

Abstract
Increasing evidence indicates that mitochondrial-associated redox signaling contributes to the pathophysiology of heart failure (HF). The mitochondrial-targeted antioxidant, mitoquinone (MitoQ), is capable of modifying mitochondrial signaling and has shown beneficial effects on HF-dependent mitochondrial dysfunction. However, the potential therapeutic impact of MitoQ-based mitochondrial therapies for HF in response to pressure overload is reliant upon demonstration of improved cardiac contractile function and suppression of deleterious cardiac remodeling. Using a new (patho)physiologically relevant model of pressure overload-induced HF we tested the hypothesis that MitoQ is capable of ameliorating cardiac contractile dysfunction and suppressing fibrosis. To test this C57BL/6J mice were subjected to left ventricular (LV) pressure overload by ascending aortic constriction (AAC) followed by MitoQ treatment (2 µmol) for 7 consecutive days. Doppler echocardiography showed that AAC caused severe LV dysfunction and hypertrophic remodeling. MitoQ attenuated pressure overload-induced apoptosis, hypertrophic remodeling, fibrosis and LV dysfunction. Profibrogenic transforming growth factor-β1 (TGF-β1) and NADPH oxidase 4 (NOX4, a major modulator of fibrosis related redox signaling) expression increased markedly after AAC. MitoQ blunted TGF-β1 and NOX4 upregulation and the downstream ACC-dependent fibrotic gene expressions. In addition, MitoQ prevented Nrf2 downregulation and activation of TGF-β1-mediated profibrogenic signaling in cardiac fibroblasts (CF). Finally, MitoQ ameliorated the dysregulation of cardiac remodeling-associated long noncoding RNAs (lncRNAs) in AAC myocardium, phenylephrine-treated cardiomyocytes, and TGF-β1-treated CF. The present study demonstrates for the first time that MitoQ improves cardiac hypertrophic remodeling, fibrosis, LV dysfunction and dysregulation of lncRNAs in pressure overload hearts, by inhibiting the interplay between TGF-β1 and mitochondrial associated redox signaling.
Nonstandard abbreviations and acronyms: MitoQ, Mitoquinone; ROS, Reactive oxygen species; AAC, Ascending aortic constriction; TGF-β, Transforming growth factor beta; NOX4, NADPH oxidase 4; SMAD, Mothers against decapentaplegic homolog; HW, Heart weight; LW, Lung weight; TL, Tibia length; LV, Left ventricle; IVSd, Interventricular septal end diastole; PWTd, Posterior wall thickness, diastole; IVSs, Interventricular septal end systole; PWTs, Posterior wall thickness, systole; CO, Cardiac output.; FS, Fractional shortening; EF, Ejection fraction; ESD, End-systolic diameter; EDD, End-diastolic diameter; ESV, End-systolic volume; EDV, End- diastolic volume; SV, Stroke volume; lncRNA, Long noncoding RNA; Nrf2, nuclear factor erythroid 2-derived factor 2; GSR, Glutathione reductase; PGC-1α, Peroxisome proliferator-activated receptor gamma coactivator 1α
Keywords: Mitoquinone, Redox signaling, Ascending aortic constriction, Cardiac remodeling, IncRNA
Graphical abstract
1. Introduction
Heart failure (HF) is a leading cause of mortality and morbidity in the United States. Regardless of etiology, HF is heralded by some common molecular, cellular, and biochemical events collectively called cardiac remodeling that manifest as alterations in size, shape, structure, and function of the heart [1]. In the case of pressure overload resulting from an aortic stenosis or systemic arterial hypertension, the heart often develops an adaptive concentric hypertrophy, characterized by increased left ventricular (LV) wall thickness with little or no change in chamber size [2]. However, as compensated hypertrophic remodeling can only temporarily sustain cardiac function, concentric hypertrophy will gradually progress to eccentric hypertrophy and eventually to systolic heart failure if the stress is sustained. During the transition from compensated hypertrophy to decompensated heart failure, severe fibrosis usually occurs concurrently with cardiomyocyte hypertrophy, cellular apoptosis, and inflammatory cell infiltration [3].
Extensive studies have demonstrated that fibrosis exerts adverse effects on cardiac structure and function [4], including increased muscle stiffness, reduced contractility, and abnormal electrical transduction. In pressure overload-induced HF, transforming growth factor-β1 (TGF-β1), a multifunctional cytokine that regulates cellular proliferation, differentiation, and extracellular matrix production, is a key mediator of fibrosis progression. Binding of TGF-β1 to its membrane receptors phosphorylates transcription factors SMAD2/3, which activates the canonical (SMAD-dependent) fibrogenic pathway [5], [6], leading to myofibroblast differentiation and fibrosis [7]. In addition, TGF-β1 can upregulate NADPH oxidase 4 (NOX4) expression and hence modulate redox signaling pathways [8]. Moreover, redox signaling mechanisms, potentially involving mitochondrial superoxide and hydrogen peroxide production, have been shown to involve in TGF-β1 activation and signaling [9]. Thus, a vicious positive feedback loop exists between TGF-β1, NOX4, and redox signaling [10], facilitating amplification of the profibrogenic effects of TGF-β1. In addition to promoting fibrosis, redox signaling has been shown to be implicated in hypertrophic remodeling and increased cellular apoptosis. Increased levels of reactive oxygen species (ROS) can stimulate signaling cascades including those involving protein kinase C (PKC), mitogen-activated protein kinase (MAPK) [11], c-Jun N-terminal kinase (JNK) [12], and GTP-binding protein Ras [13] - all implicated in hypertrophy and apoptosis (reviewed in Tsutsui et al. [14]). Thus, dysregulation of redox signaling appears to be commonly associated with deleterious cardiac remodeling events [15], suggesting that targeting redox signaling is a potential therapeutic strategy for HF treatment.
Mitoquinone (MitoQ) is a derivative of Coenzyme Q, in which Coenzyme Q is linked to a positively charged group TPP+ in which the cationic charge has been delocalized. Consequently, this lipophilic conjugated compound can readily cross biological membranes and accumulates in mitochondria at high concentrations [16]. Over the last decade, MitoQ has been proven to effectively attenuate redox related pathologies in both in vitro [17] and in vivo [18] studies including suppression of fibrosis in the liver and kidney [19], [20]. With respect to cardiac hypertrophy and HF, it has been shown that MitoQ attenuates mitochondrial dysfunction in both volume overload [21] and pressure overload [22] rat models. A perplexing aspect of these studies is the lack of effect of MitoQ in reversing or preventing the loss of cardiac function which occurs in these models. This disconnect raises three important possibilities that need to be addressed: a) the impairment of cardiac physiological function in HF is unrelated to mitochondrial activity; b) mitochondria are involved in the pathophysiology of HF but MitoQ is ineffective; and c) the mitochondrial contribution to the pathophysiology is not directly related to changes in the capacity to generate ATP but the retrograde signaling that regulates fibrosis. For mitochondrial therapeutics to be applied in a translational setting these aspects of the mechanisms need to be resolved and this is the focus of the current study.
While it is the case that studies in other organs have suggested that MitoQ is capable of suppressing the signaling that controls the development of fibrosis this has not been investigated in the failing heart. This is critical since there is a strong link between fibrosis and the pathophysiology of HF [23]. Accordingly, we tested the key mechanistic hypothesis that MitoQ can improve cardiac remodeling and LV dysfunction by suppressing the mitochondrial redox signaling that mediates the TGF-β1-NOX4-ROS vicious cycle, leading to decreased SMAD-mediated pro-fibrotic transcript regulation.
An important reason for the apparent lack of efficacy of MitoQ in the TAC rat may be due to limitations of the models used such as lack of fibrosis and sufficient ejection fraction reduction, the main criteria for HF classification [22]. In this study, we utilized an ascending aortic constriction (AAC)-induced pressure overload HF mouse model that rapidly develops severe fibrosis and profound LV dysfunction. This offers significant advantages over the traditional transverse aortic constriction (TAC) models that may take months to develop HF, if they develop HF at all. In fact, some laboratories have resorted to a “double hit” model of aortic constriction and arterial ligation [24]. By using our AAC model we can produce HF in a timely manner while address therapies without secondary and longer-term complications from models that develop failure over a longer timeframe. Our data demonstrate that MitoQ significantly improved LV hypertrophic remodeling and cardiac contractile dysfunction and attenuated development of fibrosis in failing hearts. Consistent with these observations, both in vivo animal and in vitro cardiac fibroblast experiments reveal that MitoQ suppressed TGF-β1-induced oxidative stress and upregulation of TGF-β1, NOX4, and the downstream genes involved in the fibrosis network. As a significant extension of these mechanistic studies, we show data which implicates a role for long noncoding RNAs (lncRNAs), such as cardiac hypertrophy-associated transcript (Chast), in HF-dependent fibrosis, supporting the emerging role of these factors as critical regulators of pathological cardiac remodeling [25]. Preliminary results of this work have been published in abstract form [26].
2. Methods
All data and materials supporting the findings of this study are available within the article and in the Online Data Supplement or from the corresponding authors on reasonable request.
2.1. Animals
Adult C57BL/6J mice were obtained from Jackson laboratories and housed in an animal facility at the University of Alabama at Birmingham. This study conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85‐23, revised 1996) and has been approved by the Institutional Animal Care and Use Committee at University of Alabama at Birmingham.
2.2. Ascending aortic constriction (AAC)
Nine-week-old male mice were anesthetized with 2% isoflurane and placed in the supine position on a heating pad (37 °C). After a horizontal skin incision of ~1 cm in length at the level of the suprasternal notch, a ~3 mm longitudinal cut was made in the proximal portion of the sternum. Surgery was then performed as previously described [27], with the following modifications. Specifically, minimally invasive surgery was performed in which the aorta was accessed via a small incision of the upper sternum which meant the mice did not require intubation. Following this entry an ascending aortic constriction (AAC) was performed by placing a titanium ligating clip (Teleflex, Catalog # 005200) that had been calibrated to 31-gauge needle diameter around the ascending aorta. The sham operation was performed following the same procedure without clip application. Twenty-four hours after AAC or sham surgery, 100 μL of water or MitoQ (2 µmol) was administered by oral gavage every morning at the same time for 7 days. Mouse body weight and consumption of drinking water were monitored. Tissue was harvested at the end of treatment for gravimetric and molecular measures.
2.3. Echocardiography
Transthoracic echocardiography were performed on mice to evaluate the peak pressure gradient and cardiac function using a 30 MHz RMV707B transducer and VisualSonics Vevo770 High-Resolution System (VisualSonics, Toronto, ON, Canada) as previously described [28]. In brief, mice were anesthetized with 1.5% inhaled isoflurane anesthesia (with 100% supplemental oxygen) and placed on a heated, bench-mounted adjustable rail system. The peak pressure gradient (ΔP) across the constriction was measured using pulse wave Doppler velocity (V) (ΔP = 4V2) two days after AAC surgery. One week after AAC and MitoQ treatment, two-dimensionally directed M mode images were captured from the long axis views. Left ventricular (LV) end-diastolic and end-systolic dimensions, and LV end-diastolic posterior wall thickness, were measured from the M-mode images, and LV fractional shortening was calculated with VisualSonics V1.3.8 software.
2.4. Gravimetric and histological analysis
Mice were sacrificed, and cardiac and lung tissues were collected. Weights of heart and lung and length of tibia of each mouse were measured. For histological analysis, LV tissues were fixed with formalin, embedded with paraffin, and sectioned at 5 µm thickness. Sections were then deparaffinized in CitriSolv and rehydrated through washes of descending percentage of ethanol.
2.4.1. Wheat germ agglutinin staining
To assess cardiomyocyte cross-sectional area, tissue sections were stained with Alexa Fluor 488-conjugated wheat germ agglutinin (1:1000). Slides were imaged with an Olympus FV1000 confocal microscope. Cardiomyocytes with transversely cross-sectioned and revealing thin and intact cell borders were included in size measurement. Around 60 cardiomyocytes per mouse were measured using ImageJ software to acquire a proper representation of the tissue.
2.4.2. Picrosirius red staining
Cardiac fibrosis was quantified on Picrosirius red (PSR)-stained sections. Briefly, slides were washed and treated with phosphomolybdic acid 0.2% aqueous. Sirius red was then added to the slides, followed by addition of 0.1% in saturated picric acid and then 0.01 N hydrochloric acid. Images were analyzed with Image Pro Premier 64-bit software (Media Cybernetics, Rockville, MD, USA).
2.4.3. Masson's trichrome staining
Collagen quantification was performed on Masson's trichrome-stained sections according to manufacturer's instruction. Images were analyzed with Image Pro Premier 64-bit software.
2.4.4. TUNEL assay
Apoptotic cells were assessed using the DeadEnd Fluorometric TUNEL System (Promega, Madison, WI) according to the manufacturer's instruction. Briefly, the slides were mounted with Permount mounting medium, with the nuclei counterstained with DAPI. The slides were then dried and imaged with a fluorescent microscope. Data were analyzed with ImageJ software (NIH, Bethesda, MD, USA).
2.5. RNA isolation and transcript expression analysis
Total RNA was extracted and purified from the LV tissues with RNeasy mini kit (Qiagen, Germantown, MD). cDNA was synthesized from 500 ng of RNA using QuantiTect Reverse Transcription Kit (Qiagen). Quantitative real-time PCR were performed using Select Master Mix (Thermos Fisher Scientific, Asheville, NC) in a BioRad IQ5 detection system (BioRad, Portland, ME). Briefly, 25 μL PCR master mix was prepared as follows: 5 ng cDNA template, 1× SYBR Select Master Mix, and 300 nM gene-specific primers. The PCR amplification protocol was the following: 5 min at 95 °C, 40 cycles at 95 °C for 15 s and 60 °C for 45 s. Two reference genes were selected: Gapdh and Rpl32, both of which showed constitutive expression across different groups. The expression levels of target genes were normalized to the average levels of Gapdh and Rpl32. The specificity of PCR primers (Table 1) was confirmed with 1% agarose gel electrophoresis and melt curves. The fold difference for the mRNA expression level was calculated using 2−ΔΔCt.
Table 1.
List of real-time RT-PCR primers.
| mRNA | Forward | Reserve |
|---|---|---|
| Nppa | ATGGGCTCCTTCTCCATCA | TCCAGGTGGTCTAGCAGGTT |
| Nppb | GGTGCTGTCCCAGATGATTC | GCCATTTCCTCCGACTTTTC |
| Chast | TCTGAGGTGTGCCTGTGAAC | TGCTGCTGTCTTTGGTTGTC |
| Malat-1 | CTTTGCGGGTGTTGTAGGTT | AAAACTGGGAGGTTGTGCTG |
| Mhrt | GAGAGCATTTGGGGATGGTA | GGCAACACTTTTCATTTTCCTC |
| Agt | CGAACTCAAAGCAGGAGAGG | CTGGGGGTTATTCACTCTGC |
| Pdgf | AACGCCAACTTCCTGGTGT | GGCTTCTTTCGCACAATCTC |
| Tgf-β1 | TGAGTGGCTGTCTTTTGACG | CGTGGAGTTTGTTATCTTTGCT |
| Edn1 | CCCAAAGTACCATGCAGAAAA | ACGAAAAGATGCCTTGATGC |
| Nox4 | GAAGCCCATTTGAGGAGTCA | GATGTCTCTGCACACCCAGA |
| Ctgf | CCACCCGAGTTACCAATGAC | GACAGGCTTGGCGATTTTAG |
| Acta2 | CCTGAAGAGCATCCGACACT | CAGAGGCATAGAGGGACAGC |
| Col1a1 | ACGCCATCAAGGTCTACTGC | GAATCCATCGGTCATGCTCT |
| Col1a2 | GGAGAGAAGGGAACCAAAGG | AGGGAAGCCAGTCATACCAG |
| Col3a1 | CGTAAGCACTGGTGGACAGA | CGGCTGGAAAGAAGTCTGAG |
| Gapdh | CATGGCCTTCCGTGTTCCTA | CCTGCTTCACCACCTTCTTGAT |
| Rpl32 | CTGGAGGTGCTGCTGATGT | GGGATTGGTGACTCTGATGG |
| Nfe2l2 | CTGAACTCCTGGACGGGACTA | CGGTGGGTCTCCGTAAATGG |
| Nqo1 | AGGATGGGAGGTACTCGAATC | TGCTAGAGATGACTCGGAAGG |
| Cyp1a | AAGTGCAGATGCGGTCTTCT | AAAGTAGGAGGCAGGCACAA |
| Nd1 | TCCCCTACCAATACCACACC | GGCTCGTAAAGCTCCGAATA |
| Ppargc1a | AAGGTCCCCAGGCAGTAGAT | TTTCGTGCTCATAGGCTTCA |
2.6. Immunohistochemistry
After being blocked with 10% inactivated goat serum and 1% bovine serum albumin (BSA) in PBS, tissue sections were incubated overnight at 4 °C with an anti-vimentin (1:200) and anti-4-HNE (1:100) primary antibodies. Thereafter, sections were washed with 0.1% BSA in PBS, blocked again with blocking buffer (30 min at room temperature), and incubated with an Oregon green 488 anti-rabbit IgG (1:500) and ATTO 647 N anti-mouse IgG (1:300) secondary antibodies to visualized 4-HNE and vimentin, respectively. Images were acquired using an Olympus FV1000 confocal microscope with FluoView 1000 software (Olympus, Center Valley, PA, USA).
2.7. Protein isolation and expression analysis
Western blotting was performed to analyze the levels of fibrosis-related protein expression, including TGF-β1, NOX4, total SMAD2 and p-SMAD2, and 4-HNE-protein adducts in lysate of LV tissues. Briefly, liquid nitrogen frozen ventricle tissues were homogenized with sonication in 300 μL RIPA buffer added with phosphatase inhibitor cocktail (1:100). Tissue lysates were agitated gently at 4 °C for 1 h, followed by centrifuge and supernatant collection. Protein concentration in the supernatants was determined by the BCA assay. Protein samples were loaded onto Nupage 4–12% Bis-Tris gels. After electrophoresis, proteins were electro-transferred to a PVDF membrane. The blotted membrane was then blocked with TBS containing 5% fat-free milk powder and 0.1% Tween 20 for 1 h at room temperature and incubated with specific primary antibodies overnight at 4 °C. The membrane was rinsed three times and incubated with HRP-conjugated secondary antibodies (anti rabbit or mouse depending on the primary antibody, 1:3000 for 1 h at room temperature). Membrane was treated with Luminata Forte Western HRP substrate (Millipore, Boston, MA) and then imaged with MyECL imager (Thermo Fisher Scientific). Quantification analysis of blots was performed with the ImageJ software. Targeted bands were normalized to GAPDH.
2.8. Mouse cardiac fibroblast isolation and in vitro experiments
Freshly isolated hearts from 9-week-old C57BL/6 male mice were rinsed with HBSS. Left ventricles were gently separated and cut with a sterile scalpel into small pieces. Minced tissues were digested in collagenase II (600 IU) and DNase I (enzyme solution) in a petri dish. The tissue was mechanically dissociated by pipetting up and down. The dish was incubated at 37 °C for 15 min. After that, the supernatant was collected into a 15 mL conical tube. The collagenase solution was added to any remaining tissue fragments. The dissociation step was repeated until a single cell suspension was obtained. After centrifugation and rinsing, the resulting isolated cardiac fibroblasts (CF) were cultured in DMEM/F12 medium supplemented with 15% FBS and 1% P/S antibiotics.
To study the role of MitoQ in ameliorating TGF-β1-induced profibrogenic signaling, CF cultures (within 3 passages) were starved in FBS-free DMEM medium for 24 h, followed by exposure to 10 ng/mL TGF-β with or without the presence of MitoQ (300 nmol/L). After 24 h, cells were harvested for transcript expression analysis and confocal imaging.
To further investigate the effect of MitoQ on pressure overload-induced TGF-β1 signaling activation, isolated CF were cultured on a thin poly dimethylsiloxane (PDMS) membrane within custom cell culture chambers. The cell culture chamber was then integrated within the biomimetic cardiac tissue model (BCTM) platform and subjected to cyclic stretch under different pressures as previously described [29]. While the inner wall of the ventricle experiences high pressure (>120 mmHg), the outer wall does not experience any pressure. Depending on the location of cells within the ventricular wall, the pressure can range from ventricular pressure to 0 mmHg. Experimentally we evaluated the equivalent pressure indicative of normal or pressure overload conditions for cells cultured as a monolayer within the BCTM and found that 15 mmHg and 30 mmHg correspond to normal and pressure overload conditions, respectively. After 24 h of stretch in the presence or absence of MitoQ, cells were collected for transcript expression analysis and confocal imaging.
2.9. Mouse cardiomyocyte isolation and in vitro experiments
Adult mouse cardiomyocytes were isolated from 10-week-old male C57BL/6 J mouse heart, as previously described [30]. After isolation, cardiomyocytes were cultured in EMEM medium supplement with 10% FBS, 2 mM ATP, 25 µM blebbistatin and 1% penicillin/streptomycin) for 2 h in incubator (5% CO2, 37 °C). Then, the medium was changed to EMEM medium with 0.1% BSA, 25 µM blebbistatin and 1% penicillin/streptomycin, and cultures was maintained in the incubator for experiments. To further investigate the mechanism by which MitoQ regulates cardiac remodeling-associated lncRNA expression, cardiomyocytes were treated in the presence or absence of 10 μM phenylephrine (PE) and 200 nM MitoQ for 24 h. Afterwards, cells were collected and used for RNA isolation and lncRNA expression analysis.
2.10. Confocal imaging
ROS levels in CF cultures were measured with MitoSox (5 μmol/L), an superoxide-sensitive fluorescent indicator, as described previously [21], [31]. In brief, the dish containing fluorescent dye-loaded CF was equilibrated at 37 °C with unrestricted access to atmospheric oxygen on the stage of an Olympus IX81 microscope. MitoSox images were recorded using an Olympus FV1000 confocal microscope with excitation at 488 nm and analyzed offline with the ImageJ software.
2.11. Antibodies and chemicals
Primary antibodies against Vimentin (ab24525), 4-HNE (ab46545), and NOX4 (ab133303) and HRP secondary antibodies were purchased from AbCam (Cambridge, MA, USA). Antibodies against SMAD2 (mAb #8685), p-SMAD2 (mAb # 8828), TGF-β1 (pAb #3711) were purchased from Cell Signaling Technology (Danvers, MA, USA). Secondary antibody Oregon green 488, collagenase II, DMEM, FBS, and DNase I were purchased from Thermo Fisher Scientific (Waltham, MA, USA). ATTO 647 N anti-mouse IgG were obtained from Sigma-Aldrich (St. Louis, MO, USA). MitoSox and Alexa Fluor 488-conjugated wheat germ agglutinin were purchased from Molecular Probes (Eugene, OR, USA). All other chemicals are acquired from Sigma-Aldrich.
2.12. Statistical analysis
All data are reported as mean ± SEM. Statistical comparisons among groups were performed using the two-way ANOVA Tukey's multiple comparison test. P < 0.05 was considered statistically significant.
3. Results
3.1. AAC leads to rapid cardiac hypertrophic remodeling and heart failure in mice
A new mouse HF model was developed by inducing a minimally invasive ascending aortic constriction (AAC) with a calibrated titanium clip. The rationale was that constriction of the ascending aorta, instead of the transverse aorta, leads to a much higher hemodynamic load on the heart, which causes accelerated development of cardiac hypertrophy and HF. Doppler echocardiography showed that the peak pressure gradient across the constriction was 4-fold higher in AAC mice as compared with sham (99.5 ± 9.76 mmHg vs. 16.9 ± 5.2 mmHg), indicating that AAC induced much more severe pressure overload than the commonly-performed transverse aortic constriction (TAC), which is typically less than 2-fold [27], [32]. Heart weight (HW), ventricular weight, and HW normalized to tibia length were all significantly increased in AAC mice one week after surgery (Table 2). AAC mice also exhibited a substantial increase in lung weight normalized to tibia length (Table 2), consistent with pulmonary congestion. Furthermore, AAC mice showed a significant decrease in body weight (Table 2). Wheat germ agglutinin (WGA) staining was used to assess cardiomyocyte size (Fig. 1A), which showed that consistent with cardiac enlargement, average cardiomyocyte cross sectional areas in AAC mice increased by 96% compared with sham (Fig. 1B). In addition, the transcript levels of atrial natriuretic factor (Nppa) and brain natriuretic peptide (Nppb), two cardiac hypertrophy marker genes, increased 36-fold and 5-fold, respectively, in AAC mice (P < 0.001) (Fig. 1C).
Table 2.
MitoQ improves cardiac hypertrophy induced by ascending aortic constriction (AAC) after one week of treatment.
| Sham | Sham + MitoQ | AAC | AAC + MitoQ | |
|---|---|---|---|---|
| Body Weight, g | 24 ± 0.3 | 24 ± 0.5 | 21 ± 0.8 * | 22 ± 0.6 |
| Heart Weight, mg | 89.3 ± 0.8 | 94.4 ± 5.9 | 154.2 ± 2.9 * | 124.9 ± 7.8# |
| LV Weight, mg | 72.5 ± 0.9 | 75.3 ± 3.9 | 130.8 ± 2.7 * | 104.5 ± 6.3# |
| RV Weight, mg | 12.8 ± 0.6 | 13.5 ± 0.7 | 18.1 ± 1.5 * | 16.4 ± 1.3 |
| Lung Weight, mg | 146.9 ± 9.1 | 147 ± 3.2 | 296 ± 19.6 * | 168 ± 9.5# |
| TL, mm | 16.23 ± 0.11 | 16.48 ± 0.09 | 16.34 ± 0.06 | 16.30 ± 0.09 |
| Heart Weight/TL, mg/mm | 5.50 ± 0.03 | 5.72 ± 0.34 | 9.43 ± 0.15 * | 7.65 ± 0.46# |
| Lung Weight/TL, mg/mm | 9.04 ± 0.52 | 8.92 ± 0.24 | 18.09 ± 1.16 * | 10.29 ± 0.57# |
| Number of mice | 6 | 4 | 6 | 6 |
LV: left ventricle; RV: right ventricle; TL: tibia length. Values are means ± SEM. * : P < 0.05 vs. Sham. #: P < 0.05 vs. AAC.
Fig. 1.

MitoQ reduces pressure overload-induced cardiac hypertrophy in mice. A: Representative images of WGA staining of LV myocardium; B: quantification of WGA staining (right); and C: Nppa and Nppb mRNA expression levels; **: P < 0.01 vs. Sham. #: P < 0.05 vs. AAC. ##: P < 0.01 vs. AAC. n = 4–6 each group. Scale bars in A: 40 µm. AAC: ascending aortic constriction; WGA: wheat germ agglutinin.
AAC also caused rapid development of LV dysfunction and HF, as shown by transthoracic echocardiography (Fig. 2A). In just one week, LV fractional shortening (FS) and ejection fraction (EF) were reduced 70% (Fig. 2B) and 50%, respectively, in AAV mice as compared with sham (Fig. 2C). In addition to a decline in LV function, AAC mice underwent substantial LV chamber remodeling. While there were no notable differences between the sham and AAC mice in LV interventricular septal and end-systolic posterior wall thickness, both end-systolic dimension (Fig. 2D) and volume (Table 3) markedly increased after AAC (P < 0.01). In addition, end-diastolic dimension (Fig. 2E), as well as end-diastolic posterior wall thickness, endo-diastolic volume, and stroke volume were also altered in response to pressure overload (Table 3).
Fig. 2.

MitoQ improves left ventricular (LV) contractility and chamber remodeling in AAC mice. The LV parameters were assessed using serial echocardiography one week after surgery. A: Representative images showing M mode echocardiography of mice; B: Fractional shortening (FS); C: Ejection fraction (EF); D: End-systolic diameter (ESD); and E: End-diastolic diameter (EDD). **: P < 0.01 vs. Sham. #: P < 0.05 vs. AAC. n = 4–6 each group. AAC: ascending aortic constriction.
Table 3.
MitoQ improves left ventricular chamber remodeling and contractile dysfunction induced by ascending aortic constriction (AAC) after one week of treatment.
| Sham | Sham + MitoQ | AAC | AAC+ MitoQ | |
|---|---|---|---|---|
| Heart Rate, bpm | 456 ± 13.2 | 497 ± 10.2 | 441 ± 16.4 | 495 ± 13.3 |
| LV IVSd, mm | 0.77 ± 0.06 | 0.70 ± 0.03 | 0.87 ± 0.05 | 0.82 ± 0.05 |
| LV PWTd, mm | 0.65 ± 0.04 | 0.78 ± 0.03 | 0.99 ± 0.07 * | 1.14 ± 0.06 |
| LV IVSs, mm | 0.91 ± 0.07 | 0.94 ± 0.04 | 0.92 ± 0.04 | 0.92 ± 0.03 |
| LV PWTs, mm | 0.95 ± 0.05 | 1.07 ± 0.04 | 1.00 ± 0.07 | 1.30 ± 0.07 |
| LV EDV, μL | 60.0 ± 0.97 | 56.7 ± 3.68 | 73.6 ± 1.92 * | 56.6 ± 5.89 # |
| LV ESV, μL | 23.2 ± 0.81 | 20.9 ± 1.85 | 51.6 ± 2.77 * | 29.0 ± 4.23 # |
| SV, μL | 36.7 ± 0.81 | 35.8 ± 2.02 | 21.9 ± 1.91 * | 27.6 ± 2.29 |
| CO, μL | 16,698 ± 323 | 17,851 ± 1255 | 9673 ± 885 * | 13,563 ± 975 |
| Number of mice | 6 | 4 | 6 | 6 |
LV: left ventricular; IVSd: interventricular septal end diastole; PWTd: diastolic posterior wall thickness; IVSs: interventricular septal end systole; PWTs: systolic posterior wall thickness; EDV: end-diastolic volume; ESV: end-systolic volume; SV: stroke volume; CO: cardiac output. Values are means ± SEM. * : P < 0.05 vs. Sham. #: P < 0.05 vs. AAC.
3.2. MitoQ attenuates pressure overload-induced cardiac hypertrophy, LV remodeling and HF
Previous studies have shown that MitoQ decreases ischemia-reperfusion-induced cardiac dysfunction [33] and cardiac hypertrophy [34], we thus examined whether MitoQ imposes beneficial effects in pressure overload mice. As reported in Table 2, both pathological heart and lung enlargements in AAC mice were significantly mitigated by MitoQ treatment. MitoQ also attenuated AAC-induced cardiomyocyte hypertrophy measured by WGA staining and blunted upregulation of the expressions of cardiac hypertrophic marker genes (Fig. 1). MitoQ had no effect on HW, lung weight, and cardiomyocyte size in sham mice (Table 2).
Importantly, echocardiography data showed that MitoQ treatment significantly attenuated cardiac dysfunction in mice subjected to pressure overload (Fig. 2). Both LV fractional shortening and ejection fraction were significantly higher in MitoQ-treated AAC mice than in AAC (P < 0.05). MitoQ also effectively prevented pressure overload-induced LV chamber remodeling. The end-systolic diameter and end-diastolic diameter, as well as end-diastolic volume (EDV) and end-systolic volume (ESV) in MitoQ-treated mice were essentially normalized (Fig. 2 and Table 3). Furthermore, MitoQ treatment modestly improved the reduced stroke volume in AAC mice (Table 3). As expected, MitoQ did not affect LV function and chamber size in sham mice.
3.3. MitoQ decreases cellular apoptosis and improves mitochondrial biogenesis in AAC mouse myocardium
Loss of cardiomyocytes via apoptosis is known to contribute to impaired cardiac function in response to pressure overload stress [35], [36], [37]. Consistent with previous studies, in situ TUNEL assays showed that cell death, reflected by the percentage of TUNEL positive nuclei, was significantly higher in AAC mouse myocardium in comparison to sham, which was greatly diminished by MitoQ (Fig. 3A and Fig. S1). In addition, MitoQ decreased myocardial oxidative stress in response to pressure overload, as detected by 4-hydroxynonenal (4-HNE) immunostaining (Fig. 3B) and western blotting (Fig. 3C). Finally, MitoQ increased mitochondrial copy number and biogenesis in the AAC myocardium, as measured by the ratio of mitochondrial DNA (ND1) to nuclear DNA (CYP1a1) (Fig. 3D) and the levels of the gene Ppargc1a which encodes the transcriptional coactivator PGC-1α (Fig. 3E), respectively.
Fig. 3.

Effect of MitoQ on cellular apoptosis, oxidative stress and mitochondrial biogenesis in pressure overload mouse hearts. A: Quantitative summary of TUNEL assay. Please refer to Supplemental Fig. S1 for representative DAPI and TUNEL images; B: Representative immunostaining of 4-HNE and Vimentin in LV myocardium; C: Representative western blotting of 4-HNE-protein adducts; D: Effect of MitoQ on mitochondrial copy number; and E: Effect of MitoQ on Ppargc1α mRNA expression. *: P < 0.05 vs. Sham. **: P < 0.01 vs. Sham. #: P < 0.05 vs. AAC. ##: P < 0.01 vs. AAC. n = 4–6 each group. 4-HNE: 4-hydroxynonenal; AAC: ascending aortic constriction.
3.4. MitoQ inhibits cardiac fibrosis in pressure overload-induced HF
Ventricular fibrosis is indicative of structural remodeling and typically accompanies cardiac dysfunction. To evaluate the degree of cardiac fibrosis and effect of MitoQ, Masson's trichrome and picrosirius red (PSR) staining were performed on LV tissue sections. As shown in Fig. 4, the fibrotic area increased dramatically (14–23 times) in AAC myocardium compared with sham and this was significantly attenuated by MitoQ. Fibrosis development involves the transformation of fibroblasts into active myofibroblasts, and TGF-β1 plays a major role in pathological fibrogenesis in diseased hearts, suggesting that MitoQ might affect this signaling pathway. Real time PCR revealed that there were no significant differences in the levels of angiotensinogen (Agt) and platelet-derived growth factor (Pdgf) mRNA expression among all groups (Fig. 5A). However, the levels of expression of TGF-β1 and endothelin 1 (Edn1) transcripts were significantly upregulated in AAC myocardium (Fig. 5A). The expression levels of TGF-β1 downstream genes NADPH oxidase 4 (Nox4), connective tissue growth factor (Ctgf), actin assembly-inducing protein 2 (Acta2), and collagens (Col1a1, Col1a2, and Col3a1) were all upregulated in myocardium of AAC mice (Fig. 5B), indicating activation of the TGF-β1 signaling pathway in response to pressure overload. MitoQ treatment attenuated overexpression of TGF-β1 and all of its downstream genes in AAC mice (Fig. 5B).
Fig. 4.

MitoQ attenuates cardiac fibrosis in AAC mice. A: Representative images of Masson trichrome staining. Please refer to Supplemental Fig. S2 for larger high-quality images. B: Quantification of interstitial fibrosis measured by Masson trichrome staining; C: Representative images of Picrosirius red (PSR) staining; and D: Quantification of interstitial fibrosis measured by PSR staining. **: P < 0.01 vs. Sham. #: P < 0.05 vs. AAC. ##: P < 0.01 vs. AAC. n = 4 each group. Scale bars: 200 µm. AAC: ascending aortic constriction.
Fig. 5.

MitoQ blunts activation of myocardial TGF-β1 and profibrogenic signaling in response to pressure overload. A: mRNA expression levels of Agt, Pdgf, TGF-β1 and Edn1; B: mRNA expression levels of Nox4, Ctgf, Acta2, and collagen (Col1a1, Col1a2 and Col3a1); C: Western blotting of TGF-β1, NOX4, phosphorylated SMAD2 (p-SMAD2), and total SMAD2; and D: Quantification of normalized TGF-β1 (left), NOX4 (middle) and p-SMAD2/total SMAD2 (right) protein expression. *: P < 0.05 vs. Sham. **: P < 0.01 vs. Sham. #: P < 0.05 vs. AAC. ##: P < 0.01 vs. AAC. n = 4–6 each group. Agt: angiotensinogen; Pdgf: platelet-derived growth factor; Edn1: endothelin; Nox4: NADPH oxidase 4; Ctgf: connective tissue growth factor; Acta: actin assembly-inducing protein. AAC: ascending aortic constriction.
To confirm activation of the TGF-β signaling pathway and inhibitory effects of MitoQ on fibrogenesis in AAC mice at the protein level, we measured TGF-β1, NOX4, p-SMAD2 and total SMAD2 protein levels in LV tissue extracts by western blotting (Fig. 5C). Consistent with the transcript data, AAC mice exhibited pronounced increases in TGF-β1 and NOX4 protein levels (Fig. 5D). Phosphorylation of SMAD2 was also significantly increased in AAC mouse hearts, whereas the total SMAD2 protein levels remained unchanged (Fig. 5D). With the exception of total SMAD2, MitoQ treatment in AAC mice, but not in sham mice, diminished the overexpression of those proteins (Figs. 5C and 5D).
3.5. MitoQ prevents TGF-β-induced fibrosis in cardiac fibroblast cultures
To obtain more detailed information on the anti-fibrogenic role of MitoQ in HF, we isolated and cultured cardiac fibroblasts (CF) from 9-week-old C57BL/6J mice. These CF cultures were treated with TGF-β1 for 24 h in the presence or absence of MitoQ, followed by biochemical analysis. Confocal imaging revealed that the fluorescent intensity of MitoSox in TGF-β1-treated cultures was significantly higher than in control (Fig. 6A), indicating that TGF-β1 induced mitochondrial superoxide production in CF. Associated with this finding, the expression of Nox4, Ctfg, Acta2, and Col1a2 transcripts (Fig. 6B), as well as NOX4 and p-SMAD2 proteins (Fig. 6C and D), were upregulated in CF cultures after TGF-β1 treatment, suggesting activation of fibrotic signaling. Importantly, we found that TGF-β1 transcript expression increased 110% in TGF-β1-treated CF (Fig. 6B). MitoQ treatment abolished TGF-β1-induced oxidative stress and TGF-β1 upregulation and prevented overexpression of NOX4 and the downstream fibrotic genes and SMAD2 protein phosphorylation (Fig. 6A-D). To further understand the mechanism by which MitoQ regulates TGF-β1, we examined the effect of MitoQ on the expression of nuclear factor (erythroid-derived 2) like 2 (Nrf2), a global antioxidant gene inducer, in CF exposed to TGF-β1. Real-time PCR revealed that TGF-β1 caused significant downregulation of Nrf2 and its downstream gene Nqo1 (that encodes NAD(P)H quinone dehydrogenase 1), which were prevented by MitoQ (Fig. 6E). The transcript expressions of Nrf2 and Nqo1 in control CF were not altered by MitoQ (Fig. 6E). At the tissue level, MitoQ significantly increased Nrf2 protein expression in AAC mice and slightly in sham mice (Fig. 6F).
Fig. 6.

MitoQ attenuates NOX4-induced oxidative stress and SMAD2 signaling pathway in TGF-β-treated cardiac fibroblasts (CF). A: Representative confocal images of MitoSox stained CF (left) and quantification of ROS measurement (right); B: mRNA expression of TGF-β1, Nox4, Ctgf, Acta2 and Col1a2 in CF; C: Representative western blotting of NOX4, phosphorylated SMAD2 (p-SMAD2), and total SMAD2; D: Quantification of normalized NOX4 and p-SMAD2/total SMAD2 protein expression; E: mRNA expression of Nrf2 and Nqo1 in CF; and F: Nrf2 protein expression in AAC myocardium *: P < 0.05 vs. Control. #: P < 0.05 vs. TGF-β (A-E) or AAC (F). n = 3 each group for CF (A-E) and n = 6 each group for mouse (F). NOX4: NADPH oxidase 4; Ctgf: connective tissue growth factor; Acta: actin assembly-inducing protein; Col1: collagen type I alpha 2 chain; Nrf2: nuclear factor erythroid 2-related factor 2; Nqo1: NAD(P)H quinone dehydrogenase 1. AAC: ascending aortic constriction.
3.6. MitoQ inhibits TGF-β1 signaling in cardiac fibroblasts exposed to pressure and mechanical stresses
Recently, the BCTM model has been developed that enables cultured cells to be subjected to defined hemodynamic loads to mimic critical mechanical stresses experienced by cardiac cells in vivo [29]. Using the BCTM, we examined the effect of MitoQ on CF cultures subjected to pressure overload and mechanical stress that is similar to the in vivo microenvironment in failing hearts. Confocal imaging of MitoSox showed that 24-h pressure overload (i.e. 30 mmHg) and cyclic stretch resulted in increased signal in CF cultures compared with static (i.e. 0 mmHg) and normal (i.e. 15 mmHg) pressure (Fig. 7A). The transcript levels of TGF-β1 and the downstream fibrotic genes (e.g., Nox4, Ctgf, and Col1a2) were moderately increased in CF exposed to normal pressure as compared with the static conditions (Figs. 7B-E). Consistent with the in vivo results, pressure overload resulted in a > 3-fold elevation in TGF-β1 expression (Fig. 7B). NOX4 transcript expression was also upregulated in pressure overload CF (Fig. 7C), which may underlie the increased ROS production. The activation of TGF-β1 signaling induced fibrosis in CF, which is indicated by increased Ctgf and Col1a2 transcript levels (Fig. 7D and E). MitoQ attenuated oxidative stress and overexpression of TGF-β1, Nox4, Ctgf, and Col1a2 in CF cultures in response to pressure and mechanical stresses.
Fig. 7.

MitoQ attenuates oxidative stress and TGF-β1 signaling activation in cardiac fibroblasts (CF) expose to pressure overload (PO) and mechanical stretch. A: Representative confocal images of MitoSox stained CF (left) and quantification of ROS measurement (right); B: mRNA expression of TGF-β1; C: mRNA expression of Nox4; D: mRNA expression of Ctgf; and E: mRNA expression of collagen (Col1a2). *: P < 0.05 vs. Normal. #: P < 0.05 vs. PO. n = 3 each group. Nox4: NADPH oxidase 4; Ctgf: connective tissue growth factor; Col1a2: collagen type I alpha 2 chain.
3.7. MitoQ ameliorates dysregulation of cardiac remodeling-associated lncRNA expression
Finally, we examined the effects of MitoQ on long noncoding RNAs (lncRNAs) that have been reported to be associated with cardiac remodeling in HF, including cardiac hypertrophy-associated transcript (Chast), metastasis associated in lung adenocarcinoma transcript 1 (Malat-1) and myosin heavy chain associated transcript (Mhrt). Chast expression was significantly upregulated in myocardium of AAC mice (Fig. 8A), which was consistent with the observations that AAC induced accelerated cardiac hypertrophic remodeling in mice. The expression of Malat-1, a lncRNA may regulate cardiac hypertrophy during hypoxia-induced pulmonary hypertension [38], was not significantly altered in pressure overload mice (Fig. 8B), which is in agreement with a recent study [39]. Expression of Mhrt, a cytoprotective lncRNA that is involved in the regulation of oxidative stress-induced cellular apoptosis expression, decreased substantially in AAC mice (P < 0.01) (Fig. 8C). MitoQ attenuated AAC-induced dysregulation of Chast and Mhrt and did not influence their expression in sham mice.
Fig. 8.

MitoQ ameliorates dysregulation of cardiac remodeling-associated lncRNAs. A-C: Effect of MitoQ on lncRNA Chast (A), Malat-1 (B), and Mhrt (C) expression in mouse myocardium; D-F: Effect of MitoQ on the expression of Chast (D) in TGF-β-treated CF, and Malat-1 (E) and Mhrt (F) in phenylephrine (PE)-treated mouse CM. Chast: cardiac hypertrophy-associated transcript; Malat-1: metastasis associated in lung adenocarcinoma transcript 1; and Mhrt: myosin heavy chain associated RNA transcript. *: P < 0.05 vs. Sham (in A-C) or Control (D and F); #: P < 0.05 vs. AAC (A-C) or TGF-β (D) or PE (F). n = 6 each group for A-C and n = 3 each group for D-F.
We next tested the regulatory effect of MitoQ on these lncRNAs in isolated cardiac myocytes (CM) and CF subjected to PE (phenylephrine) and TGF-β1 treatment, respectively. Twenty-four hours TGF-β1 treatment caused significant upregulation of Chast which was prevented by MitoQ (Fig. 8D). The expressions of Malat-1 were not changed by either PE or MitoQ (Fig. 8E), but Mhrt decreased significantly in PE-treated CMs, and the downregulation was blocked by MitoQ (Fig. 8F). Interestingly, real-time PCR showed that the expression of those cardiac remodeling-associated lncRNAs are cell type specific: Chast expression was barely detected in CM while Mhrt expression was very low in CF (cycle threshold values were 31–33 compared with ~17 for GAPDH).
4. Discussion
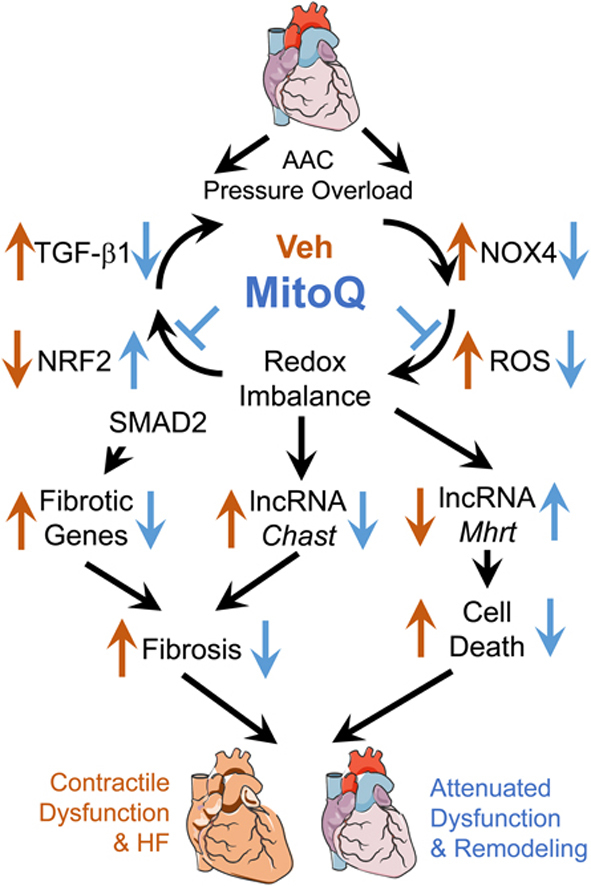
The main results of the present study, as summarized in Fig. 9, are that (1) ascending aortic constriction induces rapid development of cardiac hypertrophic remodeling and HF in mice; (2) MitoQ improves pressure overload-induced cardiac hypertrophic remodeling and LV contractile dysfunction; (3) MitoQ suppresses the TGF-β1, NOX4 and ROS positive feedback loop, leading to reduced fibrosis; and (4) MitoQ regulates expression of lncRNAs involving in cardiac remodeling. These new findings support the notion that the TGF-β1-NOX4-ROS signaling axis is critical for modulation of cardiac hypertrophy and fibrosis in the context of pressure overload-induced HF.
Fig. 9.

Summary of the novel insights of the current study beyond past research on MitoQ and heart failure. Using the AAC-induced pressure overload mouse model we demonstrate that MitoQ is capable of reducing fibrosis and LV contractile dysfunction in HF by suppressing the profibrotic TGF-β1-NOX4-ROS vicious cycle and dysregulation of the cardiac remodeling-associated lncRNAs.
Over the last few decades various animal models have been developed to explore the pathophysiology and treatment of cardiac hypertrophy and HF. While large animal models such as swine and canine are considered more clinically relevant, small animals, especially mouse, are increasingly used for mechanistic studies in HF, largely due to recent technical advances in gene editing and manipulation [40]. Currently-used mouse models of cardiac hypertrophy and HF are typically generated by genetic manipulation such as Gαq overexpression [41] and CaMKII overexpression [42], or surgical introduction such as aortic constriction [43] and left coronary ligation [44]. In the current study we developed a new minimally invasive ascending aortic constriction (AAC) mouse model that exhibits rapid and robust development of cardiac dysfunction and HF. In particular, both HW and lung weight doubled, and FS and EF dropped more than 65% in AAC mice in as short as one week. These characteristics make our model more (patho)physiologically relevant to human HF for assessing the therapeutic impact of MitoQ on pressure overload-dependent cardiac dysfunction compared to the traditional TAC models. Given those advantages, it is worth mentioning that the etiology and pathogenesis of AAC-induced HF is not exactly the same as that in human HF, which is a progressive multifactorial disease that may take years to develop. In addition, human HF represents the summation of multiple anatomic, functional, electrical and metabolic alterations which may induce some secondary complications. These limitations should be taken into account when considering the path towards translational application of these findings.
Using the AAC model, we demonstrate that MitoQ protects against pressure overload-induced cardiac remodeling and LV dysfunction in mice, as assessed by echocardiographic measurement of LV dimensions, chamber size, and contractility. Pathological changes, including organ weights, cellular sizes, and fibrosis are also ameliorated by MitoQ. The different therapeutic role of MitoQ in HF between the present AAC and the previous TAC [22] studies could due to several important factors most notably the dosing regimen and animal model. We elected to administer MitoQ by oral gavage, rather than drinking water since in initial studies we noted that the mice (both AAC and sham) consumed significantly less water over time (~3.2 mL/day/mouse without MitoQ vs. ~0.5 mL/day/mouse with MitoQ), decreasing the effective MitoQ dose. Secondly, we began to feed mice with MitoQ 24 h after AAC surgery, while in the TAC rat study MitoQ was given three days after the surgery [22]. As ROS levels and dysregulation of redox signaling may arise within a few days after aortic constriction, allowing animals to receive timely MitoQ treatment might be critical. Another important aspect is that AAC, rather than TAC, was used in this study to induce pressure overload. As discussed above, AAC caused a rapid and severe LV remodeling leading to HF in mice. In particular, we observed severe cardiac fibrosis in AAC mice, a key factor underlying LV remodeling and reduced contractility, and MitoQ effectively ameliorated cardiac fibrosis. However, it was not discussed whether cardiac fibrosis occurred in the TAC rat study. Finally, the dosage of MitoQ used in the present study was higher than that used in the TAC rat study (100 μM in drinking water). Altogether, it appears that the dosing regimen and timing of MitoQ treatment and types of animal and disease model may all contribute to the greater efficacy of MitoQ on LV dysfunction in HF observed in the present study.
Cardiac fibrosis is a late-stage irreversible process during the progression of HF, which greatly increases the stiffness of the heart wall and reduces the contractility and compliance of cardiac muscle. Fibrosis can be activated by multiple sources, including increased mechanical stretch [45], dysregulated redox signaling [41], paracrine cytokines from injured cells [46], and infiltration of circulatory immune cells [28]. Secreted cytokines from fibroblasts and cardiomyocytes, including TGF-β1, endothelin, angiotensinogen, and connective tissue growth factor, activate differentiation of fibroblast into myofibroblast and extracellular matrix remodeling. Of those growth factors, we found that TGF-β1 transcript expression in AAC myocardium nearly doubled when compared with sham. SMAD2 phosphorylation and expression of downstream genes were also upregulated, indicating activation of the canonical fibrotic pathway. In addition, our data clearly show that TGF-β1 contribute to redox signaling through NOX4, in accordance with previous studies [10]. Importantly, NOX4 modulates mitochondrial ROS production [11], [47], which together enhance TGF-β1 expression, resulting in further ROS production through a positive-feedback mechanism. Thus, TGF-β1-activated ROS production can amplify the TGF-β1-mediated fibrogenic signaling, as suggested previously [48]. We speculate that MitoQ attenuates AAC-induced cardiac fibrosis by suppressing the redox-dependent retrograde signaling. The effects of MitoQ on TGF-β1 and NOX4 expression and redox signaling are confirmed in two in vitro experiments in which isolated CF were exposed to TGF-β1 treatment or pressure overload, respectively. The direct anti-fibrosis effect of MitoQ has been reported in liver [19], [49], kidney [20], and lung [50]. The present study demonstrates that MitoQ can also retard fibrosis in hearts subjected to pressure overload. Intriguingly, we showed that TGF-β1 treatment suppressed Nrf2 and Nqo1 mRNA expression in isolated CF (Fig. 6E). While this appears potentially counterintuitive but would suggest that it is a key characteristic of the pathology of HF. For example, Xing et al. reported that expressions of cytosolic and nuclear Nrf2 proteins are reduced in myocardium of mice subjected to TAC surgery [51]. In non-cardiac tissue, it has been shown that increased TGF-β inhibits Nrf2 expression and its ability for antioxidants production in lung fibroblast, thereby leads to oxidative stress [52]. These changes may create a feed forward cycle which amplifies and increases TGF-β expression. Importantly, MitoQ increased Nrf2 expression in both the AAC myocardium and TGF-β1-treated CF, suggesting that MitoQ ameliorates TGF-β1 signaling and fibrosis likely by activating the Nrf2 signaling that suppresses the TGF-β1-NOX4-ROS redox signaling axis.
Emerging evidence show that long noncoding RNAs (lncRNAs) play prominent regulatory roles in the development of cardiovascular diseases (see recent reviews [25], [53], [54], [55]). One of the most recently identified lncRNAs associated with cardiac hypertrophic remodeling in HF is Chast. It has been shown that myocardial Chast expression is upregulated in pressure overload mice and aortic stenosis patients [56]. Similarly, we found that the Chast expression levels were significantly higher in AAC mice than in sham, accompanied by substantial cardiac hypertrophy. Another lncRNA we have examined is Mhrt that confers cytoprotective effects [57]. The target of Mhrt is Brg1, a stress-activated chromatin-remodeling factor that induces aberrant gene expression and cardiac myopathy [58]. Consistent with a recent study showing that Mhrt was downregulated in cardiac pressure overload [57], we found that myocardial Mhrt expression reduced 70% in AAC mice as compared with sham. The detailed mechanism underpinning the regulation of lncRNAs in failing hearts is poorly understood. It has been reported that lncRNAs respond to environmental and developmental conditions similarly with protein-coding genes [59], [60]. In addition, several studies showed that the promoters of lncRNAs are almost as conserved as those of protein-coding genes [61], [62]. Moreover, the transcription and processing of most lncRNAs appears to be similar to those of protein-coding genes. Altogether, we conjecture that the canonical TGF-β1-NOX4-ROS signaling, which is markedly activated in the AAC heart, is involved in the dysregulation of Chast and Mhrt expression, as suggested by our data (Fig. 8D) and several recent studies [63], [64], [65]. Intriguingly, we showed that MitoQ significantly diminished the dysregulation of those lncRNA expression in pressure overload hearts and PE/TGF-β treated CM/CF, supporting the potential role of TGF-β-NOX4-ROS redox signaling axis in lncRNA regulation. With that being said, future mechanistic studies are warranted to completely understand how MitoQ is involved in the regulation of lncRNAs. Importantly, our finding of cell type-specific expression of those lncRNAs may lead to novel targeted treatment of HF patients.
In summary, the current study provides direct evidence that the mitochondrial-targeted redox modulatory compound, MitoQ, attenuates cardiac remodeling and improves LV contractile dysfunction in pressure overload-induced HF in mice. The mechanism, at least in part, involves inhibition of interplay between TGF-β1 and mitochondrial-associated redox signaling, which ameliorates the dysregulation of profibrogenic genes, antioxidant gene inducer Nrf2, and cardiac remodeling-associated lncRNAs. It has to be noted that HF is a multifactorial syndrome, and we cannot exclude the protective effect of MitoQ on other subcellular systems that can also contribute to the improved pressure overload-induced cardiac dysfunction.
Sources of funding
This work was supported by National Institutes of Health (R01HL121206 to L.Z., R01HL133011 to A.W. and HL118067 to NSR) and University of Alabama at Birmingham VSRC core Grant P30 EY003039 and the Nathan Shock Center P30 AG 050886.
Disclosure
No relevant conflicts of interest.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2019.101100.
Appendix A. Supplementary material
Fig. S1.

Representative image of TUNEL assay and DAPI staining of Sham, Sham+MitoQ, AAC and AAC+MitoQ myocardium.
Fig. S2.

Representative images of Masson trichrome staining of Sham, Sham+MitoQ, AAC and AAC+MitoQ myocardium.
References
- 1.Cohn J.N., Ferrari R., Sharpe N. Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000;35(3):569–582. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 2.Kehat I., Molkentin J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122(25):2727–2735. doi: 10.1161/CIRCULATIONAHA.110.942268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burchfield J.S., Xie M., Hill J.A. Pathological ventricular remodeling: mechanisms: Part 1 of 2. Circulation. 2013;128(4):388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan D., Takawale A., Lee J., Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenes. Tissue Repair. 2012;5(1):15. doi: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verrecchia F., Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J. Investig. Dermatol. 2002;118(2):211–215. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 6.Khalil H., Kanisicak O., Prasad V., Correll R.N., Fu X., Schips T., Vagnozzi R.J., Liu R., Huynh T., Lee S.J., Karch J., Molkentin J.D. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017;127(10):3770–3783. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lijnen P., Petrov V., Rumilla K., Fagard R. Transforming growth factor-beta 1 promotes contraction of collagen gel by cardiac fibroblasts through their differentiation into myofibroblasts. Methods Find. Exp. Clin. Pharmacol. 2003;25(2):79–86. doi: 10.1358/mf.2003.25.2.723680. [DOI] [PubMed] [Google Scholar]
- 8.Jiang F., Liu G.S., Dusting G.J., Chan E.C. NADPH oxidase-dependent redox signaling in TGF-beta-mediated fibrotic responses. Redox Biol. 2014;2:267–272. doi: 10.1016/j.redox.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koitabashi N., Danner T., Zaiman A.L., Pinto Y.M., Rowell J., Mankowski J., Zhang D., Nakamura T., Takimoto E., Kass D.A. Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J. Clin. Investig. 2011;121(6):2301–2312. doi: 10.1172/JCI44824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cucoranu I., Clempus R., Dikalova A., Phelan P.J., Ariyan S., Dikalov S., Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005;97(9):900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 11.Kimura S., Zhang G.X., Nishiyama A., Shokoji T., Yao L., Fan Y.Y., Rahman M., Abe Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension. 2005;45(3):438–444. doi: 10.1161/01.HYP.0000157169.27818.ae. [DOI] [PubMed] [Google Scholar]
- 12.Pant I., Rao S.G., Kondaiah P. Role of areca nut induced JNK/ATF2/Jun axis in the activation of TGF-β pathway in precancerous Oral Submucous Fibrosis. Sci. Rep. 2016;6:34314. doi: 10.1038/srep34314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell L., Hobbs G.A., Aghajanian A., Campbell S.L. Redox regulation of Ras and Rho GTPases: mechanism and function. Antioxid. Redox Signal. 2013;18(3):250–258. doi: 10.1089/ars.2012.4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsutsui H., Kinugawa S., Matsushima S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011;301(6):H2181–H2190. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 15.Seddon M., Looi Y.H., Shah A.M. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007;93(8):903–907. doi: 10.1136/hrt.2005.068270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith R.A., Murphy M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N.Y. Acad. Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 17.Apostolova N., Garcia-Bou R., Hernandez-Mijares A., Herance R., Rocha M., Victor V.M. Mitochondrial antioxidants alleviate oxidative and nitrosative stress in a cellular model of sepsis. Pharm. Res. 2011;28(11):2910–2919. doi: 10.1007/s11095-011-0528-0. [DOI] [PubMed] [Google Scholar]
- 18.Dare A.J., Bolton E.A., Pettigrew G.J., Bradley J.A., Saeb-Parsy K., Murphy M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015;5:163–168. doi: 10.1016/j.redox.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rehman H., Liu Q., Krishnasamy Y., Shi Z., Ramshesh V.K., Haque K., Schnellmann R.G., Murphy M.P., Lemasters J.J., Rockey D.C., Zhong Z. The mitochondria-targeted antioxidant MitoQ attenuates liver fibrosis in mice. Int. J. Physiol. Pathophysiol. Pharmacol. 2016;8(1):14–27. [PMC free article] [PubMed] [Google Scholar]
- 20.Chacko B.K., Reily C., Srivastava A., Johnson M.S., Ye Y., Ulasova E., Agarwal A., Zinn K.R., Murphy M.P., Kalyanaraman B., Darley-Usmar V. Prevention of diabetic nephropathy in Ins2+/-AkitaJ mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 2010;432(1):9–19. doi: 10.1042/BJ20100308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yancey D.M., Guichard J.L., Ahmed M.I., Zhou L., Murphy M.P., Johnson M.S., Benavides G.A., Collawn J., Darley-Usmar V., Dell'Italia L.J. Cardiomyocyte mitochondrial oxidative stress and cytoskeletal breakdown in the heart with a primary volume overload. Am. J. Physiol. Heart Circ. Physiol. 2015;308(6):H651–H663. doi: 10.1152/ajpheart.00638.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribeiro R.F., Junior, Dabkowski E.R., Shekar K.C., KA O.C., Hecker P.A., Murphy M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018;117:18–29. doi: 10.1016/j.freeradbiomed.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Travers J.G., Kamal F.A., Robbins J., Yutzey K.E., Blaxall B.C. Cardiac fibrosis: the fibroblast awakens. Circ. Res. 2016;118(6):1021–1040. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinheimer C.J., Lai L., Kelly D.P., Kovacs A. Novel mouse model of left ventricular pressure overload and infarction causing predictable ventricular remodelling and progression to heart failure. Clin. Exp. Pharmacol. Physiol. 2015;42(1):33–40. doi: 10.1111/1440-1681.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ottaviani L., da Costa Martins P.A. Non-coding RNAs in cardiac hypertrophy. J. Physiol. 2017;595(12):4037–4050. doi: 10.1113/JP273129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goh K.Y., Wende A.R., Soorappan R.N., Halade G., Darley-Usmar V., Jinno M., Prabhu S.D., Zhou L. Effect of mitoquinone treatment on cardiac function and pathophysiology in pressure overload-induced Heart failure. FASEB J. 2017;31(1_supplement):59.7. [Google Scholar]
- 27.Wende A.R., Kim J., Holland W.L., Wayment B.E., O'Neill B.T., Tuinei J., Brahma M.K., Pepin M.E., McCrory M.A., Luptak I., Halade G.V., Litwin S.E., Abel E.D. Glucose transporter 4-deficient hearts develop maladaptive hypertrophy in response to physiological or pathological stresses. Am. J. Physiol. Heart Circ. Physiol. 2017;313(6):H1098–H1108. doi: 10.1152/ajpheart.00101.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ismahil M.A., Hamid T., Bansal S.S., Patel B., Kingery J.R., Prabhu S.D. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ. Res. 2014;114(2):266–282. doi: 10.1161/CIRCRESAHA.113.301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rogers A.J., Fast V.G., Sethu P. Biomimetic Cardiac Tissue Model Enables the Adaption of Human Induced Pluripotent Stem Cell Cardiomyocytes to Physiological Hemodynamic Loads. Anal. Chem. 2016;88(19):9862–9868. doi: 10.1021/acs.analchem.6b03105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li D., Wu J., Bai Y., Zhao X., Liu L. Isolation and culture of adult mouse cardiomyocytes for cell signaling and in vitro cardiac hypertrophy. J. Vis. Exp. 2014;87 doi: 10.3791/51357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou L., Aon M.A., Liu T., O'Rourke B. Dynamic modulation of Ca2+ sparks by mitochondrial oscillations in isolated guinea pig cardiomyocytes under oxidative stress. J. Mol. Cell. Cardiol. 2011;51(5):632–639. doi: 10.1016/j.yjmcc.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia-Menendez L., Karamanlidis G., Kolwicz S., Tian R. Substrain specific response to cardiac pressure overload in C57BL/6 mice. Am. J. Physiol. Heart Circ. Physiol. 2013;305(3):H397–H402. doi: 10.1152/ajpheart.00088.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adlam V.J., Harrison J.C., Porteous C.M., James A.M., Smith R.A., Murphy M.P., Sammut I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19(9):1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 34.Graham D., Huynh N.N., Hamilton C.A., Beattie E., Smith R.A., Cocheme H.M., Murphy M.P., Dominiczak A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54(2):322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 35.Maillet M., Lynch J.M., Sanna B., York A.J., Zheng Y., Molkentin J.D. Cdc42 is an antihypertrophic molecular switch in the mouse heart. J. Clin. Investig. 2009;119(10):3079–3088. doi: 10.1172/JCI37694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruan G., Ren H., Zhang C., Zhu X., Xu C., Wang L. Cardioprotective effects of QiShenYiQi dripping pills on transverse aortic constriction-induced heart failure in mice. Front. Physiol. 2018;9:324. doi: 10.3389/fphys.2018.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuroda J., Ago T., Matsushima S., Zhai P., Schneider M.D., Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA. 2010;107(35):15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brock M., Schuoler C., Leuenberger C., Buhlmann C., Haider T.J., Vogel J., Ulrich S., Gassmann M., Kohler M., Huber L.C. Analysis of hypoxia-induced noncoding RNAs reveals metastasis-associated lung adenocarcinoma transcript 1 as an important regulator of vascular smooth muscle cell proliferation. Exp. Biol. Med. 2017;242(5):487–496. doi: 10.1177/1535370216685434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peters T., Hermans-Beijnsberger S., Beqqali A., Bitsch N., Nakagawa S., Prasanth K.V., de Windt L.J., van Oort R.J., Heymans S., Schroen B. Long non-coding RNA Malat-1 is dispensable during pressure overload-induced cardiac remodeling and failure in mice. PLoS One. 2016;11(2):e0150236. doi: 10.1371/journal.pone.0150236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patten R.D., Hall-Porter M.R. Small animal models of heart failure: development of novel therapies, past and present. Circ. Heart Fail. 2009;2(2):138–144. doi: 10.1161/CIRCHEARTFAILURE.108.839761. [DOI] [PubMed] [Google Scholar]
- 41.Dai D.F., Johnson S.C., Villarin J.J., Chin M.T., Nieves-Cintron M., Chen T., Marcinek D.J., Dorn G.W., 2nd, Kang Y.J., Prolla T.A., Santana L.F., Rabinovitch P.S. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011;108(7):837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang T., Maier L.S., Dalton N.D., Miyamoto S., Ross J., Jr., Bers D.M., Brown J.H. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 2003;92(8):912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 43.Dai D.F., Hsieh E.J., Chen T., Menendez L.G., Basisty N.B., Tsai L., Beyer R.P., Crispin D.A., Shulman N.J., Szeto H.H., Tian R., MacCoss M.J., Rabinovitch P.S. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 2013;6(5):1067–1076. doi: 10.1161/CIRCHEARTFAILURE.113.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patten R.D., Aronovitz M.J., Deras-Mejia L., Pandian N.G., Hanak G.G., Smith J.J., Mendelsohn M.E., Konstam M.A. Ventricular remodeling in a mouse model of myocardial infarction. Am. J. Physiol. 1998;274(5 Pt 2):H1812–H1820. doi: 10.1152/ajpheart.1998.274.5.H1812. [DOI] [PubMed] [Google Scholar]
- 45.Froese A.R., Shimbori C., Bellaye P.S., Inman M., Obex S., Fatima S., Jenkins G., Gauldie J., Ask K., Kolb M. Stretch-induced activation of transforming growth factor-beta1 in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2016;194(1):84–96. doi: 10.1164/rccm.201508-1638OC. [DOI] [PubMed] [Google Scholar]
- 46.Yue Z., Zhang Y., Xie J., Jiang J., Yue L. Transient receptor potential (TRP) channels and cardiac fibrosis. Curr. Top. Med. Chem. 2013;13(3):270–282. doi: 10.2174/1568026611313030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doughan A.K., Harrison D.G., Dikalov S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008;102(4):488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 48.Purnomo Y., Piccart Y., Coenen T., Prihadi J.S., Lijnen P.J. Oxidative stress and transforming growth factor-beta1-induced cardiac fibrosis. Cardiovasc Hematol. Disord. Drug Targets. 2013;13(2):165–172. doi: 10.2174/1871529x11313020010. [DOI] [PubMed] [Google Scholar]
- 49.Gane E.J., Weilert F., Orr D.W., Keogh G.F., Gibson M., Lockhart M.M., Frampton C.M., Taylor K.M., Smith R.A., Murphy M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010;30(7):1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 50.Wiegman C.H., Michaeloudes C., Haji G., Narang P., Clarke C.J., Russell K.E., Bao W., Pavlidis S., Barnes P.J., Kanerva J., Bittner A., Rao N., Murphy M.P., Kirkham P.A., Chung K.F., Adcock I.M. Copdmap, Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015;136(3):769–780. doi: 10.1016/j.jaci.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xing Y., Niu T., Wang W., Li J., Li S., Janicki J.S., Ruiz S., Meyer C.J., Wang X.L., Tang D. Triterpenoid dihydro-CDDO-trifluoroethyl amide protects against maladaptive cardiac remodeling and dysfunction in mice: a critical role of Nrf2. PLoS One. 2012;7(9):e44899. doi: 10.1371/journal.pone.0044899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sueblinvong V., Tseng V., Smith T., Saghafi R., Mills S.T., Neujahr D.C., Guidot D.M. TGFbeta1 mediates alcohol-induced Nrf2 suppression in lung fibroblasts. Alcohol Clin. Exp. Res. 2014;38(11):2731–2742. doi: 10.1111/acer.12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dong Y., Liu C., Zhao Y., Ponnusamy M., Li P., Wang K. Role of noncoding RNAs in regulation of cardiac cell death and cardiovascular diseases. Cell Mol. Life Sci. 2018;75(2):291–300. doi: 10.1007/s00018-017-2640-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao J., Xu W., Wang J., Wang K., Li P. The role and molecular mechanism of non-coding RNAs in pathological cardiac remodeling. Int. J. Mol. Sci. 2017;18(3) doi: 10.3390/ijms18030608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang N., Jiang S., Yang Y., Liu S., Ponnusamy M., Xin H., Yu T. Noncoding RNAs as therapeutic targets in atherosclerosis with diabetes mellitus. Cardiovasc. Ther. 2018;36(4):e12436. doi: 10.1111/1755-5922.12436. [DOI] [PubMed] [Google Scholar]
- 56.Viereck J., Kumarswamy R., Foinquinos A., Xiao K., Avramopoulos P., Kunz M., Dittrich M., Maetzig T., Zimmer K., Remke J., Just A., Fendrich J., Scherf K., Bolesani E., Schambach A., Weidemann F., Zweigerdt R., de Windt L.J., Engelhardt S., Dandekar T., Batkai S., Thum T. Long noncoding RNA Chast promotes cardiac remodeling. Sci. Transl. Med. 2016;8(326):326ra22. doi: 10.1126/scitranslmed.aaf1475. [DOI] [PubMed] [Google Scholar]
- 57.Han P., Li W., Lin C.H., Yang J., Shang C., Nuernberg S.T., Jin K.K., Xu W., Lin C.Y., Lin C.J., Xiong Y., Chien H., Zhou B., Ashley E., Bernstein D., Chen P.S., Chen H.V., Quertermous T., Chang C.P. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514(7520):102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han P., Hang C.T., Yang J., Chang C.P. Chromatin remodeling in cardiovascular development and physiology. Circ. Res. 2011;108(3):378–396. doi: 10.1161/CIRCRESAHA.110.224287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cawley S., Bekiranov S., Ng H.H., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Wheeler R., Wong B., Drenkow J., Yamanaka M., Patel S., Brubaker S., Tammana H., Helt G., Struhl K., Gingeras T.R. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116(4):499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- 60.Hansen T.B., Wiklund E.D., Bramsen J.B., Villadsen S.B., Statham A.L., Clark S.J., Kjems J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011;30(21):4414–4422. doi: 10.1038/emboj.2011.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G., Lagarde J., Veeravalli L., Ruan X., Ruan Y., Lassmann T., Carninci P., Brown J.B., Lipovich L., Gonzalez J.M., Thomas M., Davis C.A., Shiekhattar R., Gingeras T.R., Hubbard T.J., Notredame C., Harrow J., Guigo R. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guttman M., Amit I., Garber M., French C., Lin M.F., Feldser D., Huarte M., Zuk O., Carey B.W., Cassady J.P., Cabili M.N., Jaenisch R., Mikkelsen T.S., Jacks T., Hacohen N., Bernstein B.E., Kellis M., Regev A., Rinn J.L., Lander E.S. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singh K.K., Matkar P.N., Quan A., Mantella L.E., Teoh H., Al-Omran M., Verma S. Investigation of TGFbeta1-induced long noncoding RNAs in endothelial cells. Int. J. Vasc. Med. 2016;2016:2459687. doi: 10.1155/2016/2459687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Janakiraman H., House R.P., Gangaraju V.K., Diehl J.A., Howe P.H., Palanisamy V. The long (lncRNA) and short (miRNA) of it: TGFbeta-mediated control of RNA-binding proteins and noncoding RNAs. Mol. Cancer Res. 2018;16(4):567–579. doi: 10.1158/1541-7786.MCR-17-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang P.M.-K., Tang P.C.-T., Chung J.Y.-F., Lan H.-Y. TGF-β1 signaling in kidney disease: from Smads to long non-coding RNAs. Non-coding RNA Res. 2017;2(1):68–73. doi: 10.1016/j.ncrna.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]