

Abstract
The underlying biology of essential tremor (ET) is poorly understood. Purkinje cell (PC) loss has been observed in some studies, although this finding remains somewhat controversial. Basket cells are interneurons whose axonal collaterals form a plexus around PC soma. When there is PC loss, this basket plexus appears empty. We used dual immunohistochemical staining for calbindin D28k and glutamic acid decarboxylase to quantify “empty baskets” as an indirect and alternative method of detecting PC loss. Microscopic analyses on 127 brains included ET and a spectrum of motor neurodegenerative diseases (50 ET, 27 spinocerebellar ataxias [SCAs], 25 Parkinson disease, 25 controls). The median percentage of empty baskets in ET patients was 1.5 times higher than controls (48.8% vs 33.5%, p < 0.001) but lower in ET than in SCA1 (59.7%, p = 0.011), SCA2 (77.5%, p = 0.003), and SCA6 (87.0%, p < 0.001). PC loss is not a feature of SCA3, and the median percentage of empty baskets (30.1%) was similar to controls (p = 0.303). These data provide support for PC loss in ET and are consistent with the notion that ET could represent a mild form of cerebellar degeneration with an intermediate degree of PC loss.
Keywords: Basket cells, Cerebellum, Essential tremor, Neurodegeneration, Parkinson disease, Purkinje cells, Spinocerebellar ataxia
INTRODUCTION
Essential tremor (ET) is the most prevalent tremor disorder and one of the most common neurological diseases (1). Despite this, its underlying patho-mechanisms are not well understood and this remains an area of active study. Recent postmortem studies have revealed a range of structural changes in the ET cerebellar cortex compared to that of controls. These changes, which are centered on the Purkinje cell (PC) and neighboring neuronal populations, include an increase in the number of torpedoes and related PC axonal changes, an increase in number of PC dendritic swellings and a truncation of the PC dendritic arbor, increased numbers of heterotopic PCs, changes at the PC-climbing fiber synaptic interface and changes at the PC-basket cell interface (2–8). In addition, PC loss has been observed in ET patients compared to controls. The latter finding has been reproduced variably across studies and, therefore, it remains a focus of controversy (5, 9–12). Therefore, additional studies and alternative approaches to the question are called for.
Basket cells are γ-aminobutyric acid (GABA)-ergic interneurons that provide inhibitory input to the PCs on both the PC body, forming a perisomal basket plexus, and around the axon initial segment (AIS), where their axonal collaterals form a pinceau structure (13–15). Basket cells and other molecular layer inhibitory interneurons serve to “sculpt” the response and output of the PCs, affecting both complex and simple spike activities as well as initiation of axon potentials at the PC AIS (16–18). Multiple neighboring basket cells provide axon collaterals converging on PCs to form the perisomal plexus, with direct “en passant” type synaptic contacts on the PC soma; in contrast, only a small subset of basket cell axons in the pinceau have direct synaptic contact with the PC AIS and the majority of terminals end freely, where they join other basket cell axons by septate-like junctions to provide an inhibitory electric field around the AIS (13, 19). In the setting of PC loss and relatively preserved basket cells, the basket plexus appears empty. Hence, “empty baskets” are a marker of PC loss. As such, the quantification of empty baskets in ET provides us an indirect and alternative method of quantifying PC loss in the disease.
Empty baskets have never been quantified in ET. Here, we quantified empty baskets in ET patients and controls (i.e. baskets in which the PC is completely missing, with no visible PC body). We also quantified empty baskets across a spectrum of cerebellar degenerative disorders characterized by variable degrees of PC loss (spinocerebellar ataxia [SCA] types 1, 2, and 6 [with marked loss] and SCA3 [with modest or no loss]) and another motor neurodegenerative disorder, Parkinson disease (PD) (20). The correlation between the empty basket pathology and PC counts across this spectrum of disorders was also studied to identify a potential interplay between these 2 neuronal types within the context of cerebellar degeneration. The overarching goal of these analyses was to advance our understanding of the basic neuropathology of ET, and furthermore, to view it not only with respect to control brains but within the context of a broader group of motor neurodegenerative conditions as well.
MATERIALS AND METHODS
Brain Repository, Study Subjects, and Clinical Assessments
ET brains were collected from the ET Centralized Brain Repository, a longstanding joint collaboration between investigators at Yale and Columbia Universities (4) to bank ET brains. This centralized repository of brains provided patients with ET in the United States. ET diagnoses were carefully confirmed or assigned by a senior movement disorder neurologist (E.D.L.) utilizing each of the following 3 sequential methods (21). First, the patients were clinically diagnosed with ET by their treating physicians. Second, each patient answered a set of semistructured clinical questionnaires to gather demographic and clinical data. All ET patients were required to submit 4 standardized hand-drawn Archimedes spirals (both right and left hand). These spirals were then assessed and tremor was rated. The following criteria were used for ET: (i) moderate or greater amplitude arm tremor (rating of 2 or higher) in at least one of the submitted Archimedes spirals; (ii) no history of PD or dystonia; and (iii) no other cause for tremor (e.g. medications and hyperthyroidism) (21). Third, ET patients underwent a detailed videotaped neurological examination to allow for assessment of tremor and other movements (E.D.L.) (22). The videotaped examination included assessments of postural tremor, kinetic tremor, rest, and intention tremor of the arms, neck, voice, and jaw. The videotape assessment also included the motor portion of the Unified PD Rating Scale that made available information on speech, facial expression, rest tremor, bradykinesia, posture, and gait and assessments of dystonia (23). Combined with the questionnaire data, the final diagnosis of each ET patient was re-examined, using previously published diagnostic criteria, which have been shown to be both reliable and valid (24). ET patients were followed prospectively, with clinical information regularly updated.
Eighteen control brains were from the New York Brain Bank. These individuals were followed prospectively at the Alzheimer’s Disease Research Center or the Washington Heights Inwood Columbia Aging Project at Columbia University. During serial neurological examinations, these individuals were clinically free of ET and other neurodegenerative disorders, including Alzheimer disease, PD, or progressive supranuclear palsy (4). An additional 7 control brains were obtained from the National Institutes of Health NeuroBioBank (University of Miami, Miami, FL and University of Maryland, Baltimore, MD).
We were able to obtain 27 SCA brains: 11 SCA1, 6 SCA2, 5 SCA3, and 5 SCA6. These were attained from multiple brain repositories, with 21 from a hereditary ataxia specimen repository maintained by a coauthor (A.H.K.) at the Veterans Affairs Medical Center in Albany, New York, 5 from the National Institutes of Health NeuroBioBank (University of Maryland, Baltimore, MD), and one from the New York Brain Bank. Tissue from the PD brains was obtained from the New York Brain Bank. During life, all study subjects agreed to sign informed consent forms approved by the respective university ethics boards.
Subject Selection
Subject selection was guided by available ET tissue. We identified 50 ET patients from the New York Brain Bank and selected 25 older controls to achieve a 2:1 age-matching scheme. We performed a power analysis that utilized data from our previous publication on PC counts in ET patients and controls (5). Assuming alpha = 0.05, and two-sided statistical tests, a sample of 40 ET patients and 20 controls would provide >80% power to detect at least a 25.0% increase in the empty basket pathology in ET patients compared to controls. Our study sample of 50 ET patients and 25 controls exceeded this required number. Cerebellar pathology in the SCA subjects was expected to be double that of ET patients. We utilized all available 27 SCA brains with a minimum number of 5 patients for each SCA type, and this provided >80% power (alpha = 0.05; 2-tailed) to detect differences between each SCA type and controls. Assuming that PD subjects would serve as disease controls, we set the number of PD brain tissue equal to controls (n = 25), preferentially selecting the oldest PD patients first so as to achieve an age distribution that approached that of our controls and ET patients. Thus, a total of 127 postmortem brains were analyzed. Clinical data were serially and prospectively collected on all ET cases and controls from the New York Brain Bank; clinical data from other patients were derived from clinical records after death.
Tissue Processing
All ET, PD, and 18 control brains had a complete neuropathological assessment at the New York Brain Bank. Brains had standardized measurements of brain weight (grams), and postmortem interval (hours between death and the completion of processing fresh brain into frozen tissues and placing remaining brain in formalin) (25). Whenever possible, 17 standardized blocks were harvested from each brain and processed, and 7-μm-thick paraffin sections were stained with Luxol fast blue/hematoxylin and eosin (LH&E) (5). In addition, selected sections were stained by the Bielschowsky method, and with mouse monoclonal antibodies to human glial fibrillary acidic protein (GFAP, clone GA5, Novocastra, Newcastle upon Tyne, UK), α-synuclein (clone KM51, Novocastra), phosphorylated tau (clone AT8, Research Diagnostics, Flanders, NJ), and β-amyloid (clone 6F/3D, Dako, Carpenteria, CA) (5). All tissues were examined microscopically by a senior neuropathologist blinded to clinical information including age and diagnosis (5). Brains had Braak and Braak Alzheimer disease staging for neurofibrillary tangles, and Consortium to Establish a Registry for Alzheimer’s disease (CERAD) ratings for neuritic plaques (26–29). Cerebellar tissues received from other brain repositories were from the same standard region as harvested at the New York Brain Bank.
Characterization of Cerebellar Pathology
Quantification of PCs, Torpedoes and Related Cerebellar Pathology
A standard 3 × 20 × 25-mm formalin-fixed tissue block from each brain was obtained from a parasagittal slice located 1–1.5 cm from the cerebellar midline and containing anterior and posterior quadrangulate lobules and the underlying dentate nucleus. Hence, we sampled the motor cerebellum (30, 31). PCs and torpedoes were quantified in a single 7-μm-thick, LH&E-stained section from that block (Fig. 1I). PCs (i.e. stained PC bodies) were quantified by counting and averaging the number of PCs across 15 100× nonoverlapping microscopic fields (12, 32). Similarly, torpedoes (i.e. fusiform swellings of the proximal portion of the PC axon [Fig. 1I, green arrow]) were counted in one entire LH&E section, as described in Refs. (4, 5). Torpedo counts were then normalized to PC layer length (i.e. divided by PC layer length) to account for any potential variations in amount of cerebellar cortex in the tissue block; this metric was referred to as n-torpedo count. We have previously shown that quantitative microscopic analysis of a single, standard section provided a sufficient representation of the pathology within that sample block (32). Using a Bielschowsky-stained section from the same block, we assigned a semiquantitative rating of the density of the basket cell plexus surrounding PC bodies (ratings = 0, 0.5, 1, 1.5, 2, 2.5, and 3), with 3 indicating dense, tangled basket cell processes, as described in Ref. (8).
FIGURE 1.

Pathologic criteria for empty and full basket cells (A–H, calbindin D28k [CB] and glutamic acid decarboxylase (GAD) dual immunohistochemical staining), and histologic identification of Purkinje cells and torpedoes (I, Luxol fast blue-hematoxylin and eosin [LH&E]). (A–H) GAD-labeled pinceau (brown) have a characteristic overall triangular shape at the molecular layer-granule cell layer junction and at the base of PC soma (red CB-labeled). Perisomal basket cell processes extend vertically into the molecular layer, surrounding the PC soma (when present), and are of variable thickness and complexity. The presence of CB-stained PC cytoplasm classifies a basket as full, even if only a small portion of PC cytoplasm is identified (e.g. white arrows in C, E, H). Black arrows point to empty baskets, with a detectable portion of the pinceau, remaining basket processes in the adjacent molecular layer and lacking any detectable PC body. Granule cell layer interneurons (blue arrows in B) also stain with GAD and are distinguished by their rounder shape, presence of nucleus, and/or shorter branching processes. Axonal torpedoes (green arrows in C, D) stain red to reddish-brown with a dense glassy appearance, and are located beneath the brown GAD-stained pinceau, seen here in the upper granule cell layer. (A) Control. Three full baskets, 2 empty baskets; the perisomal basket processes are slender in this specimen. (B) Control. Four full baskets (one on right partially cut off), 4 empty baskets. (C) ET. Four full baskets, 3 empty baskets. (D) ET. Two full baskets, 3 empty baskets. (E) PD. Two full baskets, 6 empty baskets. (F) PD. Two full baskets, 2 empty baskets. (G) SCA2. One full basket, 8 empty baskets. (H) SCA1. Two full baskets. (I) LH&E-stained paraffin section from an ET case. Three PCs are counted; from left to right, the PC body has (i) only granular Nissl rich cytoplasm, (ii) a nucleus and a prominent nucleolus, and (iii) a nucleus lacking a nucleous. The axonal torpedo (green arrow) has a fusiform shape with glassy eosinophilic axoplasm. Scale bar (in I): 50 μm.
Quantification of Empty Baskets
A trained graduate student (P.J.L.), who was blinded to clinical and postmortem data, performed dual immunohistochemical staining for calbindin D28k and glutamic acid decarboxylase (GAD). The dual staining was performed sequentially on 7-μm-thick paraffin sections to visualize basket cell plexuses (brown-GAD antibody) around PC soma (red-calbindin D28k antibody) in the PC layer (Figs. 1 and 2). The sections were preheated in the incubator at 60°C for 1 hour, rehydrated and antigen retrieval performed in Trilogy [Cell Marque, Rocklin, CA] in a steamer for 40 minutes followed by 30 minutes at room temperature. The sections were blocked with serum blocking solution (10% normal goat serum, 1% IgG-free bovine serum albumin [Jackson ImmunoResearch, West Grove, PA], 1% Triton X-100, in phosphate buffered saline) and then incubated in rabbit anticalbindin D28k primary antibody (1:1000) [Swant, Marly, Switzerland] in antibody diluent (1% IgG-free bovine serum albumin [Jackson ImmunoResearch], 1% Triton X-100, in phosphate buffered saline) overnight at 4°C. After several washes in phosphate buffered saline-0.1% Tween 20, goat antirabbit alkaline phosphatase secondary antibody (1:1000) [Fisher Scientific, Hampton, NH] was applied for 2 hours at room temperature, and the calbindin stains were developed with Immpact Vector Red alkaline phosphatase substrate according to manufacturer’s instructions [Vector Laboratories, Burlingame, CA]. Next, endogenous peroxidase activity was blocked with 3% H2O2, and mouse anti-GAD primary antibody (1:300) [MBL International Corp, Woburn, MA] was applied to the sections overnight at 4°C. The sections were washed in phosphate buffered saline-0.1% Tween 20 and then incubated with biotinylated goat antimouse secondary antibody (1:300) [Vector Laboratories] for 1 hour at room temperature, followed by 1-hour incubation with peroxidase mouse IgG substrate (Vectastain ABC kit) [Vector Laboratories]. The GAD stains were then developed with 3,3′-diaminobenzidine in DAKO DAB+ chromogen solution (Agilent Technologies, Santa Clara, CA).
P.J.L. quantified empty basket pathology at ×200–×400 magnification. “Full baskets” and “empty baskets” were identified as the plexus of basket cell axons around the PC soma with and without a detectable PC body, respectively (Fig. 1). The presence of PC cytoplasm stained by calbindin D28k antibodies is a defining criterion in our dual immunohistochemical stain; in particular, a full basket contains any portion of the PC soma and is surrounded by variably complex GAD-positive basket cell processes. The calbindin D28k antibodies will highlight small portions of a PC body (Fig. 1C, E, H, white arrows), thereby providing greater sensitivity in designating a basket as full versus empty. An empty basket is identified by GAD staining of various portions of the pinceau region located at the molecular layer-granule cell layer interface, along with remaining perisomal basket processes that extend into the molecular layer and absence of any detectable calbindin-labeled PC soma (Fig. 1A–H, black arrows). P.J.L.’s counts agreed highly (Pearson’s r = 0.98, p < 0.001) with those of a senior neuropathologist (P.L.F.), who independently quantified empty basket pathology in 15% of the sample (i.e. 20 patients) across all diagnoses (i.e. controls [n = 3]; ET [n = 6]; SCA [n = 8, 2 of each subtype]; and PD [n = 3]). Quantification of empty basket pathology was performed using a primary and a secondary method. In the primary analysis, the percentage of empty baskets was assessed. Thus, a sample of 127 brains was analyzed by the counting on a microscope of 400 basket plexuses per cerebellar tissue section. To minimize potential bias in the selection of fields, 4 randomly selected, noncontiguous regions of the cerebellar cortex were selected, each region yielding up to 100 total basket plexus counts. For each brain section, the percentage of empty baskets was calculated by taking the number of empty baskets and dividing by the total number of full and empty baskets. In the secondary analysis, the number of empty baskets was also quantified in a randomly selected sample of 47 brains (i.e. controls [n = 5]; ET patients [n = 10]; PD [n = 5]; SCA1 [n = 11]; SCA2 [n = 6]; SCA3 [n = 5]; and SCA6 [n = 5]); this was done by counting and averaging the number of empty baskets across 10 randomly selected microscopic fields at ×200 magnification.
Statistical Analysis
Clinical and pathological features of ET patients, controls, and other disease groups were compared (Table 1). Categorical or ordinal variables (e.g. gender) were compared using Chi-square tests. For the analysis of continuous variables (e.g. age, brain weight), we used parametric statistics when the variable followed a normal (i.e. Gaussian or “bell-shaped” curve) distribution, and we used nonparametric statistics when the variable did not follow a normal distribution (33, 34). We used the Kolmogorov-Smirnov test to determine whether each variable was normally distributed or not. Brain weight and postmortem interval were normally distributed (Kolmogorov-Smirnov normality test p value >0.05) and were therefore compared across groups using parametric statistics: one-way analysis of variance (ANOVA) with Tukey’s post hoc comparison tests. Other postmortem variables, including PC counts, n-torpedo counts, basket rating, and both metrics of empty baskets, were not normally distributed. Therefore, these were compared across groups using nonparametric statistics: Kruskal-Wallis tests and then Mann-Whitney U tests. We also tested whether either of the 2 empty basket metrics correlated with any clinical or pathological variables, using Spearman’s rank correlation coefficient. Furthermore, both linear and nonlinear quadratic regression models were fit, and the 2 models’ goodness of fit were compared against each other (ANOVA) to best model the relationship between PC counts and the number of empty baskets. If the models were significantly different from each other, the model with a superior fit was plotted. If there was no statistical significant difference between the models, a linear regression model was fit to explain the relationship between PC counts and empty basket pathology.
TABLE 1.
Clinical and Pathological Features by Diagnosis
| Characteristics | Controls | ET | SCAs | PD | Significance Across 4 Groups |
|---|---|---|---|---|---|
| N | 25 | 50 | 27 | 25 | NA |
| Age at death (years) | 84.5 ± 10.1 | 86.6 ± 6.3 | 59.1 ± 14.7 | 81.2 ± 3.1 | p < 0.001a |
| [84.0]NA/ns | [87.0]ns/NA | [61.0]***/*** | [80.0]*/*** | ||
| Male gender | 14 (56.0%)NA/ns | 17 (34.0%)ns/NA | 19 (70.4%)ns/** | 21 (84.0%)*/*** | p < 0.001c |
| Brain weight (grams) | 1255.1 ± 205.1NA/ns | 1182.7 ± 143.2ns/NA | 1178.3 ± 189.5ns/ns | 1320.2 ± 126.9ns/** | p = 0.003b |
| [1250.0] | [1167.8] | [1185.0] | [1308.4] | ||
| Braak Alzheimer disease score | 2.5 ± 1.3 | 2.9 ± 1.1 | ND | 2.7 ± 1.1 | p = 0.180a |
| [2.0]NA/ns | [3.0]ns/NA | [3.0]ns/ns | |||
| CERAD | 0.6 ± 0.7 | 0.8 ± 0.9 | ND | 1.1 ± 0.8 | p = 0.172a |
| [1.0]NA/ns | [1.0]ns/NA | [1.0]ns/ns | |||
| Postmortem interval (hours) | 15.5 ± 9.4NA/*** | 29.0 ± 10.0***/NA | 17.7 ± 12.2ns/*** | 18.8 ± 11.2ns/** | p < 0.001b |
| [14.2] | [26.6] | [16.5] | [16.1] | ||
| Purkinje cell counts | 11.2 ± 1.8 | 8.8 ± 1.4 | 5.7 ± 4.1 | 10.3 ± 1.9 | p < 0.001a |
| [11.4]NA/*** | [8.5]***/NA | [4.2]***/*** | [10.1]ns/** | ||
| n-Torpedo counts | 3.5 ± 3.1 | 7.2 ± 6.4 | 16.1 ± 11.6 | 3.8 ± 3.0 | p < 0.001a |
| [2.4]NA/** | [6.2]**/NA | [12.7]***/*** | [3.2]ns/** | ||
| Density of the basket plexus | 1.6 ± 0.5 | 2.0 ± 0.7 | 2.2 ± 0.8 | 1.9 ± 0.6 | p = 0.011a |
| [1.5]NA/** | [2.0]**/NA | [2.5]**/ns | [2.0]*/ns |
Values represent mean ± standard deviation, and median is reported in brackets.
ET, essential tremor; SCA, spinocerebellar ataxia; PD, Parkinson disease; NA, not applicable; ND, no data available on this metric for these brains; ns, not significant.
Kruskal-Wallis test comparing all four groups.
ANOVA with Tukey’s post hoc comparison comparing all four groups.
Chi-square test comparing all 4 groups.
p Values reported using controls as reference/p values reported using essential tremor cases as reference.
p < 0.05 when compared to the reference in Tukey’s post hoc comparisons or in Mann-Whitney test.
p < 0.01 when compared to the reference in Tukey’s post hoc comparisons or in Mann-Whitney test.
p < 0.001 when compared to the reference in Tukey’s post hoc comparisons or in Mann-Whitney test.
RESULTS
We compared all 4 groups (ET, SCAs, PD, and controls) to one another as well as ET, SCA, and PD patients to controls (our reference group). There were differences across groups in terms of age, gender, brain weight, and postmortem interval (Table 1). As expected, SCA patients were the youngest and a large proportion of PD patients were male. We also compared SCA and PD patients to ET patients, a diagnosis of primary interest in our laboratory (Table 1). ET patients and controls had similar ages, brain weight, and Braak Alzheimer disease and CERAD plaque scores (Table 1). ET patients had a longer postmortem interval than controls (p < 0.001, Table 1).
ET patients had significantly lower PC counts (p < 0.001 [LH&E]), higher normalized torpedo counts (p < 0.01 [LH&E]), and higher density of basket plexus rating (p < 0.01 [Bielschowsky]) than controls (Table 1). SCA subjects had significantly lower PC counts (p < 0.001) and higher normalized torpedo counts (p < 0.001) compared to both controls and ET patients (Table 1). PD subjects had higher PC counts (p < 0.01) and lower normalized torpedo counts (p < 0.05) compared to ET patients, and with respect to these metrics they were similar to controls (Table 1).
We used a dual light microscopic immunostaining to clearly identify the PC soma (Calbindin D28K antibody-red alkaline phosphatase chromogen) and highlight the surrounding basket cell plexus (GAD antibody-brown DAB chromogen) (Figs. 1A–H and 2). This enabled us to identify potentially small portions of a PC body in the basket plexus, as well as visualize atrophic PCs that were observed, particularly in SCA patients (Fig. 2G, white arrows in panel and inset). Thus, this procedure increased the accuracy of defining a basket plexus as “empty.” We also identified prominent empty baskets in SCA6 patients with a complex and anastomosing morphology, seen throughout the cerebellar cortex in these subjects (Fig. 2G).
FIGURE 2.

Empty basket pathology revealed by calbindin D28k (CB) and glutamic acid decarboxylase (GAD) dual immunohistochemical staining. (A) CB immunostain identifies the Purkinje cell (PC) soma (red), and the adjacent basket cell plexus is highlighted by GAD immunostain (brown) in control, (B) essential tremor (ET), (C) Parkinson disease (PD), (D) spinocerebellar ataxia (SCA) type 3, (E) SCA1, (F) SCA2, and (G) SCA6 subjects. An atrophic PC highlighted by the white arrow is magnified in the inset. Black arrows, empty baskets. White arrows (G, inset in G), atrophic PC. Scale bar (in G), 50 μm; 20 μm for inset.
Compared to controls, there was a significant increase in the percentage of empty baskets for ET (p < 0.001), all SCAs combined (p < 0.001), and PD (p = 0.010) (Fig. 3A). After separating the SCAs into their specific subtypes, the percentage of empty baskets was again compared to controls (Fig. 3B). SCA3 subjects (p = 0.303) had similar percentage of empty baskets compared to controls, while SCA1 (p < 0.001), SCA2 (p < 0.001), and SCA6 (p < 0.001) subjects all had significantly higher percentages of empty baskets (Table 2; Fig. 3B, which presents SCA groups from lowest to highest value for percentage of empty baskets). Compared to ET patients, SCA3 (p < 0.001) and PD (p = 0.002) subjects as well as controls (p < 0.001) had significantly lower percentages of empty baskets (Table 2). All other disease groups, including SCA1 (p = 0.011), SCA2 (p = 0.003), and SCA6 (p < 0.001), displayed significantly higher percentages of empty baskets than ET patients (Table 2). When the percentage of empty baskets and PC counts were plotted by disease groups, we observed an inverse relationship between the 2 variables (Fig. 4A). Overall, disease groups with higher levels of PC loss resulted in higher percentages of empty basket formation (Fig. 3). In addition, SCA6 patients commonly had highly complex and often anastomosing basket cell processes frequently devoid of detectable PC somas (Fig. 2G).
FIGURE 3.
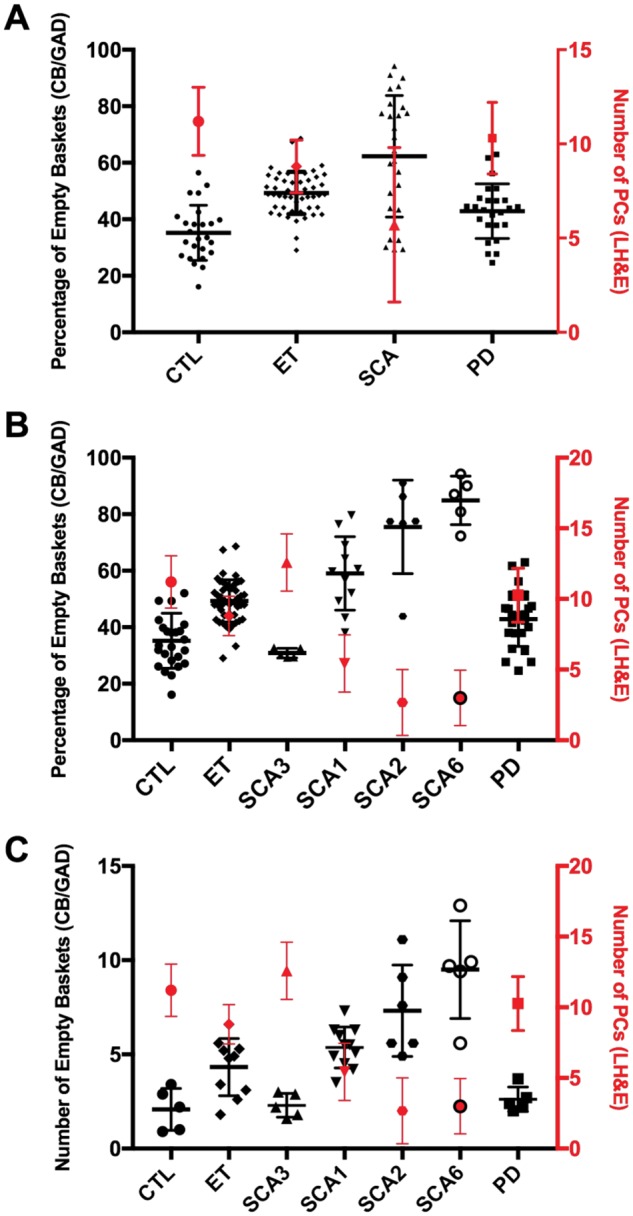
Empty basket and Purkinje cell quantification across motor neurodegenerative diseases. (A) The percentage of empty baskets (black) and number of Purkinje cells (PC) (red) across controls (CTL), essential tremor (ET) patients, all spinocerebellar ataxia (SCA) patients, and Parkinson disease (PD) patients. (B) The percentage of empty baskets (black) and number of PCs (red) across CTL, ET patients, SCAs stratified by type (SCA3, SCA1, SCA2, and SCA6; in order from least to greatest in the percentage of empty baskets), and PD patients. (C) The number of empty baskets (black) and number of PCs (red) across CTL, ET patients, SCAs stratified by type (SCA3, SCA1, SCA2, and SCA6; in order from least to greatest in the percentage of empty baskets), and PD subjects. LH&E, Luxol fast blue/hematoxylin and eosin; CB/GAD, calbindin D28k and glutamic acid decarboxylase.
TABLE 2.
Quantification Measures of Empty Baskets by Diagnosis
| Characteristics | Controls | ET | SCA1 | SCA2 | SCA3 | SCA6 | PD |
|---|---|---|---|---|---|---|---|
| n | 25 | 50 | 11 | 6 | 5 | 5 | 25 |
| Percentage of empty baskets | 35.2 ± 9.8 [33.5] | 49.3 ± 7.5 [48.8] | 59.1 ± 13.0 [59.7] | 75.5 ± 16.5 [77.5] | 30.8 ± 1.7 [30.1] | 84.9 ± 8.6 [87.0] | 42.9 ± 9.7 [43.9] |
| p Valuea | NA/<0.001 | <0.001/NA | <0.001/0.011 | <0.001/0.003 | 0.303/<0.001 | <0.001/<0.001 | 0.010/0.002 |
| N | 5 | 10 | 11 | 6 | 5 | 5 | 5 |
| Number of empty baskets per 200× microscopic field | 2.1 ± 1.1 [2.2] | 4.3 ± 1.5 [4.9] | 5.4 ± 1.1 [5.5] | 7.3 ± 2.4 [6.6] | 2.3 ± 0.6 [2.2] | 9.5 ± 2.6 [9.7] | 2.6 ± 0.7 [2.4] |
| p Valuea | NA/0.023 | 0.023/NA | 0.002/0.130 | 0.006/0.014 | 0.753/0.013 | 0.009/0.004 | 0.465/0.050 |
Values represent mean ± standard deviation, and for variables with a non-normal distribution, median is reported in brackets.
ET, essential tremor; SCA, spinocerebellar ataxia; PD, Parkinson disease; NA, not applicable.
Mann-Whitney test compared to reference group.
p Values reported using controls as the reference group/p values reported using essential tremor cases as the reference group.
FIGURE 4.
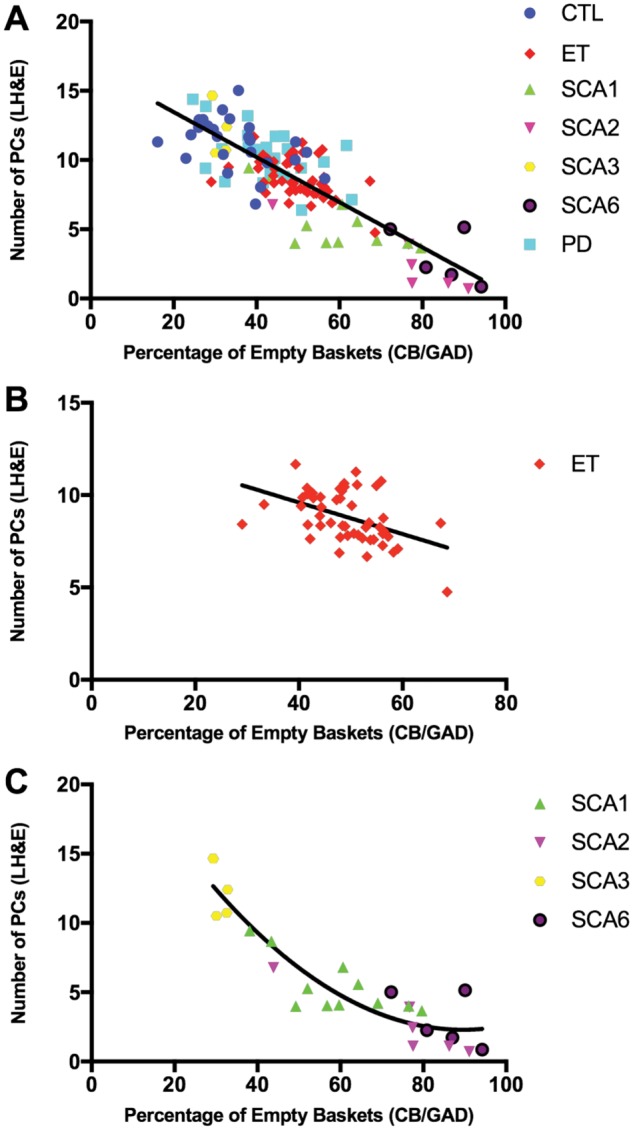
Correlation between the number of Purkinje cells and percentage of empty baskets. (A) The number of Purkinje cells (PC) [Y-axis] versus percentage of empty baskets [X-axis] in 25 normal controls (CTL; blue circle markers), 50 essential tremor patients (ET; red diamond markers), 11 spinocerebellar ataxia type 1 (SCA1; green up arrow makers), 6 SCA2 (purple down arrow markers), 5 SCA3 (yellow hexagon markers), 5 SCA6 (black circle markers), and 25 Parkinson disease (PD; cyan square markers) subjects. Spearman’s rank correlation coefficient is −0.741 with p < 0.001. The fit line is linear (i.e. y = 16.7–0.2 x with adjusted R squared = 0.678). (B) The number of PCs [Y-axis] versus percentage of empty baskets [X-axis] in 50 ET patients (red diamond markers). Spearman’s rank correlation coefficient is −0.443 with p = 0.001. The fit line is linear (i.e. y = 13.0–0.1 x with adjusted R squared = 0.194). (C) The number of PCs [Y-axis] versus percentage of empty baskets [X-axis] in 27 SCA subjects (11 SCA1 [green up arrow makers], 6 SCA2 [purple down arrow markers], 5 SCA3 [yellow hexagon markers], and 5 SCA6 [black circle markers]). Spearman’s rank correlation coefficient is −0.885 with p < 0.001. The fit line is nonlinear quadratic (i.e. y = 25.2–0.5 x–0.003 x2 with adjusted R squared = 0.850). LH&E, Luxol fast blue/hematoxylin and eosin; CB/GAD, calbindin D28k and glutamic acid decarboxylase.
The percentage of empty baskets did not correlate with age at death (Spearman’s r = −0.123; p = 0.170), gender distribution (Spearman’s r = −0.053; p = 0.551), or postmortem interval (Spearman’s r = 0.160; p = 0.086) among all 127 brains (Table 3). The n-torpedo count (Spearman’s r = 0.399; p < 0.001) and density of the basket cell plexus rating (Spearman’s r = 0.370; p < 0.001) positively correlated with the percentage of empty baskets (Table 3). As expected, PC counts (Spearman’s r = −0.741; p < 0.001) strongly and inversely correlated with the percentage of empty baskets (Table 3; Fig. 4A). The degree of this negative correlation varied moving down the fit line and across different disease states, particularly in diseases experiencing severe PC loss (Fig. 4A). The inverse correlation between PC counts and percentage of empty baskets remained significant among ET patients; however, the magnitude of the correlation coefficient decreased (Spearman’s r = −0.443; p = 0.001) (Fig. 4B). Among all SCA subjects, there was a more robust inverse correlation between the 2 variables (Spearman’s r = −0.885; p < 0.001, Fig. 4C). Both linear (adjusted R2 = 0.678, residual sum of squares = 367.5, p < 0.001) and nonlinear quadratic (adjusted R2 = 0.683, residual sum of squares = 359.5, p < 0.001) regression models were fit to characterize the complex association; however, there was no statistically significant difference (p = 0.099) in the regression fitness between the 2 models (Fig. 4A).
TABLE 3.
Correlations of Clinical and Pathological Characteristics With Percentage of Empty Baskets and Number of Empty Baskets
| Percentage of Empty Baskets (n = 127) |
Number of Empty Baskets per 200× Microscopic Field (n = 47) |
||||
|---|---|---|---|---|---|
| rsa | p Value | rsa | p Value | ||
| Age at death (years) | −0.123 | 0.170 | −0.226 | 0.128 | |
| Gender | −0.053 | 0.551 | −0.055 | 0.715 | |
| Brain weight (grams) | −0.170 | 0.061 | −0.328 | 0.028 | |
| Postmortem interval (hours) | 0.160 | 0.086 | −0.221 | 0.154 | |
| Purkinje cell counts | −0.741 | <0.001 | −0.815 | <0.001 | |
| n-Torpedo counts | 0.399 | <0.001 | 0.444 | 0.002 | |
| Density of the basket cell plexus | 0.370 | <0.001 | 0.593 | <0.001 | |
Spearman’s rank correlation coefficient is reported as rs with associated statistical significance reported in p value.
A secondary analysis was performed to better estimate the absolute number of empty baskets per 200× microscopic field. SCA3 (p = 0.753) and PD (p = 0.465) subjects had similar numbers of empty baskets compared to controls (Table 2; Fig. 3C). All the other disease groups, including ET patients (p = 0.023), SCA1 (p = 0.002), SCA2 (p = 0.006), and SCA6 subjects (p = 0.009), had higher numbers of empty baskets than controls (Table 2; Fig. 3C). Compared to ET patients, SCA2 (p = 0.014) and SCA6 (p = 0.004) subjects displayed significantly higher numbers of empty baskets while SCA3 (p = 0.013) and PD (p = 0.050) subjects had lower numbers of empty baskets (Table 2; Fig. 3C). When the number of empty baskets and PC counts were plotted across disease types, the overall trend closely mirrored that of the primary analysis (Fig. 3C).
The number of empty baskets did not correlate with age at death (Spearman’s r = −0.226; p = 0.128), postmortem interval (Spearman’s r = −0.221; p = 0.154), and gender distribution (Spearman’s r = −0.055; p = 0.715) (Table 3). However, it did correlate inversely with brain weight (Spearman’s r = −0.328; p = 0.028) (Table 3). Among pathological features, the number of empty baskets correlated inversely and robustly with PC counts (Spearman’s r = −0.815; p < 0.001), and also correlated with torpedo counts (Spearman’s r = 0.444; p = 0.002), and with the density of the basket cell plexus rating (Spearman’s r = 0.593; p < 0.001) (Table 3). The relationship between PC counts and the number of empty baskets across all diagnoses was best predicted by a nonlinear quadratic model (adjusted R2 = 0.644; residual sum of squares = 230.5; p < 0.001), which displayed a fit that was superior (p = 0.006) to that of a simple linear regression model (adjusted R2 = 0.575; residual sum of squares = 275.1; p < 0.001) (Fig. 5).
FIGURE 5.
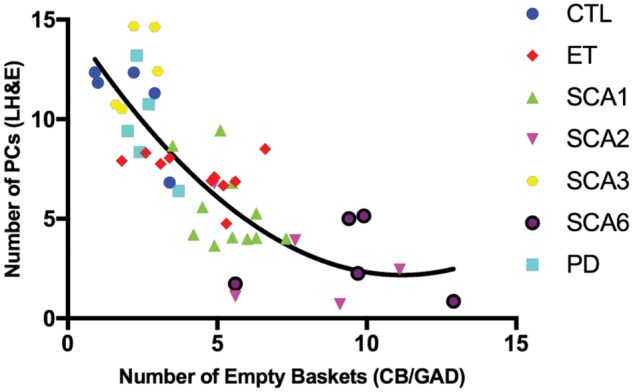
Correlation between the number of Purkinje cells and number of empty baskets. The number of Purkinje cells (PC) [Y-axis] versus number of empty baskets per 200× microscopic field [X-axis]. Correlations in all subjects, including 5 controls (CTL; blue circle markers), 10 essential tremor patients (ET; red diamond markers), 11 spinocerebellar ataxia type 1 (SCA1; green up arrow makers), 6 SCA2 (purple down arrow markers), 5 SCA3 (yellow hexagon markers), 5 SCA6 (black circle markers) and 5 Parkinson disease (PD; cyan square markers) subjects. Spearman’s rank correlation coefficient is −0.815 with p < 0.001. The fit line is nonlinear quadratic (i.e. y = 15.0–2.3 x–0.1 x2 with adjusted R squared = 0.644). LH&E, Luxol fast blue/hematoxylin and eosin; CB/GAD, calbindin D28k and glutamic acid decarboxylase.
DISCUSSION
Recent postmortem studies of ET brains have identified a range of pathological features that are centered on the PC population in the cerebellar cortex (2–6, 8, 12, 14, 21, 32). These morphological changes indicate that these PCs are experiencing stress, and the maintenance of neuronal function is challenged (21). PC dysfunction initiates either compensatory or degenerative processes in the cerebellum that also involves the neighboring basket cell interneuron population (35, 36). Specifically, a hypertrophy of basket cell axonal processes and changes in the basket cell-PC interface have been observed in ET (8, 14). In this study, we quantified basket cell plexuses lacking a PC soma in ET and control brains, and evaluated this change in the broader spectrum of motor neurodegenerative diseases, including SCAs and PD.
The percentage of empty baskets in ET patients was 1.5 times greater than that of controls. There was no significant change in the percentage of empty baskets in SCA3 subjects compared to controls. This may be due to the relative sparing of PCs in the SCA3 cerebellar cortex compared to that of SCA types 1, 2, and 6 (37, 38). The percentage of empty baskets in SCA types 1, 2, and 6 were 1.8, 2.3, and 2.6 times that of controls, respectively. The neurodegenerative processes of these SCA types cause a marked and widespread neuronal loss in the cerebellar PC layer (39). We also identified an excessively complex and anastomosing morphology of the basket plexus in SCA6 patients, demonstrating persistence of the basket plexus despite marked PC degeneration. This may reflect the often relatively late onset of disease and slow disease progression, and predominance of neuropathologic changes affecting PCs in this disorder (36, 37). Compared to controls, there was a mild but significant increase in the percentage of empty baskets in PD subjects. The role of the cerebellum in PD has been debated. Both pathological effects from dopamine neuronal loss and compensatory changes in response to tremor modulating activities in the cerebellum have been reported (40, 41).
The increase in the percentage of empty baskets observed between ET patients and controls was less marked than that observed between SCA types 1, 2, 6, and controls. In this sense, as well as others, ET seems to represent a milder form of cerebellar degeneration, with an intermediate degree of PC loss (4). Basket cell pathology has yet to be extensively quantified in human SCAs, except for one recent study demonstrating increased basket cell plexus density in both human SCA1 subjects and a mouse model for this disease, supporting a pathogenic role for increased GABAergic inhibition in disrupting PC function (36). The strong correlation between the percentage of empty baskets, PC loss, torpedo counts, and basket cell plexus density that we demonstrated in this study suggests that they are all part of the same pathophysiological cascade (i.e. individuals with more torpedoes have also greater PC loss, a greater percentage of empty baskets, and a compensatory increase in the density of the basket cell plexus).
This study should be interpreted within the context of several limitations. First, although this study was large, with a total sample of 127 brains, the number of available brains for each SCA type was small. Nevertheless, given the size of expected differences, our power analysis confirmed that the number of brains analyzed would be more than sufficient to test our hypothesis, and indeed we were able to demonstrate significant differences in basket cell pathology between SCA types 1, 2, 6, and controls. Second, the age at death of SCAs and PD subjects was lower than that of the age-matched ET patients and controls. However, the age difference could not have confounded the analyses as we demonstrated that the empty basket pathology did not correlate with age at death. Third, the analysis was restricted to one region in the cerebellum, and future analyses examining other regions of the cerebellum are warranted. Fourth, with atrophy in the setting of marked PC loss, for our secondary analysis (i.e. counting the absolute number of empty baskets per 200× microscopic field), it is conceivable that we overestimated our counts in diseases with more atrophy due to PC loss (e.g. SCA 1, 2, and 6). However, for our primary analysis (i.e. counting the percentage of empty baskets), this would not have been an issue because our primary measure was a percentage rather than an absolute number. Fifth, our study focused on an anatomical finding, the empty basket, and we are limited in terms of the physiological inferences we can make based on our data. Sixth, our findings are research findings and their diagnostic import is uncertain at this time, particularly given the overlap between diagnostic groups in terms of the degree of empty basket pathology. This study had numerous strengths. First, the research question we addressed was unique and has not been examined before. Hence, this was the first study to quantify the empty basket pathology in ET patients. Second, we provided a comparative analysis with other neurodegenerative disorders of the cerebellum as well as another degenerative motor disorder, thereby placing our findings within the larger context of such diseases. Empty baskets have been previously noted in human SCA patients, although, to our knowledge, no studies have quantified this change or examined its correlation with other cerebellar pathologies. Third, based on age, we carefully matched our ET patients to controls.
In summary, the quantification of empty baskets in ET provides an indirect and alternative method of quantifying PC loss in that disease. Here, we demonstrated that the percentage of empty baskets in ET patients was 1.5 times greater than that of age-matched controls. Although significant, this difference was less marked than that observed between SCA types 1, 2, 6, and controls. The notion that ET could be neurodegenerative is controversial; however, these data provide support for PC loss in ET and are consistent with the notion that ET could represent a mild form of cerebellar degeneration with an intermediate degree of PC loss (4). Furthermore, the presence of empty baskets indicates that the basket cell plexus persists even after the loss of PCs, demonstrating that basket cell neuronal processes are more resistant to degeneration. This is particularly evident in SCA types 1, 2, and 6 patients, where both the percent and number of empty baskets are markedly increased. The robust correlation between the percentage of empty baskets, PC loss, torpedo counts, and basket cell plexus density suggests that they are all part of the same pathophysiological cascade; thus, individuals with more torpedoes have also greater PC loss, a greater percentage of empty baskets, and a compensatory increase in the density of the basket cell plexus.
ACKNOWLEDGMENTS
We thank the National Institutes of Health NeuroBioBank (University of Miami, Miami, FL and University of Maryland, Baltimore, MD) for providing human control, SCA2, and SCA3 tissue.
Elan D. Louis has received research support from the National Institutes of Health: NINDS R01 NS086736 (principal investigator) and NINDS R01 NS088257 (principal investigator). Phyllis L. Faust has received funding from the National Institutes of Health: NINDS R21 NS077094 and NINDS R01 NS088257 (principal investigator). Arnulf H. Koeppen has received funding from the National Ataxia Foundation to expand and maintain the ataxia tissue repository at the Albany Veterans Affairs Medical Center.
The authors have no duality or conflicts of interest to declare.
REFERENCES
- 1. Louis ED, Ferreira JJ.. How common is the most common adult movement disorder? Update on the worldwide prevalence of essential tremor. Mov Disord 2010;25:534–41 [DOI] [PubMed] [Google Scholar]
- 2. Yu M, Ma K, Faust PL, et al. Increased number of Purkinje cell dendritic swellings in essential tremor. Eur J Neurol 2012;19:625–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Louis ED, Lee M, Babij R, et al. Reduced Purkinje cell dendritic arborization and loss of dendritic spines in essential tremor. Brain 2014;137:3142–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Louis ED, Kuo SH, Tate WJ, et al. Heterotopic Purkinje cells: A comparative postmortem study of essential tremor and spinocerebellar ataxias 1, 2, 3, and 6. Cerebellum 2018;17:104–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Louis ED, Faust PL, Vonsattel JP, et al. Neuropathological changes in essential tremor: 33 cases compared with 21 controls. Brain 2007;130:3297–307 [DOI] [PubMed] [Google Scholar]
- 6. Louis ED. Linking essential tremor to the cerebellum: Neuropathological evidence. Cerebellum 2016;15:235–42 [DOI] [PubMed] [Google Scholar]
- 7. Kuo SH, Lin CY, Wang J, et al. Climbing fiber-Purkinje cell synaptic pathology in tremor and cerebellar degenerative diseases. Acta Neuropathol 2017;133:121–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Erickson-Davis CR, Faust PL, Vonsattel JP, et al. “Hairy baskets” associated with degenerative Purkinje cell changes in essential tremor. J Neuropathol Exp Neurol 2010;69:262–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Symanski C, Shill HA, Dugger B, et al. Essential tremor is not associated with cerebellar Purkinje cell loss. Mov Disord 2014;29:496–500 [DOI] [PubMed] [Google Scholar]
- 10. Rajput AH, Robinson CA, Rajput ML, et al. Essential tremor is not dependent upon cerebellar Purkinje cell loss. Parkinsonism Relat Disord 2012;18:626–8 [DOI] [PubMed] [Google Scholar]
- 11. Louis ED, Babij R, Lee M, et al. Quantification of cerebellar hemispheric Purkinje cell linear density: 32 ET cases versus 16 controls. Mov Disord 2013;28:1854–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Choe M, Cortes E, Vonsattel JP, et al. Purkinje cell loss in essential tremor: Random sampling quantification and nearest neighbor analysis. Mov Disord 2016;31:393–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sotelo C. Development of “Pinceaux” formations and dendritic translocation of climbing fibers during the acquisition of the balance between glutamatergic and gamma-aminobutyric acidergic inputs in developing Purkinje cells. J Comp Neurol 2008;506:240–62 [DOI] [PubMed] [Google Scholar]
- 14. Kuo SH, Tang G, Louis ED, et al. Lingo-1 expression is increased in essential tremor cerebellum and is present in the basket cell pinceau. Acta Neuropathol 2013;125:879–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Buttermore ED, Piochon C, Wallace ML, et al. Pinceau organization in the cerebellum requires distinct functions of neurofascin in Purkinje and basket neurons during postnatal development. J Neurosci 2012;32:4724–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Donato R, Rodrigues RJ, Takahashi M, et al. GABA release by basket cells onto Purkinje cells, in rat cerebellar slices, is directly controlled by presynaptic purinergic receptors, modulating Ca2+ influx. Cell Calcium 2008;44:521–32 [DOI] [PubMed] [Google Scholar]
- 17. Korn H, Axelrad H.. Electrical inhibition of Purkinje cells in the cerebellum of the rat. Proc Natl Acad Sci USA 1980;77:6244–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lennon W, Hecht-Nielsen R, Yamazaki T.. A spiking network model of cerebellar Purkinje cells and molecular layer interneurons exhibiting irregular firing. Front Comput Neurosci 2014;8:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palay SL, Chan-Palay V.. Cerebellar Cortex: Cytology and Organization. Berlin: Springer; 1974 [Google Scholar]
- 20. Koeppen AH. The pathogenesis of spinocerebellar ataxia. Cerebellum 2005;4:62–73 [DOI] [PubMed] [Google Scholar]
- 21. Babij R, Lee M, Cortes E, et al. Purkinje cell axonal anatomy: Quantifying morphometric changes in essential tremor versus control brains. Brain 2013;136:3051–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Louis ED, Zheng W, Applegate L, et al. Blood harmane concentrations and dietary protein consumption in essential tremor. Neurology 2005;65:391–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Louis ED, Asabere N, Agnew A, et al. Rest tremor in advanced essential tremor: A post-mortem study of nine cases. J Neurol Neurosurg Psychiatry 2011;82:261–5 [DOI] [PubMed] [Google Scholar]
- 24. Louis ED, Ottman R, Ford B, et al. The Washington Heights-inwood genetic study of essential tremor: methodologic issues in essential-tremor research. Neuroepidemiology 1997;16:124–33 [DOI] [PubMed] [Google Scholar]
- 25. Louis ED, Kuo SH, Wang J, et al. Cerebellar pathology in familial vs sporadic essential tremor. Cerebellum 2017;16:786–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112:389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mirra SS. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer's disease: A commentary. Neurobiol Aging 1997;18:S91–S4 [DOI] [PubMed] [Google Scholar]
- 28. Braak H, Braak E.. Diagnostic criteria for neuropathologic assessment of Alzheimer's disease. Neurobiol Aging 1997;18:S85–S8 [DOI] [PubMed] [Google Scholar]
- 29. Kuo SH, Wang J, Tate WJ, et al. Cerebellar pathology in early onset and late onset essential tremor. Cerebellum 2017;16:473–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stoodley CJ, Schmahmann JD.. Evidence for topographic organization in the cerebellum of motor control versus cognitive and affective processing. Cortex 2010;46:831–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stoodley CJ, Schmahmann JD.. Functional topography in the human cerebellum: A meta-analysis of neuroimaging studies. Neuroimage 2009;44:489–501 [DOI] [PubMed] [Google Scholar]
- 32. Axelrad JE, Louis ED, Honig LS, et al. Reduced Purkinje cell number in essential tremor: A postmortem study. Arch Neurol 2008;65:101–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pagano M, Gauvreau K.. Principles of Biostatistics, 2nd edn Pacific Grove, CA: Duxbury; 2000 [Google Scholar]
- 34. Faraway J. Linear Models with R. Boca Raton, FL: Chapman & Hall/CRC; 2014 [Google Scholar]
- 35. Rossi F, Jankovski A, Sotelo C.. Target neuron controls the integrity of afferent axon phenotype: A study on the Purkinje cell-climbing fiber system in cerebellar mutant mice. J Neurosci 1995;15:2040–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Edamakanti CR, Do J, Didonna A, et al. Mutant ataxin1 disrupts cerebellar development in spinocerebellar ataxia type 1. J Clin Invest 2018;128:2252–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paulson HL, Shakkottai VG, Clark HB, et al. Polyglutamine spinocerebellar ataxias—From genes to potential treatments. Nat Rev Neurosci 2017;18:613–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koeppen AH, Ramirez RL, Bjork ST, et al. The reciprocal cerebellar circuitry in human hereditary ataxia. Cerebellum 2013;12:493–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rub U, Schols L, Paulson H, et al. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol 2013;104:38–66 [DOI] [PubMed] [Google Scholar]
- 40. Wu T, Hallett M.. The cerebellum in Parkinson's disease. Brain 2013;136:696–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Helmich RC, Toni I, Deuschl G, et al. The pathophysiology of essential tremor and Parkinson's tremor. Curr Neurol Neurosci Rep 2013;13:378. [DOI] [PubMed] [Google Scholar]