem) Th17 cells (30). rTem cells are thought to be retained in tissues, to become terminally differentiated and to be capable of mounting rapid responses to site-specific cues (30). These findings imply that Th17 memory cells expand locally in response to tissue-specific triggers and potentially acquire pathogenic functions. In this regard, experimental data in murine models of periodontitis points to a disease-associated microbiome being a local trigger for Th17 cell expansion and related bone loss in periodontitis.
These studies provide an opportunity to compare and contrast regulation of gingival Th17 cells in health and disease. Our previous work had investigated the physiologic regulation of oral Th17 cells in health. We found that Th17 cells naturally accumulate in the oral mucosa with age (15). This physiologic accumulation of Th17 cells was dependent on ongoing mechanical stimulation, which occurs during mastication, and triggers Th17 cell accumulation in an IL-6 and commensal independent manner (15). Our current work investigates mechanisms involved in further amplification of Th17 cells observed in periodontitis. We show that while homeostatic Th17 cells accumulate in a commensal independent manner (15), amplification of Th17 cells in periodontitis necessitates microbial triggering. Furthermore, while homeostatic Th17 are dependent solely on IL-6, disease-associated Th17 cell expansion requires both IL-6 and IL-23. IL-23 is known to be required for pathogenic Th17 cell development in other experimental disease settings (11).
We further investigated microbial triggering of Th17 cells in periodontitis. Microbial triggering of Th17 cells has been well documented at various other barrier sites, including the lower GI (31) and skin (32). In fact, barrier Th17 cells have been documented to have specificity for local commensals in the GI tract (33). Antigen-specific Th17 cell responses to commensal bacteria could explain the preferential proliferation of Th17 cells rather than other cellular sources of IL-17 in the setting of periodontitis and are consistent with Nur77eGFP+ cells expanding in LIP. Innate, antigen-independent TCRαβ IL-17+ cells have also been shown to expand at the oral barrier (specifically within the tongue) in response to Candida infection (34) and could conceivably be a contributing source of TCRαβ IL-17+ in periodontitis.
We find that dysbiotic changes in microbial communities, rather than a mere increase in total microbial load, are necessary for triggering Th17 cells to cause disease. Indeed, antibiotics which inhibited Th17 cell accumulation did not necessarily reduce microbial load or target specific high-abundance bacterial species. Rather, the protective antibiotics appeared to mediate shifts in the balance of commensal bacteria within communities. However, treatment with protective antibiotics did not reveal specific bacteria that trigger Th17 cell accumulation, but suggested that dysbiotic changes involving anaerobes mediate pathogenic inflammation. Interestingly, the human periodontal disease-associated microbiome is characterized by microbial overgrowth and dysbiotic shifts in bacterial communities, with an overgrowth of gram-negative anaerobes (35, 36). Yet, in human disease it is not practical to dissect whether increased biomass, specific constituents or global shifts in microbial communities are directly important for disease induction. The present study has succeeded in uncoupling disease initiation from non-specific biofilm accumulation and further supports dysbiotic changes as the trigger for Th17 cell accumulation and downstream bone loss. In this regard, it should be noted that periodontitis is currently thought to be driven by dysbiosis of synergistic polymicrobial communities characterized by an imbalanced growth of a subset of commensal species (also known as pathobionts) rather than by a single or select few pathogens (14, 37–39). Standard-of-care treatment of periodontal disease currently aims at removal of the periodontitis-associated microbial commensal communities (36, 40, 41) by means of mechanical dental debridement with adjunct systemic antibiotics for severe disease (42). Interestingly, metronidazole, which effectively inhibited Th17 cell accumulation, is an antibiotic of choice in human periodontitis, as it classically targets Gram-negative anaerobes.
While the link of commensal microbes to periodontal destructive inflammation is well documented clinically (41), the mechanisms by which commensal bacteria trigger pathogenic inflammation in periodontitis are poorly understood. Our work establishes Th17 cells as the missing mechanistic link between microbial stimulation and induction of inflammatory immunopathology in periodontitis. Indeed, specific genetic inhibition of Th17 cells (by knocking out Stat3 or Rorc in αβ T cells) led to inhibition of Th17 cell differentiation in gingival mucosal tissues, without interruption or augmentation of other cellular sources of IL-17. Importantly, inhibition of Th17 differentiation led to significantly reduced bone loss (50–70%), suggesting that Th17 cells constitute a major driver of periodontal immunopathology. Our genetic inhibition of Th17 cells without interrupting other sources of IL-17 could be important for maintenance of homeostatic immune protection and barrier integrity, as is the case in the GI tract (43). Similarly, in our pharmacological inhibition of RORγt, we selected a small-molecule inhibitor (GSK805) that preferentially targets Th17 cell differentiation (44) while sparing other sources of IL-17 (22). Small-molecule inhibitors of RORγt have been advocated and are currently being tested in Th17 cell-dominated diseases (45–47). Such inhibitors may be ideally suited for localized diseases, such as periodontitis, where local treatment can avoid systemic effects on thymocyte and lymph node development (46, 48).
Therapeutic inhibition of RORγt/Th17 cells in experimental periodontitis also afforded us the opportunity to investigate mechanisms whereby Th17 cells mediate periodontal immunopathology. By performing RNAseq transcriptome analysis of periodontitis lesions after five days of LIP in the presence of RORγt or vehicle, we evaluated genes inhibited during RORγt/Th17 treatment. Zeroing in on specific gene regulation, we detected a signature consistent with inhibition of IL-17 and IL-17-dependent pathways. While we detected a dominant inhibition of IL-17- related genes, we cannot completely rule out off-target effects of RORγt inhibition. Top genes inhibited included IL-17 dependent neutrophil recruitment, granulopoiesis, macrophage activation and overall activation of pro-inflammatory pathways (49). Genes inhibited also revealed an overall reduced activation of matrix and bone destruction. These findings suggest that the main mediator of Th17 cell-induced immunopathology is IL-17. Indeed, inhibition of the cytokine IL-17 significantly inhibited periodontal bone loss. To investigate mechanisms by which IL-17 mediates periodontal immunopathology, we focused on neutrophils as candidate downstream drivers of immunopathology. Top genes inhibited during small molecule Th17 cell inhibition were related to neutrophil granulopoesis and neutrophil recruitment. Neutrophil accumulation is also known as a hallmark of human periodontitis and has been long speculated as a driver of pathology (50–52). Indeed, our experimentation revealed that reduction of neutrophil accumulation protects from periodontal bone loss. Taken together, these data suggest a microbial-Th17 cell-neutrophil axis as a driver for periodontitis immunopathology. Although it is well established that neutrophils secrete tissue-destructive proteases (53), further work is needed to dissect mechanisms by which neutrophils mediate downstream osteoclast activation and bone loss. In this respect, a recent study has shown a requirement for inflammation-associated RANKL expression in osteoblasts for induction of periodontal bone loss (27).
IL-17 has previously been shown to be a primary mediator of periodontal immunopathology in settings of altered background immunity such as immune deficiency (54, 55), diabetes (56) and ageing (57). We have identified IL-23/IL-17 dysregulation in periodontitis associated with the rare genetic disorder, Leukocyte adhesion deficiency type 1 (LAD1), and have exploited that target by IL-23 blockade in humans (55). Interestingly, despite the commonality of Th17 cell amplification in both endemic and genetic forms of periodontitis, Th17 cell regulation and functionality appears to be distinct in genetic versus common forms of disease. In LAD1, defective neutrophil transmigration leads to tissue neutropenia which underlies amplification of Th17 cell responses. Furthermore, Th17 cell-mediated bone destruction occurs in the absence of tissue neutrophils in LAD1. In contrast, in common periodontitis, neutrophil accumulation is a hallmark of disease and a driver of periodontal bone loss. Collectively our previous and current work suggest distinct regulation and functionality of Th17 cells in genetic and common forms of periodontitis which nonetheless may respond to a common therapeutic target.
Our work further establishes human relevance by evaluating patient cohorts with known genetic defects leading to impaired Th17 cell differentiation (AD-HIES) (23). We document that AD-HIES patients have blunted Th17 cell mucosal responses and susceptibility to recurrent oral fungal infection, but reduced periodontal inflammation (bleeding on probing) and bone loss compared to healthy volunteers. These findings are consistent with a key role for Th17 cells in oral inflammatory bone loss and provide a unique demonstration of the distinct roles of Th17 cells within the same tissue and in the human setting.
A limitation of our current study is that the bulk of data implicating Th17 cells in periodontitis pathogenicity comes from animal experimentation. While patients with AD-HIES present with reduced periodontitis susceptibility, it is not possible to formally prove resistance to periodontitis in this setting. Future biologic treatment studies of Th17 inhibition are necessary to conclusively implicate Th17 cells in human periodontitis pathogenesis and to evaluate safety and efficacy of such Th17-based treatment in humans. In this regard, our current study not only reveals critical insights into the regulation of Th17 cells at the oral barrier, but also provides mechanistic justification for Th17 targeting in human periodontitis.
Materials and Methods
Study design
This study evaluated mechanisms of Th17 cell accumulation in the disease periodontitis and in experimental periodontitis and assessed preclinical Th17 cell targeting. Oral clinical evaluation and biopsy sampling were performed in healthy volunteers, patients with periodontitis and AD-HIES patients. Oral examinations included full mouth periodontal examination with a standardized instrument (periodontal probe Hu-Friedy UNC-15 probe). Gingival biopsies were obtained for histology and evaluation of Th17 cells by Flow Cytometry. All subjects were enrolled in an IRB-approved protocol (clinicaltrials.gov #NCT01568697, #NCT00404560) at the NIH Clinical Center and all provided written informed consent. For inclusion in this study, AD-HIES patients were diagnosed based on clinical phenotype and a positive STAT3 mutation. Healthy volunteers were systemically healthy based on medical history and select laboratory testing. Periodontitis patients presented with more than 4 sites of moderate-severe bone loss and visible signs of inflammation (Inclusion/exclusion criteria, Table S1).
For experimental periodontitis studies in mice, ligature-induced periodontitis was performed as previously described (18) in different strains of mice in the presence or absence of antibiotic treatments, RORγt inhibition, FTY720 treatment and antibody treatments for IL-17, Ly6G or isotype controls. Th17 cells within tissues were evaluated by flow cytometry. Murine oral microbiome was evaluated by 16S rRNA gene sequencing and transcriptional responses in gingiva in the presence or absence of RORγt inhibition were evaluated by RNA sequencing and analysis. Experimental approaches are further detailed in the Supplemental Materials Section.
Statistical analysis
Flow cytometry, real-time PCR and bone loss measurement data were evaluated by Shapiro-Wilk normality test. Two-tailed unpaired t test or Mann-Whitney test was used when comparing two groups. When more than two groups were compared, ANOVA or Kruskal-Wallis with post-hoc analysis was applied. P values < 0.05 were considered statistically significant (data were evaluated using Prism 7 program, GraphPad Software, CA, USA). For RNA sequencing analysis gene expression was evaluated by three independent statistical methods (DESeq2, edgeR and Limma-Voom). Differentially expressed genes (FDR ≤ 0.05) were considered for further analyses based on results from edgeR. For 16S rRNA gene sequencing analysis, AMOVA (analysis of molecular variance) was used to test for differences in community diversity and structure, as implemented in mothur (www.mothur.org).
Supplementary Material
Acknowledgments:
We thank the NIDCR Veterinary Resource core, NIDCR Combined Technical Research core, genomics core and the Microbiome and Genetics Core (CCR, NCI, NIH), NIH HPC Biowulf cluster. We thank Wuxing Yuan and Vishal Thovarai for assistance with 16s rRNA sequencing and data deposition and Dr. Erich Boger for technical expertise.
Funding: Intramural programs of NIDCR, NIAID and NCI and NIDCR grants DE024153 and DE026152 to GH. La Roche-Posay, CEDEF, fondation groupe pasteur mutualité, Société Française de Dermatologie, Philippe Foundation, Fondation pour la Recherche Médicale to CH.
Footnotes
Competing Interests: Authors have no competing interests to declare.
Data and material availability: RNA sequencing read data have been deposited in the Gene Expression Omnibus site (GSE118166). The microbiome sequencing read data have been uploaded to SRA under bioproject PRJNA484972
References
- 1.Eke PI et al. , Update on Prevalence of Periodontitis in Adults in the United States: NHANES 2009 to 2012. Journal of periodontology 86, 611–622 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajishengallis G, Periodontitis: from microbial immune subversion to systemic inflammation. Nature reviews. Immunology 15, 30–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cardoso CR et al. , Evidence of the presence of T helper type 17 cells in chronic lesions of human periodontal disease. Oral microbiology and immunology 24, 1–6 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Allam JP et al. , IL-23-producing CD68(+) macrophage-like cells predominate within an IL-17-polarized infiltrate in chronic periodontitis lesions. Journal of clinical periodontology 38, 879–886 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Abusleme L, Moutsopoulos NM, IL-17: overview and role in oral immunity and microbiome. Oral diseases, (2016). [DOI] [PMC free article] [PubMed]
- 6.Dutzan N, Konkel JE, Greenwell-Wild T, Moutsopoulos NM, Characterization of the human immune cell network at the gingival barrier. Mucosal immunology 9, 1163–1172 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohyama H et al. , The involvement of IL-23 and the Th17 pathway in periodontitis. Journal of dental research 88, 633–638 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Stockinger B, Omenetti S, The dichotomous nature of T helper 17 cells. Nature reviews. Immunology, (2017). [DOI] [PubMed]
- 9.Veldhoen M, Interleukin 17 is a chief orchestrator of immunity. Nature immunology 18, 612–621 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Lee Y et al. , Induction and molecular signature of pathogenic TH17 cells. Nature immunology 13, 991–999 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaffen SL, Jain R, Garg AV, Cua DJ, The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nature reviews. Immunology 14, 585–600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holland SM et al. , STAT3 mutations in the hyper-IgE syndrome. The New England journal of medicine 357, 1608–1619 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Puel A et al. , Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332, 65–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamont RJ, Hajishengallis G, Polymicrobial synergy and dysbiosis in inflammatory disease. Trends in molecular medicine 21, 172–183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dutzan N et al. , On-going Mechanical Damage from Mastication Drives Homeostatic Th17 Cell Responses at the Oral Barrier. Immunity 46, 133–147 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moutsopoulos NM et al. , Porphyromonas gingivalis promotes Th17 inducing pathways in chronic periodontitis. Journal of autoimmunity 39, 294–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nares S et al. , Rapid myeloid cell transcriptional and proteomic responses to periodontopathogenic Porphyromonas gingivalis. The American journal of pathology 174, 1400–1414 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abe T, Hajishengallis G, Optimization of the ligature-induced periodontitis model in mice. Journal of immunological methods 394, 49–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Lee YS, Yu CR, Egwuagu CE, Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. Journal of immunology 180, 6070–6076 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durant L et al. , Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 32, 605–615 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glowacki AJ et al. , Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes. Proceedings of the National Academy of Sciences of the United States of America 110, 18525–18530 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Withers DR et al. , Transient inhibition of ROR-gammat therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells. Nature medicine 22, 319–323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milner JD et al. , Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452, 773–776 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lionakis MS, Netea MG, Holland SM, Mendelian genetics of human susceptibility to fungal infection. Cold Spring Harbor perspectives in medicine 4, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belkaid Y, Harrison OJ, Homeostatic Immunity and the Microbiota. Immunity 46, 562–576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moutsopoulos NM, Konkel JE, Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends in immunology, (2017). [DOI] [PMC free article] [PubMed]
- 27.Tsukasaki M et al. , Host defense against oral microbiota by bone-damaging T cells. Nat Commun 9, 701 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zenobia C, Hajishengallis G, Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol 2000 69, 142–159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Al-Mossawi MH et al. , Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat Commun 8, 1510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sathaliyawala T et al. , Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 38, 187–197 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivanov II et al. , Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naik S et al. , Compartmentalized control of skin immunity by resident commensals. Science 337, 1115–1119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y et al. , Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 510, 152–156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verma AH et al. , Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci Immunol 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griffen AL et al. , Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. The ISME journal 6, 1176–1185 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abusleme L et al. , The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. The ISME journal 7, 1016–1025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diaz PI, Hoare A, Hong BY, Subgingival Microbiome Shifts and Community Dynamics in Periodontal Diseases. J Calif Dent Assoc 44, 421–435 (2016). [PubMed] [Google Scholar]
- 38.Darveau RP, Periodontitis: a polymicrobial disruption of host homeostasis. Nature reviews. Microbiology 8, 481–490 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Hajishengallis G, Lamont RJ, Dancing with the Stars: How Choreographed Bacterial Interactions Dictate Nososymbiocity and Give Rise to Keystone Pathogens, Accessory Pathogens, and Pathobionts. Trends in microbiology 24, 477–489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen RE, Research S, A. A. o. P. Therapy Committee, Position paper: periodontal maintenance. Journal of periodontology 74, 1395–1401 (2003). [DOI] [PubMed] [Google Scholar]
- 41.P. American Academy of, Comprehensive periodontal therapy: a statement by the American Academy of Periodontology *. Journal of periodontology 82, 943–949 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Slots J, Research S, Therapy C, Systemic antibiotics in periodontics. Journal of periodontology 75, 1553–1565 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Lee JS et al. , Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 43, 727–738 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiao S et al. , Small-molecule RORgammat antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity 40, 477–489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huh JR, Littman DR, Small molecule inhibitors of RORgammat: targeting Th17 cells and other applications. European journal of immunology 42, 2232–2237 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santori FR et al. , Identification of natural RORgamma ligands that regulate the development of lymphoid cells. Cell metabolism 21, 286–297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miossec P, Kolls JK, Targeting IL-17 and TH17 cells in chronic inflammation. Nature reviews. Drug discovery 11, 763–776 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Guo Y et al. , Inhibition of RORgammaT Skews TCRalpha Gene Rearrangement and Limits T Cell Repertoire Diversity. Cell reports 17, 3206–3218 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Amatya N, Garg AV, Gaffen SL, IL-17 Signaling: The Yin and the Yang. Trends in immunology 38, 310–322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hajishengallis G, Moutsopoulos NM, Hajishengallis E, Chavakis T, Immune and regulatory functions of neutrophils in inflammatory bone loss. Seminars in immunology 28, 146–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kantarci A, Oyaizu K, Van Dyke TE, Neutrophil-mediated tissue injury in periodontal disease pathogenesis: findings from localized aggressive periodontitis. Journal of periodontology 74, 66–75 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Matthews JB et al. , Neutrophil hyper-responsiveness in periodontitis. Journal of dental research 86, 718–722 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Soehnlein O, Steffens S, Hidalgo A, Weber C, Neutrophils as protagonists and targets in chronic inflammation. Nature reviews. Immunology 17, 248–261 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Moutsopoulos NM et al. , Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Science translational medicine 6, 229ra240 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moutsopoulos NM et al. , Interleukin-12 and Interleukin-23 Blockade in Leukocyte Adhesion Deficiency Type 1. The New England journal of medicine 376, 1141–1146 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiao E et al. , Diabetes Enhances IL-17 Expression and Alters the Oral Microbiome to Increase Its Pathogenicity. Cell host & microbe 22, 120–128 e124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eskan MA et al. , The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nature immunology 13, 465–473 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abusleme L, Hong B, Hoare A, Konkel JE, Diaz PI and Moutsopoulos NM, Oral Microbiome Characterization in Murine Models. Bio-protocol 7, e2655 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.