

Abstract
Cigarette smoking significantly increases the risk of cancer and cardiovascular diseases as well as premature death. Aromatic amines (AAs) such as o-toluidine, 2-aminonaphthalene and 4-aminobiphenyl are found in cigarette smoke and are well-established human bladder carcinogens presumably acting via the formation of DNA adducts. These amines may be metabolized in the liver to acetylated or glucuronidated forms or oxidized to a hydroxylamine which may react with protein and DNA to form adducts. Free, acetylated and glucuronidated AAs are excreted in urine and can be measured as exposure biomarkers. Using isotope dilution GC–MS/MS, our laboratory quantifies six urinary AAs that are known or suspected carcinogens–o-toluidine, 2,6-dimethylaniline, o-anisidine, 1-aminonaphthalene, 2-aminonaphthalene and 4-aminobiphenyl–for large population studies such as the National Health and Nutrition Examination Survey (NHANES). We also monitor two additional corresponding structural isomers–2-aminobiphenyl and 3-aminobiphenyl–to verify isomer separation. A new and improved automated sample preparation method was developed to quantify these AAs, in which, sample cleanup was done via Supported Liquid Extraction (SLE+ ISOLUTE®) on a Hamilton STAR™ workstation. This automated method increased sample throughput by reducing sample cleanup time from 8 to 4 h while maintaining precision (intra and inter-run coefficient of variation <7%) and accuracy (±17%). Recent improvements in our GC/MS method have enhanced our assay sensitivity and specificity, resulting in longer analytical column life and maintaining or reducing the limit of detection for all six analytes. Indigo ASCENT™ software (3.7.1, Indigo BioAutomation, Inc.) is used for peak integration, calibration and quantification. A streamlined sample data flow was created in parallel with the automated method, in which samples can be tracked from receiving to final laboratory information management system output with minimal human intervention, minimizing potential human error. This newly validated, automated method and sample data flow are currently applied in biomonitoring of AAs in the US noninstitutionalized population NHANES 2013–2014 cycle.
Introduction
Cigarette smoking is a significant risk factor for cancer for both smokers and nonsmokers (1). Exposure of nonsmokers to second-hand tobacco smoke (SHS) has been linked to an increased risk of cancer, coronary heart disease and respiratory illnesses in both adults and children (2–8). As SHS causes premature death and disease in nonsmokers, its presence in the environment remains a significant public health concern (9). SHS is mainly composed of gases and particulate matters generated from a mixture of sidestream smoke, which is emitted from smoldering cigarettes between puffs and from exhaled mainstream smoke. The Surgeon General concluded in 2006 that there is no risk-free level of exposure to SHS.
Smoking tobacco and inhaling SHS may be major sources of exposure to several aromatic amines (AAs) (10–12), which are suggested to be principal agents for the development of bladder cancer in humans (13). The total nitrogen content in tobacco leaves—as derived from nitrates, ammonia, amino acids, amides and alkaloids—ultimately con-tributes to the formation of AAs in tobacco smoke (14–16). Nitrate, which is introduced to the growing tobacco plant through the application of fertilizer, can be converted to ammonia, which, in turn, is converted to other nitrogenous organic compounds such as amino acids. Intermediate NH2 radicals, forming during the pyrolysis of ammonia during tobacco combustion, may react with aromatic CH groups (from compounds already present in the tobacco leaves) to form the AAs (17). In addition to the pyrosynthetic mechanism, AAs may also be transferred directly from the tobacco leaves into the smoke via thermal degradation of alkaloids and amino acids (18, 19).
In addition to tobacco smoke, other exposure sources of AAs include several chemical industry sectors such as dyes and pigments (e.g., azo dyes, indigo dyes), pharmaceuticals, pesticides, herbicides, synthetic rubber and plastics (20–24). In manufacturing these industrial chemicals, AAs are used as raw materials or intermediates, and therefore, they should not occur in the final products. As such, occupational exposure to AAs can occur by inhalation or skin contact during the production of chemicals that use AAs as raw materials or intermediates. AAs can also be found in environmental pollution such as diesel exhaust, combustion of wood chips and rubber and substances in charcoal barbequed meats and fish (25, 26). Natural occurence of AA was reported, for example, in the aroma components of black tea and certain vegetables (27, 28). Other potential sources include emissions from cooking oils (e.g., vegetable, sun-flower and refined lard oil) (29).
The International Agency for Research on Cancer has classified several AAs as carcinogenic to humans (Group 1) or possibly carcinogenic to humans (Group 2B): AAs such as o-toluidine (OTOL), 2-aminonapthalene and 4-aminobiphenyl, for example, are well-established human bladder carcinogens (13, 30, 31). Several AAs are on the FDA’s list of harmful and potentially harmful constituents since the passage of The Family Smoking Prevention and Tobacco Control Act in 2009 and the creation of the Center of Tobacco Products (32). During smoke inhalation, AAs are first carried into the bloodstream from the lungs, metabolized by the liver, then travels to the bladder to be eventually excreted out of the body through urination. The amine functional groups may be metabolized in the liver to the acetylated and/or glucuronidated forms, or they may be oxidized to a hydroxylamine form, which undergoes further conversion via an acetylation reaction to form an N-acetoxy metabolite. The N-acetoxy metabolite is able to undergo nonenzymatic break-down to yield the reactive nitrenium ion, nitrene or a free radical that can covalently bind to tissue macromolecules (proteins) and DNA to form adducts (20, 33, 34). Formation of the hydroxylamine metabolite is considered the primary pathway to AA carcinogenicity in the bladder (20), whereas the acetylated and glucuronidated forms of AAs (excreted in urine) are the products of the body’s detoxification metabolism pathway. As such, the free, acetylated and glucuronidated AAs excreted in urine allow them to be surrogate biomarkers of AA exposure.
These compounds have been traditionally measured by manual approaches involving laborious and time-consuming sample preparation steps. Intricate sample cleanup—often utilizing hydrolysis, direct liquid–liquid extraction and/or a solid-phase extraction step— is necessary due to the complexity of the matrix analyzed (e.g., smoke, wastewater, urine, serum, breast milk) and the ultralow levels of AAs (4, 5, 14, 35, 43). Acid hydrolysis—with hydrochloric or sulfuric acid—followed by basification of hydrolysate, is the most common step taken toward deconjugating AAs in urine samples; methods involving enzyme and base hydrolysis have also been validated (4, 43). As for sample analysis, gas chromatography (GC)—following a derivatization pretreatment step—is the most common separation technique, though some labs have developed various liquid chromatography (LC) methods (4, 38, 40, 41). Detection systems range from single-, triple-quadrupole or orbitrap mass analyzers to flame ionization detector (35), UV–Vis spectrophotometer (41), electron-capture detector (5) and electrochemical detector (4), where mass spectrometry is the most commonly used detection technique (36–40, 42, 43). Electron impact (EI) ionization, negative-mode chemical ionization applying methane as reactant gas (33, 36) and positive-mode electrospray ionization (ESI+) (38) are all common ionization methods applied in conjunction with the various GC-MS or LC-MS methods developed to detect AAs.
In this paper, we present a newly validated method that replaces our previous manual sample preparation method (44) with an auto-mated approach using a Hamilton STAR™ workstation. It is the first time an automated sample preparation approach is reported. In addition, we also report a new streamlined sample data flow created in parallel to the automated sample preparation method, in which samples can be tracked from receiving to final laboratory information management system (LIMS) output with minimal human intervention. In our new method, six AAs are quantified: o-toluidine, 2,6-dimethylaniline (26DM), o-anisidine (OANS), 1-aminonaphthalene (1AMN), 2-aminonaphthalene (2AMN) and 4-aminobiphenyl (4ABP). Two additional related structural isomers are monitored to ensure isomeric separation: 2-aminobiphenyl (2ABP) and 3-aminobiphenyl (3ABP) (Figure 1).
Figure 1.
Chemical structures of eight AAs. Analytes that are quantified are indicated with an asterisk.
Method and materials
Materials
Native (unlabeled) standards used to make calibration curves were purchased from Fluka, Aldrich and Sigma (Sigma-Aldrich, St. Louis, MO). OTOL-13C6, OANS-2H7, 1AMN-2H9 were purchased from Medical Isotope (Pelhem, NH); 2AMN-2H7 was purchased from CDN Isotopes (Pointe-Claire, Quebec, Canada); 4ABP-2H9 was purchased from Cambridge Isotope Laboratory (Andover, MA) and 26DM-2H6 was purchased from Toronto Research Chemical (North York, Canada). All native standards used for the second source accuracy test were purchased from Toronto Research Chemical, except for 4ABP, which was purchased from Sigma. Sodium hydroxide pellets (semiconductor grade) were purchased from Sigma. High-purity hydrochloric acid, pentafluoropropionic anhy-dride (PFPA) and trimethylamine hydrochloride (TMA-HCl) were obtained from Sigma-Aldrich. All solvents were GC2 grade, except for water, which was HPLC-grade. All solvents were purchased from Burdick and Jackson Labs (distributed by VWR, Suwanee, GA), and all gases were ultrahigh purity grade. Isolute™ support liquid extraction (SLE) cartridges were purchased from Biotage (Charlotte, NC). Vials of 4.5-mL high recovery samples were purchased from ChemGlass (Vineland, NJ). EP Scientific 10-mL silanized glass tubes were purchased from LabDepot (Dawsonville, GA). Wheaton 1-mL amber crimp GC vials with 300-μL insert and SUN-Sri 11-mm aluminum crimp caps with rubber septum were ordered from ThermoFisher Scientific (Suwanee, GA). All GC–MS/ MS supplies were purchased from Agilent Technologies (Santa Clara, CA); all filter tips for sample aliquoting in the Hamilton Microlab STAR™ Liquid Handling Workstation were purchased from the Hamilton Company (Reno, NV).
Instrument
All sample and internal standard aliquoting was performed on the Hamilton Microlab STAR™ Liquid Handling Workstation, which was customized with a recessed deck to handle the physical dimensions of high-volume cartridges (Figure 2). The Hamilton STAR was configured with four 5-mL channels and eight 1-mL channels, compression-induced O-Ring expansion (CO-RE) paddle grippers, autoload and barcode reader and four custom-built deep well vacuum chambers. The 5-mL pipetting channels used 4.5-mL CO-RE tips with filters and the eight pipetting channels utilized both 1000-and 50-μL CO-RE tips with filters.
Figure 2.

Schematic for Hamilton STAR with a customized receding deck that can accommodate high cartridge sample preparation: (a) custom 10-mL cartridge holders; (b) custom 10-mL tube holders; (c) eight 1-mL channels; (d) four 5-mL channels; (e) CO-RE paddle grippers; (f) barcode reader; (g) custom vacuum chambers.
Automated sample preparation
Urine samples were transferred from cryovials in 2 mL aliquots, along with approximately 450 pg of internal standard (45 μL of 10 pg/μL internal standard solution in ethanol) into high recovery vials by the Hamilton STAR liquid handling system. All samples were aspirated using capacitance liquid-level detection and dispensed using jet empty settings. Samples were mixed (consisting of a rapid aspirate and dispense cycle of 500 μL into the same container) thrice prior to being transferred to the high recovery vials to ensure uniformity. The Hamilton Autoload and Barcode Scanner were used to scan and decode Code 128 barcodes affixed to the original sample containers. A Microsoft Excel (Microsoft Corporation, Redmond, WA) output file of the scanned vials was generated to assure sample tracking and placement. The delivery volumes of 45 μL and 2 mL were verified gravimetrically with less than 1% error using the Hamilton Volume Verification Kit (Reno, NV) and a Mettler-Toledo High Precision Weight Module, Model WXS205SDU (Columbus, OH).
Urine samples were hydrolyzed with 50 μL of 10 M NaOH and incubated at 90°C for approximately 15 h on a hot plate (VWR). After cooling to room temperature, the samples were relocated to the Hamilton deck, where the total volume was transferred onto Isolute™ SLE cartridges. These cartridges were placed in custom 10-mL cartridge holders over the deep well vacuum chambers. Silanized glass tubes, placed in custom tube holders, were positioned under the Isolute SLE cartridges using the CO-RE paddle grippers. Analytes were then eluted with three washes of 3 mL dichloromethane and collected into the silanized glass tubes. The silanized glass tubes were then transferred to a Thermo Scientific Savant SPD2010 SpeedVac Concentrator (Holbrook, NY), where the contents were evaporated to approximately 250 μL. To the concentrated eluate, 3 μL of 1.0 M TMA (1.0 g of 98%-purity TMA-HCl dissolved in 2 mL water, neutralized with 1–3 μL of 10 M NaOH, then extracted TMA with 5 mL hexane) and 3 μL of PFPA were added and kept capped at room temperature for 30 min to complete derivatization of the AAs. Afterward, the derivatized samples were manually transferred via pipette to 1-mL amber GC vials with 300 μL insert and further evaporated to completion in the Savant. An amount of 10 μL of toluene was added to each vial to reconstitute the sample. The vials were subsequently capped and vortexed before being stored in −20°C or analyzed on the GC–MS/MS.
GC–MS/MS analysis
All analyses were performed on two Agilent 7890–7000 C GC–MS/MS. The 7890 GCs were equipped with multimode inlet and Agilent single taper liner (4 mm ID) with glass wool. The use of liners with glass wool helped minimize contamination of the column with sample residues while maintaining the signal sensitivities of all analytes (as compared to the signal sensitivities recorded from injections made on a single taper plain liner). The injection mode was pulsed splitless at 30 psi for 0.35 min to minimize diffusion and improve analyte transfer onto the analytical column. The injection volume was 1 μL, and the injection port temperature was held at 250°C for the duration of each analytical run. A two-column setup connected by a purged union was used: the analytical column used was the Agilent J&W DB-FFAP (30 m × 0.25 mm × 0.25 μm), which is composed of a nitroterephthalic-acid-modified polyethylene glycol stationary phase of high polarity; the second column in series (“postcolumn”) was the Agilent inert fused silica (1.0 m × 0.15 mm), which connected the analytical column to the MS transfer line. The GC oven was initially set to 80°C for 2 min after sample injection, then heated to 180°C at 30°C/min ramp rate, then to the final temperature of 240°C at 15°C/min ramp rate. Each analytical run was operated under a constant 1.68 mL/min flow rate on the analytical column and 1.85 mL/min on the postcolumn, using helium as the carrier gas. To remove high-boiling sample matrix components from the analytical column, reduce interferences in subsequent sample injections and increase the overall column life, a postcolumn backflushing method was included. The backflush was setup for five void volumes, keeping a −2.20 mL/min constant back flow on the analytical column at 240°C. The transfer line and MS source temperatures were both at 280°C. The MS source mode was positive EI ionization. EI mass spectra were obtained at ionization energy of 70 eV and gain was set to 20 (×105). For quantitative analysis of the AAs, multiple reaction monitoring was chosen. Ultrahigh purity nitrogen was used as the collision gas, and ultrahigh purity helium was used as the quenching gas. Details of instrument operational conditions and the ions used for quantitation are listed in Supplementary Tables S1 and S2.
Sample data flow
An automated system for tracking sample data was created in parallel with the automated sample preparation method (Figure 3). Samples are received and logged into the LIMS reporting system before they are queued for preparation and analysis. Samples to be prepared were scanned by the Hamilton STAR, which, upon completion, generated an output file. The Hamilton output file was run through an Excel macro which generated two modified sequence files: one to be imported into Agilent MassHunter and one to be uploaded to Indigo ASCENT™ once Agilent GC/MS analyses were completed. Once the raw data were acquired from the GC–MS/MS, they could either be analyzed in Agilent MassHunter Quantitative software or uploaded to Indigo ASCENT™ for automatic peak integration. A formatted output file containing the final calculated concentration data was generated and was directly uploaded to the LIMS system. Unknown samples were evaluated individually according to set quality assurance (QA) rules, including difference in retention times of internal standard and native ion transition peaks, confirmation ion ratio, internal standard peak area and concentration exceeding calibration dynamic range. Individual samples and/or analyte would be flagged for repeat or dilution if any of the QA rules were violated. Batch quality controls (QCs) were evaluated according to modified Westgard QC rules (45). A batch would be rejected and repeated if any of the QC rules were violated. In addition, the blank was examined, and the entire batch was rejected and repeated if blank level exceeded the established limit for each analyte. (The blank limit for each analyte was obtained from 50 individual runs, only one run per day, over a period of several months.) Final results that passed all QA and QC rules were exported to a final reporting system such as National Health and Nutrition Examination Survey (NHANES).
Figure 3.
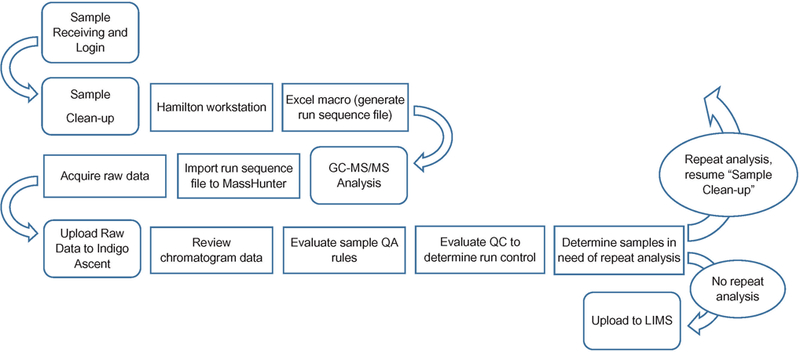
Sample data flow chart.
Results
All six quantified AAs (OTOL, 26DM, OANS, 1AMN, 2AMN and 4ABP) and corresponding structural isomers—m-toluidine (MTOL), p-toluidine (PTOL), 2,3-dimethylaniline (23DM), 2,4-dimethylaniline (24DM), 2,5-dimethylaniline (25DM), 3,4-dimethylaniline (34DM), 3,5-dimethylaniline (35DM), m-anisidine (MANS), p-anisidine (PANS), 2ABP, and 3ABP—were separated using GC. Figure 4 shows the total ion counts (TIC) of the six quantified AAs and the 11 structural isomers in a calibration standard, to validate isomeric separation from the target analytes. The TIC of the six quantified AAs and two of the monitored structural isomers (2ABP and 3ABP) in a urine sample is also shown in Figure 4. The high degree of sensitivity and specificity of this method allows detection of all six target analytes at trace levels (parts-per-trillion) in human urine samples.
Figure 4.
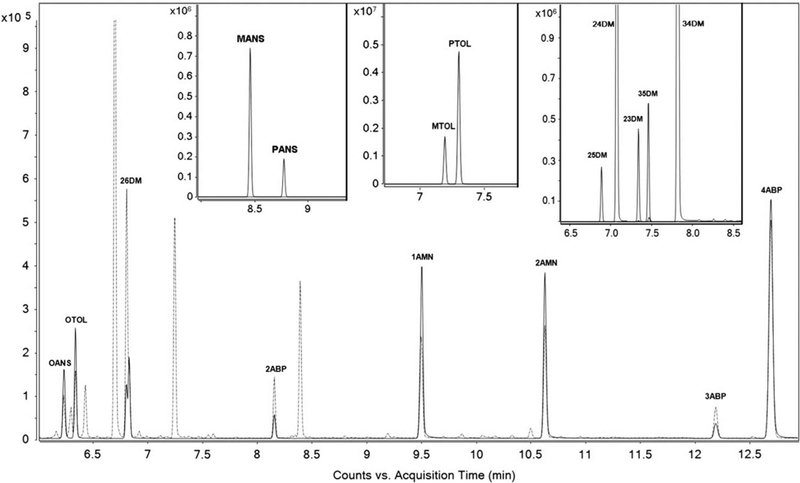
The TIC of six quantified AAs and two of the monitored isomers in an urine sample containing approximately 100 ng/L of each target analyte (repre-sented as dashed line) is overlaid on the TIC of a calibration standard containing 100 pg/μL of each target analyte (represented as solid line). The three insets show the related structural isomers of OANS, OTOL and 26DM, each spiked at 100 pg/μL.
Limit of detection
Since our previous work (44), in which the limit of detection (LoD) was estimated using the extrapolated, limiting standard deviation obtained from calibration curves, the LoD in this study was determined according to the guideline for determination of LoD by the Clinical and Laboratory Standard Institute (46) using the four LoD pools: LoD0, LoD1, LoD2 and LoD3. The pools were made using filtered nonsmoker urine and spiked at 0, 20, 40 and 60 ng/L for six analytes: OTOL, 26DM, OANS, 1AMN, 2AMN and 4ABP. The analyte LoDs (ng/L) were estimated from 50 independent analytical runs, one analytical run per day. Because OTOL is detected in the blanks, its LoD was calculated using the equation: 3σblank = LoD, where σblank is the standard deviation of calculated blank levels from 54 individual runs. The analyte LoDs are listed in Table I.
Table I.
Calculated slopes from calibration curves prepared in urine (matrix) and hexane (nonmatrix). Differences in the averages of the three slopes, obtained from each calibration set, are minimal and validates use of a nonmatrix calibration set to quantitate AAs in urine.
| Analyte | LoD (ng/L) | Hexane and urine calibration range (pg/μL) |
Hexane calibration curve, slope |
Hexane, averaged slopes |
Urine calibration curve, slope |
Urine, averaged slopes |
Difference in averaged slopes (%) |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Run 1 | Run 2 | Run 3 | Run 1 | Run 2 | Run 3 | |||||||
| OTOL | 111.2 | 0.507–1,280 | 0.01811 | 0.01673 | 0.01753 | 0.01746 | 0.01768 | 0.01688 | 0.01598 | 0.01685 | 3.49 | |
| 26DM | 15.7 | 0.502–100 | 0.01613 | 0.01567 | 0.01563 | 0.01581 | 0.01643 | 0.01573 | 0.01611 | 0.01609 | −1.76 | |
| OANS | 7.0 | 0.504–99.9 | 0.01164 | 0.01144 | 0.01160 | 0.01156 | 0.01153 | 0.01099 | 0.01141 | 0.01131 | 2.19 | |
| 1AMN | 1.5 | 0.504–99.9 | 0.03636 | 0.03736 | 0.03735 | 0.03702 | 0.03553 | 0.03610 | 0.03579 | 0.03579 | 3.33 | |
| 2AMN | 2.8 | 0.489–100 | 0.02788 | 0.02850 | 0.02849 | 0.02829 | 0.02924 | 0.02948 | 0.02932 | 0.02932 | −3.64 | |
| 4ABP | 1.8 | 0.482–98.6 | 0.00699 | 0.00698 | 0.00673 | 0.00690 | 0.00712 | 0.00716 | 0.00708 | 0.00708 | −2.62 | |
Precision
To determine intrarun and inter-run precision, two pools were used. The pools were prepared in-house from urine collected (with CDC Institutional Review Board approval) from nonsmokers and spiked with a standard solution of native (unlabeled) analytes at approximately 125 and 500 ng/L. The coefficient of variation (CV) for intraday (n = 5) and interday (n = 5) runs are calculated and listed in Table II. Intraday and interday CV values, for all AAs at each spiked concentration, were below 10%.
Table II.
Coefficient of variation (CV), %, of intraday and interday precisions, and %-bias accuracy in hexane solution (nonmatrix) and in urine (matrix).
| Analyte | Nominal concentration (ng/L) |
Intraday precision Measured concentration (ng/L), n = 5 |
CV | Interday precision Measured concentration (ng/L), n = 5 |
CV | Nominal concentration (pg/μL) |
Accuracy in solution Measured concentration (pg/μL) |
%–bias | Nominal concentration (ng/L) |
Accuracy in matrix Measured concentration (ng/L) |
%–bias |
|---|---|---|---|---|---|---|---|---|---|---|---|
| OTOL | 150.7 | 164.5 | 5.40 | 159.8 | 6.26 | 2.5 | 2.7 | 7.60 | 116.4 | 97.6 | −16.17 |
| 485.9 | 503.2 | 5.55 | 527.3 | 5.75 | 7.1 | 8.7 | 22.70 | 223.9 | 205.4 | −8.28 | |
| 26.2 | 26.8 | 2.21 | 806.0 | 772.6 | −4.14 | ||||||
| 49.3 | 50.1 | 1.62 | |||||||||
| 197.0 | 211.7 | 7.48 | |||||||||
| 26DM | 121.3 | 113.9 | 6.22 | 115.6 | 5.88 | 2.5 | 2.4 | −3.60 | 55.7 | 47.8 | −14.25 |
| 502.6 | 487.1 | 3.50 | 503.0 | 4.37 | 7.0 | 6.9 | −1.86 | 107.1 | 98.0 | −8.49 | |
| 25.7 | 25.2 | −1.79 | 385.7 | 386.1 | 0.10 | ||||||
| 40.4 | 40.8 | 1.04 | |||||||||
| 161.5 | 161.3 | −0.13 | |||||||||
| OANS | 153.4 | 149.5 | 3.61 | 157.9 | 2.62 | 2.5 | 2.8 | 13.20 | 58.5 | 52.9 | −9.47 |
| 505.8 | 506.5 | 2.78 | 526.1 | 3.44 | 7.1 | 6.8 | −4.79 | 112.5 | 99.3 | −11.74 | |
| 25.6 | 27.5 | 7.42 | 404.9 | 372.9 | −7.91 | ||||||
| 49.7 | 55.9 | 12.39 | |||||||||
| 198.7 | 231.5 | 16.53 | |||||||||
| 1AMN | 110.8 | 120.0 | 2.19 | 118.2 | 4.48 | 2.5 | 2.6 | 4.69 | 52.9 | 45.1 | −14.71 |
| 458.1 | 491.9 | 2.19 | 492.8 | 3.34 | 7.0 | 7.1 | 1.17 | 101.7 | 91.7 | −9.83 | |
| 26.4 | 25.7 | −2.69 | 366.1 | 335.0 | −8.50 | ||||||
| 42.7 | 47.9 | 12.21 | |||||||||
| 170.9 | 176.9 | 3.55 | |||||||||
| 2AMN | 111.7 | 116.7 | 1.38 | 121.0 | 4.31 | 2.4 | 2.4 | −1.25 | 57.8 | 48.0 | −16.86 |
| 479.8 | 505.6 | 2.44 | 524.4 | 3.84 | 6.8 | 6.4 | −6.47 | 111.1 | 100.9 | −9.19 | |
| 25.3 | 24.7 | −2.53 | 400.0 | 342.7 | −14.33 | ||||||
| 50.0 | 47.4 | −5.18 | |||||||||
| 200.0 | 199.8 | −0.11 | |||||||||
| 4ABP | 109.0 | 112.7 | 2.51 | 116.5 | 5.12 | 2.4 | 2.6 | 9.17 | 54.7 | 47.2 | −13.78 |
| 446.1 | 468.3 | 1.13 | 479.1 | 3.31 | 6.8 | 6.8 | −0.15 | 105.2 | 94.7 | −10.04 | |
| 26.0 | 22.9 | −11.85 | 378.9 | 353.0 | −6.83 | ||||||
| 50.5 | 54.8 | 8.44 | |||||||||
| 202.1 | 232.1 | 14.83 | |||||||||
Accuracy
Accuracy was determined by spiking known amounts of AA standard solution into hexane (accuracy in solution) and urine (accuracy in matrix). The accuracy was calculated using the equation: ((measured [AA] − nominal [AA])/nominal [AA]) × 100%. To test accuracy in solution, five levels of testing calibrators were prepared and run with the hexane calibration curve used for AA quantitation in urine samples. The native AAs used to spike the testing calibrators were purchased from vendors or lot numbers different from ones used for making the hexane calibration curve. As this evaluation is necessary to ensure accuracy of the hexane calibration curve, these testing calibrators are analyzed every time a new set of hexane calibration curve is prepared for analysis, or a new internal standard spiking solution is made. Results are listed in Table II. To test accuracy in matrix, replicates of AA-spiked urine samples, at three different concentrations, were analyzed. All spiked urine samples were prepared as unknown samples and run in triplicate over 2 days. For all samples tested, the calculated accuracy was ±17% for all analytes, and thus, accuracy in matrix tests was acceptable (Table II).
Analytical specificity
A high degree of analytical specificity was achieved for each analyte with this assay. Monitoring the correct retention times (±0.001 to 0.03 min relative to corresponding internal standards), confirmation ion ratios (which was obtained daily from the calibration standards and compared against the ratios calculated for each study sample) and precursor/product ion transitions helped ensure a high degree of specificity and minimized the influence from any potential interference(s).
Carryover
Sample carryover was examined by comparing successive pairs of injections of the highest calibrator (100 or 1,280 pg/μL) or high QC samples, with a toluene solvent blank. (The highest calibrator for OTOL was 1,280 pg/μL to accommodate for higher levels of this analyte detected in unknown samples.) No carryover was observed in the solvent blank after any injection of the highest calibrator or high QC. As a precaution, one toluene solvent blank was injected following injection of a full set of calibration standards and after each QC high sample. A toluene solvent blank was also injected at the beginning of each analytical batch to ensure no system contamination prior to standard and sample analyses. Between each individual injection, the syringe barrel was washed six times by drawing 3 μL of toluene.
Recovery
Sample matrix effects for each analyte were evaluated. Urine samples were spiked with a known amount of labeled AA internal standards and carried through the sample preparation process, as mentioned above. The percent recovery was calculated as the ratio of the response (peak area counts) of internal standards in urine samples to the responses of internal standard in the calibration standards. The average recovery was greater than 85% for all AAs.
Linearity limits
We have confirmed linear responses for all analytes (R2 ≥ 0.98) across a broad dynamic range, from 0 to 100 pg/μL (for 26DM, OANS, 1AMN, 2AMN and 4ABP) and from 0 to 1,280 pg/μL (for OTOL), with the lowest nonzero standard concentration being 0.5 pg/μL. The dynamic range for OTOL was extended in order to quantitate higher levels of OTOL in the urine samples. Regressions were calculated by plotting the quotients of the peak areas for each native analyte and that of the labeled analyte (the response ratio) as a function of the nominal concentration; a weighting factor of 1/x was used for all analytes. The calculated concentration of analytes in study samples was reported if the value was within the lowest and highest calibrators. Samples with analyte concentration exceeding the highest calibrator were repeated with appropriate dilution to bring the concentration within the validated dynamic range.
Matrix equivalence
The influence of urine matrix on the hexane (nonmatrix) calibration curve used for daily quantitation was estimated. A urine calibration curve was prepared by spiking standards of AAs at different levels in blank urine samples. The urine standards were subjected to the same sample preparation protocol as unknown urine samples (as described in the “Automated sample preparation” section above). The calibration curves built in urine matrix were run in parallel with calibration curves prepared in hexane (see Figure 5, for 4ABP). The averages of the slopes (n = 3) of each set of calibration curves were compared to assess the influence of matrix effects on AAs. As shown in Table I, the slope differences for all six analytes were within ±5%. These results indicate that a urine matrix has minimal impact on the quantitation of AAs based on calibration curves prepared in hexane.
Figure 5.
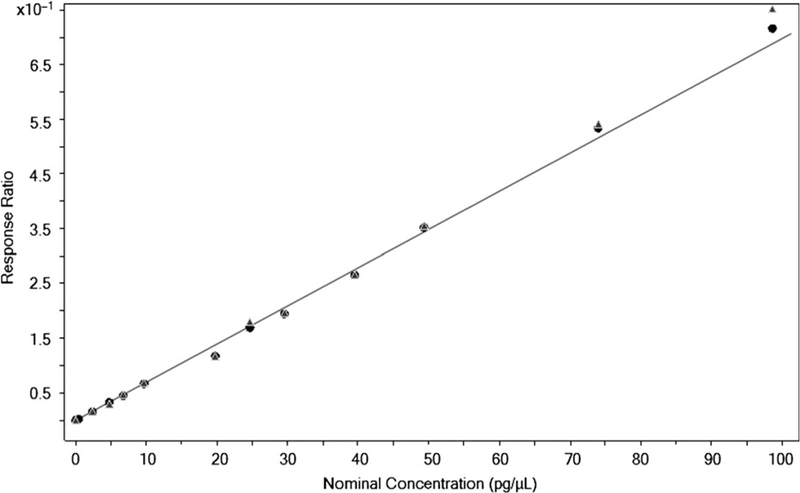
Matrix equivalence of 4ABP in urine (matrix) and hexane (nonmatrix) standards. Hexane curve (n = 1) is plotted with solid circular markers, and urine curve (n = 1) is plotted with solid triangular markers. Calibration curves, from either set of matrix, show good linearity for a broad dynamic range (0.5 to 98 pg/μL).
Thaw-refreeze and storage stability
The long-term storage stability of analytes in unprocessed samples was tested with spiked urine at low and high concentrations that had been stored at −70°C for 2 years. The results from this study indicate that long-term storage at −70°C has minimal impact on sample integrity (Table III). The effect of repeated thaw and refreeze (T/RF) cycles (−70°C – room temperature) on unprocessed urine samples was determined for each analyte. The results indicate that all six analytes are stable following at least five T/RF cycles, with average sample loss staying within the range of 5–17%. Results for individual analytes are listed in Table III.
Table III.
Short-term (24 h at 23°C), long-term (2 years at −70°C), and thaw and re-freeze (T/RF) stability of unprocessed samples; and stability of processed samples at 10°C (72 h) and −20°C (13 weeks).
| Analyte | Nominal concentration (ng/L) |
Measured concentration (ng/L) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unprocessed samples at −70°C for 2 year (%−diff) |
Unprocessed samples at 23°C for 24 h (%−diff) |
T/RF stability of unprocessed samples |
−20°C storage stability of processed samples |
10°C storage stability of processed samples |
||||||||
| Control | Five T/RF cycles (%–diff) |
Day 1 | Week 1 (%–diff) | Week 13 (%–diff) | Day 1 | Autosampler for 24 h (%–diff) |
Autosampler for 72 h (%–diff) |
|||||
| OTOL | 150.7 | 168.9 (12.10) | 218.0 | 198.2 (−9.09) | 57.0 | 60.7 (6.55) | 53.2 (−6.72) | 623.0 | 588.5 (−5.53) | 562.8 (−9.66) | ||
| 485.9 | 493.0 (1.48) | 511.3 (5.24) | 67.6 | 59.7 (−11.61) | 63.6 (−5.86) | |||||||
| 82.2 | 87.4 (6.38) | 92.3 (12.25) | ||||||||||
| 168.0 | 168.7 (0.40) | 181.1 (7.79) | ||||||||||
| 490.2 | 498.9 (1.75) | 501.3 (2.26) | ||||||||||
| 26DM | 121.3 | 109.8 (−9.46) | 157.5 | 148.9 (−5.46) | 19.8 | 17.8 (−10.28) | 17.6 (−10.92) | 459.6 | 426.9 (−7.11) | 418.3 (−8.98) | ||
| 502.6 | 492.6 (−2.01) | 519.1 (3.27) | 43.8 | 43.0 (−1.95) | 38.0 (−13.38) | |||||||
| 68.1 | 61.4 (−9.89) | 59.6 (−12.45) | ||||||||||
| 149.6 | 147.3 (−1.57) | 150.6 (0.63) | ||||||||||
| 543.4 | 480.5 (−11.58) | 499.1 (−8.16) | ||||||||||
| OANS | 153.4 | 148.0 (−3.48) | 158.2 | 144.4 (−8.71) | 44.9 | 46.4 (3.25) | 54.0 (20.39) | 427.1 | 420.2 (−1.60) | 405.1 (−5.15) | ||
| 505.8 | 498.5 (−1.44) | 488.8 (−3.37) | 70.5 | 66.4 (−5.88) | 64.1 (−9.05) | |||||||
| 92.8 | 83.0 (−10.51) | 86.4 (−6.87) | ||||||||||
| 153.5 | 137.6 (−10.39) | 141.5 (−7.81) | ||||||||||
| 544.4 | 531.9 (−2.28) | 516.4 (−5.14) | ||||||||||
| 1AMN | 110.8 | 120.0 (8.31) | 109.5 | 91.5 (−16.39) | 13.6 | 15.5 (14.37) | 14.1 (3.62) | 269.2 | 287.7 (6.87) | 303.4 (12.71) | ||
| 458.1 | 496.4 (8.36) | 434.9 (−5.08) | 34.1 | 33.7 (−1.30) | 31.8 (−6.84) | |||||||
| 43.8 | 53.1 (21.19) | 50.7 (15.84) | ||||||||||
| 94.1 | 115.7 (22.87) | 114.2 (21.25) | ||||||||||
| 449.4 | 472.3 (5.08) | 501.7 (11.64) | ||||||||||
| 2AMN | 111.7 | 116.7 (4.45) | 101.4 | 90.3 (−10.93) | 14.3 | 15.1 (5.54) | 15.2 (6.70) | 318.8 | 321.3 (0.81) | 323.7 (1.54) | ||
| 479.8 | 511.5 (6.62) | 476.7 (−0.63) | 35.4 | 38.1 (7.76) | 37.7 (6.46) | |||||||
| 53.4 | 56.8 (6.35) | 56.7 (6.27) | ||||||||||
| 96.1 | 100.7 (4.72) | 99.4 (3.42) | ||||||||||
| 528.8 | 513.3 (−2.93) | 509.1 (−3.71) | ||||||||||
| 4ABP | 109.0 | 111.9 (2.64) | 111.6 | 105.2 (−5.75) | 21.5 | 20.3 (−5.36) | 21.5 (0.00) | 347.0 | 339.9 (−2.06) | 342.0 (−1.46) | ||
| 446.1 | 467.5 (4.80) | 428.2 (−3.99) | 42.5 | 41.3 (−2.65) | 42.1 (−0.90) | |||||||
| 62.4 | 58.4 (−6.43) | 61.1 (−2.17) | ||||||||||
| 120.2 | 106.0 (−11.81) | 108.1 (−10.04) | ||||||||||
| 476.0 | 461.0 (−3.15) | 476.0 (0.00) | ||||||||||
Processed samples were either immediately analyzed on a GC– MS/MS system or provisionally stored at −20 ± 4°C until they could be analyzed. (During GC–MS/MS analysis, processed samples were stored in a cooled (10°C) autosampler.) To evaluate storage stability at −20°C, five spiked nonsmoker samples were injected after processing. The initial measurements are listed as “Day 1” in Table III. After initial injection, these samples were stored at −20°C for 1 week, then reinjected (“Week 1” measurement) and stored again at −20°C for 13 weeks then reinjected for a final time (“Week 13” measurement). The results from repeated injections of samples stored at −20°C indicate that analytes in processed samples are stable for up to 13 weeks, with a majority of the repeated measurement yielding ±10% difference from initial measurement (Table III). Likewise, the results from repeated injections of a sample stored in the autosampler indicate that analyte levels are not significantly impacted by short-term (24 and 72 h) storage conditions following the initial day (“Day 1”) sample was placed on autosampler and analyzed (Table III). The effect on analyte stability due to the length of time needed to process urine samples at room temperature (23°C ± 1°C) was also assessed. The results listed in Table III show insignificant changes in analyte levels.
Ruggedness test
To test the ruggedness of both the sample preparation and the GC– MS/MS method, QC samples were prepared and run under varied conditions in five parameters, each tested separately. Ruggedness was tested to determine which of the parameters (if any) would potentially affect assay accuracy. The parameters tested include: (i) injection port temperature; (ii) injection pulse pressure; (iii) amount of PFPA used during derivatization; (iv) sample hydrolysis duration and (v) the lot/work order of DB-FFAP column used for analysis. Samples were tested for all parameters at below, above and normal operating conditions. For all parameters tested, less than 17% difference in concentration was calculated for all analytes, with the majority at or below 10%. The complete set of result is listed in Supplementary Table S3.
Instrument cross-validation
For our assay, two Agilent GC–MS/MS systems were used to analyze processed samples. For the AA assay, five spiked and one nonspiked nonsmoker urine samples were used for instrument cross-validation. The Pearson coefficient for all six quantified analytes was within the range of 0.97–0.99.
QC characterization
The two QC pools used for this method—QC low (spiked at approximately 125 ng/L for all analytes) and QC high (spiked at approximately 500 ng/L for all analytes)—were characterized with 39 replicates from each pool, spanning almost 10 months. QC characterization statistics was subsequently used to verify methodological precision for each analytical run according to modified Westgard QC rules (45). No significant changes in concentration were observed for any analyte, in either QC pool, over the duration of the characterization period, or 16 months after the QC pools were prepared (Supplementary Table S4).
Validation of Indigo ASCENT™
Indigo ASCENT™ is a software used in our laboratory to automatically integrate chromatograms based on proprietary algorithms. To ensure the accuracy of automatic chromatographic peak integration, quantitated results from 150 samples obtained from Agilent MassHunter Workstation Quantitative Analysis and Indigo ASCENT™ were compared to ensure that the measurements of analyte concentrations were equivalent. A nonparametric test (Mann–Whitney) concluded that the calculated results obtained from Indigo ASCENT™ and MassHunter Workstation were statistically equivalent.
Discussion
Automation of sample preparation processes became a necessary part of biomonitoring measurements applied to large population studies such as NHANES. Analyzing more than 5,000 samples per 2-year cycle requires increased throughput compared with typical manual sample preparation. The automated workstation allows the analyst to increase the necessary sample throughput by reducing the time it takes to aliquot samples. Compared to our previous manual sample preparation method (44), the new automated sample preparation method effectively reduces the sample cleanup time from 8 to 4 h, while improving and maintaining the high precision of the assay.
The LoDs obtained from our method were within the range of 1.5 ng/L (26DM, OANS, 1AMN, 2AMN and 4ABP) to 111.2 ng/L (OTOL), with 1AMN, 2AMN and 4ABP exhibiting detection limits lower than 3 ng/L. With LoDs of most analytes below 16 ng/L, our new method’s detection limits are either lower than (4, 5, 38, 39, 41–43) or comparable to (35–37, 40, 44) the detection limits reported by other groups. It is important to note that the LoDs reported in the literature were estimated based on either the calibration curves used to analyze samples or through analysis of chromatographic peaks that had signal-to-noise ratios greater than 3 (37, 40, 44). For instance, the lowest detection limits for OTOL and 26DM were reported to be 5.2 and 9.3 ng/L, respectively, using aqueous standards to estimate each value (37); the lowest detection limits for 1AMN, 2AMN and 4ABP were reported to be 5.0, 3.0 and 1.5 ng/L, respectively, using solvent standards to estimate each value (40). Analyte LoDs in our assay were obtained from 50 individual runs, using three spiked levels and one nonspiked level of nonsmoker urine samples. The use of spiked urine samples to determine analyte LoDs allowed us to account for sample matrix effects directly, in contrast to other methods (4, 35, 37, 40), where nonmatrix standard(s) and information from the calibration curve were used to estimate the LoD. In obtaining relatively low analyte LoDs (particularly for 1AMN, 2AMN and 4ABP), we are able to apply our method to detect the candidate AAs at trace levels (ppb to ppt). Furthermore, as we have optimized our method toward using lower sample volumes—2 mL, compared to reported ranges of 4–20 mL (4, 5, 35, 44)—the method becomes more practicable for use in epi-demiological studies and large surveys such as the NHANES.
Typically, 30–60-m long columns, coated with phenyl-or methyl-based stationary phases (midpolar to nonpolar range, respectively), have been used for AA analysis (5, 33, 36, 37, 39). Our assay was previously developed and validated using the midpolar Agilent J&W DB-17MS column (30 m × 0.25 mm × 0.25 μm), which is composed of a (50%-phenyl)-methylpolysiloxane stationary phase (44). However, the highly polar DB-FFAP column proved to be much more rugged and specific to the analytes in the assay panel. Specifically, when analyzing urine samples with the DB-17MS column, 26DM’s quantitation ion peak almost exclusively coeluted with an interfering peak, making accurate detection and quantification of this analyte to be particularly difficult. As shown in Supplementary Figure S1, with the use of the DB-FFAP column, analytical resolution was greatly enhanced for the 26DM analyte. Furthermore, when operating with the DB-FFAP column, we were able to reduce the LoD of 4ABP in urine samples from approximately 9.0 to 1.8 ng/L. The marked improvement in 4ABP’s detection rate (with both the quantitation and confirmation ions) was largely due to the lower level of column bleed exhibited by the DB-FFAP column, at the operational temperature utilized. With better detection of both the quantitation and confirmation ions, we were more confident in identifying the 4ABP analyte from a complex matrix such as urine.
For the first time, we report the use of SLE, Isolute™ cartridges in sample cleanup of urinary AAs following hydrolysis. These cartridges have a hydrophilic frit that enables water and other aqueous components of the urine samples to absorb onto the bedding material quickly, followed by extraction of the AAs with DCM. The SLE replaced multiple steps used in our previous sample pretreatment process (44) that involved liquid–liquid extraction with hexane, back extraction with 0.1 M HCl and cleanup with a hydrophilic– lipophilic-balanced cartridge. The use of SLE cartridges can also potentially replace other labor-intensive sample cleanup steps reported in the literature (4, 5, 35–43).
The automated sample data flow reduces potential human error in sample tracking, handling and data analysis. All relevant sample information, such as the sample ID, sample volume and any dilution factors are saved with each raw data file as the samples are analyzed on the GC–MS/MS. Our current method setup allows for one of two data analyses processes to occur: one, through the Agilent MassHunter Quantitative Analysis software; and the other was through Indigo Biosystems ASCENT™ platform. Although results obtained from either software platforms were proven to be statistically equivalent, we currently use Indigo ASCENT™, exclusively, to analyze all sample data. Through the use of Indigo ASCENT, numerous custom QA rules could be imbedded to ensure consistent evaluation of each analyte on a sample-to-sample, batch-by-batch basis.
Conclusion
Overall, the new automated method is time-efficient and precise. At least two batches of 32 samples can be prepared each day when higher sample throughput is needed (compared to 1 batch of 43 samples). Laboratory automation has improved the overall accuracy and precision of the analytical method, whereas the newly implemented sample data flow has improved sample tracking and data analysis, including evaluation of individual sample QA and run QC. The complete sample data flow, from sample receiving to final result reporting, is efficient and minimizes potential source(s) of human errors.
Supplementary Material
Acknowledgments
This work was funded by the US Food and Drug Administration and the Centers for Disease Control and Prevention, Department of Health and Human Services. The authors would like to acknowledge Sam P. Caudill, Rey Decastro and Yao Li for their help with statistical analyses. The authors would like to acknowledge former team members Elizabeth Cowan, Susan Pyatt, Aaron Danberry, Kyle Emer and James Alex Hodgson for their help.
Footnotes
The findings and conclusions in this report are those of the author(s) and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
References
- 1.IARC. (1986) Tobacco Smoking. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 38, 1–432. [PMC free article] [PubMed] [Google Scholar]
- 2.Silverman DT, Hartge P, Morrison AS, Devesa SS (1992) Epidemiology of bladder cancer. Hematology/Oncology Clinics of North America, 6, 1–30. [PubMed] [Google Scholar]
- 3.Hammond SK, Coghlin J, Gann PH, Paul M, Taghizadeh K, Skipper PL, Tannenbaum SR (1993) Relationship between environmental tobacco smoke exposure and carcinogen-hemoglobin adduct levels in nonsmokers. Journal of the National Cancer Institute, 85, 474–477. [DOI] [PubMed] [Google Scholar]
- 4.Teass AW, DeBord DG, Brown KK, Cheever LE, Stettler LE, Savage RE, Weigel WW, Dankovic D, Ward E (1993) Biological monitoring for occupational exposures to o-toluidine and aniline. International Archives of Occupational and Environmental Health, 65, S115–118. [DOI] [PubMed] [Google Scholar]
- 5.Riffelmann M, Muller G, Schmieding W, Popp W, Norpoth K (1995) Biomonitoring of urinary aromatic amines and arylamine hemoglobin adducts in exposed workers and nonexposed control persons. International Archives of Occupational and Environmental Health, 68, 36–43. [DOI] [PubMed] [Google Scholar]
- 6.Tang D, Warburton D, Tannenbaum SR, Skipper PL, Santella RM, Cerijido GS, Crawford FG, Perera FP (1999) Molecular and Genetic Damage from Environmental Tobacco Smoke in Young Children. Cancer Epidemiology, Biomakers & Prevention, 8, 427–431. [PubMed] [Google Scholar]
- 7.Skipper PL, Tannenbaum SR, Ross RK, Yu MC (2003) Nonsmoking-related Arylamine Exposure and Bladder Cancer Risk. Cancer Epidemiology, Biomakers & Prevention, 12, 503–507. [PubMed] [Google Scholar]
- 8.IARC. (2004) Tobacco Smoke and Involuntary Smoking. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 83, 1–1473. [PMC free article] [PubMed] [Google Scholar]
- 9.USHHS. The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General–Executive Summary. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Heath: Atlanta, GA, 2006. [PubMed] [Google Scholar]
- 10.Bryant MS, Vineis P, Skipper PL, Tannenbaum SR (1988) Hemoglobin adducts of aromatic amines: Associations with smoking status and type of tobacco. Proceedings of the National Academy of Sciences, 85, 9788–9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castelao JE, Yuan J-M, Skipper PL, Tannenbaum SR, Gago-Dominguez M, Crowder JS, Ross RK, Yu MC (2001) Gender-and Smoking-Related Bladder Cancer Risk. Journal of the National Cancer Institute, 93, 538–545. [DOI] [PubMed] [Google Scholar]
- 12.Vineis P (1994) Epidemiology of cancer from exposure to arylamines. Environmental Health Perspectives, 102, 7–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.IARC. (2010) Some aromatic amines, organic dyes, and related exposures. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 99, 1–678. [PMC free article] [PubMed] [Google Scholar]
- 14.Stabbert R, Schäfer KH, Biefel C, Rustemeier K (2003) Analysis of aromatic amines in cigarette smoke. Rapid Communications in Mass Spectrometry, 17, 2125–2132. [DOI] [PubMed] [Google Scholar]
- 15.Fischer P Chapter 11A Tobacco Blending. In: Davis DL, Nielson MT (eds). Tobacco: Production, Chemistry and Technology Oxford; Malden, MA: Blackwell Science, 1999; pp. 348. [Google Scholar]
- 16.Dawson RF (1952) Chemistry and biochemistry of green tobacco. Industrial and Engineering Chemistry, 44, 266–270. [Google Scholar]
- 17.Patrianakos C, Hoffmann D (1979) Chemical studies on tobacco smoke LXIV. On the analysis of aromatic amines in cigarette smoke. Journal of Anlaytical Toxicology, 3, 150–154. [Google Scholar]
- 18.Schmeltz I, Hoffmann D (1977) Nitrogen-containing compounds in tobacco and tobacco smoke. Chemical Reviews, 77, 295–311. [Google Scholar]
- 19.Heckman RA, Best FW (1981) An investigation of the lipophilic bases of cigarette smoke condensate. Tobacco Science, 25, 33–39. [Google Scholar]
- 20.IARC. (2000) ortho-TOLUIDINE. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 77, 267–322. [PMC free article] [PubMed] [Google Scholar]
- 21.IARC. (1993) 2,6-DIMETHYLANILINE (2,6-XYLIDINE). IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 57, 323–335. [PMC free article] [PubMed] [Google Scholar]
- 22.IARC. (2010) 4-AMINOBIPHENYL. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 99, 71–109. [PMC free article] [PubMed] [Google Scholar]
- 23.IARC. (2010) 2-NAPHTHYLANIME. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 99, 369–405. [PMC free article] [PubMed] [Google Scholar]
- 24.IARC. (1999) ortho-ANISIDINE. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 73, 49–58. [PMC free article] [PubMed] [Google Scholar]
- 25.DeBruin LS, Pawliszyn JB, Josephy PD (1999) Detection of mono-cyclic aromatic amines, possible mammary carcinogens, in human milk. Chemical Research in Toxicology, 12, 78–82. [DOI] [PubMed] [Google Scholar]
- 26.Pereira L, Mondal PK, Alves M Aromatic amines sources, environmental impact and remediation. In: Lichtfouse E, Schwarzbauer J, Robert D (eds.). Pollutants in Buildings, Water and Living Organisms Cham, Switzerland: Springer, 2015; pp. 297–346. [Google Scholar]
- 27.Vitzthum OG, Werkhoff P, Hubert P (1975) New volatile constituents of black tea aroma. Journal of Agricultural and Food Chemistry, 23, 999–1003. [Google Scholar]
- 28.Neurath GB, Dünger M, Pein FG, Ambrosius D, Schreiber O (1977) Primary and secondary amines in the human environment. Food and Cosmetics Toxicology, 15, 275–282. [DOI] [PubMed] [Google Scholar]
- 29.Chiang TA, Pei-Fen W, Ying LS, Wang LF, Ko YC (1999) Mutagenicity and aromatic amine content of fumes from heated cooking oils produced in Taiwan. Food and Chemical Toxicology, 37, 125–134. [DOI] [PubMed] [Google Scholar]
- 30.Bartsch H, Caporaso N, Coda M, Kadlubar F, Malaveille C, Skipper PL, Talaska G, Tannenbaum SR, Vineis P (1990) Carcinogen hemoglobin adducts, urinary mutagenicity, and metabolic phenotype in active and passive cigarette smokers. Journal of the National Cancer Institute, 82, 1826–1831. [DOI] [PubMed] [Google Scholar]
- 31.Bryant MS, Skipper PL, Tannenbaum SR, Maclure M (1987) Hemoglobin adducts of 4-aminobiphenyl in smokers and nonsmokers. Cancer Research, 47, 602–208. [PubMed] [Google Scholar]
- 32.USHHS. Reporting Harmful and Potentially Harmful Constituents in Tobacco Products and Tobacco Smoke Under Section 904(a)(3) of the Federal Food, Drug, and Cosmetic Act. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Tobacco Products (CTP): Silver Spring, MD, 2012. [Google Scholar]
- 33.Riedel K, Scherer G, Engl J, Hagedorn HW, Tricker AR (2006) Determination of three carcinogenic aromatic amines in urines of smokers and nonsmokers. Journal of Analytical Toxicology, 30, 187–195. [DOI] [PubMed] [Google Scholar]
- 34.Talaska G, Zoughool MA (2003) Aromatic amine and biomarkers of human exposure. Journal of Environmental Science and Health. Part C, Environmental Carcinogenesis & Ecotoxicology Reviews, 21, 133–164. [DOI] [PubMed] [Google Scholar]
- 35.Amiri A, Baghayeri M, Nori S (2015) Magnetic solid-phase extraction using poly(para-phenylenediamine) modified with magnetic nanoparticles as adsorbent for analysis of monocyclic aromatic amines in water and urine samples. Journal of Chromatography. A, 1415, 20–26. [DOI] [PubMed] [Google Scholar]
- 36.Labat L, Thomas J, Dehon B, Humbert L, Leleu B, Nisse C, Lhermitte M (2006) Evaluation of a profession exposition of ortho-toluidine by gas phase chromatography coupled with mass spectrometry. Acta Clinica Belgica, 61, 63–67. [PubMed] [Google Scholar]
- 37.Lamani X, Horst S, Zimmermann T, Schmidt TC (2015) Determination of aromatic amines in human urine using comprehensive multi-dimensional gas chromatography mass spectrometry (GCxGC-qMS). Analytical and Bioanalytical Chemistry, 407, 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muz M, Ost N, Kühne R, Schüürmann G, Brack W, Krauss M (2017) Nontargeted detection and identification of (aromatic) amines in environmental samples based on diagnostic derivatization and LC-high resolution mass spectrometry. Chemosphere, 166, 300–310. [DOI] [PubMed] [Google Scholar]
- 39.Weiss T, Angerer J (2002) Simultaneous determination of various aromatic amines and metabolites of aromatic nitro compounds in urine for low level exposure using gas chromatography-mass spectrometry. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences, 778, 179–192. [DOI] [PubMed] [Google Scholar]
- 40.Yu J, Wang S, Zhao G, Wang B, Ding L, Zhang X, Xie J, Xie F (2014) Determination of urinary aromatic amines in smokers and non-smokers using a MIPs-SPE coupled with LC-MS/MS method. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences, 958, 130–135. [DOI] [PubMed] [Google Scholar]
- 41.Jiang C, Sun Y, Yu X, Gao Y, Zhang L, Wang Y, Zhang H, Song D (2014) Application of C18-functional magnetic nanoparticles for extraction of aromatic amines from human urine. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences, 947–948, 49–56. [DOI] [PubMed] [Google Scholar]
- 42.DeBruin LS, Josephy PD, Pawliszyn JB (1998) Solid-phase micro-extraction of monocyclic aromatic amines from biological fluids. Analytical Chemistry, 70, 1986–1992. [DOI] [PubMed] [Google Scholar]
- 43.Grimmer G, Dettbarn G, Seidel A, Jacob J (2000) Detection of carci-nogenic aromatic amines in the urine of non-smokers. Science of the Total Environment, 247, 81–90. [DOI] [PubMed] [Google Scholar]
- 44.Seyler TH, Bernert JT (2011) Analysis of 4-aminobiphenyl in smoker’s and nonsmoker’s urine by tandem mass spectrometry. Biomarkers: Biochemical Indicators of Exposure, Response, and Susceptibility to Chemicals, 16, 212–221. [DOI] [PubMed] [Google Scholar]
- 45.Caudill SP, Schleicher RL, Pirkle JL (2008) Multi-rule quality control for the age-related eye disease study. Statistics in Medicine, 27, 4094–4106. [DOI] [PubMed] [Google Scholar]
- 46.CLSI. (2004) Protocols for determination of limits of detection and limits of quantitation: approved guideline. CLSI document EP17-A, 24, 1–52. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.