

Abstract
The development of chemical strategies for site-specific protein modification now enable researchers to PEGylate a protein drug at one or more specific locations. However, aside from avoiding enzyme active sites or protein-binding interfaces-specific PEGylation, distinguishing the optimal PEGylation site from the available alternatives has conventionally been a matter of trial and error. As part of a continuing effort to develop guidelines for identifying optimal PEGylation sites within proteins, we show here that the impact of PEGylation at various sites within the β-sheet model protein WW depends strongly on the identity of the PEG-protein linker. PEGylation of Gln or of azidohomoalanine has a similar impact on WW conformational stability as does Asn- PEGylation, whereas PEGylation of propargyloxyphenylalanine is substantially stabilizing at locations where Asn-PEGylation was destabilizing. Importantly, we find that at least one of these three site-specific PEGylation strategies leads to substantial PEG-based stabilization at each of the positions investigated, highlighting the importance of considering conjugation strategy as an important variable in selecting optimal PEGylation sites. We demonstrate that using a branched PEG oligomer intensifies the impact of PEGylation on WW conformational stability and also show that PEG-based increases to conformational are strongly associated with corresponding increases in proteolytic stability.
Graphical Abstract:
Introduction
PEGylation has been used for more than thirty years as a general strategy for improving the stability and pharmacokinetic properties of protein drugs.1–4 The beneficial effects of PEGylation are thought to derive from the large hydrodynamic radius of PEG, which can shield a protein from proteolysis and antibody recognition, and can prolong its serum half-life by limiting renal filtration.5, 6 Early efforts involved non-specific PEGylation of protein surface nucleophiles with a reactive PEG-linked electrophile, resulting in a heterogeneous mixture of PEGylated protein isoforms that differed in the number and location of PEGylation sites.3, 4 More recently, chemists have devised a number of strategies for site-specific protein PEGylation, including conjugation of PEG- azide or alkyne to propargyloxyphenylalanine7 or azidohomoalanine8, respectively, via the copper (I)-catalyzed azide/alkyne cycloaddition, conjugation of a PEG amide to a glutamine residue catalyzed by the enzyme transglutaminase;9, 10 and many varieties of conjugating a moderately reactive PEG electrophile with the nucleophilic Cys thiol.11
With these site-specific PEGylation strategies in hand, chemists are now poised to ask whether some PEGylation sites or conjugation strategies provide better stability and pharmacokinetic properties than others. Aside from avoiding active sites and binding interfaces, most site-specific protein PEGylation studies have relied on combinatorial trial and error approaches to select the optimal PEGylation site from a list of possible alternatives.12, 13 Though useful, this approach is time- and resource-intensive. We seek general structure-based criteria for identifying the subset of possible PEGylation sites that are most likely to result in optimal enhancements to protein stability and pharmacokinetic properties.
We recently showed that modifying the side chain amide nitrogen of an Asn residue at certain positions within the WW domain of the human protein Pin 1 with a 190 Da monomethoxyPEG (i.e., four ethylene oxide units) substantially increases WW conformational and proteolytic stability, with optimal increases in proteolytic stability correlated with large increases in protein conformational stability.14 We chose the WW domain as a model system because its well-characterized two-state folding behavior facilitates detailed analysis of its thermodynamic properties.15–17 Based on a scan of this PEGylated Asn residue at other positions within WW, we developed a set of structure-based guidelines for identifying stabilizing PEGylation sites and used these guidelines successfully to identify a stabilizing PEGylation site within a second β-sheet protein (i.e., the Src SH3 domain).18 However, we subsequently found that using alternative PEGylation strategies at position 19 can dramatically alter the impact of PEG on WW conformational stability, depending on the identity of the PEG-protein linker. For example, modifying a propargyloxyphenylalanine residue (PrF) at position 19 with a PEG azide via the copper- catalyzed azide-alkyne cycloaddition only slightly stabilizes WW relative to the non-PEGylated variant. In contrast, PEGylation of azidohomoalanine (Aha) with the corresponding PEG alkyne or of Gln with the corresponding PEG amine is substantially more stabilizing (ΔΔGf = −0.29 ± 0.03 kcal/mol or −0.40 ± 0.02 kcal/mol, respectively), though neither imparts the same level of stabilization as does Asn-PEGylation.14
Despite their inferiority to Asn-PEG at position 19, Gln-PEG, Aha-PEG, and PrF-PEG are more straightforward to incorporate into expressed proteins than is Asn-PEG because their non-PEGylated counterparts are encodable (Gln without any reengineering of cellular translational machinery19; PrF via amber suppression7) or can be incorporated in lieu of existing amino acids in auxotrophic strains (Aha is a methionine surrogate8). Moreover, Gln, Aha, and PrF can be functionalized with PEG in the context of a native unprotected protein, via well-known chemoselective strategies (i.e. transglutaminase chemistry for Gln;10, 19 azide-alkyne cycloaddition for Aha and PrF14). We wondered whether Gln-PEG, Aha-PEG, and PrF-PEG would have a similarly smaller impact on WW stability relative to Asn-PEG at other prospective sites within WW, and whether our structure- based guidelines for identifying stabilizing Asn-PEGylation sites in WW would be useful in the context of these alternative PEGylation methods.
Results and Discussion
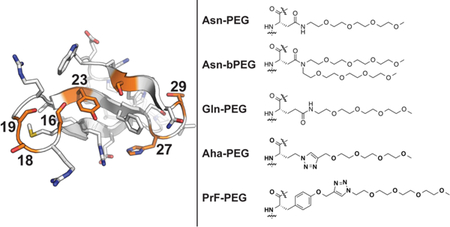
We prepared several WW variants in which we incorporated Gln vs. Gln-PEG; Aha vs. Aha-PEG; and PrF vs. PrF-PEG at positions 16, 18, 23, 27, 29, or 32 (where we previously incorporated Asn vs. Asn-PEG,18 see Figure 1). The names and amino acid sequences of these WW variants appear in Table 1. For example, Gln and Gln-PEG occupy position 16 in variants 16Q and 16Qp, respectively; Aha and Aha-PEG occupy position 16 in variants 16X and 16Xp, respectively; whereas PrF and PrF-PEG occupy position 16 in variants 16Z and 16Zp. Variants in which each of these residues occupy positions 18, 19, 23, 27, 29, and 32 are named via analogous conventions (variants at position 19 were prepared and characterized previously14).
Figure 1.
Structures of PEG-protein linkages involving Asn, Gln, azidohomoalanine (Aha), and propargyloxyphenylalanine (PrF), incorporated at the indicated sites within the WW domain of the human protein Pin 1.
Table 1.
Sequences, melting temperatures, folding free energies, and apparent proteolysis rate constants for PEGylated WW variants vs. their non-PEGylated counterpartsa
| Peptide | Sequence | Tm(°C) | ∆∆Gf (kcal/mol) | k (s−1) × 103 | ln(r) |
|---|---|---|---|---|---|
| 16Q | H2N-KLPPGWEKRMQRSSGRVYYFNHITNASQFERPSG-COOH | 56.6 ±0.1 | 8.9 ±0.2 | ||
| 16Qp | H2N-KLPPGWEKRMQRSSGRVYYFNHITNASQFERPSG-COOH | 63.5 ±0.1 | −0.67 ±0.02 | 3.2 ±0.2 | −1.0 ±0.3 |
| 16X | H2N-KLPPGWEKRMXRSSGRVYYFNHITNASQFERPSG-COOH | 52.8 ±0.3 | 15.0 ± 1.7 | ||
| 16Xp | H2N-KLPPGWEKRMXRSSGRVYYFNHITNASQFERPSG-COOH | 56.9 ±0.3 | −0.39 ±0.04 | 7.6 ±0.6 | −0.7 ±0.4 |
| 16Z | H2N-KLPPGWEKRMZRSSGRVYYFNHITNASQFERPSG-COOH | 60.3 ±0.2 | 1.4 ±0.4 | ||
| 16Zp | H2N-KLPPGWEKRMZRSSGRVYYFNHITNASQFERPSG-COOH | 59.6 ±0.5 | 0.07 ±0.05 | 5.0 ±0.3 | 1.3 ±0.6 |
| 16N | H2N-KLPPGWEKRMNRSSGRVYYFNHITNASQFERPSG-COOH | 50.6 ±0.2 | 9.4 ±0.4 | ||
| 16Np | H2N-KLPPGWEKRMNRSSGRVYYFNHITNASQFERPSG-COOH | 60.7 ±0.3 | −0.90 ±0.04 | 3.7 ±0.5 | −0.9 ±0.4 |
| 16Nbp | H2N-KLPPGWEKRMNRSSGRVYYFNHITNASQFERPSG-COOH | 70.3 ±0.1 | −1.70 ±0.03 | 0.87 ±0.05 | −2.4 ±0.3 |
| 18Q | H2N-KLPPGWEKRMSRQSGRVYYFNHITNASQFERPSG-COOH | 58.5 ±0.2 | 7.8 ±0.4 | ||
| 18Qp | H2N-KLPPGWEKRMSRQSGRVYYFNHITNASQFERPSG-COOH | 59.8 ±0.2 | −0.12 ±0.02 | 6.1 ±0.6 | −0.3 ±0.3 |
| 18X | H2N-KLPPGWEKRMSRXSGRVYYFNHITNASQFERPSG-COOH | 56.1 ±0.1 | 7.9 ±0.1 | ||
| 18Xp | H2N-KLPPGWEKRMSRXSGRVYYFNHITNASQFERPSG-COOH | 56.1 ±0.2 | 0.00 ±0.02 | 6.7 ±0.5 | −0.2 ±0.3 |
| 18Z | H2N-KLPPGWEKRMSRZSGRVYYFNHITNASQFERPSG-COOH | 52.9 ±0.2 | 11.9 ±2.0 | ||
| 18Zp | H2N-KLPPGWEKRMSRZSGRVYYFNHITNASQFERPSG-COOH | 53.5 ±0.4 | −0.05 ±0.04 | 11.4 ± 1.7 | 0.0 ±0.5 |
| 18N | H2N-KLPPGWEKRMSRNSGRVYYFNHITNASQFERPSG-COOH | 57.5 ±0.1 | 3.7 ± 1.1 | ||
| 18Np | H2N-KLPPGWEKRMSRNSGRVYYFNHITNASQFERPSG-COOH | 58.3 ±0.1 | −0.08 ±0.02 | 8.1 ±0.5 | 0.8 ±0.5 |
| 18Nbp | H2N-KLPPGWEKRMSRNSGRVYYFNHITNASQFERPSG-COOH | 57.1 ±0.1 | 0.03 ±0.01 | 5.7 ±0.1 | 0.4 ±0.5 |
| 19Q | H2N-KLPPGWEKRMSRSQGRVYYFNHITNASQFERPSG-COOH | 54.2 ±0.2 | 28.7 ±2.8 | ||
| 19Qp | H2N-KLPPGWEKRMSRSQGRVYYFNHITNASQFERPSG-COOH | 58.7 ±0.2 | −0.40 ±0.02 | 7.3 ±0.4 | −1.4 ±0.3 |
| 19X | H2N-KLPPGWEKRMSRSXGRVYYFNHITNASQFERPSG-COOH | 54.0 ±0.2 | 12.8 ±0.3 | ||
| 19Xp | H2N-KLPPGWEKRMSRSXGRVYYFNHITNASQFERPSG-COOH | 57.7 ±0.2 | −0.29 ±0.03 | 8.91 ±0.03 | −0.4 ±0.2 |
| 19Z | H2N-KLPPGWEKRMSRSZGRVYYFNHITNASQFERPSG-COOH | 52.3 ±0.3 | 17.6 ±0.4 | ||
| 19Zp | H2N-KLPPGWEKRMSRSZGRVYYFNHITNASQFERPSG-COOH | 53.7 ±0.2 | −0.13 ±0.04 | 6.0 ±0.6 | −1.1 ±0.3 |
| 19N | H2N-KLPPGWEKRMSRSNGRVYYFNHITNASQFERPSG-COOH | 56.1 ± 0.2 | 10.9 ± 1.9 | ||
| 19Np | H2N-KLPPGWEKRMSRSNGRVYYFNHITNASQFERPSG-COOH | 63.8 ± 0.1 | −0.69 ± 0.02 | 1.9 ±0.1 | −1.8 ±0.4 |
| 19Nbp | H2N-KLPPGWEKRMSRSNGRVYYFNHITNASQFERPSG-COOH | 68.1 ± 0.1 | −1.19 ± 0.03 | 1.1 ±0.1 | −2.3 ±0.5 |
| 23Q | H2N-KLPPGWEKRMSRSSGRVQYFNHITNASQFERPSG-COOH | 39.4 ±0.2 | 46.9 ±5.3 | ||
| 23Qp | H2N-KLPPGWEKRMSRSSGRVQYFNHITNASQFERPSG-COOH | 34.5 ±0.3 | 0.23 ± 0.04 | 49.7 ±2.2 | 0.1 ±0.3 |
| 23X | H2N-KLPPGWEKRMSRSSGRVXYFNHITNASQFERPSG-COOH | 44.7 ±0.3 | 57.5 ±2.5 | ||
| 23Xp | H2N-KLPPGWEKRMSRSSGRVXYFNHITNASQFERPSG-COOH | 39.5 ±0.9 | 0.43 ± 0.16 | 40.4 ±2.5 | −0.4 ±0.3 |
| 23Z | H2N-KLPPGWEKRMSRSSGRVZYFNHITNASQFERPSG-COOH | 52.1 ±0.2 | 7.9 ±0.2 | ||
| 23Zp | H2N-KLPPGWEKRMSRSSGRVZYFNHITNASQFERPSG-COOH | 55.6 ±0.2 | −0.29 ± 0.03 | 5.9 ±2.5 | −0.3 ±0.2 |
| 23N | H2N-KLPPGWEKRMSRSSGRVNYFNHITNASQFERPSG-COOH | 27.7 ±0.3 | 116.8± 1.5 | ||
| 23Np | H2N-KLPPGWEKRMSRSSGRVNYFNHITNASQFERPSG-COOH | 20.5 ±0.2 | 0.47 ±0.11 | 60.4 ±9.7 | −0.7 ±0.4 |
| 23Nbp | H2N-KLPPGWEKRMSRSSGRVNYFNHITNASQFERPSG-COOH | 24.9 ±0.3 | 0.18 ±0.10 | 12.8±0.9 | −2.2 ±0.3 |
| 27Q | H2N-KLPPGWEKRMSRSSGRVYYFNQITNASQFERPSG-COOH | 56.4 ±0.2 | 10.8 ±0.2 | ||
| 27Qp | H2N-KLPPGWEKRMSRSSGRVYYFNQITNASQFERPSG-COOH | 53.1 ±0.1 | 0.27 ±0.02 | 14.0 ±0.9 | 0.3 ±0.3 |
| 27X | H2N-KLPPGWEKRMSRSSGRVYYFNXITNASQFERPSG-COOH | 60.7 ±0.4 | 15.3 ±0.7 | ||
| 27Xp | H2N-KLPPGWEKRMSRSSGRVYYFNXITNASQFERPSG-COOH | 52.1 ±0.2 | 0.68 ±0.04 | 27.8 ±3.4 | 0.6 ±0.4 |
| 27Z | H2N-KLPPGWEKRMSRSSGRVYYFNZITNASQFERPSG-COOH | 52.0 ±0.2 | 7.9 ±0.2 | ||
| 27Zp | H2N-KLPPGWEKRMSRSSGRVYYFNZITNASQFERPSG-COOH | 59.8 ±0.3 | −0.62 ±0.03 | 2.1 ±0.4 | −1.3 ±0.4 |
| 27N | H2N-KLPPGWEKRMSRSSGRVYYFNNITNASQFERPSG-COOH | 54.9 ±0.1 | 8.4 ± 1.8 | ||
| 27Np | H2N-KLPPGWEKRMSRSSGRVYYFNNITNASQFERPSG-COOH | 52.6 ±0.2 | 0.20 ±0.02 | 7.2 ±0.5 | −0.2 ±0.5 |
| 27Nbp | H2N-KLPPGWEKRMSRSSGRVYYFNNITNASQFERPSG-COOH | 41.5 ±0.3 | 1.26 ±0.06 | 23.2 ± 1.0 | 1.0 ±0.5 |
| 29Q | H2N-KLPPGWEKRMSRSSGRVYYFNHIQNASQFERPSG-COOH | 45.3 ±0.5 | 47.2 ±2.6 | ||
| 29Qp | H2N-KLPPGWEKRMSRSSGRVYYFNHIQNASQFERPSG-COOH | 43.3 ±0.3 | 0.16 ± 0.06 | 37.3 ±2.6 | −0.2 ±0.3 |
| 29X | H2N-KLPPGWEKRMSRSSGRVYYFNHIXNASQFERPSG-COOH | 47.6 ±0.3 | 47.4 ± 1.4 | ||
| 29Xp | H2N-KLPPGWEKRMSRSSGRVYYFNHIXNASQFERPSG-COOH | 42.2 ±0.9 | 0.32 ±0.07 | 36.8 ±2.6 | −0.3 ±0.3 |
| 29Z | H2N-KLPPGWEKRMSRSSGRVYYFNHIZNASQFERPSG-COOH | 41.1 ±0.2 | 49.0 ± 1.5 | ||
| 29Zp | H2N-KLPPGWEKRMSRSSGRVYYFNHIZNASQFERPSG-COOH | 43.1 ±0.2 | −0.17 ±0.06 | 43.8 ±6.3 | −0.1 ±0.4 |
| 29N | H2N-KLPPGWEKRMSRSSGRVYYFNHINNASQFERPSG-COOH | 50.0 ±0.1 | 38.4 ±4.7 | ||
| 29Np | H2N-KLPPGWEKRMSRSSGRVYYFNHINNASQFERPSG-COOH | 56.8 ±0.1 | −0.57 ±0.01 | 14.2 ±2.9 | −1.0 ±0.5 |
| 29Nbp | H2N-KLPPGWEKRMSRSSGRVYYFNHINNASQFERPSG-COOH | 58.0 ±0.1 | −0.67 ±0.02 | 5.2 ±0.1 | −2.0 ±0.4 |
| 32Q | H2N-KLPPGWEKRMSRSSGRVYYFNHITNAQQFERPSG-COOH | 58.3 ±0.2 | 8.0 ±0.3 | ||
| 32Qp | H2N-KLPPGWEKRMSRSSGRVYYFNHITNAQQFERPSG-COOH | 63.0 ±0.2 | −0.44 ±0.02 | 2.7 ±0.2 | −1.1 ±0.3 |
| 32X | H2N-KLPPGWEKRMSRSSGRVYYFNHITNAXQFERPSG-COOH | 62.9 ±0.3 | 8.69 ±0.02 | ||
| 32XP | H2N-KLPPGWEKRMSRSSGRVYYFNHITNAXQFERPSG-COOH | 62.8 ±0.3 | 0.01 ±0.03 | 6.9 ±0.2 | −0.2 ±0.2 |
| 32Z | H2N-KLPPGWEKRMSRSSGRVYYFNHITNAZQFERPSG-COOH | 58.8 ±0.2 | 5.8 ±1.0 | ||
| 32ZP | H2N-KLPPGWEKRMSRSSGRVYYFNHITNAZQFERPSG-COOH | 63.8 ±0.3 | −0.33 ±0.03 | 5.1 ±0.6 | −0.1 ±0.5 |
| 32N | H2N-KLPPGWEKRMSRSSGRVYYFNHITNANQFERPSG-COOH | 45.1 ±0.2 | 32.1 ±2.5 | ||
| 32Np | H2N-KLPPGWEKRMSRSSGRVYYFNHITNANQFERPSG-COOH | 50.3 ±0.2 | −0.45 ±0.03 | 16.1 ±0.3 | −0.7 ±0.3 |
| 32Nbp | H2N-KLPPGWEKRMSRSSGRVYYFNHITNANQFERPSG-COOH | 55.2 ±0.1 | −0.93 ±0.03 | 7.5 ±0.8 | −1.5 ±0.4 |
N = Asn-PEG; N = Asn-branched PEG; Q = Gln-PEG; X = Aha; X = Aha-PEG; Z = PrF; Z = PrF-PEG. Melting temperatures determined by variable temperature CD experiments at 50 μM protein concentration in 20 mM sodium phosphate buffer, pH 7, except for 16N, 16Np, 16Nbp, 18N, 18Np, 18Nbp, 32N, 32Np, and 32Nbp, which were performed at 100 μM protein concentration. PEG-based differences in folding free energy are given at the melting temperature of the corresponding non-PEGylated protein. Apparent proteolysis rate constants k and rate constant ratios r were derived from an HPLC-based proteolysis assay at room temperature, as described in the supporting information.
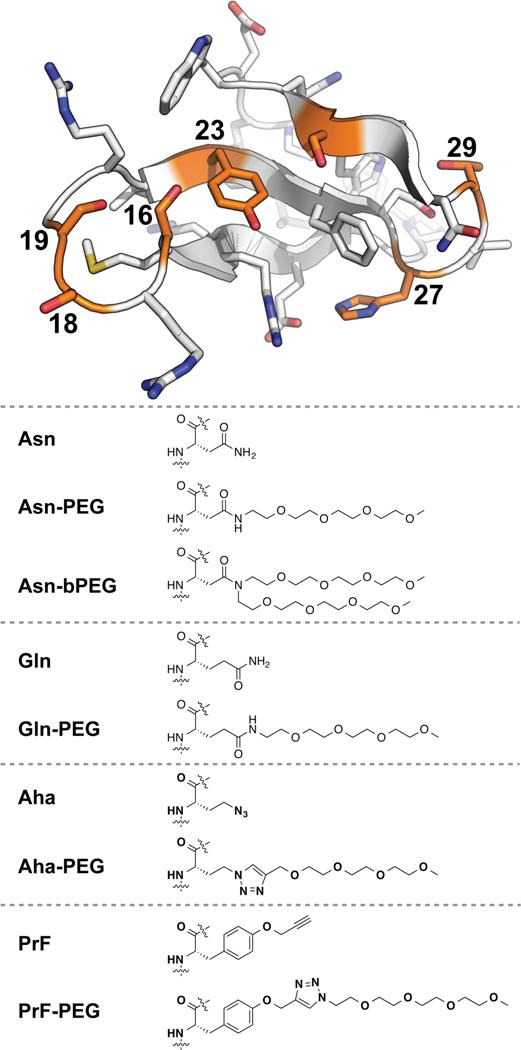
We used variable temperature circular dichroism (CD) experiments to assess the conformational stability of each variant relative to its non-PEGylated counterpart. The results of this analysis appear in Table 1, which lists the melting temperatures (Tm) of each variant and folding free energies (ΔΔGf) of each PEGylated variant relative to its non-PEGylated counterpart at the melting temperature of the non-PEGylated protein. To assess how well Gln-, Aha-, and PrF-PEGylation mimic the impact of Asn-PEGylation on WW stability, we plotted the ΔΔGf associated with Gln-, Aha-, and PrF-PEGylation vs. the ΔΔGf associated with Asn-PEGylation at each of the positions described above, using least-squares regression to fit each data set to a linear equation. The results of this analysis are shown in Figure 2.
Figure 2.
Plots of the impact (ΔΔGf) of Gln-, Aha-, and PrF-PEGylation vs. the impact of Asn-PEGylation (ΔΔGf) on the conformational stability of WW at positions 16, 18, 19, 23, 27, 29, and 32.
The impact of Gln-PEGylation on WW stability correlates relatively well with that of Asn-PEGylation (R2 = 0.62, with slope = 0.59 ± 0.21 and intercept = 0.03 ± 0.11), though Gln-PEGylation is moderately less stabilizing than Asn-PEGylation. Similarly, the impact of Aha-PEGylation on WW stability correlates similarly well with that of Asn-PEGylation (R2 = 0.63, with slope = 0.62 ± 0.21 and intercept = 0.29 ± 0.11), though as with Gln- PEGylation, Aha-PEGylation is less stabilizing. In contrast, the impact of PrF-PEGylation correlates weakly and inversely with that of Asn-PEGylation (R2 = 0.40, with slope = −0.28 ± 0.15 and intercept = −0.30 ± 0.08). Despite the poor correlation between PrF- and Asn-PEGylation, it is interesting that PrF-PEGylation substantially stabilizes WW at positions 23 (ΔΔGf = −0.29 ± 0.03 kcal/mol) and 27 (ΔΔGf = −0.62 ± 0.03 kcal/mol), locations where Asn-PEGylation was substantially destabilizing (ΔΔGf = 0.47 ± 0.11 kcal/mol at position 23; 0.20 ± 0.02 kcal/mol at position 27). Indeed, it is encouraging that for each position we investigated, at least one option among Gln-, Aha-, or PrF-PEGylation is substantially stabilizing, suggesting that matching the appropriate linker to a given site could result in PEG-based stabilization at most positions in WW, and perhaps in other proteins as well.
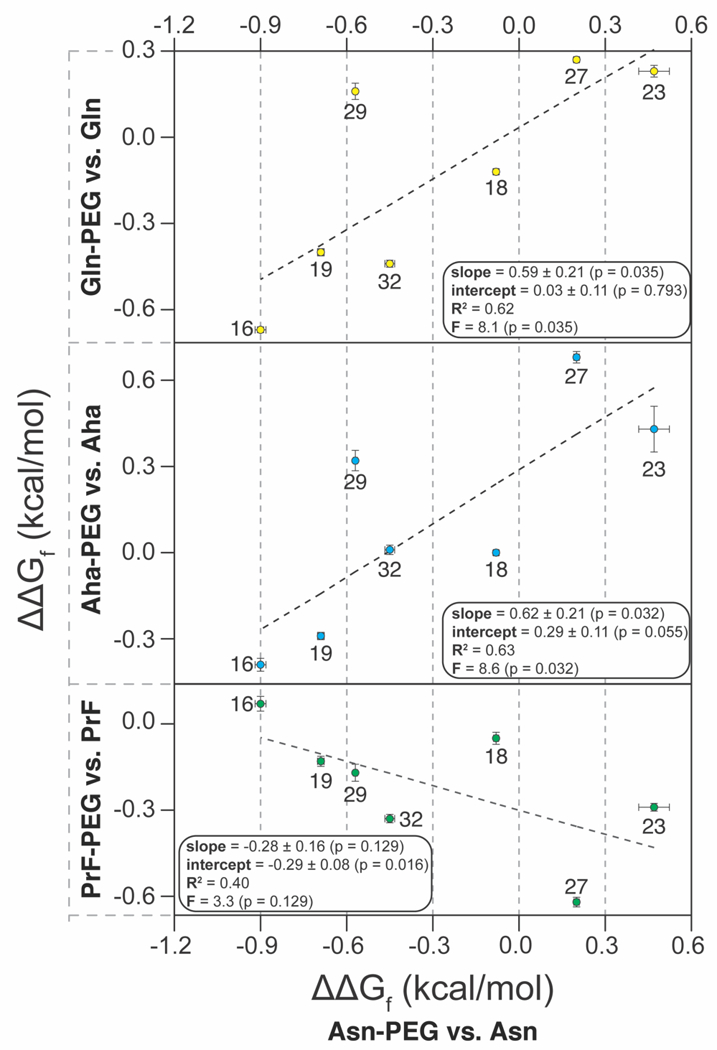
Another important determinant of PEG-based stabilization is the structure of PEG oligomer. We recently found that the stabilizing impact of Asn-PEGylation of WW at position 19 increases as the PEG oligomer lengthens from one to four ethylene oxide units; increasing PEG length beyond this point does not substantially change the observed PEG-based stabilization.20 However, alkylating the side-chain amide nitrogen in Asn-PEG with a second four-unit PEG (i.e. branched PEGylation) results in a branched PEG-Asn conjugate (i.e., Asn- bPEG, Figure 1) that has a much more stabilizing impact on WW at position 19 than its linear Asn-PEGylated counterpart.14 We wondered whether branched Asn-bPEG would have a similarly enhanced effect on WW conformational stability at the other positions we investigated previously for linear Asn-PEG (i.e. positions 16, 18, 23, 27, 29, and 32).
To explore this possibility, we prepared variants 16Nbp, 18Nbp, 23Nbp, 27Nbp, 29Nbp, and 32Nbp, in which Asn-bPEG occupies positions 16, 18, 23, 27, 29, and 32, respectively (variant 19Nbp was prepared and characterized previously14). Melting temperatures and folding free energies relative to the corresponding non- PEGylated compound are shown in Table 1. The correlation between the ΔΔGf associated with Asn-bPEG vs. the ΔΔGf associated with Asn-PEG is strong (R2 = 0.77, with slope = 1.8 ± 0.4; see Figure 3), indicating that Asn- bPEG generally has a more pronounced effect on WW stability than Asn-PEG. At locations where Asn-PEG is stabilizing, Asn-bPEG is substantially more stabilizing; at locations where Asn-PEG is destabilizing, Asn-bPEG is substantially more destabilizing.
Figure 3.
Plot of the impact of branched Asn-bPEGylation vs. linear Asn-PEGylation on the conformational stability of WW at positions 16, 18, 19, 23, 27, 29, and 32.
We previously showed that linear Asn-PEGylation at one or more positions better protects WW from proteolysis when it also increases protein conformational stability.18 We wondered whether the same would be true for Gln-, Aha-, PrF-, and branched Asn-PEGylation. To explore this possibility, we used HPLC to monitor the amount of each WW variant remaining in solution after being exposed to proteinase K (see supporting information for details). We fit the resulting data for each variant to a monoexponential function, from which we extracted the apparent proteolysis rate constants (k, in units of s−1) shown in Table 1. The apparent proteolytic rate constant for each variant is related to half-life (t1/2, in units of s) according to the following relationship: t50 = (ln 2)/k. Consequently, WW variants with smaller apparent proteolytic rate constants have correspondingly higher half-lives (i.e., they survive longer in the presence of proteinase K). In general, we find that WW variants with higher melting temperatures have lower proteolysis rate constants (Figure 4), independent of whether or not they are PEGylated, suggesting that conformational stability is the major determinant the proteolytic stability of each WW variant in the presence of proteinase K.
Figure 4.
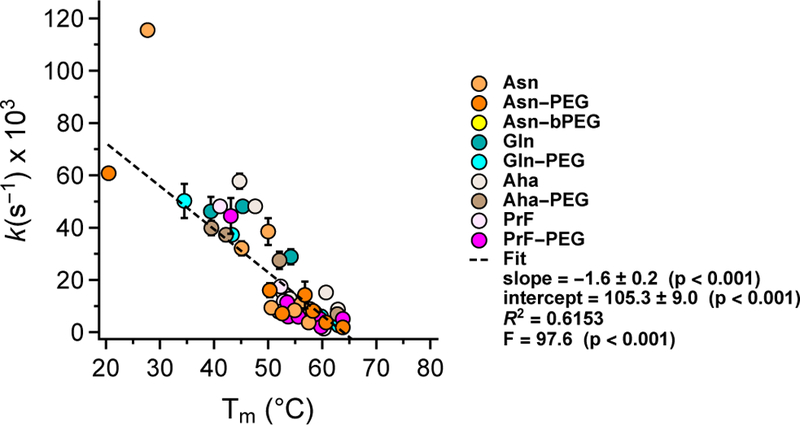
Relatioship between apparent proteolysis rate constants and melting temperatures for the PEGylated WW variants described above, along with their non-PEGylated counterparts, across all positions investigated.
We compared the apparent proteolytic rate constant of each PEGylated WW variant (k) to that of its non- PEGylated counterpart (k°) using the dimensionless rate constant ratio r (defined as r = k / k°). PEGylation accelerates proteolysis when r is greater than 1 (i.e., k > k°), and slows proteolysis when r is less than 1 (i.e., k < k°). We originally envisioned exploring the relationship between r and the observed PEG-based changes in conformational stability (ΔΔGf) for each sequenced matched pair of PEGylated vs. non-PEGylated WW variants. However, rate constants (and rate constant ratios) generally have a logarithmic relationship with changes in free energy; ΔΔGf should be more directly related to ln(r) than to r. Therefore, we used ln(r) as an alternative indicator of the proteolytic stability of each PEGylated variant relative to its non-PEGylated counterpart: PEGylation increases proteolytic stability when ln(r) is negative, and decreases proteolytic stability when and ln(r) is positive.
Figure 5A shows the combined ln(r) vs. ΔΔGf data for all PEGylation sites and methods described above. We fit these combined data to a linear equation via least-squares regression; the resulting slopes and intercepts are given in Figure 5A ± standard error, with R2, F, and p-values as indicated. The negative y-intercept from this combined analysis suggests that PEGylation generally enhances WW proteolytic stability by a small baseline amount even when ΔΔGf = 0. However, the extent of this favorable baseline change in proteolytic stability varies substantially from one PEGylation method to another, (see Figures 5B–F, in which the ln(r) vs. ΔΔGf data for each PEGylation method are analyzed separately. In some cases, baseline changes in proteolytic stability are unfavorable or are indistinguishable from zero (note the large standard errors and unacceptably high p-values for the y-intercepts in Figures 5B,E,F). For example, PrF-PEGylation at position 16 accelerates proteolysis by a factor of 3.6, even though it has almost no impact on WW conformational stability (ΔΔGf = −0.07 ± 0.05 kcal/mol; compare 16Z and 16Zp in Table 1).
Figure 5.
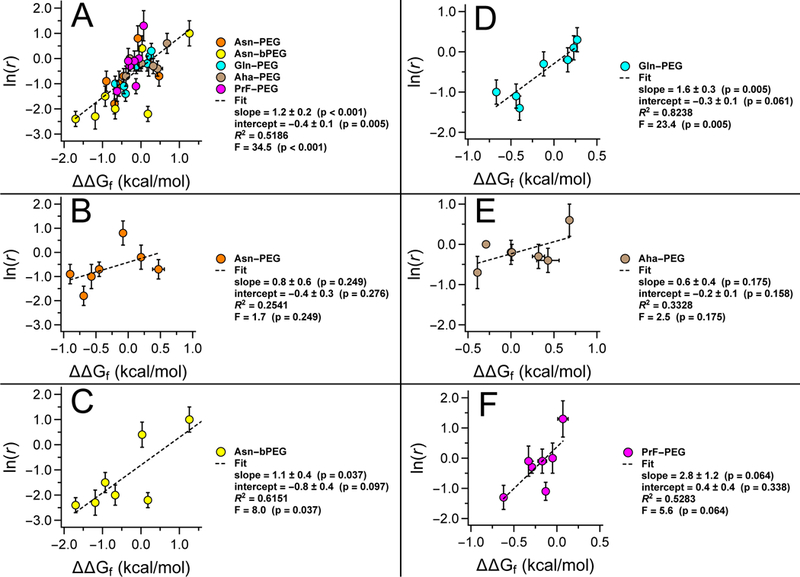
Plots of the natural logarithm of r (i.e., the ratio of apparent proteolysis rate constants for PEGylated vs. non- PEGylated WW variants) vs. PEG-based changes in WW conformational stability (ΔΔGf) for (A) all linker types, and for each individual linker type, including (B) linear Asn-PEGylation, (C) branched Asn-PEGylation, (D) Gln-PEGylation, (E) Aha-PEGylation, and (F) PrF-PEGylation.
The slope for the combined ln(r) vs. ΔΔGf data set is positive (Figure 5A): ln(r) tends to be negative when ΔΔGf is negative and positive when ΔΔGf is positive, indicating that PEG-based changes in WW conformational and proteolytic stability are tightly coupled. We observer greater PEG-based increases to proteolytic stability in tandem with larger increases in conformational stability. For example, branched Asn-PEGylation at position 16 stabilizes WW by −1.70 ± 0.03 kcal/mol and slows proteolysis by a factor of 10, the highest level of proteolytic protection we have observed (compare 16Nbp vs. 16N in Table 1). In contrast, linear Asn-PEGylation at the same position only stabilizes WW by −0.90 ± 0.04 kcal/mol; the associated increase in proteolytic stability is correspondingly smaller (compare 16Nbp vs. 16N in Table 1). PEG-based decreases in conformational stability and proteolytic stability are similarly coupled: branched Asn-PEGylation at position 27 destabilizes WW by 1.26 ± 0.06 kcal/mol and accelerates proteolysis by a factor of 2.8 (compare 27Nbp vs. 27N in Table 1). Slopes obtained from separate analyses of the ln(r) vs. ΔΔGf data for each PEGylation method lead to qualitatively similar conclusions, though the small number of points and the substantial variability within each data set make quantitative comparisons impossible (see Figures 5B–F).
Conclusion
We have shown here that the impacts of Aha-, Gln-, and branched Asn-PEGylation on WW conformational stability are correlated reasonably well with that of linear Asn-PEGylation, suggesting that the structure-based criteria developed previously for identifying stabilizing linear Asn-PEGylation sites18 should be useful for identifying stabilizing Aha-, Gln-, and branched Asn-PEGylation sites. In contrast, the impact of PrF-PEGylation on WW conformational stability does not correlate well with that of Asn-PEGylation. However, we note with interest that two destabilizing Asn-PEGylation sites are substantially stabilized by PrF-PEGylation, suggesting the intriguing possibility that most prospective PEGylation sites can experience PEG-based increases to conformational stability, when matched with the appropriate PEG-protein linker.
We have also shown that the impact of PEGylation on the proteolytic stability of each WW variant depends strongly on how PEG affects conformational stability for each PEG-protein linker explored here. The largest PEG-based reductions in proteolysis rate are strongly associated with the largest PEG-based increases in conformational stability. These results demonstrate that choosing the appropriate PEGylation site and PEG- protein linker is critical for attaining optimal PEG-based increases to conformational and proteolytic stability in the model protein WW, highlighting the continuing need for predictive structure-based tools to guide such choices. Efforts to apply this knowledge to proteins of therapeutic interest are underway.
Experimental Procedures
Proteins 16N, 16Np, 18N, 18Np, 19Q, 19Qp, 19X, 19Xp, 19Z, 19Zp, 19N, 19Np, 19Nbp, 23N, 23Np, 27N, 27Np, 29N, 29Np, 32N, and 32Np (see Table 1) were synthesized previously.14, 18 The remaining WW variants shown in Table 1 were synthesized as C-terminal acids by Fmoc-based microwave-assisted solid-phase peptide synthesis as described previously,14 using the following reagents:
Fmoc-Gly-loaded Novasyn Wang resin (EMD Biosciences)
Standard Fmoc-protected α-amino acids with acid-labile side-chain protecting groups (EMD Biosciences or Advanced ChemTech)
Previously synthesized Fmoc-L-GlnPEG4-OH [18-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-15-oxo- 2,5,8,11-tetraoxa-14-azanonadecan-19-oic acid],14 used to prepare proteins 16Qp, 18Qp, 23Qp, 27Qp, 29Qp, and 32Qp
Commercially available Fmoc-L-4-azidohomoalanine, used to prepare proteins 16X, 18X, 23X, 27X, 29X, and 32X
Previously synthesized PEG-alkyne 2,5,8,11-tetraoxatetradec-13-yne21 used to prepare 16Xp, 18Xp, 23Xp, 27Xp, 29Xp, and 32Xp from proteins 16X, 18X, 23X, 27X, 29X, and 32X via the copper (I) catalyzed azide- alkyne cycloaddition14 on resin
Previously synthesized Fmoc-L-PrF-OH N-[(9H-Fluoren-9-ylmethoxy)-O-2-propyn-1yl-l-tyrosine22, used to prepare proteins 16Z, 18Z, 23Z, 27Z, 29Z, and 32Z
Commercially available PEG-azide 13-azido-2,5,8,11-tetraoxatridecane, used to prepare proteins 16Zp, 18Zp, 23Zp, 27Zp, 29Zp, and 32Zp from proteins 18Z, 23Z, 27Z, 29Z, and 32Z via the copper (I) catalyzed azide-alkyne cycloaddition14 on resin
Previously synthesized Fmoc-L-(AsnPEG4)2-OH [(S)-17-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-15- oxo-14-(2,5,8,11-tetraoxatridecan-13-yl)-2,5,8,11-tetraoxa-14-azaoctadecan-18-oic acid],14 used to prepare proteins 16Nbp, 18Nbp, 23Nbp, 27Nbp, 29Nbp, and 32Nbp
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and N-hydroxybenzotriazole hydrate (HOBt) from Advanced ChemTech for amino acid activation
20% piperidine in N,N-dimethylformamide for removal of the Fmoc protecting group from the N-terminal α- amine
A solution of a solution of phenol (0.0625 g), water (62.5 μL), thioanisole (62.5 μL), ethanedithiol (31 μL) and triisopropylsilane (12.5 μL) in trifluoroacetic acid (TFA, 1 mL) for cleaving the protein from resin and globally removing acid-labile side-chain protecting groups
Proteins were purified by preparative reverse-phase high performance liquid chromatography (HPLC) on a C18 column using a linear gradient of water in acetonitrile with 0.1% v/v TFA. Fractions containing the desired protein product were pooled, frozen, and lyophilized. Proteins were identified by electrospray ionization time of flight mass spectrometry (ESI-TOF) as described previously18; protein purity was assessed by analytical HPLC. CD data were obtained on an Aviv 420 circular dichroism spectropolarimeter. For additional details, see the supporting information.
Supplementary Material
Acknowledgment
This work was supported by NIH R15 GM116055–01.
Footnotes
Supporting Information
Complete experimental materials and methods; ESI-TOF MS and analytical HPLC data; variable temperature CD data; and proteolysis data.
References
- (1).Veronese FM (2009) in Milestones in Drug Therapy (Parnham MJ, and Bruinvels J, Eds.), Birkhauser Verlag, Basel. [Google Scholar]
- (2).Frokjaer S, and Otzen DE (2005) Protein drug stability: a formulation challenge. Nat. Rev. Drug. Discov 4, 298–306. [DOI] [PubMed] [Google Scholar]
- (3).Abuchowski A, Mccoy JR, Palczuk NC, Vanes T, and Davis FF (1977) Effect of Covalent Attachment of Polyethylene-Glycol on Immunogenicity and Circulating Life of Bovine Liver Catalase. J. Biol. Chem 252, 3582–3586. [PubMed] [Google Scholar]
- (4).Abuchowski A, Vanes T, Palczuk NC, and Davis FF (1977) Alteration of Immunological Properties of Bovine Serum-Albumin by Covalent Attachment of Polyethylene-Glycol. J. Biol. Chem 252, 3578–3581. [PubMed] [Google Scholar]
- (5).Harris JM, Martin NE, and Modi M (2001) Pegylation - A novel process for modifying pharmacokinetics. Clin. Pharmacokinet 40, 539–551. [DOI] [PubMed] [Google Scholar]
- (6).Harris JM, and Chess RB (2003) Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov 2, 214–221. [DOI] [PubMed] [Google Scholar]
- (7).Bundy BC, and Swartz JR (2010) Site-specific incorporation of p-propargyloxyphenylalanine in a cell-free environment for direct protein-protein click conjugation. Bioconjugate Chem 21, 255–63. [DOI] [PubMed] [Google Scholar]
- (8).Link AJ, and Tirrell DA (2003) Cell surface labeling of Escherichia coli via copper(I)-catalyzed [3+2] cycloaddition. J. Am. Chem. Soc 125, 11164–11165. [DOI] [PubMed] [Google Scholar]
- (9).Fontana A, Spolaore B, Mero A, and Veronese FM (2008) Site-specific modification and PEGylation of pharmaceutical proteins mediated by transglutaminase. Adv. Drug Deliv. Rev 60, 13–28. [DOI] [PubMed] [Google Scholar]
- (10).Mero A, Schiavon M, Veronese FM, and Pasut G (2011) A new method to increase selectivity of transglutaminase mediated PEGylation of salmon calcitonin and human growth hormone. J. Control. Release 154, 27–34. [DOI] [PubMed] [Google Scholar]
- (11).Chalker JM, Bernardes GJL, Lin YA, and Davis BG (2009) Chemical Modification of Proteins at Cysteine: Opportunities in Chemistry and Biology. Chem. Asian J 4, 630–640. [DOI] [PubMed] [Google Scholar]
- (12).Deiters A, Cropp TA, Summerer D, Mukherji M, and Schultz PG (2004) Site-specific PEGylation of proteins containing unnatural amino acids. Bioorg. Med. Chem. Lett 14, 5743–5745. [DOI] [PubMed] [Google Scholar]
- (13).Cho H, Daniel T, Buechler YJ, Litzinger DC, Maio Z, Putnam AM, Kraynov VS, Sim BC, Bussell S, Javahishvili T, et al. (2011) Optimized clinical performance of growth hormone with an expanded genetic code. Proc. Natl. Acad. Sci. USA 108, 9060–9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lawrence PB, Billings WM, Miller MB, Pandey BK, Stephens AR, Langlois MI, and Price JL (2016) Conjugation Strategy Strongly Impacts the Conformational Stability of a PEG-Protein Conjugate. ACS Chem. Biol 11, 1805–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jäger M, Dendle M, and Kelly JW (2009) Sequence determinants of thermodynamic stability in a WW domain---An all-β-sheet protein. Protein Sci 18, 1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jäger M, Nguyen H, Crane JC, Kelly JW, and Gruebele M (2001) The Folding Mechanism of a β- sheet: The WW Domain. J. Mol. Biol 311, 373–393. [DOI] [PubMed] [Google Scholar]
- (17).Jäger M, Zhang Y, Bieschke J, Nguyen H, Dendle M, Bowman ME, Noel JP, Gruebele M, and Kelly JW (2006) Structure-function-folding relationship in a WW domain. Proc. Natl. Acad. Sci. USA 103, 10648–10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lawrence PB, Gavrilov Y, Matthews SS, Langlois MI, Shental-Bechor D, Greenblatt HM, Pandey BK, Smith MS, Paxman R, Torgerson CD, et al. (2014) Criteria for Selecting PEGylation Sites on Proteins for Higher Thermodynamic and Proteolytic Stability. J. Am. Chem. Soc 136, 17547–17560. [DOI] [PubMed] [Google Scholar]
- (19).Fontana A, Spolaore B, Mero A, and Veronese F (2008) Site-specific modification and PEGylation of pharmaceutical proteins mediated by transglutaminase. Adv. Drug Deliv. Rev 60, 13–28. [DOI] [PubMed] [Google Scholar]
- (20).Pandey BK, Smith MS, Torgerson C, Lawrence PB, Matthews SS, Watkins E, Groves ML, Prigozhin MB, and Price JL (2013) Impact of Site-Specific PEGylation on the Conformational Stability and Folding Rate of the Pin WW Domain Depends Strongly on PEG Oligomer Length. Bioconjugate Chem 24, 796–802. [DOI] [PubMed] [Google Scholar]
- (21).Garofalo A, Parat A, Bordeianu C, Ghobril C, Kueny-Stotz M, Walter A, Jouhannaud J, Begin-Colin S, and Felder-Flesch D (2014) Efficient synthesis of small-sized phosphonated dendrons: potential organic coatings of iron oxide nanoparticles. New J. Chem 38, 5226–5239. [Google Scholar]
- (22).Deiters A, Cropp TA, Mukherji M, Chin JW, Anderson JC, and Schultz PG (2003) Adding Amino Acids with Novel Reactivity to the Genetic Code of Saccharomyces Cerevisiae. J. Am. Chem. Soc 125, 11782–11783. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.