

Abstract
Volatile anesthetics are widely used in human medicine and generally considered to be safe in healthy individuals. In recent years, the safety of volatile anesthesia in pediatric patients has been questioned following reports of anesthetic induced neurotoxicity in pre-clinical studies. These studies in mice, rats, and primates have demonstrated that exposure to anesthetic agents during early post-natal periods can cause acute neurotoxicity, as well as later-life cognitive defects including deficits in learning and memory. In recent years, the focus of many pre-clinical studies has been on identifying candidate pathways or potential therapeutic targets through intervention trials. These reports have shed light on the mechanisms underlying anesthesia induced neurotoxicity as well as highlighting the challenges of pre-clinical modeling of anesthesia induced neurotoxicity in mice. Here, we summarize the data derived from intervention studies in neonatal mouse models of anesthetic exposure and provide an overview of mechanisms proposed to mediate anesthesia induced neurotoxicity in mice based on these reports. The majority of these studies implicate one of three mechanisms: reactive oxygen species (ROS) mediated stress and signaling, growth/nutrient signaling, or direct neuronal modulation.
Introduction
Volatile anesthetics are widely used in human medicine, and routine anesthesia is generally considered to be safe in healthy individuals. In recent years, the use of anesthetic in neonates and children has been questioned following reports of anesthetic induced neurotoxicity (AIN) in pre-clinical studies, as discussed in various reviews and commentaries (for representative examples, see (Lin et al., 2017; Vutskits and Davidson, 2017; Walters and Paule, 2017)). These studies in mice, rats, and primates have demonstrated that exposure to anesthetic agents during early post-natal periods can lead to acute neurotoxicity and later-life defects in learning and memory. Defects in subtler behavioral outcomes, such as fear responses and social interactions, have also been reported in both rodents and primates (Alvarado et al., 2017; Coleman et al., 2017; Lei et al., 2013; Raper et al., 2015; Shi et al., 2017). Acute effects of AIN have generally been quantified by staining for neurodegeneration and apoptotic cells, and cell death appears to occur in both neurons and glia.
The precise period of neonatal sensitivity to anesthesia is controversial. Some rodent studies suggest that neuronal progenitor cells remain sensitive to anesthesia throughout life, and that the period of neonatal sensitivity is primarily defined by the relative fraction of progenitors involved in rapid brain post-natal rodent brain development (Hofacer et al., 2013). However, the period of neonatal rodent hypersensitivity is generally thought to peak around post-natal day 7, with anesthetics causing little or no degeneration or behavioral defects when administered after post-natal day 10 (Jevtovic-Todorovic, 2012; Yon et al., 2005). The putative window of sensitivity is less clear in non-human primates, and there is no consensus in humans.
It is worth noting that AIN appears to occur at both extremes of life – in neonates and in the elderly (Figure 1). In contrast with pediatric studies, neurocognitive complications are well-documented in geriatric patients, involving a range of symptoms collectively referred to as ‘post-operative cognitive dysfunction’, POCD (Johnson et al., 2002). The mechanisms of POCD are unclear, but they appear to be at least partly distinct from those in the very young (Canet et al., 2003; Moller et al., 1998; Newman et al., 2001; Rasmussen et al., 2003). This review will focus on data from models of pediatric AIN models in mice.
Figure 1: Neurotoxicity of anesthesia appears to occur at both extremes of age.
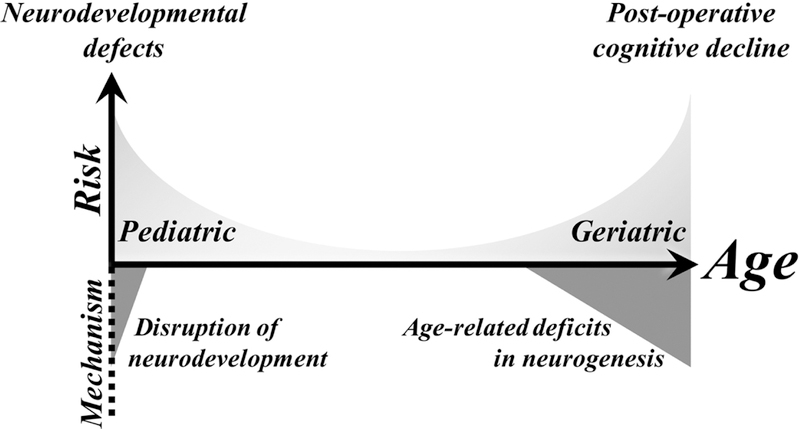
Anesthetics are associated with neuronal death and adverse cognitive effects in both pediatric and geriatric populations. While the precise mechanisms of anesthesia induced neurotoxicity are unclear, data suggests that anesthetics have some neurotoxicity at all ages. In pediatric patients, this neurotoxicity disrupts normal neurodevelopment, and neonates are highly sensitive to as a result of their relatively high number of young neurons. Conversely, sensitivity to anesthesia in geriatric patients appears to result from age-related deficits in neurogenesis which exacerbate the functional impact of neuron loss due to anesthesia exposure.
The substantial literature surrounding the phenomenon of AIN involves mouse, rat, non-human primate, and invertebrate models of exposure. This literature is the subject of multiple reviews which outline the evidence for or against the hypothesis that exposure to anesthetics during early post-natal development has the potential for acute neurotoxicity and long-term adverse cognitive effects in humans (Lin et al., 2017; Vutskits and Davidson, 2017; Walters and Paule, 2017). Here, we summarize intervention studies in neonatal mouse models of anesthetic exposure, i.e. studies where pharmacological, dietary, or genetic manipulations were used to target putative pathways of AIN, providing an overview of proposed mechanisms underlying AIN in mice based on these reports.
Neurotoxicity of Anesthetics - Mechanisms Implicated through Rodent Intervention Studies
Early reports describing the neurotoxic effects of anesthesia were primarily descriptive, but current literature in rodents is now dominated by intervention studies. Mechanisms of AIN are inferred through manipulation of candidate pathways (see Table 1, Figure 2). The majority of putative targets fall into one of three general categories: 1) Reactive oxygen species (ROS) mediated stress and signaling; 2) growth/nutrient signaling; and 3) direct neuronal modulation. A comprehensive assessment of available rodent data provides a landscape of putative mechanisms of AIN.
Table 1 –
AIN Interventions Tested In Vivo in Mice
| Compound | Dose | Mechanism(s)* Known (k), reported (r), or putative (p) | Report(s) |
|---|---|---|---|
| Apocynin | 50 mg/kg | NADPH oxidase inhibitor (k); reduces oxidative stress (r) | (Sun, Z. et al., 2016) |
| Carbon Monoxide | 5 ppm; 100 ppm | Reduced oxidative stress (r) | (Cheng and Levy, 2014; Cheng, Y. et al., 2015; Wang, L. et al., 2017a) |
| Coenzyme Q10 | 50 mg/kg | Electron transport chain component (k);antioxidant (r) | (Xu et al., 2017) |
| Curcumin | 20 mg/kg; 40 mg/kg |
Antioxidant (p) | (Ji et al., 2015) |
| Dexmedetomidine | 15 g/kg; 25 g/kg |
2 adrenergic receptor agonist (k) | (Qian et al., 2015) |
| Environmental enrichment | 2 hrs/day from P7 to P31 | Increased neurogenesis (p) | (Zheng et al., 2013) |
| ‘Hydrogen rich saline’ | n/a | Antioxidant (p) | (Li et al., 2017) |
| Lithium Chloride | 100 mg/kg | AKT/GSK3/ERK activator (p) | (Tao et al., 2016) |
| Memantine | 1 mg/kg | NMDA glutamate inhibitor (k) | (Han et al., 2015) |
| Roscovitine | 25 mg/kg/day | CDK5 inhibitor, action through ERK (p) | (Liu et al., 2017a) |
| Rutin | 25 mg/kg 50 mg/kg | Unknown; nutraceutical, putative antioxidant (p) | (Man et al., 2015) |
| Suberanilohydroxamic acid (SAHA) | 25 mg/kg | Histone deacetylase inhibitor (k) | (Lin et al., 2014) |
| Vitamin C | 80 mg/kg | Multiple; antioxidant (k,p) | (Cheng, B. et al., 2015; Xu, K.X. et al., 2015) |
| Contraindicated (worsens AIN) | Dose | Mechanism(s) | Report(s) |
| Caffeine | 80 mg/kg | Adenosine receptor agonist (k) | (Cabrera et al., 2017) |
| High-fat diet | Induced by reducing litter size and maternal high fat chow | Unknown in this paradigm | (Xu, K.X. et al., 2015) |
| Genetic | Manipulations | Gene Product Function | Report |
| FAS or FASL | KO | Ligand and receptor for extrinsic apoptosis (k) | (Song et al., 2015) |
| IL-1B | KO | Inflammatory cytokine (k) | (Cao et al., 2012) |
| miR-124 | Knockdown | Unknown; most highly expressed miRNA in neurons | (Xu, H. et al., 2015) |
| miR-34a | Knockdown | Regulates FGFR1 (p) | (Jiang et al., 2014) |
Known – generally accepted function supported by robust published pharmacological data; reported – mechanism of action proposed in the setting of AIN if distinct from known mechanisms of action (or if none known); putative – the mechanism suggested by the authors when little mechanistic data is available (includes commonly reported compounds with poorly described mechanisms of action, for example nutraceuticals).
Figure 2: Interventions and mechanisms reported in mouse models of AIN.
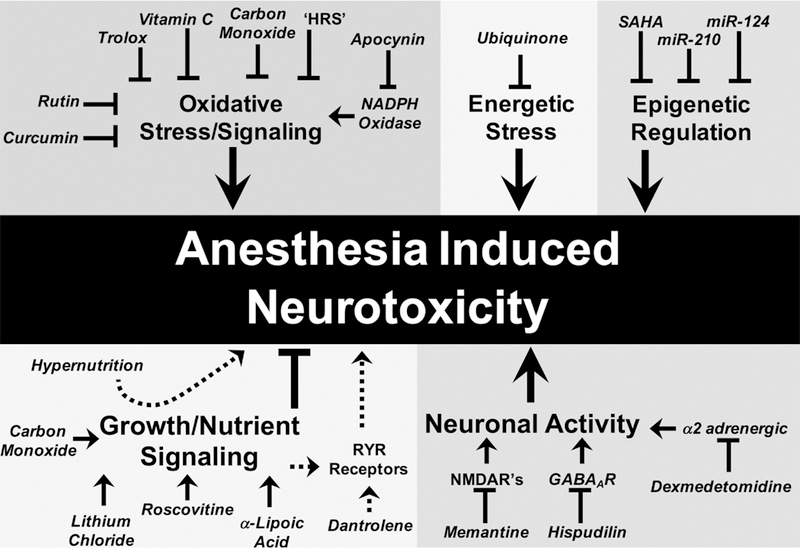
Intervention studies in mouse AIN models have identified a wide variety of candidate targets and compounds. The majority of these can be grouped into one of three major categories: oxidative stress, ROS signaling, and energetics; growth/nutrient signaling; and direct modulation of neuronal activity. Additional candidates, such as epigenetic factors, are less clearly defined.
Oxidative Stress and ROS Signaling
Oxidative stress and ROS signaling are the most widely reported therapeutic targets in rodent models of AIN. The most recognizable chemical antioxidant applied to this paradigm is ascorbic acid (vitamin C), which has been shown to attenuate many cellular, molecular, and behavioral endpoints of AIN in vitro and in vivo, including induction of cleaved caspase 3, increased ROS, mitochondrial permeability transition pore (MPTP) opening (a step in mitochondria mediated cell death), reduced ATP levels, and ‘freezing time’ in a fear conditioning assay, a cognitive behavioral phenotype (Cheng, B. et al., 2015; Xu, K.X. et al., 2015). Similarly, Trolox, a water soluble vitamin E analog, prevented neuron death in vitro in a study implicating mitochondrial ROS induced apoptosis in AIN (Bai et al., 2013).
Attenuation of AIN has also been reported in mice exposed to carbon monoxide (CO) or ‘hydrogen rich saline’ (HRS), two treatments shown to have general antioxidant effects (Cheng and Levy, 2014; Li et al., 2017). CO has been found to reduce mitochondrial ROS production in vitro, reportedly by binding to and inhibiting the peroxidase activity of cytochrome C, a source of mitochondrial ROS that is reportedly enhanced by isoflurane (Cheng and Levy, 2014; Kapetanaki et al., 2009). CO provided a dose dependent attenuation of molecular endpoints associated with anesthesia, with 5 ppm attenuating, and 100 ppm fully abrogating acute molecular and cellular markers of AIN. In agreement with these in vitro effects, CO treatment also partially attenuated behavioral defects induced by isoflurane (Wang, L. et al., 2017a). Similarly, HRS has been found to act as a potent antioxidant in vivo with, apparently, little in the way of off-target effects on redox or signaling (reviewed in Ohta, 2015 (Ohta, 2015)). The CO data are fascinating but have yet to be independently reproduced, while others have shown low dose CO can lead to neurodevelopmental defects in mice which are reminiscent of AIN (Trentini et al., 2016). Clearly, more work is needed to identify the ideal concentrations and exposure times of CO to alleviate AIN, and further characterize the mechanisms underlying the benefits of CO.
Curcumin and rutin, ‘nutraceutical’ compounds associated with many putative bioactive functions, have been used to prevent AIN in vivo (Ji et al., 2015; Man et al., 2015). IP injection of curcumin prior to sevoflurane exposure was reported to attenuate an array of outcomes including induction of cleaved caspase 3, expression of NADPH Oxidase 2 (Nox2, involved in cell non-autonomous ROS signaling), expression of brain derived neurotrophic factor (BDNF), expression of tumor necrosis factor alpha (TNFα), and prevented fear response defects. Similarly, rutin, provided orally, was found to prevent induction of cleaved caspase 3, circulating S100B, and Morris Water Maze defects. While the purported bioactive functions of these compounds are diverse, the putative antioxidant effects were implicated as mediating their benefits in the setting of AIN.
Each of these studies reported attenuation of AIN using general approaches to targeting oxidative stress through modulation of ROS levels. In contrast, a recent report by Makita et al. found that the specific NADPH oxidase inhibitor apocynin protected against AIN, as measured by the lipid peroxidation marker 4-HNE, the ROS indicator dye DHE, cleavage of caspase 3, and behavioral defects (Sun, Z. et al., 2016). NAPDH oxidases are membrane bound enzymes which produce superoxide in neutrophils and in cells involved in ROS mediated signaling, such as vascular smooth muscle cells (Garcia-Redondo et al., 2016; Prieto-Bermejo and Hernandez-Hernandez, 2017). The apocynin results implicate ROS signaling, rather than oxidative stress, in mediating the benefits of antioxidants in AIN models, as inhibition of membrane bound NADPH oxidases would not be expected to impact intracellular ROS.
Attenuation of ROS levels is generally thought to protect cellular energetic status by preventing ROS induced damage to mitochondrial macromolecules, such as subunits the electron transport chain. Targeting energetics more directly has also been attempted. Ubiquinone (coenzyme Q10 or CoQ10), a vitamin-like cofactor which carries electrons from complexes I and II to III in the mitochondrial electron transport chain, rescued ATP levels, mitochondrial membrane potential, and Morris Water Maze performance defects resulting from anesthesia exposure. Ubiquinone did not, however, attenuate increased ROS levels induced by sevoflurane exposure (Xu et al., 2017). The authors concluded energetics directly mediate the benefits of CoQ10 in this paradigm.
Studies involving ROS are mired with several caveats. Methodological approaches to measuring ROS production, or even net ROS damage, are technically challenging, and prone to false positive findings (Egea et al., 2017; Gorlach et al., 2015; Griendling et al., 2016). Compounds with antioxidant effects demonstrated via chemical analysis do not necessarily act as antioxidants in vivo, and antioxidant therapies have proven ineffective in multiple clinical settings where ROS were thought to play a causal role (Goszcz et al., 2015; Sawyer, 2011). In recent years it has become apparent that ROS act as potent intracellular and extracellular signaling molecules, and modulation of ROS levels or production can also lead to unexpected changes in signaling. Finally, even when changes to ROS production and/or oxidative damage are clearly demonstrated, causality is extremely difficult to establish in the context of ROS. For these reasons, the role of ROS in AIN must be assessed with great caution. These pitfalls of ROS assays are reviewed extensively elsewhere (Egea et al., 2017; Gorlach et al., 2015; Griendling et al., 2016) and should be carefully considered in designing experiments aimed at determining the role of ROS in AIN. Even in the best designed study, it is extremely difficult to demonstrate causality between ROS and disease. State of the art methodologies such as single cell RNA sequencing, in situ RNA sequencing, and in vivo detection of oxidative stress and redox status may provide new temporal and spatial evidence linking ROS and CNS apoptosis (Bacic et al., 2016; Lee, 2017a, b; Zhu et al., 2017). In addition, future experiments where both the timing and cellular location of antioxidants are carefully controlled may help resolve the current uncertainty in the roles of ROS in causing AIN.
Growth and nutrient sensing signaling
Growth, differentiation, viability, and survival of neurons depends on the interactions of numerous extra-and intra-cellular signaling cascades, together often referred to as growth signaling or nutrient sensing signaling (see Figure 3). Extracellular growth factors include insulin, insulin-like growth factor 1 (IGF-1), growth hormone (GH), epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), and platelet derived growth factor (PDGF). Many of these systemic factors are directly or indirectly regulated by nutritional status, and each acts on one or more cell surface receptor tyrosine kinases (RTK’s) or G-protein coupled receptors (GPCR’s) (Lemmon and Schlessinger, 2010; Wauson et al., 2013). Transduction to intracellular signaling is mediated by membrane associated factors such as phosphatidyl inositol 3 kinase (PI3K), which activates intracellular mediators of growth/nutrient signaling (Paez and Sellers, 2003; Piwien-Pilipuk et al., 2002). The mechanistic target of rapamycin (mTOR) is a hallmark intracellular mediator of nutrient growth signaling (reviewed in (Saxton and Sabatini, 2017)). mTOR activation tunes up anabolic processes, such as mRNA translation, and dampens catabolic processes, such as autophagy. mTOR is positively regulated by cell receptor/PI3K signaling via AKT mediated inactivation of the mTOR inhibitors TSC1/TSC2.
Figure 3: Growth and nutrient sensing signaling pathways at the interface between intra- and extra-cellular stimulus.
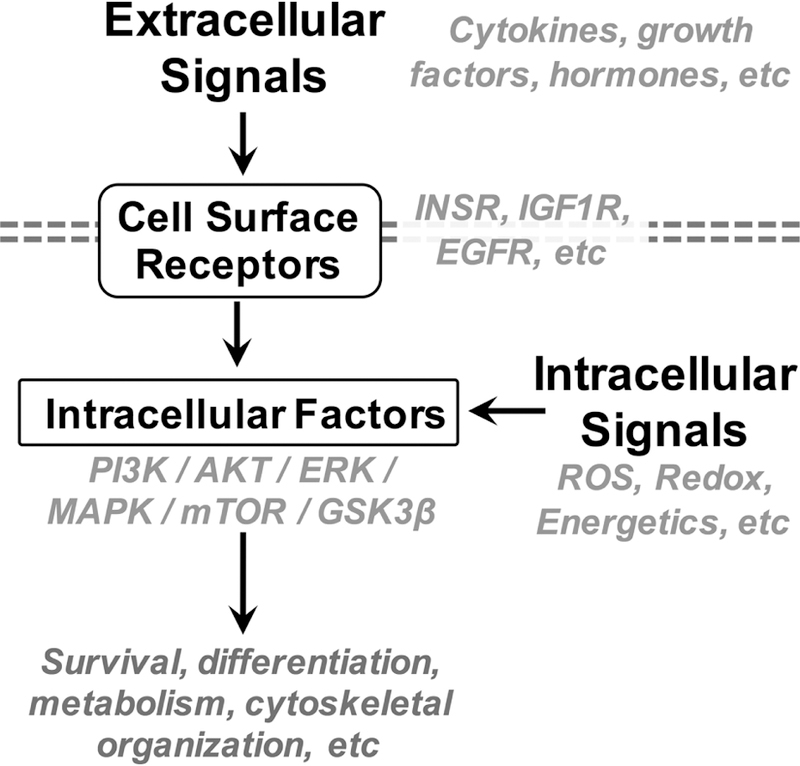
Growth and nutrient signaling pathways involve soluble signaling factors, membrane bound receptors at the cell surface, intracellular sensors, and intracellular kinases which mediate signal transduction and amplification. Growth and nutrient signaling pathways, such as the canonical PI3K/AKT/mTOR pathway, play critical roles in neuron survival, differentiation, metabolism, and cellular organization.
Growth/nutrient signaling pathways are also highly regulated intracellularly. AMP activated kinase (AMPK) is an mTOR inhibitor that is activated by low cellular ATP status. REDD1/2 are hypoxia sensors that inhibit mTOR in low oxygen conditions. mTOR is also regulated by amino acid levels at the lysosome and modulated by ribosome status, linking mTOR activity to functional capacity for mRNA translation. Together, the intra- and extra-cellular regulation of growth/nutrient sensing signaling provides for cellular adaptation to a variety of stressful conditions. Various reviews detail the role of growth/nutrient signaling in neuronal development (Switon et al., 2017; Takei and Nawa, 2014).
Nutrient sensing and signaling pathways are critical to neurogenesis, synaptogenesis, and neuron development, migration, and survival (Alsina et al., 2012; Lee, 2015; Nieto-Estevez et al., 2016). Genetic defects in growth/nutrient signaling cause overt neurological conditions including epilepsy, while subtler abnormalities in nutrient signaling are implicated in neurological disorders ranging from autism to Alzheimers’s (Adachi et al., 2018; Bedse et al., 2015; Borrie et al., 2017; Wang, L. et al., 2017b). Given their importance to neurodevelopment, it is unsurprising that these pathways have also been implicated in the pathogenesis of AIN through both in-vivo and in-vitro studies. Here, we focus on in-vitro evidence supporting a role for nutrient signaling in AIN (in vitro models are reviewed elsewhere, for example see (Wang, 2012; Wang and Slikker, 2008; Wang, C. et al., 2017)).
In some studies, isoflurane has shown to reduce levels of activating phosphorylation on AKT, GSK3B, and ERK through unknown mechanisms in the setting of both neonatal (Tao et al., 2016; Wang et al., 2012) and adulthood (Liu et al., 2014) exposures in mice. In mouse neonates, treating isoflurane anesthetized mice with lithium chloride (a poorly understood but broadly neuroactive compound) attenuated isoflurane induced decreases in AKT and GSK3B phosphorylation, and prevented neuronal death and learning/memory defects (Morris Water Maze, MWM) associated with AIN in neonatal mice (Tao et al., 2016; Wang et al., 2012). Roscovitine, an inhibitor of the cell-cycle regulator cyclin dependent kinase 5 (CDK5), was recently reported to prevent AIN through activation of the ERK pathway (Liu et al., 2017a). The reported benefits of CO have been ascribed to antioxidant effects (as mentioned above) (Levy, 2017) as well as to inhibitory effects on intracellular signaling. Specifically, CO has been found to modulate cAMP, p38 MAPK, and PKB/AKT signaling, increasing mitochondrial biogenesis, inhibiting inflammation, and preventing anesthesia induced apoptosis. The mechanistic underpinnings of these ‘signaling’ functions are yet to be defined, but the authors suggest that CO prevents anesthesia related damage at least partly through these intracellular signaling modulations.
The observation that anesthetics alter signaling is consistent with reports finding modest or no increase in cell death following anesthesia but observing behavioral defects nonetheless. Changes to signaling pathways could modify neuronal development and function independently of cell loss. Signaling may also provide a reasonable model for neonatal hypersensitivity to AIN (compared to adult animals) given the role of PI3K/AKT pathways in nervous system development, as discussed. It has been suggested that volatile anesthetic induced alterations in signaling impact neurogenesis and reduce neural stem cell pools in young, but not adult, animals. This is suggested by observed reductions in BrdU/NeuN positive (proliferating) and Sox-2/GFAP (neural progenitor) cells in young rodents exposed to volatile anesthetics (Zhu et al., 2010). A similar model supported by Hofacer et al suggests that the age of neurons, not the age of the organism, underlies sensitivity to volatile anesthetic toxicity, and that young animals are more sensitive overall simply because they have a greater pool of ‘young’ neurons (Hofacer et al., 2013). Voluntary exercise and environmental enrichment are both neurogenesis promoting interventions which have been reported to function through growth signaling, and both have been shown to attenuate AIN in mice (Zheng et al., 2013).
A number of in vitro studies of cultured neurons have found that anesthetic exposure alters neuronal survival, morphology, and function through activation of the neurotrophin receptor (p75NTR) and subsequent downstream activation of the actin cytoskeleton regulating kinase RhoA. In addition, inhibition of p75NTR can prevent apoptotic neuron death and cytoskeletal depolymerization resulting from anesthetics (Head et al., 2009; Lemkuil et al., 2011; Schallner et al., 2014). Inhibition of this p75NTR/RhoA pathway in vivo with the p75NTR inhibitor TAT—Pep5 did not attenuate memory defects induced by anesthesia exposure, suggesting an uncoupling of this apoptosis-mediating pathway from cognitive effects (Schilling et al., 2017). While intriguing, this finding is difficult to interpret, however, due to the absence of molecular data or a positive control, specifically the lack of apoptosis data derived from the in vivo model.
The precise role of growth and nutrient signaling remains controversial, however, as some reports suggest signaling activation, not inhibition, by anesthetic exposure (Liu et al., 2015). In line with this view, neonatal hyper-nutrition (accomplished through a neonatal ‘high-fat’ paradigm) has been reported to worsen AIN, although the authors attributed the effects to oxidative stress in the ‘obese’ pups (Xu, K.X. et al., 2015). In mouse models of Alzheimer’s disease, anesthetics have been reported to increase the phosphorylation of various signaling factors including PI3K, AKT, MAPK, and JNK, with some work suggesting that anesthetics directly inhibit the phosphatase PP2A, thereby increasing phosphorylation status among PP2A targets. Such a change would broadly disrupting nutrient signaling through inhibition of a negative regulator of activation (Le Freche et al., 2012; Tao et al., 2014). Other reports indicate that ER calcium release induced by ryanodine receptors (RYR’s), downstream of PI3K, mediates the neurotoxicity of anesthetics (Liang et al., 2010; Wang et al., 2014). Blocking RYR’s with the antagonist Dantrolene was found to reduce cell death and ER stress induction in cultured mouse brain slices treated with isoflurane. ER stress has also been shown to mediate isoflurane induced neurotoxicity during developmental stages in the nematode C. elegans in an mTOR dependent pathway (Na et al., 2017).
Conflicts in reported pathway directionality may be at least partly the result of the transient nature of intracellular signaling cascades and the timing of measurement. For example, one study indicates that GSK3 and AKT phosphorylation is increased by short term exposure to volatile anesthetics but decreased by long term exposure (Zhang et al., 2014). The variability is also likely to be at least partly a result of differing anesthetic protocols (see Figure 4). Accordingly, while nutrient signaling appears critically important, new approaches with careful experimental design, execution, and interpretation are needed to resolve these controversies. Most pressing, perhaps, are the need for standardization of anesthetic conditions in neonatal mouse AIN models and an effort to carefully model clinical exposures. Careful temporal dissection of the impact of anesthetics on nutrient signaling in rigorously monitored anesthetic exposures may provide a unifying model for the role of signaling in AIN. As in the case of ROS, state of the art techniques assessing single cell changes in signaling may provide direct causal evidence linking nutrient signaling to apoptotic death or altered neuronal function. Finally, experiments aimed at defining mechanistic links between nutrient signaling and other putative pathways of AIN (ie ROS, neuronal modulation, etc) will clarify the relationship between nutrient signaling and other pathways of damage in relation to the overall pathogenesis of AIN.
Figure 4: Sources of variability in pre-clinical anesthesia induced neurotoxicity literature.
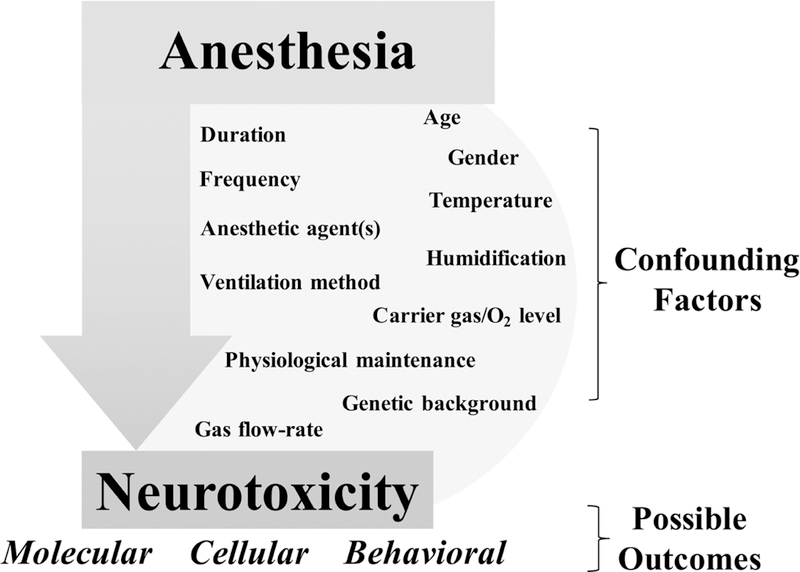
Pre-clinical models of anesthesia induced neurotoxicity have identified a variety of pathways involved in AIN, but a global assessment of these studies is hampered by the high variability in experimental conditions. Confounding factors include anesthetic dose, frequency, duration, and various aspects of physiological maintenance. In addition, measured outcomes vary widely between studies. Future work in pre-clinical AIN would benefit from standardization of anesthetic protocols and experimental methods used to assess AIN.
Additional Targets
Direct neuronal modulation
AIN has also been attenuated through direct modulators of neuronal activity, though much of the data comes from non-neonatal settings. In one study, acute treatment with dexmedetomidine, an alpha-2 agonist, prevented anesthesia induced inflammation and cognitive defects in geriatric mice (Qian et al., 2015). Attenuation of AIN by dexmedetomidine implicates excitotoxicity in anesthesia related neurotoxicity, suggesting that select non-volatile anesthetics may actually prevent the neurotoxicity of volatile agents. Consistent with this idea, Hispudilin, a nutraceutical with ‘potent anti-antiepileptic activity in rats’ and putative a GABAAR agonist, attenuated anesthetic neurotoxicity in vitro (see also above) (Niu et al., 2014). The most directly supportive data is provided by memantine, a clinically approved drug for Alzheimer’s disease which targets excitotoxicity through uncompetitive inhibition of glutamatergic AMPA. Memantine was found to attenuate the induction of an epigenetic biomarker associated with anesthesia induced neurotoxicity in P7 neonatal mice sedated with sevoflurane (Han et al., 2015). Direct modulation of neuronal signaling can also exacerbate AIN; caffeine, an adenosine receptor antagonist, substantially worsened AIN phenotypes, although the paradigm (P3 mouse pups), the exceptionally high dose of caffeine (80 mg/kg, roughly 50% of the LD50 for subcutaneous caffeine in adult mice according to Caymen chemicals), and lack of a caffeine-only control cohort greatly limit the interpretability of this report (Cabrera et al., 2017).
Central nervous system metabolism and peripheral metabolism are both acutely altered by some anesthetics, but the precise impact of any given anesthetic on individual metabolic pathways and the role of metabolic shifts in AIN are unclear (Yamada et al., 2009). Serum metabolites have, remarkably, been generally ignored in AIN studies. When circulating metabolites have been considered, hypoglycemia has been repeatedly identified as a physiological consequence of volatile general anesthetic use. 3% isoflurane induces substantial hypoglycemia by 90 minutes in 10 day old neonatal mice, and extended (6 hour) exposure to 1.5% isoflurane induces hypoglycemia in P7 mice (Loepke et al., 2009; Loepke et al., 2006). Furthermore, 6 hours of equipotent 1.5% isoflurane, 2.9% sevoflurane, or 7.4% desflurane all resulted in significantly reduced blood glucose in neonates compared to fasted controls in CD1 x C57Bl/6 hybrid mice, a background specifically selected for its ‘low mortality’ (of 20%) during 6 hours of treatment (Istaphanous et al., 2011). One report has suggested that dextrose administration fails to prevent neuronal apoptosis or learning and memory defects in isoflurane anesthetized pups. The same report presented data showing that 6 hours of 0.75% isoflurane induced neuronal apoptosis and cognitive defects without leading to marked hypoglycemia (Johnson et al., 2008). Circulating metabolites therefore appear important, but glucose alone may not fully explain AIN in mice. It is worth noting that no study to date has provideed a rigorous assessment of the physiological impact of extended anesthesia in neonatal mice.
Inflammation
Genetic deletion of FAS and FASL, two factors involved in extrinsic apoptotic signaling, prevented induction of apoptosis related to anesthesia exposure in neonatal mice (Song et al., 2015). This result suggests that the majority of AIN related apoptosis results from extrinsic, rather than intrinsic (mitochondrial), apoptotic pathways. Knockout of the receptor for IL-1B, a major cytokine involved in inflammatory signaling, was shown to prevent post-operative cognitive dysfunction in mice (Cao et al., 2012; Cibelli et al., 2010). Together, these findings suggest that neuronal apoptosis and behavioral defects associated with volatile anesthetic exposure require, and can be greatly attenuated by targeting, cell-extrinsic factors. These data are at odds with studies reporting that apoptosis results from cell intrinsic inducers such as mitochondrial oxidative stress, reduced mitochondrial membrane potential, or cellular energetic stress, and discussed above.
Epigenetics
Epigenetic and miRNA targets have also been studied in the context of AIN, though the results are difficult to interpret. Knockdown of miR-124 and miR-210 have been reported to attenuate anesthesia induced neuronal death in vitro or in vivo (Wang et al., 2016; Xu, H. et al., 2015). miR-124 was shown to activate the PKC/ERK pathway in brain and upregulate AMPA receptor phosphorylation, but direct targets and a clear mechanism were not identified. Daily oral administration of SAHA (suberanilohydroxamic acid), a histone deacetylase inhibitor with broad and non-specific actions on epigenetic regulation through histone deacetylation, was reported to modify Morris Water Maze performance in mice exposed to sevoflurane as neonates (Lin et al., 2014). While intriguing, each of these studies lack clear rationale for their epigenetic target of interest, and with such broadly acting regulatory factors, particularly histone acetylation, it is entirely unclear what the mechanisms or off-target effects of these approaches might be.
Limitations of Pre-Clinical Studies
Any discussion of the phenomena of AIN would be incomplete without a critical evaluation of the pre-clinical literature. While, as stated above, it is clear that anesthetics can cause neurotoxicity under various experimental conditions, a multitude of factors have muddied the AIN field. AIN studies tend to lack sufficiently detailed methodological reporting, often have inappropriately low sample numbers, and are missing appropriate controls. Furthermore, the high variability in anesthetic protocols and conditions largely precludes comparisons between individual reports (see Table 2). These issues have led to highly discordant body of research that is not easily extrapolated to the clinical setting. Finally, it remains unclear whether AIN models in mice are relevant to human anesthesia exposure. As noted elsewhere, mouse exposures are poorly monitored when compared to human anesthesia, and a detailed assessment of the physiological impact of anesthesia on neonatal mice is lacking (Van Biesen et al., 2015).
Table 2 –
AIN Paradigms in Neonatal Mice
| Age | Background/Gender | Drug(s) | Dose and time | Other Conditions | Endpoints | Report |
|---|---|---|---|---|---|---|
| P6–8 | C57Bl/6; gender not indicated | Sevoflurane | 3% 2hr/day for 3 days | 60% oxygen | Morris water maze at P30–34; assays for apoptosis following treatment | (Song et al., 2015) |
| P7 | C57Bl/6; both genders | Propofol | 30 or 60 mg/kg IP injected | 37 degrees C | Apoptosis and neurogenesis at P8 or P17; AKT/ERK signaling | (Huang et al., 2016) |
| P6 | C57Bl/6; male only for behavior, both genders for molecular endpoints | Sevoflurane | 3% for 6 hours | 40% oxygen, 38 degrees C | Apoptosis related endpoints immediately after treatment | (Sun, Z. et al., 2016) |
| P7 | C57Bl/6; both genders | Isoflurane; Lidocaine; Lidocaine plus midazolam | Isoflurane: 0.75% for 6 hours; Lidocaine: 4 mg/kg subcutaneous; midazolam: 9 mg/kg subcutaneous | Warmed with heat lamp, agitated as needed to increase respiratory rate | Apoptosis related endpoints from samples 6 hours after exposure | (Lee et al., 2014) |
| P7 | (F) C57Bl/6 x (M) CD-1 hybrid offspring; gender not indicated | Isoflurane | 1.5% for 6 hours | 30% oxygen, 35.5 degrees C | Apoptosis related endpoints after exposure; Morris water maze and spontaneous activity at P70 | (Loepke et al., 2009) |
| P5–7 | C57Bl/6, gender not indicated | Isoflurane; isoflurane plus midazolam | 0.75% 4 hours 0.75% plus midazolam 6 hrs; 1.5% 2 hours; 2% 1 hour | 30 degrees C | Apoptosis related endpoints | (Johnson et al., 2008) |
| P14 | C57Bl/6; males | Isoflurane | 1.7%, 35 min/day for 4 days | 50% oxygen, 37 deg C | Apoptosis related endpoints; neurogenesis | (Zhu et al., 2010) |
| P6–8 | C57Bl/6; both genders | Sevoflurane | 3% isoflurane, 2 hours/day for 3 days | 60% oxygen, 37 degrees C | pGSK3, pAKT | (Zhang et al., 2014) |
| P6 | C57Bl/6; both genders | Sevoflurane | 2.2%, 2 hours per day for 3 days | 37–38 deg C | Morris water maze at P40 | (Liu et al., 2017a) |
| P6 | C57Bl/6; both genders | Isoflurane, desflurane | 2% isoflurane or 8% desflurane (0.7 MAC) 2 hrs/day for 3 days | 60% oxygen, 37 deg C | pAKT, pGSK3 after exposure; Morris water maze P31–37 | (Tao et al., 2016) |
| P10 | 129T2/SvEvMsJ x C57BL6/J F1 hybrid, both genders | Isoflurane | 3% isoflurane, 90 minutes | Not specified; mechanical ventilation for some at 300 breaths/min | Metabolic parameters | (Loepke et al., 2006) |
| P6 | C57Bl/6; both genders | Sevoflurane | 2%, 6 hours | 40% oxygen, 1L/min flow, humidified | Apoptosis related parameters | (Satomoto et al., 2016) |
| P7 | C57Bl/6; both genders | Isoflurane; propofol | 1.5% isoflurane or 150 mg/kg propofol | Isoflurane: in 30% oxygen | Apoptosis related parameters; Morris water maze at P39 | (Yang et al., 2014) |
| P6-P8 | C57Bl/6; males | Sevoflurane | 3% 2 hours/day for 3 days | 40% oxygen, 37 degrees C | Apoptosis related parameters after exposure | (Ji et al., 2015) |
| P7 | C57Bl/6; both genders | Sevoflurane; propofol | 2.9% sevoflurane 6 hours or 150 mg/kg propofol | Sevoflurane in 30% oxygen, 38 deg C, humidified | Apoptosis related parameters after exposure; morris water maze at P31–34 | (Man et al., 2015) |
| P7 | C57Bl/6; not indicated | Sevoflurane; sevoflurane plus propofol or thiopental | 3% for 6 hours with or without 5 mg/kg thiopental or 10 mg/kg propofol | Not available | Apoptosis related parameters after treatment | (Tagawa et al., 2014) |
| P6–8 | C57Bl/6 | Sevoflurane | 3%, 2 hours daily for 3 days | 60% oxygen, 37 degrees | Apoptosis related parameters; Morris water maze at P31–37 | (Lin et al., 2014) |
| P7 | C57Bl/6; both genders | Sevoflurane, isoflurane, desflurane | Isoflurane: 1.5% 6 hours; sevoflurane 2.9% 6 hours; desflurane: 7.4% 6 hours | 30% oxygen, 38 degrees, humidified | Apoptosis related parameters; various behavior tests at P35 | (Xu, K.X. et al., 2015) |
| P7 | C57Bl/6CR; both genders | Isoflurane, sevoflurane | 0.75% isoflurane for 6 hours; 1.1% sevoflurane 6 hours | 30% oxygen, 38 degrees C, humidified | Apoptosis related parameters at 2 hours post exposure; Morris water maze at P42 | (Liang et al., 2010) |
| P7 | C57Bl/6; male mice | Sevoflurane | 1.5%, 2 hours | Air oxygen, 37 degrees C | Protein/signaling changes | (Han et al., 2015) |
| P6–8 | C57Bl/6J; both genders | Sevoflurane | 3%, 2 hours daily for 3 days | 60% oxygen, 37 degrees C | Synaptic protein levels; Morris water maze at P31–37 | (Xu et al., 2017) |
| P14–21 | C57Bl6; gender not indicated | Ketamine | 50 mg/kg/day for 6 days | n/a | Morris water maze at 2 months; molecular endpoints | (Xu, H. et al., 2015) |
| P7 | CD-1, male | Isoflurane | 2%, 1 hour | Air oxygen | Retinal cell apoptosis immediately and 5 hours after anesthesia | (Cheng, Y. et al., 2015) |
| P7 | CD-1, males | Isoflurane | 2%, 1 hour | Air oxygen | Apoptosis related endpoints | (Cheng and Levy, 2014) |
| P6–7 | C57Bl/6J; both genders for biochemical endpoints, males only for behavior | Sevoflurane | Titrated dose for 6 hours: 3.5% for 90 min, 3% for 90 min, then 2.5% for the final 3 hours. | 30% oxygen | Autism related behaviors, memory assessed by fear related assays; apoptosis related endpoints | (Chung et al., 2015) |
| P7–9 | C57Bl/6 | Isoflurane,sevoflurane | 1.5% isoflurane or 2.2% sevoflurane, 2 hours/day for 3 days | 37–38 degC | Morris water maze; apoptosis related endpoints; expression of BDNF and synaptic proteins | (Liu et al., 2017b) |
| P7–8 | CD-1 male x C57Bl/6 hybrid, both genders | Isoflurane, sevoflurane, desflurane | 6hrs 1.5% isoflurane, 2.9% sevoflurane, or 7.4% desflurane | 30% oxygen, 35.5 deg C | Apoptosis related endpoints | (Istaphanous et al., 2011) |
| P7, P21, or P49 | C57Bl/6J, both genders | Isoflurane | 1.5%, 6 hours | 30% oxygen, 35.5 deg C | Apoptosis related endpoints, neurogenesis | (Hofacer et al., 2013) |
| P7 | C57Bl/6CR, both genders | Isoflurane, sevoflurane | 0.75% isoflurane or 1.1% sevoflurane, 6 hours | 30% oxygen, 38 deg C | Apoptosis, serum S100B, Morris water maze at P42 | (Liang et al., 2010) |
| Prenatal – dams at gestation day 14 | C57Bl/6J; pregnant females, both genders of pups used | Sevoflurane | 2.5%, 2 hours | 100% oxygen | Apoptotic, inflammatory, and synaptic markers; Morris water maze at P31 | (Zheng et al., 2013) |
The lack of standardization in anesthetic protocols and conditions is arguably one of the greatest limitations of the AIN field. Even among only neonatal mouse studies it is immediately clear that there are is no standardization in pre-clinical models (Table 2, Figure 4). Chosen anesthetic agents include volatile compounds such as isoflurane, desflurane, and sevoflurane, as well as injectable drugs compounds like midozalam, ketamine, and propofol, and AIN studies sometimes include complex combinations of drugs. There is no consensus on effective or clinically relevant doses or exposure time, even in studies using single agents. Regarding duration, some laboratories report AIN phenotypes only when anesthetic exposure is long enough to induce significant mortality from cardio-respiratory failure, a situation which seems unlikely to model human clinical anesthesia. Others suggest that even very short term anesthesia can have overt effects on neuron viability and lasting effects on learning and memory. There is disagreement over whether single or repeat exposures are necessary to induce behavioral outcomes, a question of particular significance to the interpretation of human clinical trials.
The variation in timing, dose, and anesthetic choice is mirrored by variability in other conditions. Oxygen levels, temperature, humidification, flow rate, ventilation method, animal genetic background, age, gender, and physiological maintenance (i.e. dextrose infusion) may all be critically important to the outcome, but no consensus exists. In fact, many studies simply ignore these factors in their methodology, data, and discussions, leaving the reader to guess at how the animals were maintained. Finally, there is little overlap between molecular, cellular, and behavioral endpoints; even commonly used behavioral studies such as Morris water maze very widely when the details of implementation are examined. Simply put, the extraordinary number of known variables which are uncontrolled between published datasets completely precludes any broad cross comparisons or large-scale data reduction efforts.
The complexity of Figure 2 may be reasonable if each pathway or process played a partial role in the overall outcome, but published reports tend to show complete or nearly complete prevention in almost every case. Because of this, it is difficult to imagine a cohesive model for published findings in AIN. Published data can only be judged on statistical rigor, appropriate controls, and cautious interpretation of results. Some discrepancies may result from unstated caveats. For example, antibody based detection methods provide powerful tools but as reagents they vary greatly in quality and benefit from the inclusion of additional controls, such as secondary-only staining in IHC (see ref’s (Gorr and Vogel, 2015; Ivell et al., 2014; McDonough et al., 2015)). Similarly, litter-effects are of particular importance in neonatal AIN research. Litter-effects are known to invalidate standard statistical assumptions of sample independence and normal distribution and have recently been shown to be a frequent cause of false positives in mouse behavioral studies (see (Williams et al., 2017)). Population variability should be carefully assessed, with considerations given to both total sample size and litter distribution between treatments. These issues are not unique to the AIN field, but may be contributing to the complexity of the pre-clinical AIN literature.
Current perspective and additional avenues
Recent evidence from large-scale clinical trials now indicates that short duration routine anesthetic exposure in pediatrics is generally safe and without overt neurological risks, but the risks of multiple or long-term exposures remain unclear (Chinn et al., 2016; Davidson et al., 2016; Ing et al., 2016; Miller et al., 2016; Sun, L.S. et al., 2016; Vutskits and Davidson, 2017). While these unanswered questions warrant further study, the continued relevance of pre-clinical research on AIN will depend on how researchers model these clinical paradigms and whether the quality and comparability issues in the AIN field can be resolved. One of the most pressing issues in pre-clinical AIN studies is arguably the need for some form of standardization in experimental approaches, as discussed above. The intervention literature has provided a variety of intriguing targets for attenuating off-target toxic effects of anesthetics, but until these studies are convincingly validated they are unlikely to impact clinical practice.
It is clear that anesthetic agents, as any chemical compound, can cause cell death under certain conditions. While the potential for anesthetic exposures to cause damage to the neonatal brain is well-supported, the relevance of pre-clinical models to patient care remains unclear. While pathways underlying the molecular, cellular, and behavioral outcomes in pre-clinical models have been extensively probed. Data derived from neonatal mouse models of AIN strongly implicate ROS, nutrient/growth signaling, and neuronal activity in the pathogenesis of neurotoxicity resulting from anesthetic exposure in neonates, but individual reports have varied widely and the relative importance of each of these mechanisms is unclear. The risks of anesthesia in pre-clinical models is supported by a variety of data, but it is not yet clear which animal model approaches best reflect or advise the use of anesthesia in human patients. These models generally utilize long duration or repeated exposure to anesthetics under only partially controlled experimental conditions, a setting which are unlikely to fully mirror human pediatric clinical exposures. Addressing these questions will require a critical re-evaluation of the primary pre-clinical AIN literature in addition to carefully constructed clinical trials.
Highlights.
Anesthetic exposure has been reproducibly shown to induce central nervous system cell death in neonatal mouse models
Anesthesia exposure in neonatal mice is also reported to result in long-term neurocognitive defects, such as defects in learning and memory
Intervention studies in the neonatal mouse model have identified multiple putative mechanistic pathways underlying anesthesia induced neurotoxicity; the major mechanistic targets implicated are oxidative stress, altered nutrient/growth signaling, and direct neuromodulation
A lack of standardization between studies and technical issues surrounding the study of anesthesia induced neurotoxicity in neonatal rodents complicate interpretation of the this field
Acknowledgments
Funding – this work was funded by NIH/NIGMS R01 5R01GM118514-02 award to Dr. Morgan, NIH/NIGMS K99 1K99GM126147–01 awarded to Dr. Johnson. Additional funds were provided by the Northwest Mitochondrial Research Guild.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
Ethics – this work did not involve animal or human research.
Disclosures – we have no conflicts of interest to disclose.
References
- Adachi T, Takigawa H, Nomura T, Watanabe Y, Kowa H, 2018. Cowden Syndrome with a Novel PTEN Mutation Presenting with Partial Epilepsy Related to Focal Cortical Dysplasia. Intern Med 57(1), 97–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsina FC, Ledda F, Paratcha G, 2012. New insights into the control of neurotrophic growth factor receptor signaling: implications for nervous system development and repair. J Neurochem 123(5), 652–661. [DOI] [PubMed] [Google Scholar]
- Alvarado MC, Murphy KL, Baxter MG, 2017. Visual recognition memory is impaired in rhesus monkeys repeatedly exposed to sevoflurane in infancy. Br J Anaesth 119(3), 517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacic G, Pavicevic A, Peyrot F, 2016. In vivo evaluation of different alterations of redox status by studying pharmacokinetics of nitroxides using magnetic resonance techniques. Redox Biol 8, 226–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Yan Y, Canfield S, Muravyeva MY, Kikuchi C, Zaja I, Corbett JA, Bosnjak ZJ, 2013. Ketamine enhances human neural stem cell proliferation and induces neuronal apoptosis via reactive oxygen species-mediated mitochondrial pathway. Anesth Analg 116(4), 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedse G, Di Domenico F, Serviddio G, Cassano T, 2015. Aberrant insulin signaling in Alzheimer’s disease: current knowledge. Front Neurosci 9, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrie SC, Brems H, Legius E, Bagni C, 2017. Cognitive Dysfunctions in Intellectual Disabilities: The Contributions of the Ras-MAPK and PI3K-AKT-mTOR Pathways. Annu Rev Genomics Hum Genet 18, 115–142. [DOI] [PubMed] [Google Scholar]
- Cabrera OH, O’Connor SD, Swiney BS, Salinas-Contreras P, Manzella FM, Taylor GT, Noguchi KK, 2017. Caffeine combined with sedative/anesthetic drugs triggers widespread neuroapoptosis in a mouse model of prematurity. J Matern Fetal Neonatal Med 30(22), 2734–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canet J, Raeder J, Rasmussen LS, Enlund M, Kuipers HM, Hanning CD, Jolles J, Korttila K, Siersma VD, Dodds C, Abildstrom H, Sneyd JR, Vila P, Johnson T, Munoz Corsini L, Silverstein JH, Nielsen IK, Moller JT, investigators I, 2003. Cognitive dysfunction after minor surgery in the elderly. Acta Anaesthesiol Scand 47(10), 1204–1210. [DOI] [PubMed] [Google Scholar]
- Cao L, Li L, Lin D, Zuo Z, 2012. Isoflurane induces learning impairment that is mediated by interleukin 1beta in rodents. PLoS One 7(12), e51431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Zhang Y, Wang A, Dong Y, Xie Z, 2015. Vitamin C Attenuates Isoflurane-Induced Caspase-3 Activation and Cognitive Impairment. Mol Neurobiol 52(3), 1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Levy RJ, 2014. Subclinical carbon monoxide limits apoptosis in the developing brain after isoflurane exposure. Anesth Analg 118(6), 1284–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Mitchell-Flack MJ, Wang A, Levy RJ, 2015. Carbon monoxide modulates cytochrome oxidase activity and oxidative stress in the developing murine brain during isoflurane exposure. Free Radic Biol Med 86, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinn GA, Sasaki Russell JM, Sall JW, 2016. Is a short anesthetic exposure in children safe? Time will tell: a focused commentary of the GAS and PANDA trials. Ann Transl Med 4(20), 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W, Park S, Hong J, Park S, Lee S, Heo J, Kim D, Ko Y, 2015. Sevoflurane exposure during the neonatal period induces long-term memory impairment but not autism-like behaviors. Paediatr Anaesth 25(10), 1033–1045. [DOI] [PubMed] [Google Scholar]
- Cibelli M, Fidalgo AR, Terrando N, Ma D, Monaco C, Feldmann M, Takata M, Lever IJ, Nanchahal J, Fanselow MS, Maze M, 2010. Role of interleukin-1beta in postoperative cognitive dysfunction. Ann Neurol 68(3), 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman K, Robertson ND, Dissen GA, Neuringer MD, Martin LD, Cuzon Carlson VC, Kroenke C, Fair D, Brambrink AM, 2017. Isoflurane Anesthesia Has Long-term Consequences on Motor and Behavioral Development in Infant Rhesus Macaques. Anesthesiology 126(1), 74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AJ, Disma N, de Graaff JC, Withington DE, Dorris L, Bell G, Stargatt R, Bellinger DC, Schuster T, Arnup SJ, Hardy P, Hunt RW, Takagi MJ, Giribaldi G, Hartmann PL, Salvo I, Morton NS, von Ungern Sternberg BS, Locatelli BG, Wilton N, Lynn A, Thomas JJ, Polaner D, Bagshaw O, Szmuk P, Absalom AR, Frawley G, Berde C, Ormond GD, Marmor J, McCann ME, consortium GAS, 2016. Neurodevelopmental outcome at 2 years of age after general anaesthesia and awake-regional anaesthesia in infancy (GAS): an international multicentre, randomised controlled trial. Lancet 387(10015), 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea J, Fabregat I, Frapart YM, Ghezzi P, Gorlach A, Kietzmann T, Kubaichuk K, Knaus UG, Lopez MG, Olaso-Gonzalez G, Petry A, Schulz R, Vina J, Winyard P, Abbas K, Ademowo OS, Afonso CB, Andreadou I, Antelmann H, Antunes F, Aslan M, Bachschmid MM, Barbosa RM, Belousov V, Berndt C, Bernlohr D, Bertran E, Bindoli A, Bottari SP, Brito PM, Carrara G, Casas AI, Chatzi A, Chondrogianni N, Conrad M, Cooke MS, Costa JG, Cuadrado A, My-Chan Dang P, De Smet B, Debelec-Butuner B, Dias IHK, Dunn JD, Edson AJ, El Assar M, El-Benna J, Ferdinandy P, Fernandes AS, Fladmark KE, Forstermann U, Giniatullin R, Giricz Z, Gorbe A, Griffiths H, Hampl V, Hanf A, Herget J, Hernansanz-Agustin P, Hillion M, Huang J, Ilikay S, Jansen-Durr P, Jaquet V, Joles JA, Kalyanaraman B, Kaminskyy D, Karbaschi M, Kleanthous M, Klotz LO, Korac B, Korkmaz KS, Koziel R, Kracun D, Krause KH, Kren V, Krieg T, Laranjinha J, Lazou A, Li H, Martinez-Ruiz A, Matsui R, McBean GJ, Meredith SP, Messens J, Miguel V, Mikhed Y, Milisav I, Milkovic L, Miranda-Vizuete A, Mojovic M, Monsalve M, Mouthuy PA, Mulvey J, Munzel T, Muzykantov V, Nguyen ITN, Oelze M, Oliveira NG, Palmeira CM, Papaevgeniou N, Pavicevic A, Pedre B, Peyrot F, Phylactides M, Pircalabioru GG, Pitt AR, Poulsen HE, Prieto I, Rigobello MP, Robledinos-Anton N, Rodriguez-Manas L, Rolo AP, Rousset F, Ruskovska T, Saraiva N, Sasson S, Schroder K, Semen K, Seredenina T, Shakirzyanova A, Smith GL, Soldati T, Sousa BC, Spickett CM, Stancic A, Stasia MJ, Steinbrenner H, Stepanic V, Steven S, Tokatlidis K, Tuncay E, Turan B, Ursini F, Vacek J, Vajnerova O, Valentova K, Van Breusegem F, Varisli L, Veal EA, Yalcin AS, Yelisyeyeva O, Zarkovic N, Zatloukalova M, Zielonka J, Touyz RM, Papapetropoulos A, Grune T, Lamas S, Schmidt H, Di Lisa F, Daiber A, 2017. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol 13, 94–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Redondo AB, Aguado A, Briones AM, Salaices M, 2016. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharmacol Res 114, 110–120. [DOI] [PubMed] [Google Scholar]
- Gorlach A, Dimova EY, Petry A, Martinez-Ruiz A, Hernansanz-Agustin P, Rolo AP, Palmeira CM, Kietzmann T, 2015. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol 6, 372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorr TA, Vogel J, 2015. Western blotting revisited: critical perusal of underappreciated technical issues. Proteomics Clin Appl 9(3–4), 396–405. [DOI] [PubMed] [Google Scholar]
- Goszcz K, Deakin SJ, Duthie GG, Stewart D, Leslie SJ, Megson IL, 2015. Antioxidants in Cardiovascular Therapy: Panacea or False Hope? Front Cardiovasc Med 2, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, Harrison DG, Bhatnagar A, American Heart Association Council on Basic Cardiovascular, S., 2016. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement From the American Heart Association. Circ Res 119(5), e39–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han XD, Li M, Zhang XG, Xue ZG, Cang J, 2015. Single sevoflurane exposure increases methyl-CpG island binding protein 2 phosphorylation in the hippocampus of developing mice. Mol Med Rep 11(1), 226–230. [DOI] [PubMed] [Google Scholar]
- Head BP, Patel HH, Niesman IR, Drummond JC, Roth DM, Patel PM, 2009. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology 110(4), 813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacer RD, Deng M, Ward CG, Joseph B, Hughes EA, Jiang C, Danzer SC, Loepke AW, 2013. Cell age-specific vulnerability of neurons to anesthetic toxicity. Ann Neurol 73(6), 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Jing S, Chen X, Bao X, Du Z, Li H, Yang T, Fan X, 2016. Propofol Administration During Early Postnatal Life Suppresses Hippocampal Neurogenesis. Mol Neurobiol 53(2), 1031–1044. [DOI] [PubMed] [Google Scholar]
- Ing C, Rauh VA, Warner DO, Sun LS, 2016. What Next After GAS and PANDA? J Neurosurg Anesthesiol [DOI] [PMC free article] [PubMed]
- Istaphanous GK, Howard J, Nan X, Hughes EA, McCann JC, McAuliffe JJ, Danzer SC, Loepke AW, 2011. Comparison of the neuroapoptotic properties of equipotent anesthetic concentrations of desflurane, isoflurane, or sevoflurane in neonatal mice. Anesthesiology 114(3), 578–587. [DOI] [PubMed] [Google Scholar]
- Ivell R, Teerds K, Hoffman GE, 2014. Proper application of antibodies for immunohistochemical detection: antibody crimes and how to prevent them. Endocrinology 155(3), 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, 2012. Developmental synaptogenesis and general anesthesia: a kiss of death? Curr Pharm Des 18(38), 6225–6231. [DOI] [PubMed] [Google Scholar]
- Ji MH, Qiu LL, Yang JJ, Zhang H, Sun XR, Zhu SH, Li WY, Yang JJ, 2015. Pre-administration of curcumin prevents neonatal sevoflurane exposure-induced neurobehavioral abnormalities in mice. Neurotoxicology 46, 155–164. [DOI] [PubMed] [Google Scholar]
- Jiang XL, Du BX, Chen J, Liu L, Shao WB, Song J, 2014. MicroRNA-34a negatively regulates anesthesia-induced hippocampal apoptosis and memory impairment through FGFR1. Int J Clin Exp Pathol 7(10), 6760–6767. [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Young C, Olney JW, 2008. Isoflurane-induced neuroapoptosis in the developing brain of nonhypoglycemic mice. J Neurosurg Anesthesiol 20(1), 21–28. [DOI] [PubMed] [Google Scholar]
- Johnson T, Monk T, Rasmussen LS, Abildstrom H, Houx P, Korttila K, Kuipers HM, Hanning CD, Siersma VD, Kristensen D, Canet J, Ibanaz MT, Moller JT, Investigators I, 2002. Postoperative cognitive dysfunction in middle-aged patients. Anesthesiology 96(6), 1351–1357. [DOI] [PubMed] [Google Scholar]
- Kapetanaki SM, Silkstone G, Husu I, Liebl U, Wilson MT, Vos MH, 2009. Interaction of carbon monoxide with the apoptosis-inducing cytochrome c-cardiolipin complex. Biochemistry 48(7), 1613–1619. [DOI] [PubMed] [Google Scholar]
- Le Freche H, Brouillette J, Fernandez-Gomez FJ, Patin P, Caillierez R, Zommer N, Sergeant N, Buee-Scherrer V, Lebuffe G, Blum D, Buee L, 2012. Tau phosphorylation and sevoflurane anesthesia: an association to postoperative cognitive impairment. Anesthesiology 116(4), 779–787. [DOI] [PubMed] [Google Scholar]
- Lee DY, 2015. Roles of mTOR Signaling in Brain Development. Exp Neurobiol 24(3), 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, 2017a. De Novo Gene Expression Reconstruction in Space. Trends Mol Med 23(7), 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, 2017b. Quantitative approaches for investigating the spatial context of gene expression. Wiley Interdiscip Rev Syst Biol Med 9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Park YH, Song HG, Park HP, Kim HS, Kim CS, Kim JT, 2014. The effect of lidocaine on apoptotic neurodegeneration in the developing mouse brain. Korean J Anesthesiol 67(5), 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X, Zhang W, Liu T, Xiao H, Liang W, Xia W, Zhang J, 2013. Perinatal supplementation with omega-3 polyunsaturated fatty acids improves sevoflurane-induced neurodegeneration and memory impairment in neonatal rats. PLoS One 8(8), e70645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkuil BP, Head BP, Pearn ML, Patel HH, Drummond JC, Patel PM, 2011. Isoflurane neurotoxicity is mediated by p75NTR-RhoA activation and actin depolymerization. Anesthesiology 114(1), 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J, 2010. Cell signaling by receptor tyrosine kinases. Cell 141(7), 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy RJ, 2017. Carbon monoxide and anesthesia-induced neurotoxicity. Neurotoxicol Teratol 60, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Hou L, Chen D, Lin F, Chang T, Li M, Zhang L, Niu X, Wang H, Fu S, Zheng J, 2017. Hydrogen-rich saline attenuates isoflurane-induced caspase-3 activation and cognitive impairment via inhibition of isoflurane-induced oxidative stress, mitochondrial dysfunction, and reduction in ATP levels. Am J Transl Res 9(3), 1162–1172. [PMC free article] [PubMed] [Google Scholar]
- Liang G, Ward C, Peng J, Zhao Y, Huang B, Wei H, 2010. Isoflurane causes greater neurodegeneration than an equivalent exposure of sevoflurane in the developing brain of neonatal mice. Anesthesiology 112(6), 1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EP, Lee JR, Lee CS, Deng M, Loepke AW, 2017. Do anesthetics harm the developing human brain? An integrative analysis of animal and human studies. Neurotoxicol Teratol 60, 117–128. [DOI] [PubMed] [Google Scholar]
- Lin XF, Han YQ, Li HL, Zhao YP, Fei XJ, Sheng JX, Lu HH, Liu S, Zhang L, 2014. SAHA attenuates sevoflurane-induced learning and memory impairments in fetal mice. Genet Mol Res 13(4), 10769–10778. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang P, Zhang X, Zhang W, Gu G, 2014. Effects of different concentration and duration time of isoflurane on acute and long-term neurocognitive function of young adult C57BL/6 mouse. Int J Clin Exp Pathol 7(9), 5828–5836. [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yang J, Xu Y, Guo G, Cai L, Wu H, Zhao Y, Zhang X, 2017a. Roscovitine, a CDK5 Inhibitor, Alleviates Sevoflurane-Induced Cognitive Dysfunction via Regulation Tau/GSK3beta and ERK/PPARgamma/CREB Signaling. Cell Physiol Biochem 44(2), 423–435. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang X, Zhang W, Gu G, Wang P, 2015. Effects of Sevoflurane on Young Male Adult C57BL/6 Mice Spatial Cognition. PLoS One 10(8), e0134217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhao Y, Yang J, Zhang X, Zhang W, Wang P, 2017b. Neonatal Repeated Exposure to Isoflurane not Sevoflurane in Mice Reversibly Impaired Spatial Cognition at Juvenile-Age. Neurochem Res 42(2), 595–605. [DOI] [PubMed] [Google Scholar]
- Loepke AW, Istaphanous GK, McAuliffe JJ 3rd, Miles L, Hughes EA, McCann JC, Harlow KE, Kurth CD, Williams MT, Vorhees CV, Danzer SC, 2009. The effects of neonatal isoflurane exposure in mice on brain cell viability, adult behavior, learning, and memory. Anesth Analg 108(1), 90–104. [DOI] [PubMed] [Google Scholar]
- Loepke AW, McCann JC, Kurth CD, McAuliffe JJ, 2006. The physiologic effects of isoflurane anesthesia in neonatal mice. Anesth Analg 102(1), 75–80. [DOI] [PubMed] [Google Scholar]
- Man YG, Zhou RG, Zhao B, 2015. Efficacy of rutin in inhibiting neuronal apoptosis and cognitive disturbances in sevoflurane or propofol exposed neonatal mice. Int J Clin Exp Med 8(8), 14397–14409. [PMC free article] [PubMed] [Google Scholar]
- McDonough AA, Veiras LC, Minas JN, Ralph DL, 2015. Considerations when quantitating protein abundance by immunoblot. Am J Physiol Cell Physiol 308(6), C426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TL, Park R, Sun LS, 2016. Report on the Fifth PANDA Symposium on “Anesthesia and Neurodevelopment in Children”. J Neurosurg Anesthesiol 28(4), 350–355. [DOI] [PubMed] [Google Scholar]
- Moller JT, Cluitmans P, Rasmussen LS, Houx P, Rasmussen H, Canet J, Rabbitt P, Jolles J, Larsen K, Hanning CD, Langeron O, Johnson T, Lauven PM, Kristensen PA, Biedler A, van Beem H, Fraidakis O, Silverstein JH, Beneken JE, Gravenstein JS, 1998. Long-term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post-Operative Cognitive Dysfunction. Lancet 351(9106), 857–861. [DOI] [PubMed] [Google Scholar]
- Na HS, Brockway NL, Gentry KR, Opheim E, Sedensky MM, Morgan PG, 2017. The genetics of isoflurane-induced developmental neurotoxicity. Neurotoxicol Teratol 60, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman MF, Kirchner JL, Phillips-Bute B, Gaver V, Grocott H, Jones RH, Mark DB, Reves JG, Blumenthal JA, Neurological Outcome Research G, the Cardiothoracic Anesthesiology Research Endeavors, I., 2001. Longitudinal assessment of neurocognitive function after coronary-artery bypass surgery. N Engl J Med 344(6), 395–402. [DOI] [PubMed] [Google Scholar]
- Nieto-Estevez V, Defterali C, Vicario-Abejon C, 2016. IGF-I: A Key Growth Factor that Regulates Neurogenesis and Synaptogenesis from Embryonic to Adult Stages of the Brain. Front Neurosci 10, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu X, Chen J, Wang P, Zhou H, Li S, Zhang M, 2014. The effects of hispidulin on bupivacaine-induced neurotoxicity: role of AMPK signaling pathway. Cell Biochem Biophys 70(1), 241–249. [DOI] [PubMed] [Google Scholar]
- Ohta S, 2015. Molecular hydrogen as a novel antioxidant: overview of the advantages of hydrogen for medical applications. Methods Enzymol 555, 289–317. [DOI] [PubMed] [Google Scholar]
- Paez J, Sellers WR, 2003. PI3K/PTEN/AKT pathway. A critical mediator of oncogenic signaling. Cancer Treat Res 115, 145–167. [PubMed] [Google Scholar]
- Piwien-Pilipuk G, Huo JS, Schwartz J, 2002. Growth hormone signal transduction. J Pediatr Endocrinol Metab 15(6), 771–786. [DOI] [PubMed] [Google Scholar]
- Prieto-Bermejo R, Hernandez-Hernandez A, 2017. The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis. Antioxidants (Basel) 6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian XL, Zhang W, Liu MZ, Zhou YB, Zhang JM, Han L, Peng YM, Jiang JH, Wang QD, 2015. Dexmedetomidine improves early postoperative cognitive dysfunction in aged mice. Eur J Pharmacol 746, 206–212. [DOI] [PubMed] [Google Scholar]
- Raper J, Alvarado MC, Murphy KL, Baxter MG, 2015. Multiple Anesthetic Exposure in Infant Monkeys Alters Emotional Reactivity to an Acute Stressor. Anesthesiology 123(5), 1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen LS, Johnson T, Kuipers HM, Kristensen D, Siersma VD, Vila P, Jolles J, Papaioannou A, Abildstrom H, Silverstein JH, Bonal JA, Raeder J, Nielsen IK, Korttila K, Munoz L, Dodds C, Hanning CD, Moller JT, Investigators I, 2003. Does anaesthesia cause postoperative cognitive dysfunction? A randomised study of regional versus general anaesthesia in 438 elderly patients. Acta Anaesthesiol Scand 47(3), 260–266. [DOI] [PubMed] [Google Scholar]
- Satomoto M, Sun Z, Adachi YU, Makita K, 2016. Sugammadex-Enhanced Neuronal Apoptosis following Neonatal Sevoflurane Exposure in Mice. Anesthesiol Res Pract 2016, 9682703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer DB, 2011. Oxidative stress in heart failure: what are we missing? Am J Med Sci 342(2), 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM, 2017. mTOR Signaling in Growth, Metabolism, and Disease. Cell 168(6), 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallner N, Ulbrich F, Engelstaedter H, Biermann J, Auwaerter V, Loop T, Goebel U, 2014. Isoflurane but not sevoflurane or desflurane aggravates injury to neurons in vitro and in vivo via p75NTR-NF-kB activation. Anesth Analg 119(6), 1429–1441. [DOI] [PubMed] [Google Scholar]
- Schilling JM, Kassan A, Mandyam C, Pearn ML, Voong A, Grogman GG, Risbrough VB, Niesman IR, Patel HH, Patel PM, Head BP, 2017. Inhibition of p75 neurotrophin receptor does not rescue cognitive impairment in adulthood after isoflurane exposure in neonatal mice. Br J Anaesth 119(3), 465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Wang G, Li J, Yu W, 2017. Hydrogen gas attenuates sevoflurane neurotoxicity through inhibiting nuclear factor kappa-light-chain-enhancer of activated B cells signaling and proinflammatory cytokine release in neonatal rats. Neuroreport 28(17), 1170–1175. [DOI] [PubMed] [Google Scholar]
- Song Q, Ma YL, Song JQ, Chen Q, Xia GS, Ma JY, Feng F, Fei XJ, Wang QM, 2015. Sevoflurane induces neurotoxicity in young mice through FAS/FASL signaling. Genet Mol Res 14(4), 18059–18068. [DOI] [PubMed] [Google Scholar]
- Sun LS, Li G, Miller TL, Salorio C, Byrne MW, Bellinger DC, Ing C, Park R, Radcliffe J, Hays SR, DiMaggio CJ, Cooper TJ, Rauh V, Maxwell LG, Youn A, McGowan FX, 2016. Association Between a Single General Anesthesia Exposure Before Age 36 Months and Neurocognitive Outcomes in Later Childhood. JAMA 315(21), 2312–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Satomoto M, Adachi YU, Kinoshita H, Makita K, 2016. Inhibiting NADPH oxidase protects against long-term memory impairment induced by neonatal sevoflurane exposure in mice. Br J Anaesth 117(1), 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J, 2017. Molecular neurobiology of mTOR. Neuroscience 341, 112–153. [DOI] [PubMed] [Google Scholar]
- Tagawa T, Sakuraba S, Kimura K, Mizoguchi A, 2014. Sevoflurane in combination with propofol, not thiopental, induces a more robust neuroapoptosis than sevoflurane alone in the neonatal mouse brain. J Anesth 28(6), 815–820. [DOI] [PubMed] [Google Scholar]
- Takei N, Nawa H, 2014. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao G, Xue Q, Luo Y, Li G, Xia Y, Yu B, 2016. Isoflurane Is More Deleterious to Developing Brain Than Desflurane: The Role of the Akt/GSK3beta Signaling Pathway. Biomed Res Int 2016, 7919640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao G, Zhang J, Zhang L, Dong Y, Yu B, Crosby G, Culley DJ, Zhang Y, Xie Z, 2014. Sevoflurane induces tau phosphorylation and glycogen synthase kinase 3beta activation in young mice. Anesthesiology 121(3), 510–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trentini JF, O’Neill JT, Poluch S, Juliano SL, 2016. Prenatal carbon monoxide impairs migration of interneurons into the cerebral cortex. Neurotoxicology 53, 31–44. [DOI] [PubMed] [Google Scholar]
- Van Biesen S, Van de Velde M, Rex S, 2015. Anesthesia and neurotoxicity in the developing brain: A non-systematic review. Acta Anaesthesiol Belg 66(3), 67–79. [PubMed] [Google Scholar]
- Vutskits L, Davidson A, 2017. Update on developmental anesthesia neurotoxicity. Curr Opin Anaesthesiol 30(3), 337–342. [DOI] [PubMed] [Google Scholar]
- Walters JL, Paule MG, 2017. Review of preclinical studies on pediatric general anesthesia-induced developmental neurotoxicity. Neurotoxicol Teratol 60, 2–23. [DOI] [PubMed] [Google Scholar]
- Wang C, 2012. Advanced pre-clinical research approaches and models to studying pediatric anesthetic neurotoxicity. Front Neurol 3, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Slikker W Jr., 2008. Strategies and experimental models for evaluating anesthetics: effects on the developing nervous system. Anesth Analg 106(6), 1643–1658. [DOI] [PubMed] [Google Scholar]
- Wang C, Zhang X, Liu F, 2017. Application of advanced preclinical models and methods in anesthetic neurotoxicity research. Neurotoxicol Teratol 61, 1–6. [DOI] [PubMed] [Google Scholar]
- Wang H, Dong Y, Zhang J, Xu Z, Wang G, Swain CA, Zhang Y, Xie Z, 2014. Isoflurane induces endoplasmic reticulum stress and caspase activation through ryanodine receptors. Br J Anaesth 113(4), 695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang A, Supplee WW, Koffler K, Cheng Y, Quezado ZMN, Levy RJ, 2017a. Carbon monoxide incompletely prevents isoflurane-induced defects in murine neurodevelopment. Neurotoxicol Teratol 61, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhou K, Fu Z, Yu D, Huang H, Zang X, Mo X, 2017b. Brain Development and Akt Signaling: the Crossroads of Signaling Pathway and Neurodevelopmental Diseases. J Mol Neurosci 61(3), 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ni H, Zhang W, Wang X, Zhang H, 2016. Downregulation of miR-210 protected bupivacaine-induced neurotoxicity in dorsal root ganglion. Exp Brain Res 234(4), 1057–1065. [DOI] [PubMed] [Google Scholar]
- Wang Z, Shen J, Wang J, Lu T, Li C, Zhang X, Liu L, Ding Z, 2012. Lithium attenuates bupivacaine-induced neurotoxicity in vitro through phosphatidylinositol-3-kinase/threonine-serine protein kinase B- and extracellular signal-regulated kinase-dependent mechanisms. Neuroscience 206, 190–200. [DOI] [PubMed] [Google Scholar]
- Wauson EM, Lorente-Rodriguez A, Cobb MH, 2013. Minireview: Nutrient sensing by G protein-coupled receptors. Mol Endocrinol 27(8), 1188–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DR, Carlsson R, Burkner PC, 2017. Between-litter variation in developmental studies of hormones and behavior: Inflated false positives and diminished power. Front Neuroendocrinol 47, 154–166. [DOI] [PubMed] [Google Scholar]
- Xu G, Lu H, Dong Y, Shapoval D, Soriano SG, Liu X, Zhang Y, Xie Z, 2017. Coenzyme Q10 reduces sevoflurane-induced cognitive deficiency in young mice. Br J Anaesth 119(3), 481–491. [DOI] [PubMed] [Google Scholar]
- Xu H, Zhang J, Zhou W, Feng Y, Teng S, Song X, 2015. The role of miR-124 in modulating hippocampal neurotoxicity induced by ketamine anesthesia. Int J Neurosci 125(3), 213–220. [DOI] [PubMed] [Google Scholar]
- Xu KX, Tao J, Zhang N, Wang JZ, 2015. Neuroprotective properties of vitamin C on equipotent anesthetic concentrations of desflurane, isoflurane, or sevoflurane in high fat diet fed neonatal mice. Int J Clin Exp Med 8(7), 10444–10458. [PMC free article] [PubMed] [Google Scholar]
- Yamada A, Yamamoto K, Imamoto N, Momosaki S, Hosoi R, Yamaguchi M, Inoue O, 2009. Lactate is an alternative energy fuel to glucose in neurons under anesthesia. Neuroreport 20(17), 1538–1542. [DOI] [PubMed] [Google Scholar]
- Yang B, Liang G, Khojasteh S, Wu Z, Yang W, Joseph D, Wei H, 2014. Comparison of neurodegeneration and cognitive impairment in neonatal mice exposed to propofol or isoflurane. PLoS One 9(6), e99171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V, 2005. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience 135(3), 815–827. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang J, Dong Y, Swain CA, Zhang Y, Xie Z, 2014. The potential dual effects of sevoflurane on AKT/GSK3beta signaling pathway. Med Gas Res 4(1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Dong Y, Xu Z, Crosby G, Culley DJ, Zhang Y, Xie Z, 2013. Sevoflurane anesthesia in pregnant mice induces neurotoxicity in fetal and offspring mice. Anesthesiology 118(3), 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Gao J, Karlsson N, Li Q, Zhang Y, Huang Z, Li H, Kuhn HG, Blomgren K, 2010. Isoflurane anesthesia induced persistent, progressive memory impairment, caused a loss of neural stem cells, and reduced neurogenesis in young, but not adult, rodents. J Cereb Blood Flow Metab 30(5), 1017–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Qing T, Zheng Y, Jin L, Shi L, 2017. Advances in single-cell RNA sequencing and its applications in cancer research. Oncotarget 8(32), 53763–53779. [DOI] [PMC free article] [PubMed] [Google Scholar]