

Abstract
There are reports that astrocyte perivascular endfeet are damaged in some cases of multiple sclerosis (MS). This study was designed to determine the origin and outcome of astrocyte damage in acute, resolving, and inactive plaques. Ten acute plaques from 10 early MS cases and 14 plaques of differing histological age from 9 subacute and chronic cases were examined immunohistochemically. Also examined were nonnecrotic early lesions in 3 patients with neuromyelitis optica (NMO). Plaques from 3 MS cases were examined electron microscopically. The edge zones in each of the 10 acute MS lesions revealed a complete loss of astrocyte cell bodies and their pericapillary, perineuronal, and perivascular foot processes. Dendrophagocytosis of degenerate astrocytes was observed. Astrocyte precursors, similar to those that replace destroyed astrocytes in nonnecrotic NMO lesions, were present in areas depleted of astrocytes. Resolving plaques were repopulated initially by stellate astrocytes that stained negatively for the water channel molecule aquaporin4 (AQP4). In older lesions, astrocytes were predominantly AQP4-positive. Loss and recovery of astrocytes in new MS lesions may be as important as myelin loss as a cause of conduction block responsible for symptoms in patients with relapsing and remitting and secondary progressive MS.
Keywords: Astrocytes, Aquaporin4, Blood-brain barrier, Conduction block, KIR4.1, Neuromyelitis optica, Secondary progressive MS
INTRODUCTION
Multiple sclerosis (MS) is a disease characterized in the beginning by discrete clinical attacks—relapses—that are abrupt in onset and that remit, often completely, over the course of a few weeks, each clinical episode the result of a new, sharply circumscribed perivascular or plaque of myelin loss. The conduction block responsible for the onset of symptoms is attributed to a loss of myelin from still intact axons, the return in conduction and remission of symptoms to remyelination or the establishment of conduction in demyelinated axons.
That there might be a clinically significant astrocyte lesion in MS was raised by a report by Lennon et al of a serum antibody directed against aquaporin4 (AQP4) in patients with neuromyelitis optica (NMO), a perivascular inflammatory demyelinating disease traditionally regarded as a form of MS (1). AQP4 is a pore-forming integral membrane protein located in astrocyte endfeet that mediates water flow and the ionic environment within the central nervous system (CNS). Lennon’s study led to the discovery of an NMO lesion unique in human neuropathology, namely, perivascular demyelination where myelin loss from axons is preceded by a loss of staining of the tissue for AQP4 (2, 3).
The possibility that there may exist a similar astrocyte lesion in MS has been the subject of a number of investigations, in most instances with no evidence reported of any sort of astrocyte lesion in early or late MS lesions (2, 4–10). Contrary to these reports, however, in our own study comparing early nonnecrotic NMO lesions and early MS lesions (11), we reported immunohistochemical and electron microscopical evidence of damage to astrocyte perivascular endfeet in early MS lesions as well as the presence in some MS lesions of glial fibrillary acidic protein (GFAP)-positive astrocytes that are unreactive for AQP4, a type of astrocyte considered to be the key pathological feature of early NMO lesions. Sharma et al (12) and Brosnan and Raine (13) also report electron microscopical and immunohistochemical evidence of damage to astrocyte perivascular endfeet in MS, and Kira et al noted the presence of GFAP-positive AQP4-negative stellate astrocytes in some MS lesions and in lesions in the Balo form of the disease (14).
The present study was designed to obtain a more complete picture of the pathology and fine structural pathology of astrocyte endfeet in early MS lesions, and whether such changes in astrocyte endfeet persist in remyelinating and other inactive chronic MS lesions.
MATERIALS AND METHODS
Design
Astrocyte foot processes in human autopsy tissue appear as spaces containing AQP4-positive membranous remnants that correspond to ruptured astrocyte endfeet shown by electron microscopy to be expanded electron-lucent processes of perivascular and perineuronal astrocytes (15). For purposes of the present study, astrocytes and astrocyte foot processes in 10 newly forming acute plaques from 10 autopsy cases of early MS and a series of MS lesions of increasing histological age in a second group of MS patients were examined using routinely stained sections and GFAP and AQP4 immunohistochemistry. Following the discovery during the course of the study of astrocyte precursors in newly forming MS plaques, an antibody was added that recognizes GFAPδ, a novel GFAP isoform expressed during maturation of precursor cells into astrocytes in the developing and adult human brain (16).
Astrocyte foot processes were examined by electron microscopy in 3 other cases. Blood-brain barrier function was assessed in sections immunostained for IgM, and the presence of inflammatory cells examined in sections stained for CD45RO-positive memory and effector T cells. Developing lesions in 3 cases of NMO were examined immunohistochemically.
Patient Selection
Between 1969 and 2009 neuropathologists in the US, Australia, Canada, New Zealand, and the United Kingdom were contacted for archival paraffin-embedded autopsy tissue obtained from patients with acute forms of MS who died shortly after the onset of the illness. The resulting pool of 40 cases of early fatal MS and 16 cases of NMO (11) provided the 18 cases of MS and 3 NMO cases selected for the present immunohistochemical investigation. The clinical features of the 3 typical cases of NMO are described elsewhere (Cases 3, 5, and 12 in Reference [11]). Ten early newly forming plaques were obtained from 10 MS cases—6 female and 4 male subjects aged 14–70, duration of the fatal illness 17 hours to 8 months (median 18 days; Table 1). Fourteen plaques of increasing histological age were obtained from a second series of MS cases—5 female and 4 male subjects age 23–55 years, disease duration 29 days to 13 years, median 3 years (Table 2). None of the MS or NMO cases was examined for AQP4 autoantibodies. The study complies with the requirements of the Human Ethics Committee of The University of Sydney.
TABLE 1.
The 10 Acute MS Plaques
| Case | Plaque | Sex/Age | Duration of Illness/Terminal Illness | Site | Neuropathologist | Institution |
|---|---|---|---|---|---|---|
| 1 | 1-1 | F 31 | 3 months/7 days | Brainstem | C.W.M. Adams | Guy’s Hospital Medical School, London, UK |
| 2 | 2-1 | F 20 | 2 weeks | Periventricular | C.W.M. Adams | Guy’s Hospital Medical School, London, UK |
| 3 | 3-1 | M 25 | 29 days | Cerebral hemisphere | S. Pogacar | Brown University Medical School, Providence, RI, |
| 4 | 4-1 | F 42 (40) | 2.5 months/2 weeks | Cerebellum | R.D. Terry | Albert Einstein College of Medicine, The Bronx, NY |
| 5 | 5-1 | M 14 | 18 days | Cerebral hemisphere | J. McLaughlin | Royal Free Hospital, Hampstead, London, UK |
| 6 | 6-1 | F 70 | 21 days | Cerebral hemisphere | S. Love | The National Hospital, Queen Square, London, UK |
| 7 | 7-1 | M 32 | 10 months/8 months | Periventricular | R. Doshi | The Maudsley Institute of Psychiatry, London, UK |
| 8 | 8-1 | F 48 | 18 days | Cerebral hemisphere | C.G. Harper | Royal Perth Hospital, Perth, WA, Australia |
| 9 | 9-1 | F 14 | 9 months/17 hours | Medulla | G.N. Budzilovich | New York University Medical Center, NY |
| 10 | 10-1 | M 36 | 3 years/not known | Medulla | D.M. Boehme | VA Hospital, East Orange, NJ |
Remyelinating lesions in the same section or elsewhere: Cases 1, 3, 4, 9, 10. Received corticosteroids: Cases 3, 7, 9, 10. Received azathioprine: Case 3. Prominent concentric bands: Case 8. Neurogenic pulmonary edema: Cases 9, 10. Herniation: Cases 3, 8.
TABLE 2.
Astrocytes in MS Plaques of Increasing Histological Age
| Case | Plaque | Sex/Age | Disease Duration | Lipid Macrophages | AQP4-positive Astrocytes | GFAP-positive Astrocytes |
|---|---|---|---|---|---|---|
| 11 | 11-1 | F 23 | 5 weeks | +++ | o | ± Present but rare |
| 12 | 12-1 | F 23 | 30 days/60 hoursa | +++ | o | ± Present but rare |
| 13 | 13-1 | F 27 | 32 months | +++ | o | o |
| 3 | 3-2 | M 25 | 29 days | ++ | ± | + |
| 3 | 3-3 | M 25 | 29 days | ++ | ± | +–++ |
| 14 | 14-1 | F 29 | 12 months | ++ | ± | +–++ |
| 13 | 13-2 | F 27 | 32 months | ++ | + | +–++ |
| 15 | 15-1 | F 27 | 49 months | ++ | + | + |
| 16 | 16-1 | M 34 | 3 years 11 months | ++ | + | +–++ |
| 17 | 17-1 | M 43 | 13 years | ++ | + | +–++ |
| 15 | 15-2 | F 27 | 49 months | ± | ++ | ++ |
| 10 | 10-2 | M 36 | 3 years | ± | ++ | ++ |
| 18 | 18-1 | M 55 | “Long standing” | ± | ++ | ++ |
| 18 | 18-2 | M55 | “Long standing” | ± | ++ | ++ |
Cell counts in demyelinated edge zones: o, none; ±, few or none; +, small numbers; ++, moderate numbers; +++, large numbers. Case 12, the third patient in the present study with fatal neurogenic pulmonary edema, died 60 hours after the onset of a third relapse. No corticosteroids or other immunosuppressive medication received.
Although clinically atypical, the autopsy findings in each of the 10 acute MS cases were definitive. First, disseminated focal demyelinating lesions with characteristic morphology were present in each case, with lesions of different histological ages in 6 of the 10. Second, there was no evidence of any other multifocal demyelinating disease such as acute disseminated encephalomyelitis or NMO (17–19). Regarding NMO, this diagnosis was excluded based on the absence in any of the 18 MS cases in the study of nonnecrotic still-myelinated AQP4-negative disseminated focal lesions.
Plaque Selection
Ten acute plaques judged to be exceptionally early lesions were selected based on the presence of changes in surrounding tissues indicating that the area of myelin loss was expanding at the time of death, an event usually lasting probably no more than a few days. Such evidence consists of the presence of “prephagocytic” changes in normally myelinated tissue bordering areas of active demyelination (partially demyelinated tissue infiltrated by macrophages containing LFB-positive particles of myelin) thought to presage impending extension of myelin loss into surrounding tissues. Previously identified “prephagocytic” indications of imminent myelin breakdown include early myelin ball formation associated with the presence of IgG-positive microglia/macrophages, the presence of oligodendrocytes with pyknotic (“apoptotic”) nuclei, oligodendrocyte loss, swollen oligodendrocytes immunoreactive for activated complement, myelin sheaths immunoreactive for activated complement, intramyelinic edema, and loss of myelin-associated glycoprotein immunoreactivity (20–30) (Supplementary Data Figs. S1–S4).
Immunohistochemistry
Areas of different histological ages examined immunohistochemically for astrocyte changes were defined as follows: Myelinated prephagocytic areas bordering bands or patches of active demyelination, “immediate” postphagocytic zones (LFB-positive macrophages in completely demyelinated tissue), postphagocytic zones (LFB-negative lipid macrophages in demyelinated tissue), and “late” postphagocytic areas (lipid macrophages numerous but located chiefly in perivascular spaces) (Supplementary DataFig. S4).
Paraffin sections were stained for myelin (Luxol fast blue-periodic acid-Schiff [LFB-PAS], axons [Bodian’s silver stain], and using hematoxylin and eosin [H&E]). LFB-PAS also stains eosinophils and was used for this purpose in the examination of NMO lesions. To correlate astrocyte pathology and macrophage activity, paraffin sections were stained for myelin using LFB-PAS followed by an astrocyte immunohistochemical stain.
Paraffin sections were stained immunohistochemically using antibodies and procedures listed in Table 3 and as described before (11, 28, 31). Primary antibodies comprised GFAP (clone GF12-24, Chemicon, Boronia, Australia), GFAP polyclonal (DakoCytomation, Carpinteria, CA), GFAPδ polyclonal (EMD Millipore Corp, Temecula, CA), AQP4 polyclonal (Millipore Corp, Billerica, MA), S-100 polyclonal (Abcam, Cambridge, UK), IgM (DakoCytomation), CD45RO (clone UCHL1, Novocastra, Newcastle-Upon-Tyne, UK). Following incubation with the primary antibody, sections were treated with a biotinylated or polymer-based horseradish peroxidase labelled second antibody (Vector ABC Elite Kit, Vector Laboratories, Burlingame, CA; Dako EnVision+ and LSAB+ Kits, DakoCytomation) with diaminobenzidine as the chromogen. Staining was enhanced by reapplying the primary and second antibody layers (32). Harris’s hematoxylin was used as a nuclear counterstain.
TABLE 3.
Primary Antibodies
| Antigen | Clone | Dilution | Antigen Retrieval | Source |
|---|---|---|---|---|
| GFAP | GF12-24 | 1:1000 | Microwave/citrate | Chemicon Australia, Boronia, Australia |
| GFAP | Polyclonal | 1:2500 | Microwave/citrate | DakoCytomation Inc., Carpinteria, CA |
| GFAP delta | Polyclonal | 1:1000 | Microwave/citrate | EMD Millipore Corporation, Temecula, CA (Cat. AB9598) |
| AQP4 | Polyclonal | 1:500–1000 | Microwave/citrate | Millipore Corp., Billerica, MA |
| S-100 | Polyclonal | 1:500 | Microwave/citrate | Abcam, Cambridge, UK |
| IgM | Polyclonal | 1:8000 | Proteinase K | DakoCytomation Inc. |
| CD45RO | UCHL1 | 1:150 | Microwave/citrate | Novocastra, Newcastle-Upon-Tyne, UK |
Microwave/citrate, microwave antigen retrieval in a citrate buffer pH6.
Extensive use was made of immunostained sections prepared during previous immunohistochemical studies of most of the same plaques examined in the present study. These included sections immunostained for IgG and other serum proteins, kappa and lambda light chains, MRP-14 (monocyte marker), HAM56 (mature human macrophages), CD4, CD8, LCA (CD45), UCHL1 (CD45RO—activated and memory T cells), CD209 (dendritic cells), MHC2, myelin proteins, CNP (oligodendrocyte/myelin marker), HNK-1 (immature oligodendrocytes and type 2 astrocytes) using colloidal gold-silver staining (Goldmark Biologicals, Phillipsburg, NJ), activated caspase-3, GFAP, AQP4, RCA-1 lectin (endothelial cells and macrophages), and complement proteins (C3d, monoclonal B7 antiMAC 5b-9).
Electron Microscopy
Electron micrographs from previous EM studies of a number of plaques from 3 MS patients were reviewed. In one autopsy case, the plaques examined were chronic plaques with few or no lipid macrophages (33). A second autopsy case provided 2 hypercellular postphagocytic plaques with numerous lipid macrophages (33). A biopsy specimen of a plaque with numerous lipid macrophages and numerous nonmyelinating oligodendrocytes described previously by Raine and Scheinberg was from the third case (34).
RESULTS
General Description of the 10 Acute Plaques
In 4 patients the 4 acute lesions as well as other lesions available for study were early actively demyelinating lesions of the same histological age (Cases 2, 5, 6, 8). Six patients had lesions of different histological ages as well as the 6 acute lesions. Remyelinating shadow plaques were present adjacent to the acute lesion or in other blocks in Cases 1, 3, 4, 9, and 10. One of the 10 acute plaques had several concentric bands of myelin loss (Case 8). Neurogenic pulmonary edema was present in 2 cases (Cases 9 and 10). Most of the 10 acute plaques had postphagocytic areas with numerous LFB-PAS-negative lipid macrophages together with small to moderate numbers of GFAP-positive stellate astrocytes and HNK-I—positive oligodendrocytes. No case had necrotic spinal cord lesions. Two of the 10 cases had autopsy evidence of herniation (Cases 3 and 8). Four cases were receiving hydrocortisone at the time of death (Cases 3, 7, 9, 10) and one azathioprine (Case 3). CD45RO T cells, in small perivascular cuffs and in the parenchyma, were present in all 10 acute plaques (Fig. 1; Supplementary Data Figs. S1–S3).
FIGURE 1.
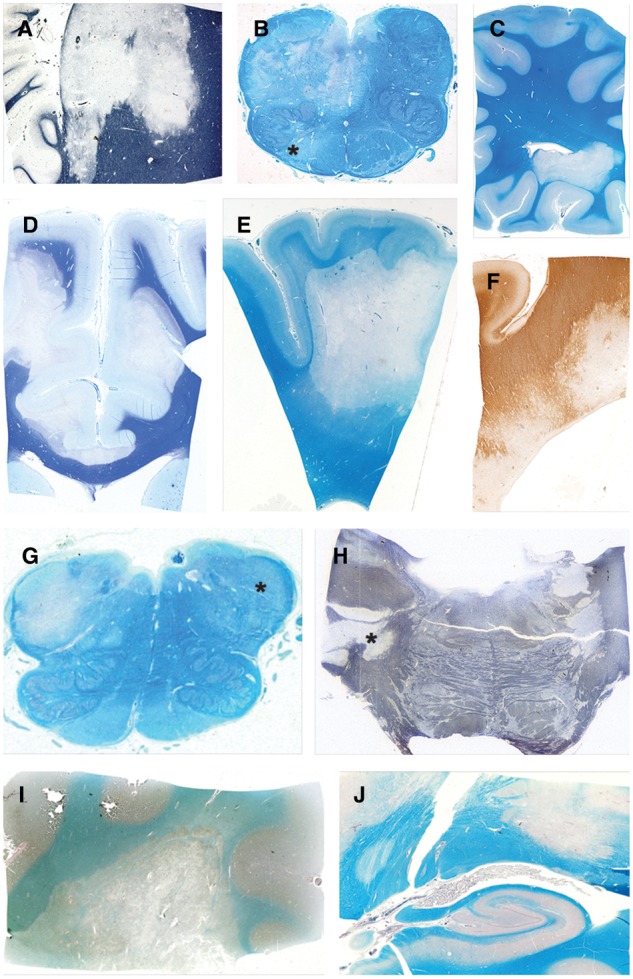
Myelin-stained sections of the 10 acute plaques. (A) Case 4, plaque 1. (B) Case 9, plaque 1. (C) Case 2, plaque 1. (D) Case 5, plaque 1. (E) Case 6, plaque 1. (F) Case 7, plaque 1. (G) Case 10, plaque 1. (H) Case 1, plaque 1. (I) Case 8, plaque 1. (J) Case 3, plaque 1. Asterisks indicate remyelinating shadow plaques. (A, D, H: LFB; B, C, E, G, I, J: LFB-PAS; F: myelin oligodendrocyte glycoprotein [MOG]).
Oligodendrocytes
Oligodendrocytes with pyknotic nuclei were detected in normally myelinated tissue bordering areas of active demyelination in 7 of the 10 acute lesions (the exceptions plaques in Cases 5, 6, and 8). The cytoplasm appeared compacted, with affected cells lacking the halo normally present in oligodendrocytes examined in routinely obtained autopsy tissue. Small microglia with activated morphology were frequent accompaniments. Phagocytosis of degenerate (pyknotic) oligodendrocytes in intact tissue was observed in Case 9-plaque 1 and in an acute plaque in Case 4. Prephagocytic deposition of complement on myelin was observed in tissue bordering 4 plaques (Cases 2, 3, 5, and 10) (Supplementary Data Figs. S1–S5).
In areas of active demyelination all 10 acute lesions had reduced numbers of oligodendrocytes, while in postphagocytic areas, oligodendrocytes were relatively numerous. Oligodendrocytes in demyelinated postphagocytic tissue stained positively for the antigen HNK-1. Oligodendrocytes in surrounding tissues were unreactive for HNK-1. In the few instances where degenerate oligodendrocyte cell bodies were observed, the cell body was swollen, with a pyknotic or more often a pale, poorly defined nucleus. Swollen oligodendrocytes immunoreactive for complement were detected in one case.
Astrocytes in Bordering and Remote Tissues
Numerous AQP4-positive GFAP-positive S100-positive reactive stellate astrocytes with typical morphology were present in myelinated tissue bordering each of the 10 acute lesions. Swollen astrocyte feet ensheathing capillaries, perivascular spaces and cell bodies of neurons stained positively for AQP4 and—rather less often—for GFAP. Protoplasmic astrocytes in the cerebral cortex usually stained strongly for AQP4.
FIGURE 2.

Reactive astrocytes and astrocyte endfeet in intact tissue. (A, B) GFAP-positive AQP4-positive reactive stellate astrocytes in intact myelinated tissue bordering an actively demyelinating plaque. (B) The wall of a capillary stains positively for AQP4. The spaces around oligodendrocytes (“halos”) are a postmortem artefact. (C) The space around the vessel is a common post mortem artefact. It lies outside the true perivascular space and is formed by grossly swollen, partly ruptured, astrocyte foot processes, leaving AQP4-positive remnants in the space. (D) A vessel located in a postphagocytic plaque shows an absence of GFAP-positive astrocyte foot processes within the artefactual perivascular space. (E) Perfusion-fixed rat central nervous system tissue. The tissue is compact with no spaces around neurons or other cells. The only “spaces” visible are capillary lumens. (F) Intact periplaque tissue. Perineuronal spaces, another autolytic artefact formed by swollen ruptured astrocyte foot processes, are lined with AQP4-positive membranes. (A: GFAP, ×250; B: AQP4, ×250; C: AQP4, ×125; D: GFAP, ×250; E: 1-µm-thick epoxy section, toluidine blue, ×375; F: AQP4, ×250).
Astrocytes in Areas of Active Demyelination
In sections stained for AQP4, there was a complete absence of AQP4 immunoreactivity towards the edge of the lesion. This did not extend into bordering myelinated tissue as seen in early NMO lesions. In sections stained for GFAP, there was a complete or almost complete absence of GFAP-positive stellate astrocytes in and adjacent to the zone of active demyelination. At greater magnifications, astrocytes, by all staining methods including H&E and LFB-PAS, were absent or present in very small numbers in such areas. Also absent were AQP4-positive pericapillary, perineuronal, and perivascular space astrocyte feet (Figs. 3–6A; Supplementary DataFigs. S6 and S7).
FIGURE 3.
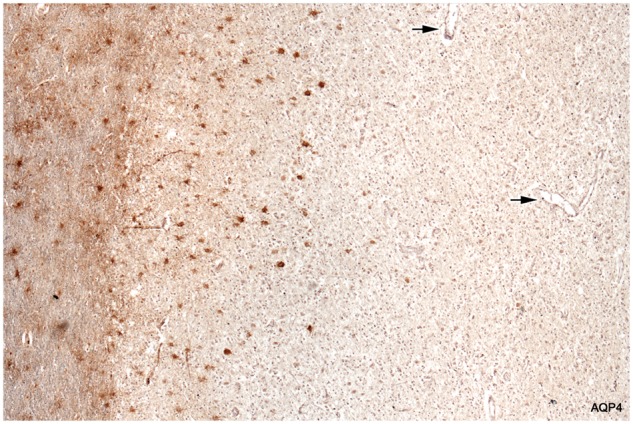
Loss of AQP4-positive astrocytes. Intact myelinated tissue bordering the lesion stains diffusely for AQP4 (astrocyte processes). Cell bodies of stellate reactive astrocytes in the intact border also stain normally for AQP4. The plaque itself is largely AQP4-negative. Degenerate AQP4-positive astrocytes are present at the edge of the plaque but are absent elsewhere. Arrows point to vessels inside the plaque that lack AQP4-positive astrocyte foot processes. The fine linear profiles towards the edge of the plaque are AQP4-positive capillaries. (Case 5, AQP4, ×60).
FIGURE 4.
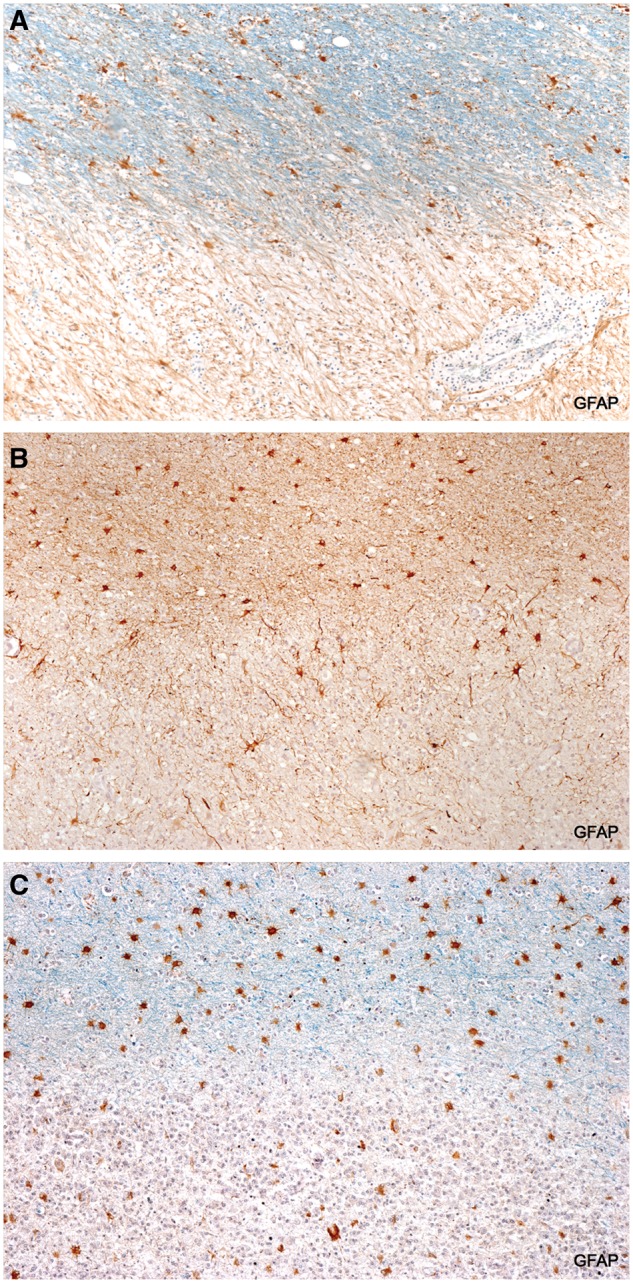
Loss of GFAP-positive astrocytes. GFAP-positive astrocytes are present in intact myelinated tissue but are absent or scant in demyelinated plaque tissue. (A: Case 1, LFB-GFAP, ×80; B: Case 6, GFAP, ×80; C: Case 15, LFB-GFAP, ×80).
Plaques in the Cerebral Cortex
The few plaques in the cerebral cortex that were examined were actively demyelinating lesions extending into subcortical white matter. Reactive fibrous astrocytes were lost in areas of ongoing demyelination (Fig. 5).
FIGURE 5.

Astrocyte loss: A cortical plaque centered on a sulcus is seen expanding into subcortical white matter. There is an abrupt reduction in fibrous astrocyte numbers in demyelinated tissue infiltrated by macrophages at the plaque margin (arrows). The black angular particles in panel C are congealed myelin lipids. (Case 15, Table 2. Preparation courtesy of Dr. T.H. Moss, Frenchay Hospital, Bristol, UK. Golgi silver technique. A: ×3.5; B: ×8; C: ×120).
Dendrophagocytosis
Macrophages clustered around large GFAP-positive AQP4-positive astrocytes with fragmenting processes located at the edge of the plaque were observed in 5 of the 10 plaques. In 2 plaques, AQP4-positive macrophages were present in perivascular spaces and in the subarachnoid space. This appearance, termed in the older literature “dendrophagocytosis,” is a form of astrocyte degeneration where the cell fragments in the presence of a cluster of microglia/macrophages. In the MS lesions dendrophagocytosis was restricted to large GFAP-positive AQP4-positive astrocytes at plaque margins, never in older parts of the plaque or involving AQP4-negative stellate astrocytes which were especially common in early postphagocytic lesional areas (Fig. 6B–E).
FIGURE 6.

Dendrophagocytosis. (A) Fragmenting residual AQP4-positive astrocytes are present towards the edge of the plaque (arrows). (B, C) Disrupted AQP4-positive astrocytes are contacted by macrophages. (D, E) Remnants of large GFAP-positive astrocytes are closely associated with macrophages. (A: Case 9, AQP4, ×100; B: Case 8, AQP4, ×580; C: Case 7, AQP4, ×580; D, E: Case 15, GFAP, ×810).
Clasmatodendrosis
A nodular breakdown of astrocyte cell bodies and processes with preservation of oligodendrocytes and myelin, a common agonal and postmortem autolytic change unrelated to dendrophagocytosis, was widespread in white and grey matter in 4 of the acute cases (Cases 1, 2, 4, and 8) (Supplementary DataFig. S8).
GFAP-Positive AQP4-Negative Astrocyte Precursors
GFAP staining revealed the presence among macrophages in demyelinated tissue depleted of astrocytes a population of small bipolar GFAP-positive AQP4-negative astrocyte precursors similar in appearance to GFAP-positive AQP4-negative astrocyte precursors present in early NMO Lesions (11). They were also positive for GFAPδ and sometimes HNK-1. They were most common near blood vessels and not infrequently occurred in pairs. Mitotic figures were observed in both GFAP-positive and GFAPδ-positive small bipolar precursors as well as in rare, large, rounded (no processes) GFAP-positive astrocytes present at plaque margins in 2 cases (Cases 2 and 7; Figs. 7–9).
FIGURE 7.

GFAPδ-positive astrocyte precursors. Small bipolar cells with scant cytoplasm are present amongst numerous lipid macrophages in demyelinated postphagocytic tissue. No GFAP-positive or GFAPδ-positive stellate astrocytes were present in this region of the plaque. (Case 4, GFAPδ, ×840).
FIGURE 8.

Astrocyte precursors. Pairs of matching small bipolar astrocytes located amongst lipid macrophages in a postphagocytic plaque. (Case 3, a postphagocytic plaque, GFAP, ×670).
FIGURE 9.

Mitotic precursors. Dividing bipolar GFAP-positive (AQP4-negative) astrocyte precursors. (A: Case 2, plaque 1, GFAP, ×870; B–D: Case 4, plaque 1, GFAPδ, ×1020).
Resolving and Chronic Plaques
A semiquantitative assessment of astrocyte populations in 14 plaques of different histological ages from 10 patients revealed that postphagocytic lesions with very large numbers of lipid macrophages had no or rare GFAP-positive or AQP4-positive astrocytes present. Gliosed and remyelinating plaques, on the other hand, had numerous GFAP-positive and AQP4-positive stellate astrocytes present together with few or no lipid macrophages. Between these 2 extremes were plaques with moderate numbers of GFAP-positive stellate astrocytes, relatively numerous lipid macrophages, and importantly, varying numbers of AQP4-negative GFAP-positive stellate astrocytes, the proportion of astrocytes unreactive for AQP4 ranging from about a third to most of those present. These data show that the proportion of newly generated stellate GFAP-positive astrocytes that are also AQP4-positive increases with time. Plaque astrocytes in longstanding inactive chronic plaques react strongly for GFAP and AQP4 (Figs. 10–12; Table 2).
FIGURE 10.

AQP4-negative stellate astrocytes. (A) An inactive hypercellular postphagocytic plaque (numerous lipid macrophages, no LFB-positive macrophages) stains negatively for AQP4. Large process-bearing AQP4-positive astrocytes are present in intact bordering white matter. Rare AQP4-positive large astrocytes and remnants of “original” GFAP-positive AQP4-positive astrocytes can be seen towards the edge of the plaque (arrows). (B) A greater magnification showing a preponderance of the numerous large astrocytes present are AQP4-negative gemistocytic astrocytes (there are 4 astrocytes in the field, one a stellate AQP4-positive astrocyte and 3 AQP4-negative gemistocytic astrocytes). (Case 14, AQP4, A: ×140; B: ×900.).
FIGURE 11.

AQP4-negative GFAP-positive astrocytes. Skip serial sections of an inactive late postphagocytic plaque (no LFB-positive or MBP-positive macrophages) in a patient with fulminant early MS (Case 3). (A, B) None of the numerous GFAP-positive stellate astrocytes present in the plaque is reactive for AQP4. (C, D) IgG-positive (C) and IgM-positive (D) astrocytes are indicative of an open blood-brain barrier. (E, F) MH2-positive inflammatory cells and macrophages are restricted largely to the perivascular cuff (E). This, and the paucity of MRP-positive monocytes (F), indicate a waning inflammatory milieu. (An inactive plaque in Case 3, disease duration 29 days). (A: GFAP; B: AQP4; C: IgG; D: IgM; E: MHC2; F: MRP14. A–F: ×115).
FIGURE 12.
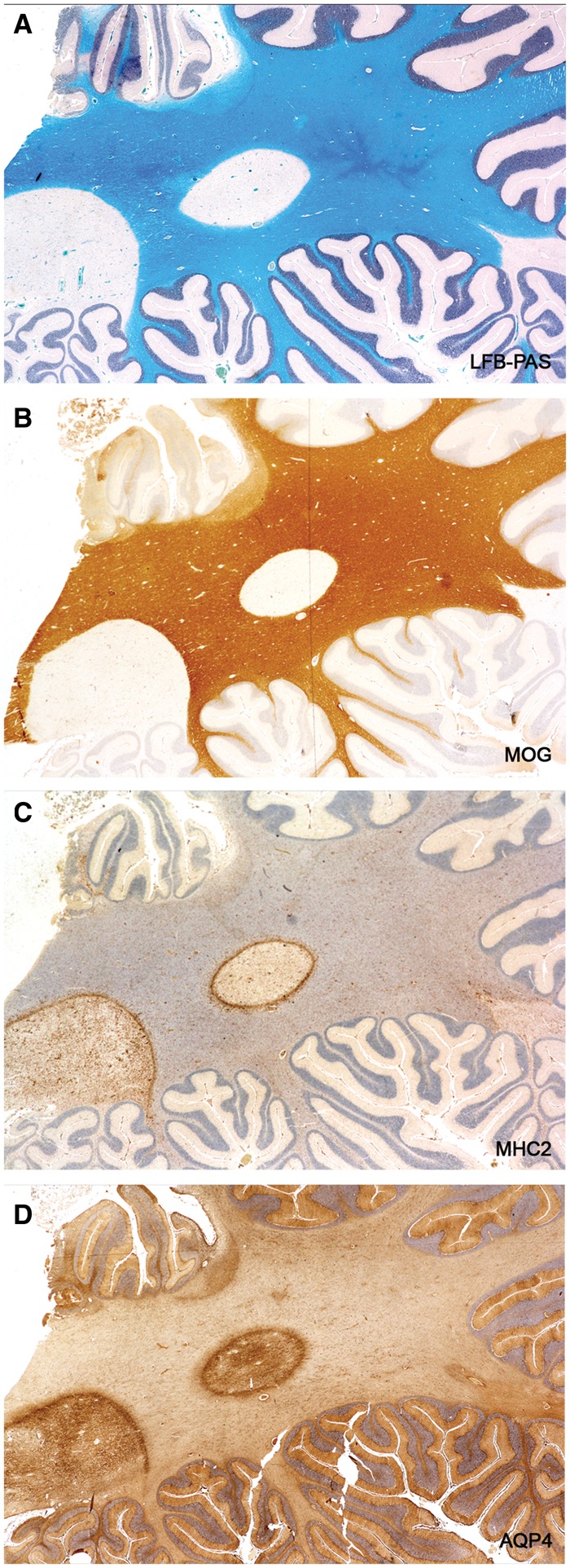
Recovering AQP4 immunoreactivity. An old gliosed plaque with no evidence of recent myelin breakdown (no MOG-positive macrophages, no lipid macrophages) stains positively for AQP4. Astrocytes inside the plaque stain more intensely for AQP4 than those located in the surrounding intact white matter. (Case 18 disease duration “long standing” [not known]). (A: LFB-PAS; B: MOG; C: MHC2; D: AQP4. A–D: ×3.3).
Astrocyte Loss and Regeneration in Patients with Classical Less Acute Disease
A female subject, age 27, with secondary progressive MS of 4 years and 1-month duration is of interest for 2 reasons (Table 2, Case 15). First, findings in this case indicated that astrocyte loss and recovery is not restricted to uncommon acute forms of the disease but occurs also in patients with classical chronic forms of the disease. Sections of the cerebellum revealed 9 plaques of different histological ages, one a postphagocytic plaque with a narrow band of ongoing demyelination associated with macrophages containing complement-reactive (C3d) particles of myelin around the whole circumference of the lesion (Fig. 13). While stellate astrocytes within this band were largely absent, small bipolar astrocytes precursors were present in large numbers (Fig. 14). The case also says something about the unexpectedness of the finding of an astrocyte lesion in MS, in that this particular plaque had been examined immunohistochemically many times in the past without notice of any astrocyte pathology (Figs. 1 and 8 [30]; Case 20 [35]; Case 51 [36]).
FIGURE 13.

Astrocyte destruction and regeneration in secondary progressive multiple sclerosis. Sections of the cerebellum showing 9 plaques with different degrees of gliosis and edge activity in a patient with progressive disease and a disease duration of 4 years and 1 month (Case 15, Table 2). Higher magnifications of the edge zone of the newly forming lesion (center left) show numerous small bipolar astrocyte precursors in areas with no large stellate astrocytes (Fig. 14). (A, B: Case 15, Table 2. A: LFB-PAS followed by GFAP immunostaining; B: Immunostained for macrophages [ricinus communis agglutinin-1(RIC)]. A, B: Original magnification: ×2). Reproduced with permission, Prineas JW, Parratt JD. Oligodendrocytes and the early multiple sclerosis lesion. Ann Neurol 2012;72:18–31.
FIGURE 14.
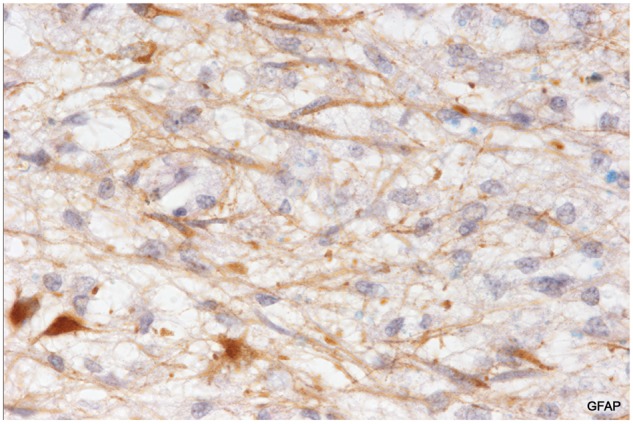
Astrocyte precursors in a patient with classical progressive MS. A magnified view of the edge of the newly forming plaque described and illustrated in Figure 13. Astrocyte precursors are present (together with C3d positive macrophages and myelin sheaths immunoreactive for activated complement) at the extreme edge of the lesion. (Case 15, Table 2, GFAP, ×700).
Electron Microscopy
Twenty blood vessels in 2 postphagocytic lesions were examined (Fig. 15). Every vessel showed disrupted astrocyte foot processes leaving bare stretches of glia limitans basement membrane. In contrast, the glia limitans of 31 capillaries and larger vessels examined in chronic gliosed plaques showed intact astrocyte feet with both cytoplasm and the attached plasma membrane of normal appearance. This specialized plasma membrane with periodic densities was observed only on astrocyte feet attached to capillaries and the outer wall of perivascular spaces, not on astrocyte plasma membranes elsewhere.
FIGURE 15.
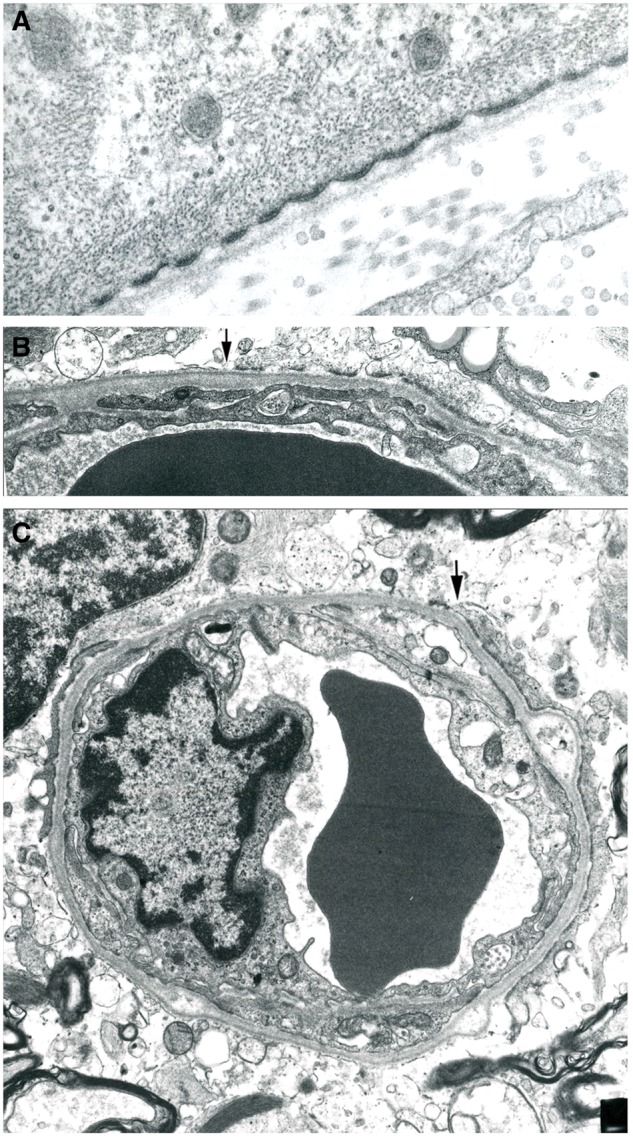
Perivascular astrocyte foot processes. (A) An astrocyte foot process forms part of the glia limitans of a blood vessel at the edge of an inactive chronic plaque in a patient with secondary progressive MS. The plasma membrane with periodic densities (the location of the water channel molecule AQP4) abutting the basement membrane is of normal appearance. The cytoplasm also is of normal appearance, with contained mitochondria, glial filaments and cytoplasmic tubules. (B, C) Two small blood vessels located at the edge of typical postphagocytic plaques. Both show marked thickening of the basement membrane component of the glia limitans and an absence of astrocyte foot processes to the left of the arrows. The loss involves most of the circumference of each capillary. (Electron micrographs. A: ×40,000; B: ×16,000; C: ×10,000).
Neuromyelitis optica
In the 3 NMO cases examined the earliest nonnecrotic perivascular lesions and the edge zone of shelving necrotic lesions with intact sometime pale myelin showed a complete absence by all staining methods of stellate astrocytes. Lipid macrophages were present but few in number and oligodendrocytes were either absent or had pyknotic nuclei. Perivascular eosinophils were present, sometimes in large numbers. In subacute demyelinated lesions that stained negatively for AQP4, scattered large AQP4-positive deformed astrocytes were present towards the edge of the lesion. However, in none of the several very early lesions examined was it possible to determine the pathological changes in astrocytes that precede their disappearance, in particular whether or not dendrophagocytosis occurred. That macrophages were involved, however, in the destruction and/or removal of AQP4-positive astrocytes was indicated by the presence of macrophages whose contents stained positively for AQP4. In still older demyelinated lesions there were varying numbers of small bipolar AQP4-negative astrocyte precursors and sometimes large numbers of AQP4-negative GFAP-positive stellate astrocytes. On the basis of the tissue available for study we are unable to comment on reports that AQP4-negative astrocytes in NMO lesions do not regain AQP4 immunoreactivity which would be a key difference between evolving NMO and MS lesions (2) (Fig. 16; Supplementary DataFigs. S9 and S10).
FIGURE 16.
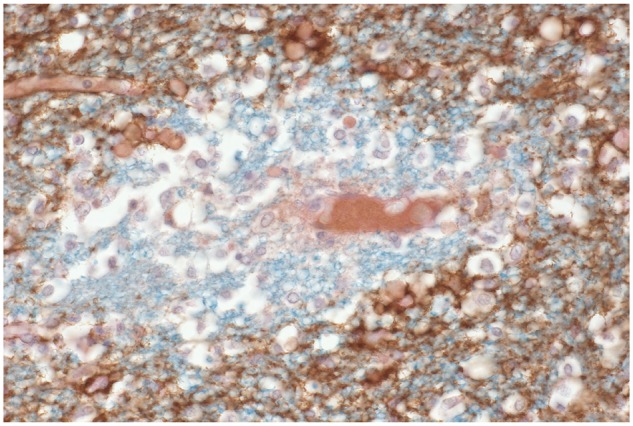
Neuromyelitis optica. A section double stained for myelin and aquaporin4 shows a complete loss of aquaporin4 immunoreactivity affecting intact nonnecrotic perivascular myelinated tissue. (LFB-PAS, AQP4, ×460).
Cerebral Cortex
A patchy sometimes laminar loss of AQP4 immunoreactivity due to a reduction in number of individual protoplasmic astrocytes was observed in 3 MS cases. Affected astrocytes showed a progressive loosening and loss of definition of cell processes. In sections of these areas stained for microglia/macrophages it was apparent that the loss of astrocytes was unrelated to the activity of macrophages. Whether the astrocyte loss was an agonal autolytic event or originated in some other way was not investigated (Figs. 17 and 18).
FIGURE 17.

Cerebral cortex—loss of protoplasmic astrocytes. (A, B) Parts of the cerebral cortex show a laminar loss of aquaporin4 immunoreactivity. The loss is not diffuse but appears to result from a drop out of individual protoplasmic astrocytes. (Case 2, AQP4, A: ×6; B: ×80).
FIGURE 18.

Cerebral cortex—loss of protoplasmic astrocytes. (A) A magnified view of the lesion illustrated in Figure 17. Individual protoplasmic astrocytes show a loss of definition of their processes together with a reduction in AQP4 immunoreactivity. (B) The same area stained for microglia/macrophages shows small, ramified microglia present but no evidence of macrophage participation in the loss of protoplasmic astrocytes. (Case 2, A: AQP4, ×160; B: CD45, ×160).
DISCUSSION
The present study is the first report that astrocytes, like oligodendrocytes, are destroyed together with myelin in newly forming MS lesions, and that precursors of both cell types proliferate to repopulate resolving lesions with astrocytes and oligodendrocytes. The study also shows that the numerous large astrocytes typically seen together with lipid macrophages in early postphagocytic MS plaques are not normal reactive astrocytes but newly generated AQP4-negative gemistocytic astrocytes, and that these abnormal (still not fully differentiated) astrocytes are the progeny of a small bipolar AQP4-negative, GFAPδ-positive HNK-1-positive astrocyte precursor similar in appearance to the precursors that repopulate nonnecrotic early NMO lesions (11). Gemistocytes are described in the older literature as a type of astrocyte “with voluminous homogeneous milk-like cytoplasm which appear above all where nervous tissue has gone through extreme but circumscribed degeneration” (39).
Our findings contrast sharply with prevailing current opinions regarding astrocyte pathology in MS that make no mention of any regressive change in astrocytes in newly forming plaques (2, 4–10). For example, in the Roemer study comparing NMO and MS lesions, in which 57 MS plaques from 11 patients were examined (45 classified as Type 1–4 actively demyelinating lesions), the authors report an increased expression of AQP4 in actively demyelinating MS lesions (2).
AQP4-Negative Astrocytes
Such astrocytes, first identified in early NMO lesions, are still considered the defining astrocyte lesion in nonnecrotic early NMO lesions based on experimental evidence that these are astrocytes that have lost AQP4 immunoreactivity following sublethal injury caused by exposure to anti-AQP4 antibody (2–6, 37). However, our own study of evolving nonnecrotic NMO lesions revealed an initial complete loss of astrocytes followed by the later appearance, in subacute lesions, of an AQP4-negative astrocyte progenitor and AQP4-negative GFAP-positive stellate astrocytes (11). In other words, AQP4-negative GFAP-positive stellate astrocytes in NMO originate—as they do in the early MS lesion—from a small bipolar AQP4-negative astrocyte progenitor, not by downregulating AQP4 expression by existing astrocytes.
Recovery of AQP4 Immunoreactivity
There are reports that AQP4 expression is reduced or undetectable in inactive gliosed MS lesions (2, 6, 8, 19). However, as reported by Sinclair et al (38), the present study shows that astrocytes in chronic inactive lesions exhibit enhanced immunoreactivity for AQP4. This might be related to the fact that perivascular astrocyte feet with normal fine structural features are present in old lesions but not in postphagocytic lesions which is in keeping with experimental evidence that interaction of progenitors with blood vessels induces astrocyte differentiation and the expression of astrocyte markers.
Pathogenesis
Dendrophagocytosis of astrocytes is a nonspecific form of macrophage-mediated astrocyte injury seen in the vicinity of thermal and other traumatic lesions, abscesses and tumors (39, 40). In the evolving MS lesion macrophage activity appeared to be selective, involving only AQP4-positive GFAP-positive astrocytes and not AQP4-negative astrocytes in the same location. Whether this microglia/macrophage activity represents scavenging activity directed at cells injured nonspecifically or at cells targeted by innate or adaptive immune mechanisms is unknown. The same unanswered questions apply to mechanisms underlying microglial/macrophage phagocytosis of pyknotic oligodendrocytes and disrupted myelin in newly forming MS perivascular lesions and plaques (41, 42).
Auto-antibodies
AntiKIR4.1 antibodies have been detected in sera in some MS cases. That the antibodies may be pathogenic is suggested by a report that in actively demyelinating MS lesions there are GFAP-positive astrocytes with reduced KIR4.1 immunoreactivity together with phagocytes containing KIR4.1-positive particles, with “recovery” of KIR4.1 immunoreactivity in inactive lesions (43). The present findings suggest an alternative, namely that astrocytes with reduced KIR4.1 immunoreactivity are not astrocytes that have lost KIR4.1 but are regenerated astrocytes with delayed expression of both AQP4 and KIR4.1. The same can be said of AQP4-negative GFAP-positive astrocytes that populate subacute and chronic nonnecrotic NMO plaques, namely it is likely that these regenerated astrocytes also under express KIR4.1.
Bystander Injury
The fact that oligodendrocytes and astrocytes are damaged and regenerate at about the same time at the expanding edge of a new lesion may indicate a “bystander” form of injury to oligodendrocytes, myelin and astrocytes from a nonspecific mechanism (44), with phagocytosis by IgG-positive microglia/macrophages of degenerate oligodendrocytes, degenerate myelin and degenerate astrocytes a reaction to the presence of injured cells.
Functional Consequences
The role of astrocytes in maintaining blood-brain barrier integrity and the water, ionic, and neurotransmitter state of the adult CNS is well established (10, 45, 46). It is likely that the early abrupt loss of astrocytes followed by repopulation of the plaque by astrocytes initially underexpressing AQP4 (and probably KIR4.1) will contribute not only to changes in nerve conduction that accompany the onset of a clinical relapse but also the recovery of nerve conduction associated with remission of a clinical relapse. Support for an astrocyte lesion contributing directly to disturbed conduction is a study by Schreiner et al who report that experimentally induced loss of astrocyte function leads to microglial activation, neuronal cell loss and severe motor deficits (47). The fact that inactive plaques frequently exhibit evidence of a chronically impaired blood-brain barrier (48, 49) indicates that the astrocyte lesion may also be a contributing factor in patients in whom the disease worsens without remission, that is, in patients with secondary progressive MS. Whether the regressive changes in astrocytes observed in the cerebral cortex are of significance is unclear.
Summary and Conclusions
MS is the second perivascular inflammatory demyelinating disease described where astrocytes are destroyed together with myelin in actively expanding new lesions. Beginning around a vessel or group of vessels (50), microglia/macrophages at the edge of the enlarging area of myelin loss phagocytose reactive astrocytes, oligodendrocytes and myelin, leaving axons relatively intact. This is consistent with reports of raised levels of CSF GFAP chiefly in cases of clinically active disease (51). Trailing early postphagocytic tissue is populated initially by AQP4-negative GFAP-positive gemistocytic astrocytes and HNK-1-positive oligodendrocytes derived from different precursors that appear almost immediately in recently demyelinated tissue. With time new astrocytes reform specialized contacts with capillaries and perivascular spaces and begin to express AQP4 and probably also KIR4.1. From the beginning, serum proteins, T lymphocytes and other inflammatory cells are present in small numbers in the parenchyma and perivascular spaces. Repair of the lesion is accompanied not only by regeneration or attempted regeneration of oligodendrocytes and myelin but also reappearance in the tissue of competent astrocytes. It is likely that maturation of the astrocyte response contributes to remission of symptoms in patients with relapsing and remitting MS. Also possible is a role for the astrocyte lesion in patients in whom the disease worsens without remission, namely in patients with secondary progressive MS.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. B. Paul Morgan for a monoclonal antiMAC antibody B7, Dr. C.S. Raine and Dr. B.A. Kakulas for autopsy tissue from patients with early MS, and Dr. K.J. Smith for helpful discussion.
Funding was received from Multiple Sclerosis Research Australia, The Nerve Research Foundation University of Sydney, The National Multiple Sclerosis Society (JWP RG 2731-A-B), and the NSW Ministry for Science and Medical Research. Control tissue was received from the Australian Brain Donor Programs NSW Tissue Resource Centre which is supported by The University of Sydney and the National Health and Medical Research Council of Australia, The National Neurological Research Specimen Bank, VAMC Wadsworth Division, Los Angeles, California, which is sponsored by NINDS/NIMH, National Multiple Sclerosis Society, Hereditary Disease Research Foundation, Comprehensive Epilepsy Program, Tourette Syndrome Association, Dystonia Medical Research Foundation, and Veterans Health Services and Research Administration, Department of Veterans Affairs.
The authors have no duality or conflicts of interest to declare.
Supplementary Data can be found at academic.oup.com/jnen.
REFERENCES
- 1. Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202:473–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roemer SF, Parisi JE, Lennon VA, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 2007;130:1194–205 [DOI] [PubMed] [Google Scholar]
- 3. Misu T, Fujihara K, Kakita A, et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007;130:1224–34 [DOI] [PubMed] [Google Scholar]
- 4. Fujihara K, Misu T, Nakashima I, et al. Neuromyelitis optica should be classified as an astrocytopathic disease rather than a demyelinating disease. Clin Exp Neuroimmunol 2012;3:58–73 [Google Scholar]
- 5. Misu T, Hoftberger R, Fujihara K, et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol 2013;125:815–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lucchinetti CF, Guo Y, Popescu BF, et al. The pathology of an autoimmune astrocytopathy: Lessons learned from neuromyelitis optica. Brain Pathol 2014;24:83–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Correale J, Farez MF.. The role of astrocytes in multiple sclerosis progression. Front Neurol 2015;6:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ludwin SK, Rao V, Moore CS, et al. Astrocytes in multiple sclerosis. Mult Scler 2016;22:1114–24 [DOI] [PubMed] [Google Scholar]
- 9. Reich DS, Lucchinetti CF, Calabresi PA.. Multiple sclerosis. N Engl J Med 2018;378:169–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ponath G, Park C, Pitt D.. The role of astrocytes in multiple sclerosis. Front Immunol 2018;9:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parratt JD, Prineas JW.. Neuromyelitis optica: A demyelinating disease characterized by acute destruction and regeneration of perivascular astrocytes. Mult Scler 2010;16:1156–72 [DOI] [PubMed] [Google Scholar]
- 12. Sharma R, Fischer MT, Bauer J, et al. Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathol 2010;120:223–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brosnan CF, Raine CS.. The astrocyte in multiple sclerosis revisited. Glia 2013;61:453–65 [DOI] [PubMed] [Google Scholar]
- 14. Kira JI. Astrocytopathy in Balo’s disease. Mult Scler 2011;17:770–8 [DOI] [PubMed] [Google Scholar]
- 15. Brown AW, Brierley JB.. Anoxic-ischaemic cell change in rat brain. Light microscopic and fine-structural observations. J Neurol Sci 1972;16:59–84 [DOI] [PubMed] [Google Scholar]
- 16. Middeldorp J, Hol EM.. GFAP in health and disease. Prog Neurobiol 2011;93:421–43 [DOI] [PubMed] [Google Scholar]
- 17. Prineas JW, McDonald WI, Franklin RJM.. Demyelinating diseases In: Graham DI, Lantos PL, eds. Greenfield’s Neuropathology, 7th Edition, Vol. 2. London, UK: Arnold; 2002:471–535 [Google Scholar]
- 18. Lassmann H. Pathology of multiple sclerosis In: A Compston, G Ebers, H Lassmann, I McDonald, B Mathews, H Wekerle, eds. McAlpines's Multiple Sclerosis. 3rd ed London, Edinburgh, New York, Philadelphia, Sydney, Toronto: Churchill Livingstone; 1998:323–58 [Google Scholar]
- 19. Moore GRW, Stadelmann-Nessler C.. Demyelinating diseases In: Love S, Budka H, Ironside JW, Perry A, eds. Greenfield's Neuropathology, 9th Edition, Vol. 2. Florida: CRC Press, Taylor and Francis Group; 2015:1297–1412 [Google Scholar]
- 20. Adams RD, Kubik CS.. The morbid anatomy of the demyelinating diseases. Am J Med 1952;12:510–46 [DOI] [PubMed] [Google Scholar]
- 21. Itoyama Y, Sterhberger NH, Webster HDF, et al. Immunocytochemical observations on the distribution of myelin-associated glycoprotein and myelin basic protein in multiple sclerosis lesions. Ann Neurol 1980;7:167–77 [DOI] [PubMed] [Google Scholar]
- 22. Prineas JW, Graham JS.. Multiple sclerosis: Capping of surface immunoglobulin G on macrophages engaged in myelin breakdown. Ann Neurol 1981;10:149–58 [DOI] [PubMed] [Google Scholar]
- 23. Prineas JW, Kwon EE, Goldenberg PZ, et al. Multiple sclerosis. Oligodendrocyte proliferation and differentiation in fresh lesions. Lab Invest 1989;61:489–503 [PubMed] [Google Scholar]
- 24. Prineas JW, Barnard RO, Kwon EE, et al. Multiple sclerosis: Remyelination of nascent lesions. Ann Neurol 1993;33:137–51 [DOI] [PubMed] [Google Scholar]
- 25. Ozawa K, Suchanek G, Breitschopf H, et al. Patterns of oligodendroglia pathology in multiple sclerosis. Brain 1994;117:1311–22 [DOI] [PubMed] [Google Scholar]
- 26. Gay FW, Drye TJ, Dick GW, et al. The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis. Identification and characterization of the primary demyelinating lesion. Brain 1997;120:1461–83 [DOI] [PubMed] [Google Scholar]
- 27. Lucchinetti C, Bruck W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann Neurol 2000;47:707–17 [DOI] [PubMed] [Google Scholar]
- 28. Barnett MH, Prineas JW.. Relapsing and remitting multiple sclerosis: Pathology of the newly forming lesion. Ann Neurol 2004;55:458–68 [DOI] [PubMed] [Google Scholar]
- 29. Breij EC, Brink BP, Veerhuis R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol 2008;63:16–25 [DOI] [PubMed] [Google Scholar]
- 30. Prineas JW, Parratt JD.. Oligodendrocytes and the early multiple sclerosis lesion. Ann Neurol 2012;72:18–31 [DOI] [PubMed] [Google Scholar]
- 31. Henderson AP, Barnett MH, Parratt JD, et al. Multiple sclerosis: Distribution of inflammatory cells in newly forming lesions. Ann Neurol 2009;66:739–53 [DOI] [PubMed] [Google Scholar]
- 32. Van Norden S. Problems and solutions In: Beesley JE, ed. Immunocytochemistry, A Practical Approach. Oxford: IRL Press; 1993:207–39 [Google Scholar]
- 33. Barnett MH, Parratt JD, Cho ES, et al. Immunoglobulins and complement in post-mortem multiple sclerosis tissue. Ann Neurol 2009;65:32–46 [DOI] [PubMed] [Google Scholar]
- 34. Prineas JW, Parratt JD, Kirwan PD.. Fibrosis of the choroid plexus filtration membrane. J Neuropathol Exp Neurol 2016;75:855–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prineas JW. Multiple sclerosis: Presence of lymphatic capillaries and lymphoid tissue in the brain and spinal cord. Science 1979;203:1123–5 [DOI] [PubMed] [Google Scholar]
- 36. Raine CS, Scheinberg L, Waltz JM.. Multiple sclerosis. Oligodendrocyte survival and proliferation in an active established lesion. Lab Invest 1981;45:534–46 [PubMed] [Google Scholar]
- 37. Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007;69:2221–31 [DOI] [PubMed] [Google Scholar]
- 38. Sinclair C, Kirk J, Herron B, et al. Absence of aquaporin-4 expression in lesions of neuromyelitis optica but increased expression in multiple sclerosis lesions and normal-appearing white matter. Acta Neuropathol 2007;113:187–94 [DOI] [PubMed] [Google Scholar]
- 39. Penfield W. Neuroglia. Normal and pathological In: Wilder Penfield, ed. Cytology and Cellular Pathology of the Nervous System, Vol. 2. New York: Paul B Hoeber Inc; 1932:421–80 [Google Scholar]
- 40. Blakemore WF. The ultrastructural appearance of astrocytes following thermal lesions of the rat cortex. J Neurol Sci 1971;12:319–32 [DOI] [PubMed] [Google Scholar]
- 41. Ulvestad E, Williams K, Matre R, et al. Fc receptors for IgG on cultured human microglia mediate cytotoxicity and phagocytosis of antibody-coated targets. J Neuropathol Exp Neurol 1994;53:27–36 [DOI] [PubMed] [Google Scholar]
- 42. Boven LA, Van Meurs M, Van Zwam M, et al. Myelin-laden macrophages are anti-inflammatory, consistent with foam cells in multiple sclerosis. Brain 2006;129:517–26 [DOI] [PubMed] [Google Scholar]
- 43. Schirmer L, Srivastava R, Kalluri SR, et al. Differential loss of KIR4.1 immunoreactivity in multiple sclerosis lesions. Ann Neurol 2014;75:810–28 [DOI] [PubMed] [Google Scholar]
- 44. Mahad DH, Trapp BD, Lassmann H.. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 2015;14:183–93 [DOI] [PubMed] [Google Scholar]
- 45. Nicchia GP, Nico B, Camassa LMA, et al. The role of aquaporin-4 in the blood–brain barrier development and integrity: Studies in animals and cell culture models. Neuroscience 2004;129:935–45 [DOI] [PubMed] [Google Scholar]
- 46. Vinters HV, Kleinschmidt-DeMasters BK.. General pathology of the central nervous system In: S Love, DN Louis, DW Ellison, eds. Greenfield’s Neuropathology, 8th edition Vol. 1. London: Hodder Arnold; 2008:1–56 [Google Scholar]
- 47. Schreiner B, Romanelli E, Liberski P, et al. Astrocyte depletion impairs redox homeostasis and triggers neuronal loss in the adult CNS. Cell Rep 2015;12:1377–84 [DOI] [PubMed] [Google Scholar]
- 48. Kwon EE, Prineas JW.. Blood-brain barrier abnormalities in longstanding multiple sclerosis lesions. An immunohistochemical study. J Neuropathol Exp Neurol 1994;53:625–36 [DOI] [PubMed] [Google Scholar]
- 49. Kirk J, Plumb J, Mirakhur M, et al. Tight junctional abnormality in multiple sclerosis white matter affects all calibers of vessel and is associated with blood-brain barrier leakage and active demyelination. J Pathol 2003;201:319–27 [DOI] [PubMed] [Google Scholar]
- 50. Gaitan MI, Shea CD, Evangelou IE, et al. Evolution of the blood-brain barrier in newly forming multiple sclerosis lesions. Ann Neurol 2011;70:22–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kassubek R, Gorges M, Schocke M, et al. GFAP in early multiple sclerosis: A biomarker for inflammation. Neurosci Lett 2017;657:166–70 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.