

Abstract
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a rare hematodermic myeloid malignancy that is known to be derived from plasmacytoid dendritic cells which are characterized by expression of CD4, CD56, and more specific markers such as CD123. Here, the authors present three cases of BPDCN diagnosed in the past two years and address different available diagnostic modalities such as morphology, immunohistochemistry, flow cytometry, and cytogenetics. Overall, we believe that although BPDCN is a rare diagnosis, it should not be left unchecked. Currently, available immunophenotyping markers are of great help, but the main clue to figure out the problem of BPDCN is clinicopathologic suspicion.
Keywords: Leukemia , Dendritic cells , Iran
What’s Known
Although blastic plasmacytoid dendritic cell neoplasm (BPDCN) is rather a new term, its immunophenotyping and overall clinical course is known.
What’s New
To our knowledge, this is the first case series of this rare disease in Iran. We presented patients with different clinical courses and discussed the available diagnostic modalities. This can be a practicable educational source.
Little is known about cytogenetics of BPDCN. We added two cases to the available literature
Introduction
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a rare myeloid malignancy accounting for less than 1% of all acute leukemias. Before its first introduction in 2008 in the World Health Organization (WHO) classification of hematolymphoid neoplasms, it was designated as a blastic natural killer cell leukemia/lymphoma. However, it is now known to be derived from the recently recognized subtype of dendritic cells named plasmacytoid dendritic cells (PDC) which are characterized by expression of CD4, CD56, and more PDC-specific markers such as CD123 and CD303.1-4
The disease typically presents indolent cutaneous manifestations, although it subsequently progresses byrapid spreading to the bone marrow in its clinical course. This two-faced demeanor makes BPDCN a diagnostic dispute. Furthermore, BPDCN shares significant morphological and immunophenotypic features with other hematolymphoid neoplasms, especially mature NK/T cell lymphomas, rendering it a diagnostic challenge for pathologists. BPDCN is also a treatment problem since no standard optimal chemotherapy regimen is currently available and the systemic dissemination of disease harbors a poor long-term prognosis.4,5
The authors herein present three cases of BPDCN diagnosed in the past two years and, especially, state the different available diagnostic modalities. To date, this is the first report from Iran.
Case Presentation
This retrospective case series was assembled from patients who had referred to the Department of Hematopathology, Molecular Pathology, and Cytogenetics at Shiraz University of Medical Sciences, Shiraz, Iran. The study was directed in conformity with the declaration of Helsinki. Written informed consents were obtained.
Case 1
In February 2017, a 71-year-old man presented with bicytopenia and huge splenomegaly. Laboratory data showed a hemoglobin level of 6.7 g/dL, white blood cell count of 6.5×103/µL, and platelet count of 25×103/µL. On peripheral blood film examination, about 40% blasts were present, accompanied by moderate hypochromic anemia, anisopoikilocytosis, and nucleated red blood cells (10 per 100 white blood cells). Moreover, physical examination revealed a skin ulcer on his chest. No lymphadenopathy was detected (figures 1 and 2).
Figure1.
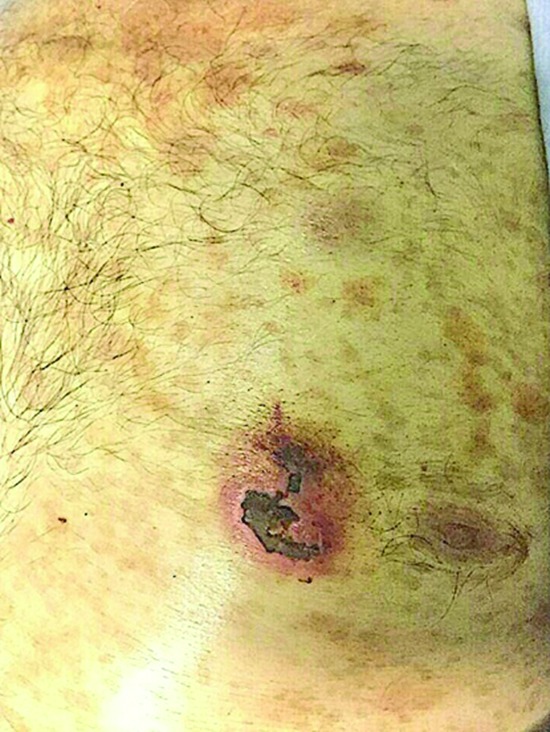
The figure shows ulcerated skin nodule on the chest wall of case 1. The other two patients also had similar skin lesions.
Figure2.
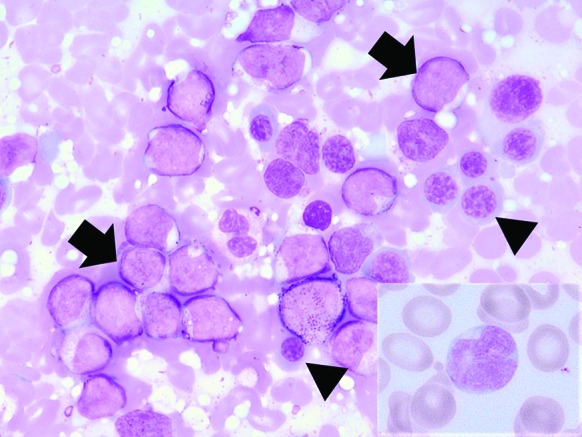
Multiple blasts (arrows) admixed with erythroid precursor cells (arrow heads) (bone marrow imprint, Wright-Giemsa stain, ×1000). Insert shows a blast in the concurrent peripheral blood (Wright-Giemsa stain, original magnification: ×1000). The blasts are medium-sized cells with irregular nuclei and scant cytoplasms which cannot be morphologically recognized as neoplastic dendritic cells. These images are related to case 1.
Bone marrow aspiration and biopsy were consistent with peripheral blood and demonstrated hypercellularity with about 41% blast cells (figure 2). Immunophenotyping by flow cytometry showed the blast cells to express LCA, CD2, CD4, human leukocyte antigen (HLA)-DR, and CD56. Lineage-specific markers, including myeloid (CD13, CD33, CD117, MPO), monocytic (CD11b, CD14, CD64), megakaryocytic (CD61), B-cell (CD19, CD10, CD20) and T-cell (CD3) antigens, were negative. CD34 and TdT immaturity markers were also negative.
A diagnosis of T lymphoproliferative disorder would have been made based on these findings, although further complementary immunophenotyping by immunohistochemistry proved the blast cells to be also positive for CD123 which is more restricted to plasmacytoid dendritic cell marker. Moreover, immunohistochemistry showed reactivity for CD99 while the blasts were negative for plasma cell marker, CD138, Langerhans cell marker, CD1a, cytotoxic T cell marker, perforin, pan-B marker, and pax5.
The conventional cytogenetic study showed normal 46, XY karyotype.
The patient responded to treatment although he experienced a period of relapse seven months later. Further treatment was only fortunate for three months, and he is in another course of relapse these days.
Case 2
A 61-year-old male presented in September, 2016, with a single persistent subcutaneous chest wall nodule located on his left breast, accompanied by cervical and inguinal lymphadenopathy.
An excisional biopsy revealed diffuse monomorphic dermal infiltrate of small- to medium-sized atypical tumor cells which was separated from the overlying epidermis by a distinct Grenz zone (figure 3a and 3b).
Figure3.
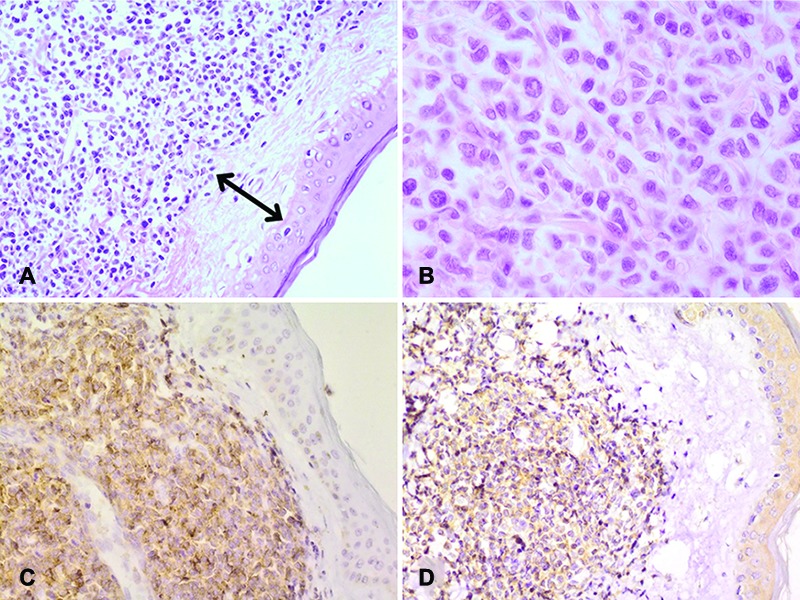
Blast cells are separated from the epidermis by a well-defined Grenz zone (double-arrow) (upper left, H&E, ×200)(A); Monomorphic medium-sized blast cells with irregular nuclei (upper right, H&E, ×400)(B); Positive membranous CD56 immunostaining (lower left, ×200)(C); Positive cytoplasmic CD123 immunostaining (lower right, ×200)(D). Since CD123 is a specific marker for plasmacytoid dendritic cells, this positive result is a key finding without which an accurate diagnosis cannot be made. These images are related to case 2.
Based on immunohistochemistry, the cells were discovered to express leukocyte common antigen (LCA), CD4, CD43, CD56, terminal deoxynucleotidyl transferase (TdT), S100, and CD123. In spite of CD4, they were negative for other T-cell markers such as CD3, CD5, CD7, and CD8. They also did not react with B-cell (CD20 and pax5), myeloid (myeloid peroxidase and CD117), or immaturity (CD34) markers. In addition, the Ki-67 proliferation index was 40% (Figure 3c and 3d).
Peripheral blood showed slightly decreased white blood cell count (4×103/µL), thrombocytopenia (platelet count, 80×103/µL), mild hypochromia, some nucleated red blood cells (2 per 100 white blood cells), and occasional abnormal cleaved cells. No obvious involvement was present. However, the concurrent bone marrow was otherwise occupied by about 90% blast cells.
Flow cytometry results were similar to case 1. Figure 4 illustrates the key flow cytometry findings.
Figure4.

Flow cytometry characteristics of BPDCN in a patient with about 90% blasts in bone marrow. The blasts are positive for CD45, CD4, CD56, HLA-DR, and negative for CD3, B-markers (CD19 and CD10), CD13, and CD14. Selecting a pertinent panel needs high suspicion for this neoplasm. These images are related to case 2.
Conventionalcytogenetics revealed48,XY,+mar×2[1]/46,XY,-13,-17,+mar×2[1]/48,XY,+12,-13,-17,+mar×3[1]/47,XY,-8,+12,-14,+mar×2[1]/46,XY[14].
The patient’s response to systemic chemotherapy was promising and he is in remission after thirteen months from his first diagnosis.
Case 3
A 57-year-old man presented in October, 2015, with a few months history of multiple persistent skin nodules located on his trunk.
Immunohistochemical stains were similar to case 2. However, S100 was negative and ki67 proliferation index was higher (about 60-70%).
Histopathological and flow cytometric examination of bone marrow aspiration and biopsy showed active normocellular marrow without neoplastic involvement. Unfortunately, the patient passed away within a month from the diagnosis.
Discussion
For the first time, we reported a series of Iranian patients with blastic plasmacytoid dendritic cell neoplasm and presented our current practice at Shiraz University of Medical Sciences. Table 1 summarizes the cases.
Table 1.
Main clinical features, immunophenotype, and outcome of patients
| Case# | Sex | Age | First presentation | Site of skin involvement | Hematologic involvement | Immunophenotyping (combination of FC and IHC) | Outcome |
|---|---|---|---|---|---|---|---|
| 1 | Male | 71 | Bicytopenia | Chest wall | Present | LCA, CD2, CD4, HLA-DR, CD56 | 10 months (alive, but in relapse) |
| 2 | Male | 61 | Single skin lesion | Chest wall | Present | LCA, CD4, CD43, CD56, TdT, S100 | 13 months (alive, in remission) |
| 3 | Male | 57 | Multiple skin lesions | Trunk | Absent | LCA, CD4, CD43, CD56, TdT, S100 | 1 month (dead) |
FC, flow cytometry; HLA, human leukocyte antigen; IHC, immunohistochemistry; LCA, leukocyte common antigen; TdT, terminal deoxynucleotidyl transferase
BPDCN is generally known as a rare and clinically aggressive hematodermic neoplasm occurring in the elderly (median age: 66 years old) with a male-to-female ratio of 3:1. However, its exact epidemiological information, such as incidence and prevalence, is not precisely known due to ever-changing terminology and diagnostic criteria especially before the introduction of the 2008 WHO classification. Overall, the estimated incidence is less than 1% of all acute leukemias and a recent systematic review has shown that only 356 cases have been reported to date. Interestingly, the major published investigations on BPDCN treatment modalities include only an average patient number of 22 (range: 6-41 patients).6-8 The three presented cases showed up during two years and as our department offers diagnostic services to approximately two hundred new cases of acute leukemia annually, an incidence of 0.75% is estimated for BPDCN, which is comparable to the generally accepted incidence.
All of our patients were elderly (with a mean age of 63 years), were men and had skin involvement. These findings are expected from BPDCN, although both pediatric and non-cutaneous forms are also on record.8,9 Nguyen et al reported in their literature review that pediatric BPDCN is similar to adult form according to clinical, morphological, and immunophenotypic features. However, the pediatric BPDCN is more associated with favorable prognostic factors such as initial response to treatment. Afterward, Kim et al. have confirmed these statements by their systematic review.8,10
Morphologically, it is impossible to differentiate BPDCN from other acute leukemias. Blasts look like either lymphoblasts or myeloblasts and present as monomorphic medium-sized cells with irregular nuclei, one or more prominent nucleoli, and scant cytoplasms. Moreover, they lack cytoplasmic granules or Auer rods.2 Our cases were in no way exceptional and all had the same blast morphological characteristics.
BPDCN shares significant immunophenotypic characteristics with T/NK cell lymphoma/leukemia, and the definitive dual reactivity for CD4 and CD56 in BPDCN cannot sufficiently distinguish these two entities. At the point, more specific markers for blastic plasmacytoid dendritic cells, such as CD123, TCL1, CD303, CD2AP, BCL11a, and SPIB, are helpful in this differentiation. We used CD123 as well, otherwise; we could not reach definite diagnoses. CD123, which is the interleukin-3 receptor alpha chain, is positive in most BPDCNs. However, it is not specific and can also be expressed in myeloid leukemias, hairy cell leukemia, and Langerhans cell histiocytosis.6,7 Therefore, a panel of antibodies, as well as careful clinical correlation, is required to diagnose BPDCN.
BPDCN is a diagnosis of suspicion and can be easily missed approaching the patients with acute leukemia. Case 1 is an example who would have been falsely diagnosed as a T lymphoproliferative disorderby flow cytometry using routine antibodies, but wisely, later immunohistochemistry showed CD123 reactivity and changed the preliminary diagnosis to BPDCN. This is also true while dealing with cutaneous hematopoietic infiltrates where a correct diagnosis of BPDCN is important because of the high probability of future bone marrow dissemination.
No specific karyotypic abnormality is yet known to be associated with BPDCN, but six recurrent cytogenetic alterations involving 5q (72%), 12p (64%), 13q (64%), 6q (50%), 15q (43%), and monosomy 9 (28%) have been previously proposed by Leroux et al.11 Two of our patients had bone marrow involvement and their specimens have been cultured. One showed normal 46, XY, but the other patient revealed marker chromosomes of unknown origin, trisomy 12, and monosomies of 8, 13, 14, and 17. Since these abnormalities do not fulfill the minimal criteria for clonality (three cells for losses and two cells for gains), we cannot confidently call them true cytogenetic abnormalities. In fact, there is always a possibility that such findings may have happened artefactually during invitro cell divisions. However, we prefer not to ignore them, especially in rare disorders like BPDCN, because they may help in future minimal residual disease assessments. Moreover, the conventional cytogenetic findings of such rare diseases are not yet fully known and any report may aid other investigators to consider them in their own studies.
No optimal treatment is yet standardized for BPDCN although it has been demonstrated that acute lymphoblastic leukemia-oriented chemotherapy regime is more efficient than other treatment options. Regrettably, the patients present poor prognosis with a median overall survival varying from 12 to 16 months. One of our patients died in one month. The other, although alive after ten months, has not yet reached the expected median survival and is currently in a course of relapse. However, the third patient is in remission after thirteen months from diagnosis. Alas, our data and most other available reports cannot precisely express the survival of BPDCN patients due to small sample sizes.4,5,7
Overall, we believe that BPDCN is a rare diagnosis that should not be missed. Currently available immunohistochemical and flow cytometric markers are of great aid, but the key to solve the problem of BPDCN is clinicopathologic suspicion.
Footnotes
Conflict of Interest: None declared.
References
- 1.Laribi K, Denizon N, Besancon A, Farhi J, Lemaire P, Sandrini J, et al. Blastic Plasmacytoid Dendritic Cell Neoplasm: From Origin of the Cell to Targeted Therapies. Biol Blood Marrow Transplant. 2016;22:1357–67. doi: 10.1016/j.bbmt.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 2.Facchetti F, Cigognetti M, Fisogni S, Rossi G, Lonardi S, Vermi W. Neoplasms derived from plasmacytoid dendritic cells. Mod Pathol. 2016;29:98–111. doi: 10.1038/modpathol.2015. [DOI] [PubMed] [Google Scholar]
- 3.Galati D, Corazzelli G, De Filippi R, Pinto A. Dendritic cells in hematological malignancies. Crit Rev Oncol Hematol. 2016;108:86–96. doi: 10.1016/j.critrevonc.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Wilson CS, Medeiros LJ. Extramedullary Manifestations of Myeloid Neoplasms. Am J Clin Pathol. 2015;144:219–39. doi: 10.1309/AJCPO58YWIBUBESX. [DOI] [PubMed] [Google Scholar]
- 5.Riaz W, Zhang L, Horna P, Sokol L. Blastic plasmacytoid dendritic cell neoplasm: update on molecular biology, diagnosis, and therapy. Cancer Control. 2014;21:279–89. doi: 10.1177/107327481402100404. [DOI] [PubMed] [Google Scholar]
- 6.Facchetti F, Fisogni S. Blastic plasmacytoid dendritic cell neoplasm. In: Jaffe ES, Arber DA, Campo E, Harris NL, Quintanilla-Fend L, edithors. Hematopathology. 2th ed. New York: Elsevier; 2016. pp. 943–53. [Google Scholar]
- 7.Pagano L, Valentini CG, Grammatico S, Pulsoni A. Blastic plasmacytoid dendritic cell neoplasm: diagnostic criteria and therapeutical approaches. Br J Haematol. 2016;174:188–202. doi: 10.1111/bjh.14146. [DOI] [PubMed] [Google Scholar]
- 8.Kim MJ, Nasr A, Kabir B, de Nanassy J, Tang K, Menzies-Toman D, et al. Pediatric Blastic Plasmacytoid Dendritic Cell Neoplasm: A Systematic Literature Review. J Pediatr Hematol Oncol. 2017;39:528–37. doi: 10.1097/MPH.0000000000000964. [DOI] [PubMed] [Google Scholar]
- 9.Paluri R, Nabell L, Borak S, Peker D. Unique presentation of blastic plasmacytoid dendritic cell neoplasm: a single-center experience and literature review. Hematol Oncol. 2015;33:206–11. doi: 10.1002/hon.2147. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen CM, Stuart L, Skupsky H, Lee YS, Tsuchiya A, Cassarino DS. Blastic Plasmacytoid Dendritic Cell Neoplasm in the Pediatric Population: A Case Series and Review of the Literature. Am J Dermatopathol. 2015;37:924–8. doi: 10.1097/DAD.0000000000000348. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leroux D, Mugneret F, Callanan M, Radford-Weiss I, Dastugue N, Feuillard J, et al. CD4(+), CD56(+) DC2 acute leukemia is characterized by recurrent clonal chromosomal changes affecting 6 major targets: a study of 21 cases by the Groupe Francais de Cytogenetique Hematologique. Blood. 2002;99:4154–9. doi: 10.1182/blood.v99.11.4154. [DOI] [PubMed] [Google Scholar]