

Summary
The Populus shoot undergoes primary growth (longitudinal growth) followed by secondary growth (radial growth), which produces biomass that is an important source of energy worldwide. We adopted joint PacBio Iso‐Seq and RNA‐seq analysis to identify differentially expressed transcripts along a developmental gradient from the shoot apex to the fifth internode of Populus Nanlin895. We obtained 87 150 full‐length transcripts, including 2081 new isoforms and 62 058 new alternatively spliced isoforms, most of which were produced by intron retention, that were used to update the Populus annotation. Among these novel isoforms, there are 1187 long non‐coding RNAs and 356 fusion genes. Using this annotation, we found 15 838 differentially expressed transcripts along the shoot developmental gradient, of which 1216 were transcription factors (TFs). Only a few of these genes were reported previously. The differential expression of these TFs suggests that they may play important roles in primary and secondary growth. AP2, ARF, YABBY and GRF TFs are highly expressed in the apex, whereas NAC, bZIP, PLATZ and HSF TFs are likely to be important for secondary growth. Overall, our findings provide evidence that long‐read sequencing can complement short‐read sequencing for cataloguing and quantifying eukaryotic transcripts and increase our understanding of the vital and dynamic process of shoot development.
Keywords: PacBio Iso‐Seq, stem cell maintenance, primary growth, secondary growth, transcription factor
Introduction
All woody plants and some non‐woody trees undergo primary and secondary growth. During primary growth, the primary meristems, including the shoot apical meristem (SAM) and procambium, which were already established during embryogenesis, maintain apical growth by providing stem cells to the primary growth zone (Aichinger et al., 2012; Miyashima et al., 2013; Ohashi‐Ito and Fukuda, 2010; Weigel and Jurgens, 2002). Once the primary vascular tissues that are derived from procambium are established, the procambium develops into the cambium, which produces cells that develop into vascular tissues. This stage indicates the beginning of secondary growth where the shoot begins to undergo radial growth that results in increased girth. In trees, the biomass produced by secondary growth is a source of biofuel, and understanding how the transition from primary to secondary growth is regulated can aid efforts to improve biomass production.
Several genes involved in the regulation of primary growth from the SAM, which provides cells for apical growth and lateral organogenesis, have been identified. The SAM consists of a central zone (CZ), a surrounding peripheral zone (PZ) and an underlying rib zone (Aichinger et al., 2012). In the CZ, pluripotent stem cells and the organizing centre (OC) together form a stem cell niche. In Arabidopsis thaliana, a regulatory loop between the CLAVATA (CLV) and WUSCHEL (WUS) genes maintains stem cell fate in the SAM (Schoof et al., 2000). SHOOT MERISTEMLESS (STM) also competes with CLV to initiate and maintain the SAM in Arabidopsis (Clark et al., 1996). The CUP‐SHAPED COTYLEDON (CUC)/STM regulatory pathway is associated with the establishment of organ boundaries and the initiation of the SAM (Aida et al., 1999; Long and Barton, 1998).
Different sets of genes regulate lateral organ development from the PZ, which is located at the tip of the meristem. In these regions, small groups of cells show signs of differention based on upregulation of markers for asymmetric cell division and differentiation in Arabidopsis, ZWILLE/PINHEAD (ZWI), PIN‐FORMED1 (PIN1), and REVOLUTA (REV) (Aida et al., 2002; Carraro et al., 2006; Lynn et al., 1999; Otsuga et al., 2001). AINTEGUMENTA (ANT) and LEAFY (LFY) are subsequently activated, but in a symmetric manner.
The following section of primary growth along the longitudinal direction is the procambium. The procambium asymmetrically gives rise to xylem and phloem precursor cells, which ultimately differentiate into the centripetal xylem and centrifugal phloem to form vascular bundles that are arranged along a ring passing through the ground tissue (Baucher et al., 2007; Carlsbecker and Helariutta, 2005). In Arabidopsis embryos and leaves, the polar auxin efflux protein PIN1 is activated by the HD‐ZIPIII protein Arabidopsis thaliana Homeobox Gene 8 (ATHB8), which may in turn be activated by MONOPTEROS (MP)/AUXIN RESPONSE FACTOR5 (ARF5) (Aida et al., 2002; Donner et al., 2009; Ohashi‐Ito and Fukuda, 2010). These processes form a positive regulatory loop from auxin to MP‐ATHB8‐PIN1, and PIN1 also participates in auxin flow (Miyashima et al., 2013). But the regulatory mechanism in poplar is unclear.
Secondary growth takes place when the vascular cambium initials differentiate from the procambium within the vascular bundles (fascicular cambium) and from parenchymatous rays in the interfascicular regions (interfascicular cambium) (Baucher et al., 2007). Similar to primary growth, most of the genes involved in secondary growth have been identified based on molecular genetic studies in Arabidopsis, which has a well‐annotated genome sequence available. But research has mainly focused on the Arabidopsis root. In Arabidopsis, DOF5.6/HCA2 contributes to the regulation of interfascicular cambium formation and vascular tissue development (Guo et al., 2009). The MYB transcription factor (TF) ALTERED PHLOEM DEVELOPMENT (APL) is required for phloem differentiation and inhibition of xylem differentiation in the phloem positions in Arabidopsis roots (Bonke et al., 2003). The NAC domain TFs, SECONDARY WALL‐ASSOCIATED NAC DOMAIN PROTEIN1 (SND1), VASCULAR‐RELATED NAC‐DOMAIN6 (VND6) and VND7, play essential roles in xylem vessel differentiation in Arabidopsis (Kubo et al., 2005; Zhong et al., 2006). Each activates the expression of 12 downstream TF genes, which are mostly MYBs, or directly affects the transcription of genes associated with lignin, cellulose and hemicellulose biosynthesis (Wang and Dixon, 2012). The feedback mechanism between PXY‐CLE41 can affect vascular cell division and determine the orientation of cell division in aspen and Arabidopsis (Etchells and Turner, 2010; Fisher and Turner, 2007).
Some of the pathways that regulate secondary growth in Arabidopsis are conserved in trees. For example, PXY‐CLE41 signalling acts to regulate both the rate of cambial cell division and secondary growth in aspen (Populus tremuloides) (Etchells et al., 2015). In Populus, the same type of regulatory mechanism involving the ARBORKNOX1 (ARK1, ortholog of STM) genes has been shown to control cell fate in the vascular cambium (Du et al., 2009; Groover et al., 2006; Liu et al., 2015). PtaLBD1 promotes the expression of APL and restricts the expression of ARK1 and ARK2, which are involved in maintaining the cambium meristem (Yordanov et al., 2010).
Populus is used as a model system for secondary growth because it has better wood formation than the model plant Arabidopsis and is an important source of biofuel (Jansson and Douglas, 2007). However, secondary growth in the Populus shoot is quite different from that in the Arabidopsis root and it is therefore necessary to identify the genes that regulate secondary growth in poplar (Du and Groover, 2010). Since the release of the black cottonwood tree, Populus trichocarpa, draft genome (Tuskan et al., 2006), more genes have been discovered. But not all the functional transcripts have been identified. Microarray and RNA‐seq technologies have also been extensively used to analyze Populus cambium development, vascular tissue formation and phloem and xylem differentiation during secondary growth (Aspeborg et al., 2005; Hertzberg et al., 2001; Ko et al., 2012a; Schrader et al., 2004; Sundell et al., 2017). In one study, 271 differentially expressed fragments that represent genes that may be involved in the regulation of secondary growth were sequenced from different stem segments (Prassinos et al., 2005). In a microarray study, 3016 genes were found to be differentially expressed during stem development (Dharmawardhana et al., 2010). But due to technical limitations, these transcripts represent far less than the 28 294 annotated genes that have been found to be expressed during cambial growth and wood formation by RNA‐seq methods (Sundell et al., 2017).
Short‐read sequencing using the Illumina platform, is a powerful method for quantifying gene expression. However, the limitations of short‐read sequencing lead to a number of computational challenges and hamper transcript reconstruction and the detection of splice events. In the last few years, an increasing number of Pac‐Bio full‐length transcriptomes have been generated. The PacBio Iso‐Seq (isoform sequencing) platform provides long reads—often up to 10 kb, making it possible to accurately reconstruct full‐length splice variants. These studies have led to the discovery of thousands of novel genes and alternatively spliced isoforms in several species, including human, sorghum, maize, yeast, moso bamboo and cotton (Abdel‐Ghany et al., 2016; Wang et al., 2016, 2017, 2018). Also, third‐generation sequencing of human embryonic stem cells combined with previously generated EST and RNA‐seq databases uncovered 2103 novel isoforms, ~10% of which were derived from unannotated genetic loci, indicating that full‐length isoform identification and quantification can generate a high‐confidence isoform dataset (Au et al., 2013). Overall, the above studies provide evidence that long‐read sequencing complements short‐read sequencing in cataloguing and quantifying eukaryotic transcripts and can aid in finding more alternatively spliced isoforms.
Here, we adopted joint PacBio full‐length sequencing and RNA‐seq analysis to identify specific genes and pathways involved in the transition from primary to secondary growth in Populus. We obtained 15 838 differentially expressed transcripts (DETs) from the apex and different sections of the stem, including 1216 TFs. The differentially expressed transcripts may be related to primary and secondary growth.
Results and discussion
Anatomic analysis of the Populus stem
To determine key factors that control the transition from primary growth to secondary growth, we used PacBio to sequence the transcriptome of P. deltoides × P. euramericana cv. ‘Nanlin895’ stems undergoing these transitions. Because it is widely planted in south China, grows quickly and is easily transformed (Song et al., 2011; Zhao et al., 2013; Zhu et al., 2013), we used the ‘Nanlin895’ cultivar for this study. To determine which plant materials to combine for PacBio sequencing, we performed anatomical analysis of the shoot apex (hereafter referred to as apex) and successive internodes below the apex, referred to as plastochron indices IN1, IN2, IN3, IN4 and IN5, respectively, as described in a previous study (Larson and Isebrands, 1971). The apex consists of an apical dome zone of stem cells and a peripheral zone where leaf primordia initiate. The cells located in the SAM have a relatively flat shape (Figure 1). And the procambium, which gives rise to primary phloem and xylem, initiates acropetally through the residual meristem beneath the SAM. The primary phloem and xylem are dominant in IN1, IN2 and IN3, as described previously (Dharmawardhana et al., 2010). The first signs of interfascicular parenchyma cells that differentiate into a full ring of cambium initials are visible in IN3, indicating the beginning of radial secondary stem growth. IN5 has well‐developed secondary phloem tissue and secondary xylem vessels, as well as fibres with well‐lignified secondary walls (Figure 1). With the development of the vascular cambium and initiation of secondary growth in IN4‐5, stem elongation ceases while the diameter of the stem girth continues to increase. From these observations, IN4‐5 represents secondary growth. Therefore, we selected these internodes to represent the transition from primary growth to secondary growth.
Figure 1.

Anatomic analysis of the poplar apex and stem. (a) Cross section of the shoot apex. (b) Transverse section of internode 1. (c) Transverse section of internode 2. (d) Transverse section of internode 3. (e) Transverse section of internode 4. (f) Transverse section of internode 5. SAM: shoot apical meristem, PC: procambium, MC: metacambium, PP: protophloem, MP: metaphloem, Xy: xylem, Ph: phloem, FC: fascicular cambium, IC: interfascular, C: cambium. Scale bars, 50 μm.
Sequencing of the Populus transcriptome using the PacBio Iso‐Seq platform
To identify genes that regulate the SAM and the transition from primary to secondary growth in ‘Nanlin 895’, we sequenced the transcriptomes of different regions of the Populus stem using the PacBio Iso‐Seq (isoform sequencing) platform. This platform provides long reads of complete isoforms. To identify transcripts that are as long as possible, high‐quality RNA was used for Iso‐Seq (see Methods for details). Total RNA was extracted from three regions of the Populus Nanlin895 stem: (i) apex, (ii) a mixture of IN1‐3, and (iii) a mixture of IN4‐5. To avoid loading bias, which is the preferential sequencing of shorter transcripts, multiple size‐fractionated libraries including 0–1, 1–2, 2–3 and 3–10 kb libraries were made using a SageELF device. Libraries were sequenced on the PacBio RS II platform using the latest P6–C4 chemistry with 27 SMRT cells, yielding 1 613 676 Reads of Inserts (ROI), of which 887 877 (55%) were full‐length (containing the 5′ barcoded primer, 3′ barcoded primer and the poly (A) tail) (Data S1).
We used an isoform‐level clustering algorithm, Iterative Clustering for Error Correction (ICE) (Gordon et al., 2015), to improve consensus accuracy and polished full‐length consensus sequences from ICE using RS_IsoSeq (v2.3.0). We obtained 308 549 consensus isoforms, of which 234 687 were high‐quality consensus transcript sequences. High‐quality transcript sequences were mapped to the Populus genome (Phytozome, Populus trichocarpa Pt_v3.0) using GMAP. Short‐read Illumina sequencing of the same samples was done to quantify the Iso‐Seq non‐redundant isoforms. ToFu (Gordon et al., 2015) processing yielded 87 150 non‐redundant isoforms (Table 1). Of the three tissues, the apex had the highest proportion of tissue‐specific isoforms (22 146; 26.2%), followed by IN4‐5 (18 464; 21.6%), and IN1‐3 (17 017; 19.9%) (Figure 2a). Longer isoforms were identified from Iso‐Seq than from the reference database (Pt_v3.0) and more exons were found in this study (Figure 2b,c). The non‐redundant transcript isoforms were used in subsequent analyses (Figure S1).
Table 1.
PacBio Iso‐Seq output statistics
| Samples | Number of SMRT cells | Polymerase reads | Number of insert reads | Number of insert base reads | Number of five prime reads | Number of three prime reads | Number of poly‐A reads | Number of full‐length reads | Number of full‐length non‐chimeric reads | Average full‐length non‐chimeric read length | Full‐length percentage (FL%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Apex | 9 | 1 352 628 | 604 385 | 1 191 087 341 | 379 572 | 407 917 | 400 207 | 320 351 | 315 844 | 7822 | 53.00 |
| IN1‐3 | 9 | 1 352 628 | 560 769 | 1 058 569 542 | 362 406 | 387 945 | 380 417 | 310 043 | 306 526 | 7743 | 55.29 |
| IN4‐5 | 9 | 1 352 628 | 448 522 | 853 915 854 | 304 979 | 322 000 | 317 875 | 268 186 | 265 507 | 8221 | 59.79 |
Figure 2.

Comparison of Populus trichocarpa v3.0 (Reference) and PacBio Iso‐Seq data isoform annotations. (a) Identified isoforms in three samples (Apex, IN1‐3 and IN4‐5). (b) Distribution of the percentage transcripts with different exon numbers for reference and PacBio Iso‐Seq data. (c) Comparison of gene model and PacBio Iso‐seq isoform length. (d) A pie chart showing the percentage of PacBio Iso‐Seq isoforms that are the same as existing gene models, novel isoforms of known genes and novel isoforms of novel genes.
Isoform detection and tissue‐specific alternative spliced isoforms
We compared the 87 150 isoforms against the Populus genome gene set (Pt_v3.0), which contains 71 013 isoforms from 41 335 genetic loci. We classified isoforms into three groups as follows: (i) 25 092 known isoforms detected by Iso‐Seq and mapped to the gene set, (ii) 59 977 new isoforms of annotated genes and (iii) 2081 isoforms, which belong to 1575 likely novel genetic loci, that do not overlap with any annotated gene (Figure 2d). In a study where the maize transcriptome was obtained by single‐molecule long‐read sequencing, out of 111 151 isoforms identified, 62 547 (57%) were new isoforms and 2803 encoded 2253 new genes. In a sorghum study, 27 860 isoforms were identified, including 11 342 (40.7%) new isoforms and 3141 isoforms encoding new genes. The high percentage of new isoforms identified by us and other groups demonstrates that PacBio full‐length sequencing can provide a more comprehensive set of isoforms than next‐generation sequencing.
To determine if the 1575 putative novel Populus genes are present in other plants, we conducted BLASTX searches against Swiss‐Prot (E‐value ≤e−10; see Methods). In total, 965 (46.3%) of these isoforms were annotated in the Swiss‐Prot database, and the remaining isoforms were unannotated. We integrated the 2081 transcripts from new genes and the 59 977 new isoforms of known genes to improve the Populus reference genome annotation (Data S2). By combining our newly identified isoforms with the 71 013 existing isoforms in the Populus gene set, we generated an updated Populus reference sequence database containing 135 071 isoforms of 42 910 genetic loci.
We used AStalavista (Foissac and Sammeth, 2007) to determine the number of transcripts in the apex, IN1‐3 and IN4‐5 generated by each of the five main types of alternative splicing (intron retention, exon skipping, alternative 3′ splice site, alternative 5′ splice site and mutually exclusive exon). Among all alternative splicing events, intron retention predominated, accounting for 58.15–64.2% of alternatively spliced transcripts (Figure 3a, Data S3). There were more alternatively spliced transcripts found among Iso‐Seq transcripts compared with previously annotated transcripts (Figure 3b). We found that several genes reported to be important for plant development had more isoforms than previously annotated, such as sucrose synthase Pt‐SUS2.2 (Potri.002G202300, 23 isoforms) and auxin response factor PtARF2 (Potri.003G163600, 18 isoforms) (Figure 3c). We selected three genes ARF8 (PB.6368), SUTR11 (PB.8036) and CYP707A4 (PB.13883) and verified the production of the isoforms by RT‐PCR (Figure S2) and found that there was more than one isoform of these genes. Interestingly, different isoforms of CYP707A4 were expressed during primary and secondary growth.
Figure 3.
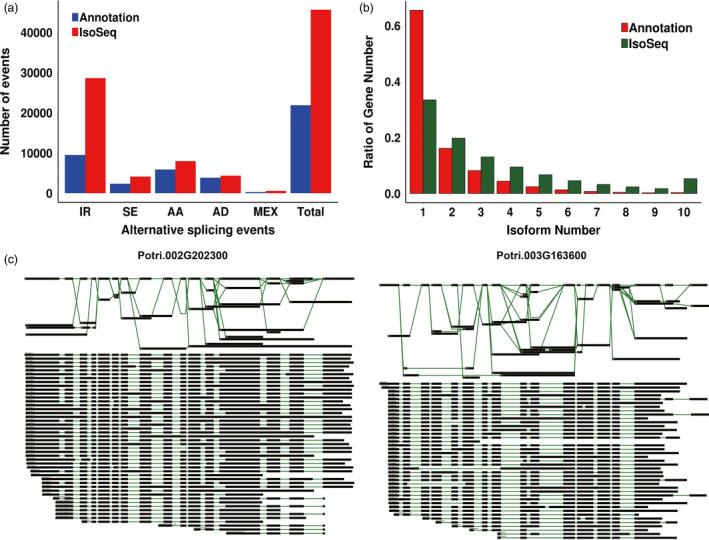
Alternative splicing events and different isoforms from Iso‐seq. (a) The total number of alternative splicing (AS) events in genes based on Iso‐Seq data and annotated gene models. Annotation, AS events in genes based on gene models; Iso‐Seq, AS events in genes based on Iso‐Seq reads. IR, intron retention. SE, exon skipping. AA, alternative 3′ splice site. AD, alternative 5′ splice site. MEX, mutually exclusive exon. (b) The number of genes with one or more splice isoforms based on gene annotation and Iso‐Seq data. (c) Different isoforms of Potri.002G202300 and Potri.003G163600.
Long non‐coding RNA identification
Long non‐coding RNAs (lncRNAs) constitute an important class of regulators of gene expression in most eukaryotes (Flynn and Chang, 2014). We used four computational approaches (see Methods) to identify lncRNAs from the 87 150 PacBio Iso‐Seq isoforms. By filtering and excluding transcripts with an open reading frame (ORF) > 300 bp (Clamp et al., 2007), we finally obtained 1187 lncRNAs (493 in the apex, 413 in IN1‐3 and 467 in IN4‐5). Only 33 lncRNAs were found in all three samples (Data S4).
The length of lncRNAs varied from 273 to 5560 bp, with the majority (>63%) having a length ≤1000 bp. The mean length was 1052 bp, which was much shorter than the mean length of all 87 150 isoforms (2417 bp). In Populus, lncRNAs contain an average of 1.77 exons compared with 7.10 for mRNAs. Mapping lncRNAs to chromosomes revealed that they have a distribution similar to that of mRNAs; both are enriched outside of pericentromeric regions (Figure 4). By comparing the 1187 lncRNAs to Populus lncRNAs (PNRD database), 553 were identified. However, only 31 were previously annotated, which indicates that more transcripts can be identified with PacBio Iso‐Seq than with other methods.
Figure 4.
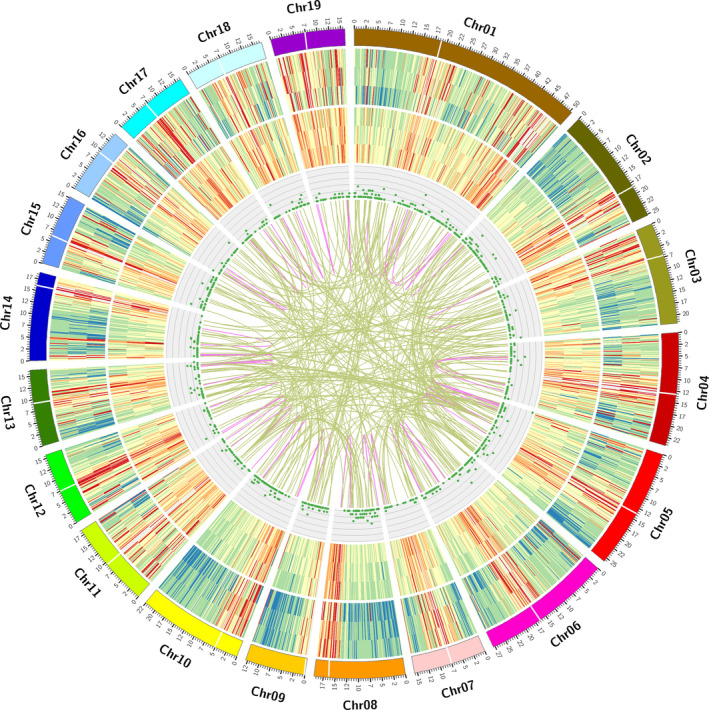
CIRCOS visualization of genomic and transcriptomic features. (a) Poplar chromosomes. (b) Gene density of different samples (Apex, IN1‐3, and IN4‐5). (c) Transcript density of different samples (Apex, IN1‐3, and IN4‐5). (d) Long non‐coding RNA (IncRNA) distribution. (e) Fusion transcript distribution: intra‐chromosome (purple); inter‐chromosome (yellow). The location of centromere regions is labelled according to previous research (Pinosio et al., 2016).
Fusion transcript identification
A fusion transcript is a chimeric RNA encoded by a single fusion gene or by two different genes that are subsequently joined by trans‐splicing. Generation of fusion transcripts involves the splicing machinery, suggesting either trans‐splicing of distinct genes or splicing of chimeric genes formed by somatic chromosomal rearrangements. Gene fusion, which is caused by somatic chromosomal rearrangement, is a common feature in humans, but only a few examples of gene fusion have been described in plants. Long‐distance movement of a homeobox fusion from mutants to wild‐type scions in grafting experiments causes changes in leaf morphology (Kim et al., 2001; Wang et al., 2016). Here we identified 356 full‐length transcripts that map to two or more loci in the genome and thus are considered fusion transcripts (Data S5). The highest number of tissue‐specific fusion transcripts was found in the apex (119), followed by IN4‐5 (117) and IN1‐3 (78). Only 14 fusion transcripts were found in all three samples. The fusion transcripts were validated using 18 transcriptome datasets (described below). Specifically, fusion transcripts were required to have at least one uniquely mapping paired‐end read using bowtie 2 (Langmead and Salzberg, 2012), and 25 transcript fusions met this criterion. Gene ontology (GO) analysis of fusion transcripts revealed that most were associated with ATP binding (22 genes), transmembrane transport (10 genes) and proteolysis (5 genes).
PacBio full‐length sequencing extends the Populus annotation and increases the accuracy of transcript quantification
The P. trichocarpa genome has been sequenced and annotated, but due to technical limitations, transcript sequences and annotations continue to be updated as more data become available. For example, the latest annotation (Pt_v3) contains more annotations than the previous versions because RNA‐seq data have been integrated (Liu et al., 2014). The P. trichocarpa genome is also used as a reference for other Populus species, but due to species divergence, low mapping ratios are observed when mapping transcriptomes to the current annotation (Chen et al., 2015; Liu et al., 2014). In order to compare the expression level determined by RNA‐seq based on PacBio reads, eighteen mRNA samples from six different tissues (Apex, IN1‐IN5) were subjected to 2 × 150 bp paired‐end sequencing using the HiSeq platform. In total, 414.2 million trimmed reads were actually mapped to the reference genome, of which 388.8 million reads (88.8%) mapped within known exons in the updated Nanlin895 annotation database (Data S6). In total, we quantified 126 690 isoforms from all samples. The mapped RNA‐seq reads were used to quantify all Pac‐Bio Iso‐seq isoforms from each sample based on the fragments per kilobase of exon per million fragments mapped (FPKM) values. Hierarchical clustering analysis of RNA‐Seq data showed that the data from biological repeats clustered together (Figure S3).
Different regulatory mechanisms have been adopted for primary and secondary growth
To identify key factors involved in the transition from primary growth to secondary growth, we identified 15 838 DETs from 9950 differentially expressed genes (DEGs) based on RNA‐seq data, which accounted for 18.17% of all isoforms identified in this study (Data S7). Among all transcripts differentially expressed between two adjacent stem regions, most (2747) were identified as being differentially expressed between the apex and IN1, and the smallest number of DETs (183) were identified between IN4 and IN5. Using the K‐means clustering algorithm, 15 834 DETs were grouped into 9 clusters (K1‐9), which represent 99.97% of all DETs (Figure 5a, Data S8). Transcripts from the K6, K4, K8, K1, K5 and K9 clusters were predominantly expressed in the apex and IN1‐5, respectively. Expression levels of transcripts from the K2 cluster increased gradually from the apex to IN5, and the expression level was highest in IN4‐5, which indicates that these transcripts are potentially involved in the transition to secondary growth. Expression levels of transcripts from the K7 cluster increased gradually from the apex to IN3 and reached a steady state in IN4‐5, which was similar to the expression pattern of K2 transcripts. In contrast, the expression levels of transcripts from the K3 cluster decreased from the apex to IN3 and reached a steady state in IN4‐5. These transcripts are potentially involved in primary growth.
Figure 5.

K‐means clustering and enrichment analysis of transcripts differentially expressed between the apex and IN1‐5. (a) K‐means clustering showing the transcriptome expression profiles. Nine clusters were identified based on expression levels in six developmental zones (Apex, IN1, IN2, IN3, IN4 and IN5). (b) Gene Ontology enrichment among the nine clusters. Yellow to red, significant enrichment; white, not significant.
To better understand the functions of the genes expressed in different regions of the stem, we performed GO enrichment analysis with the updated annotation as the background (Figure 5b, Data S9). Most enriched GO terms were found for the transcripts differentially expressed during the transition from the apex to IN1 (K6), which is consistent with this transition having the most differentially expressed transcripts. Previous studies have reported that histone modification, DNA methylation or demethylation, and chromatin silencing by small RNAs are important for plant development (Feng et al., 2010), and in this study, genes involved in these processes were enriched in the apex. These genes include Decrease in DNA Methylation 1 (DDM1), DDM2, chromomethylase 3 (CMT3), AGO4, AGO6, ATXR6/SDG34, ASHH3/SDG7, ASHR3/SDG4 and CURLY LEAF (CLF/SDG1). DDM1, DDM2 and CMT3 are required to maintain CG and CHG methylation in Arabidopsis and rice (Gendrel et al., 2002; Habu et al., 2015; Jackson et al., 2002; Numa et al., 2015; Ronemus et al., 1996). SDG1 and SDG34 are responsible for methylation of H3K27, which is required for transcriptional repression (Jacob et al., 2009; Shafiq et al., 2014; Wood et al., 2006). ASHH3/SDG7 and ASHR3/SDG4 are members of the trithorax group (TrxG) family, but their function is unknown (Baumbusch et al., 2001). AGO4 and AGO6, which were previously reported to be expressed in the SAM of Arabidopsis (Havecker et al., 2010), may act sequentially to mediate RNA‐directed DNA methylation (Duan et al., 2015). High expression of these epigenetic‐related genes in the Populus apex, suggests that they may control the expression of a large number of different genes in the SAM.
Transcripts that were predominantly expressed in the apex (K3 and K6) and IN4‐5 (K2) function in similar pathways, such as DNA replication, cell cycle, cell division, cytokinesis and cytoskeleton organization, but genes from different families of cyclin and cyclin‐dependent kinases, E2Fs, and different members of the microtubule‐associated protein (MAP) family were found in each cluster. Expression levels of cyclin A, B, D and cyclin‐dependent kinases A and B, E2F3, E2F TARGET GENE 1 (ETG1) and DP‐E2F‐LIKE 1 (DEL1) were highest in the apex. The genes that were highly expressed in the apex are canonical cell cycle genes, except for DEL1, which was previously reported to be a repressor of E2FA and to be regulated by light in Arabidopsis (Berckmans et al., 2011; Inzé and Veylder, 2006). Peak expression of P‐type cyclins (CYCPs) was observed in IN4‐5. CYCP2;1 was reported to promote meristem cell division mediated by STIP/WOX9 and sucrose signals in Arabidopsis (Peng et al., 2014). In our study, the expression patterns of CYCPs suggest that they may mainly regulate secondary growth. Different MAPs can affect the organization and functions of microtubule arrays, which can control the direction of cell division and expansion during plant morphogenesis and development (Hamada, 2014). For example, MAP70‐5 was found to be upregulated upon xylem tracheary element differentiation and to regulate secondary wall patterning in Arabidopsis wood cells (Pesquet et al., 2010). Consistent with different functions of MAPs during primary and secondary growth, we found that MAP65‐6, MAP65‐8 and MAP70‐5 were expressed primarily in IN4‐5, but MAP65‐1, MAP65‐3, MAP65‐5 and MAP70‐1 were expressed primarily in the apex.
The expression patterns of cell cycle‐ and cell division‐associated DETs may give insight into their roles in the regulation of cell division during primary and secondary growth. The SAM can be divided into distinct cell layers; cells from the epidermal layer L1 and subepidermal layer L2 undergo anticlinal cell divisions, while cells from the corpus or L3 layer divide in many planes (Meyerowitz, 1997). In contrast, during secondary growth, the vascular cambium increases the stem diameter and circumference by undergoing periclinal divisions and anticlinal divisions, respectively, resulting in a developmental continuum of secondary phloem and xylem (Chaffey, 1999). Thus, transcripts differentially expressed during primary and secondary growth may play different roles in anticlinal and periclinal divisions.
Primary and secondary cell wall‐associated transcripts are predominantly expressed in different regions of the stem
During Populus shoot growth, primary cell walls play a vital role in the expansion of actively growing cells, and secondary cell walls play a larger role in water transport and mechanical support. Consistent with the different roles of primary and secondary cell walls, GO enrichment analysis indicated that expression of genes involved in cell wall organization or biogenesis peaked in IN1 (K4) and IN4‐5 (K2). Cell wall modification and pectin catabolic process genes were expressed in IN1, and secondary cell wall biogenesis genes were expressed in IN4‐5, which was also enriched in genes involved in phloem or xylem histogenesis.
In total, the expression levels of 469 protein‐coding transcripts were found to differ between regions of the stem. Among these, 18 were novel isoforms of annotated genes and 187 were new genes (Data S10). For example, cellulose synthases (CESAs) CESA1, CESA3 and CESA6, which are required for cellulose synthesis in primary cell walls in Arabidopsis (Desprez et al., 2007), were expressed predominantly in the apex (K6), apex‐IN2 (K3), IN1 (K4) and IN2 (K8). In the apex to IN2, where cells are undergoing cell expansion, expansins and cellulose synthases were highly expressed (Data S10). The high levels of expansins gene expression in IN1‐IN2 suggest that cell walls are loosening and cells are expanding during this developmental stage (Cosgrove, 2015). Although expansins were annotated in the first version of the Populus genome (Pt_v1), their expression patterns have not been described (Cosgrove, 2015; Sampedro et al., 2006). We identified two transcripts encoding CSLD5 that are predominantly expressed in the apex and IN1‐2, although CSLD5 was previously reported to play roles late in the cell cycle and to contribute to wall formation in new cells in the Arabidopsis shoot apex (Yang et al., 2016). The expression levels of CESA9, CELLULOSE SYNTHASE‐LIKE A2 (CSLA2), CSLA5 and CSLA9 also peaked in the apex, IN1 or IN2.
Expression of CESA4, CESA7 and CESA8 (Taylor et al., 2003), which are involved in cellulose synthesis in the secondary cell wall in Arabidopsis, peaked in IN4‐5. We identified three new alternatively spliced isoforms encoding CESA8. Apart from cellulose synthase, two major alternatively spliced COBRA‐like 4 (COBL4) transcripts were expressed predominantly in IN4‐5; COBL4 has been shown to affect cellulose microfibril crystallinity in rice (Liu et al., 2013). It seems that the expression patterns of these genes are consistent with their known functions in secondary cell wall synthesis. But the functions of CSLA09, GH9B3 and BXL1 in secondary growth have not been reported previously and need to be studied in the future.
Transcription factor dynamics during shoot development
TFs play important roles in developmental transitions. In addition, many TFs that are important for shoot development in Populus have been identified (Dharmawardhana et al., 2010; Prassinos et al., 2005). Therefore, we investigated the expression patterns of TFs during the transition from primary to secondary growth. Of the 8347 TFs expressed during Populus shoot development, 1216 were differentially expressed along a developmental gradient (Figure 6a). To determine the co‐expression and correlation networks of all differentially expressed TFs, weighted correlation network analysis (WGCNA) was conducted. Five modules of highly correlated TFs that are highly expressed during different stages of Populus shoot development (coloured turquoise, blue, brown, yellow and grey in Figure 6b; Figure S4 and Data S11) were identified. Most TFs (403) belong to the turquoise module, in which peak expression of most TFs is in the apex. This suggests that TFs play an important role in stem cell maintenance and lateral meristem cell differentiation. TFs belonging to the brown (94) and blue (319) modules have decreased and increased expression from the apex to IN5, respectively. The yellow module contains the smallest number of TFs (58), which are most highly expressed in IN1‐IN2.
Figure 6.

Weighted Gene Co‐expression Network Analysis (WGCNA) of transcription factors. (a) Cluster dendrogram of transcription factors based on expression levels in the six developmental zones (Apex, IN1, IN2, IN3, IN4 and IN5). Each branch represents a gene and each colour below represents a gene co‐expression module. Dynamic tree cut indicates the modules divided based on the gene clustering results. Merged dynamic indicates the modules divided by combining modules with similar expression patterns. (b) Heatmap of transcription factor expression patterns in different samples (Apex, IN1, IN2, IN3, IN4 and IN5). The expression patterns of five modules are shown by the heatmap. The colour bar indicates expression levels from low (blue) to high (red). (c) Distribution of transcription factor families in different WGCNA modules. Each colour represents a co‐expression module, and the numbers indicate the number of transcription factors in the module. (d) Transcription factors highly expressed in the apex, IN1‐3 and IN4‐5.
Using the Arabidopsis TF database TFDB 3.0 (Jin et al., 2014) and the TF prediction algorithm HMMER 3.0 (Finn et al., 2011), TFs were classified into 58 families. Family‐specific expression was observed in different regions of the shoot (Figure 6c). Expression of most AP2 (AP2‐ERF family), ARF (B3 family), YABBY (C2C2 family) and GRF genes peaked in the apex. Expression of most NAC, bZIP, PLATZ and HSF TFs peaked in IN4 and IN5. Expression of most ERF (AP2‐ERF family), G2‐like (GARP family), GRAS and BELL (HB family), LOB, MYB, MYB‐related, TCP, WRKY and zf‐HD TFs peaked in both the apex and IN4 or IN5 (Figure 6d).
Based on a previous report, MYB and NAC family TFs play important roles in secondary growth in Arabidopsis (Zhong et al., 2010). Consistent with this, in this study, different MYB family genes were expressed in the apex and IN4‐5. For example, AS1/ATPHAN, which maintains meristem activity and is required for growth and dorsoventrality of lateral organs in Antirrhinum (Waites et al., 1998) was predominantly expressed in the apex. But PtrMYB206, whose orthologue MYB59 may be involved in cell division in Arabidopsis (Mu et al., 2009), was predominantly expressed during secondary growth. NAC family genes were also expressed predominantly in IN4‐5, including NST1, SND1, ATAF1, ATNAC2 and ATNAC3. NSTs can directly activate MYB46 and its close homologs (Zhao and Dixon, 2011), and SND1 directly regulates MYB46, which is involved in secondary cell wall biosynthesis in Arabidopsis (Ko et al., 2012b; Zhong et al., 2007). MYB46 orthologues are also expressed in IN4‐5 (Data S11).
Genes responsible for Arabidopsis shoot and root meristem development, such as SCR, YABBY2, YABBY5, ASL4/LOB, and so on, were expressed in the apex. In Arabidopsis, SCR is expressed in ground tissue in both the root and shoot during embryogenesis as well as postembryonically, and is required for distal determination of the root quiescent centre (Sabatini et al., 2003; Wysocka‐Diller et al., 2000). Other TFs, such as YABBY2, YABBY5 and ASL4/LOB, have previously been shown to play roles in lateral organ initiation in meristems in Arabidopsis (Byrne et al., 2000; Rast and Simon, 2008; Sun et al., 2002), and the high expression levels of these genes in the apex suggests that they have similar functions in Populus.
Many hormone‐associated receptors that are also TFs, such as CRF2, PtrERF43, ARF18 and MP, were expressed in the apex, including SHN1 and ATERF‐1 in IN1‐3, and ERF062, RAP2.10 and RAP2.3 in IN4‐5. Many of these genes, including MP and ARF18, have previously been found to play roles in embryo and procambium development in Arabidopsis (Miyashima et al., 2013; Weigel and Jurgens, 2002). And new isoforms of MP (PB9131.3) and ARF18 (PB.19400.8) were identified in this study. In contrast to previous studies in Arabidopsis, which showed that CRF2 is induced in response to cytokinin in whole seedlings and is important for root apical meristem development and lateral root initiation after germination (Jeon et al., 2016; Raines et al., 2016), we found that CRF2 was predominantly expressed in the SAM in Populus. The expression of WAX INDUCER1 (WIN1)/SHN1 in the apex and the expression of long‐chain acyl‐CoA synthetase (LACS2) in the apex and IN1 is consistent with a previous finding that WIN1/SHN1 modulates cuticle permeability in Arabidopsis by regulating genes encoding cutin pathway enzymes, such as LACS2 (Kannangara et al., 2007). Real‐time PCR analysis of AFO, MP, SHN1 and ARK1b expression indicated that AFO and MP were expressed predominantly in the apex, while SHN1 and ARK1b were expressed predominantly in IN1‐2 and IN4‐5, respectively (Figure S5). We observed ARK1b expression in IN4‐5; this gene was previously reported to regulate the SAM, vascular cambium, and wood formation in Populus (Groover et al., 2006; Liu et al., 2015).
Conclusions
In this study, we used Pac‐Bio Iso‐Seq and RNA‐Seq to profile changes in gene expression during the transition to primary and secondary growth in Populus. We identified 87 150 transcripts, including new alternatively spliced isoforms of previously annotated genes as well as novel genetic loci, and used this information to update the Populus annotation. GO enrichment analysis of genes differentially expressed during Populus stem development revealed that although similar processes, such as cell division and oxidation‐reduction, are important during both primary and secondary growth, different genes participate in these processes in different stages of development. We also found that different types of TFs tend to be expressed at different stages. For example, AP2, ARF, YABBY and GRF TFs are highly expressed in the apex, whereas NAC, bZIP, PLATZ and HSF TFs are likely to be important for the transition to secondary growth. There are many transcripts expressed predominantly in primary and secondary growth zone according to our study, but their functions are still unknown. These genes are a potential resource to study primary and secondary growth, especially the key transition from primary to secondary growth.
Materials and methods
Sample collection and anatomic analysis
Populus deltoides × P. euramericana cv ‘Nanlin895’ was grown in a phytotron with a light and dark cycle of 16 and 8 h at 23 °C. Apical and stem samples were collected from apex, the first internode (IN1), the second internode (IN2), the third internode (IN3), the fourth internode (IN4) and the fifth internode (IN5) from plants as shown in Figure 1. Each sample was pooled from 15 plants, and 3 biological repeats were performed for a total of 45 plants for each region of the stem. Three mixed samples from 45 plants were collected for PacBio full‐length sequencing.
Stem samples were cut by knife blade, fixed in FAA, and then dehydrated through a gradient of ethanol and embedded in LR White resin (London Resin Company Ltd). One‐micrometre‐thick sections were cut with a Leica EM UC7 microtome (Leica Microsystems IR GmbH, Wetzlar, Germany), stained with toluidine blue O (Sigma‐Aldrich, St. Louis), and viewed with an optical microscope.
RNA quantification and assessment of quality
RNA degradation and contamination were assessed on 1% agarose gels. RNA purity was checked using a NanoPhotometer spectrophotometer (IMPLEN, CA). RNA concentration was measured using the Qubit RNA Assay Kit and a Qubit 2.0 Fluorometer (Life Technologies, CA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit and the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA).
Illumina transcriptome library preparation and sequencing
A total of 1 μg RNA per sample was used as input material to generate sequencing libraries using the NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB) following the manufacturer's recommendations, and index codes were added to attribute sequences to a specific sample. Briefly, mRNA was purified from total RNA using poly‐T oligo‐attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in the NEBNext First Strand Synthesis Reaction Buffer (5×). First strand cDNA was synthesized using random hexamer primers and M‐MuLV Reverse Transcriptase (RNase H‐). Second strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3′ ends of DNA fragments, NEBNext Adaptors with hairpin loop structures were ligated to prepare for hybridization. In order to select cDNA fragments 200–250 bp in length, the library fragments were purified with the AMPure XP system (Beckman Coulter, Beverly). Then 3 μL USER Enzyme (NEB) was incubated with size‐selected, adaptor‐ligated cDNA at 37 °C for 15 min followed by 5 min at 95 °C. Then PCR was performed with Phusion High‐Fidelity DNA polymerase, Universal PCR primers and Index (X) Primer. Finally, PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system. The clustering of index‐coded samples was performed on a cBot Cluster Generation System using the TruSeq PE Cluster Kit v4‐cBot‐HS (Illumina) according to the manufacturer's instructions. After cluster generation, the libraries were sequenced on an Illumina Hiseq X Ten platform, and paired‐end reads were generated.
PacBio Iso‐Seq library preparation and sequencing
The sequencing library was prepared according to the Iso‐Seq protocol as described by Pacific Biosciences (P/N100‐377‐100‐05 and P/N100‐377‐100‐04). The SMARTer PCR cDNA Synthesis Kit was used to synthesize cDNA from the same RNA samples used for Illumina sequencing. After 23 cycles of PCR amplification, products were size selected using the BluePippin Size Selection System with the following bins for each sample: 1–2, 2–3 and 3–10 kb. The amplified cDNA products were used to generate SMRTbell Template libraries according to the Iso‐Seq protocol. Libraries were prepared for sequencing by annealing a sequencing primer and adding polymerase to the primer‐annealed template. The polymerase‐bound template was bound to MagBeads and sequencing was performed on a PacBio RSII instrument.
Illumina data analysis
Raw data (raw reads) in fastq format were first processed using in‐house perl scripts. In this step, clean data (clean reads) were obtained by removing reads containing adapters, reads containing poly‐N and low‐quality reads. These clean reads were then mapped to the reference genome sequence using Tophat2. Only reads with a perfect match or one mismatch were further analyzed and annotated based on the reference genome. Gene expression levels were estimated by FPKM. Differential expression analysis between two conditions/groups was performed using the DESeq R package (1.10.1) (Anders, 2010). The resulting P values were adjusted using the Benjamini and Hochberg's approach for controlling the false discovery rate (Anders, 2010). Genes identified by DESeq with FDR ≤0.01 and FC ≥2 were defined as differentially expressed. K‐means clustering was conducted based on Pearson correlation of gene expression profiles (Walvoort et al., 2010).
PacBio data analysis
SMRT‐Analysis software package v3.0 (https://github.com/ben-lerch/IsoSeq-3.0/blob/master/README.md) was used for Iso‐Seq data analysis. First, ROIs were generated using the minimum filtering requirement of 0 or greater passes of the insert and a minimum read quality of 75. Then, FLNC (full‐length non‐chimeric) reads containing the 5′ and 3′ adapters used in the library preparation as well as the poly (A) tail were identified.
lncRNA identification from PacBio sequences
Transcripts were screened for putative lncRNAs using four computational approaches, including coding‐non‐coding index (CNCI), coding potential calculator (CPC), coding potential assessment tool (CPAT), and Pfam database to identify non‐protein coding RNA candidates and putative protein‐coding RNAs from the unknown transcripts. Putative protein‐coding RNAs were filtered out using a minimum exon length and number threshold. Transcripts with lengths greater than 200 nt and with more than two exons were selected as lncRNA candidates. This threshold was chosen in order to filter abundant low‐expression, low‐confidence and single‐exon transcripts (Bogu et al., 2016; Peng et al., 2018; Yang et al., 2017). The putative lncRNAs were further screened using CPC, CNCI, CPAT and Pfam, which have the power to distinguish protein‐coding genes from non‐coding genes.
Fusion transcript identification from PacBio sequences
The following criteria were used to define fusion transcripts: (a) full‐length transcripts map to two or more loci in the genome; (b) each mapped locus must align with at least 5% of the transcript; (c) the combined alignment coverage must be at least 95%; and (d) the mapped loci must be at least 10 kb apart. Furthermore, Illumina short reads generated from the HiSeqX Ten platform were used to validate candidate fusion transcripts. After mapping using bowtie2, junction‐spanning reads and discordant read pairs were found to support the candidate fusion transcripts.
Functional annotation of transcripts
Transcripts were annotated by conducting blastx searches against public databases, including the NCBI non‐redundant protein database (Nr), NCBI non‐redundant nucleotide database (Nt), Swiss‐Prot, Protein Family (Pfam), Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG), with an E‐value threshold of 10−5.
Arabidopsis homologous genes searches
Transcripts were used as queries in translated nucleotide BLAST (BLASTX) searches against Arabidopsis protein sequences in the Arabidopsis Information Resource 10 database to obtain the closest Arabidopsis homolog (E‐value ≤e−10).
GO enrichment analysis
GO enrichment analysis was performed using topGO (Alexa et al., 2006) with Fisher's exact test, and the negative log10 transformed P values were visualized using heatmaps as previously described (Xue et al., 2013).
Transcription factor identification and analysis
The set of Arabidopsis TFs in Plant TFDB 3.0 (Jin et al., 2014) was used as the reference TF database. The Transcription Factor Prediction algorithm HMMER 3.0 (Finn et al., 2011) was used to identify TFs and assign genes to different families. The best BLAST hits had maximal E‐values of 1e−10. The WGCNA (v1.42) package (Langfelder and Horvath, 2008) in R was used to construct co‐expression networks. Genes with FPKM values >1 were used for WGCNA co‐expression network analysis. The modules were obtained using the automatic network construction function blockwiseModules with default settings.
Isoform identification and quantitative real‐time polymerase chain reaction (qRT‐PCR)
Primers were designed from specific PacBio‐seq isoform sequences (Table S1). Isoform‐specific primers were designed to distinguish between different alternative splicing events. Reverse transcription of 3 μg mRNA was used done using the M‐MLV first strand kit (ThermoFisher Scientific, MA). QRT‐PCR reactions were performed using SYBR Premix Ex Taq (Takara, Shiga, Japan) on a LightCycler 480 system (Roche, Basel, Switzerland). The amplification program consisted of 30 s of initial denaturation at 95 °C, followed by 30 cycles of 10 s at 95 °C, 20 s at 60 °C and 20 s at 72 °C, and ended with a final extension step at 72 °C for 20 s. All samples were normalized to the reference gene tubulin from Populus to analyze the isoform expression level. The final relative expression level was calculated using the formula F = 2−ΔΔCt.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Figure S1 Schematic of the computational and bioinformatics pipeline for joint analysis of PacBio Iso‐Seq and RNA‐seq reads.
Figure S2 RT‐PCR verification of alternative splicing events for three genes.
Figure S3 Analysis of expression correlation between samples.
Figure S4 Heatmaps of different transcription factor WGCNA modules.
Figure S5 Expression verification of isoforms in different sections of the Populus stem.
Table S1 Sequences of primers used for alternative splicing and gene expression verification
Data S1 PacBio Iso‐Seq output analysis
Data S2 Transcripts from new genes
Data S3 Alternative splicing events
Data S4 Long non‐coding RNAs
Data S5 Fusion transcripts
Data S6 Mapping ratios of RNA‐seq sequences
Data S7 Differently expressed transcripts
Data S8 K‐means clusters
Data S9 GO enrichment analysis
Data S10 Cell wall‐associated transcripts
Data S11 Transcription factors
Acknowledgements
We thank Lai‐geng Li for providing ‘Nanlin895’ and Hui Zhang for his excellent technical assistance on the optical microscope. This study was supported by the National High Technology Research and Development Program of China (grant no. 2013AA102701). Dong Zhang is affiliated with Biomarker Technologies Corporation.
Accession numbers: These sequence data have been submitted to the GenBank database under accession number BioProject PRJNA471819. Addresses are as follows: http://www.ncbi.nlm.nih.gov.
References
- Abdel‐Ghany, S.E. , Hamilton, M. , Jacobi, J.L. , Ngam, P. , Devitt, N. , Schilkey, F. , Ben‐Hur, A. et al. (2016) A survey of the sorghum transcriptome using single‐molecule long reads. Nat. Commun. 7, 11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aichinger, E. , Kornet, N. , Friedrich, T. and Laux, T. (2012) Plant stem cell niches. Annu. Rev. Plant Biol. 63, 615–636. [DOI] [PubMed] [Google Scholar]
- Aida, M. , Ishida, T. and Tasaka, M. (1999) Shoot apical meristem and cotyledon formation during Arabidopsis embryogenesis: interaction among the CUP‐SHAPED COTYLEDON and SHOOT MERISTEMLESS genes. Development, 126, 1563–1570. [DOI] [PubMed] [Google Scholar]
- Aida, M. , Vernoux, T. , Furutani, M. , Traas, J. and Tasaka, M. (2002) Roles of PIN‐FORMED1 and MONOPTEROS in pattern formation of the apical region of the Arabidopsis embryo. Development, 129, 3965–3974. [DOI] [PubMed] [Google Scholar]
- Alexa, A. , Rahnenführer, J. and Lengauer, T. (2006) Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics, 22, 1600–1607. [DOI] [PubMed] [Google Scholar]
- Anders, S. (2010) Analysing RNA‐Seq data with the DESeq package. Mol. Biol. 43, 1–17. [Google Scholar]
- Aspeborg, H. , Schrader, J. , Coutinho, P.M. , Stam, M. , Kallas, Å. , Djerbi, S. , Nilsson, P. et al. (2005) Carbohydrate‐active enzymes involved in the secondary cell wall biogenesis in hybrid aspen. Plant Physiol. 137, 983–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au, K.F. , Sebastiano, V. , Afshar, P.T. , Durruthy, J.D. , Lee, L. , Williams, B.A. , van Bakel, H. et al. (2013) Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl Acad. Sci. 110, E4821–E4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucher, M. , El Jaziri, M. and Vandeputte, O. (2007) From primary to secondary growth: origin and development of the vascular system. J. Exp. Bot. 58, 3485–3501. [DOI] [PubMed] [Google Scholar]
- Baumbusch, L.O. , Thorstensen, T. , Krauss, V. , Fischer, A. , Naumann, K. , Assalkhou, R. , Schulz, I. et al. (2001) The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucleic Acids Res. 29, 4319–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berckmans, B. , Lammens, T. , Van Den Daele, H. , Magyar, Z. , Bögre, L. and De Veylder, L. (2011) Light‐dependent regulation of DEL1 is determined by the antagonistic action of E2Fb and E2Fc. Plant Physiol. 157, 1440–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogu, G.K. , Vizán, P. , Stanton, L.W. , Beato, M. , Di Croce, L. and Marti‐Renom, M.A. (2016) Chromatin and RNA maps reveal regulatory long noncoding RNAs in mouse. Mol. Cell. Biol. 36, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonke, M. , Thitamadee, S. , Mahonen, A.P. , Hauser, M.‐T. and Helariutta, Y. (2003) APL regulates vascular tissue identity in Arabidopsis . Nature, 426, 181–186. [DOI] [PubMed] [Google Scholar]
- Byrne, M.E. , Barley, R. , Curtis, M. and Arroyo, J.M. (2000) Asymmetric leaves1 mediates leaf patterning and stem cell function in Arabidopsis . Nature, 408, 967. [DOI] [PubMed] [Google Scholar]
- Carlsbecker, A. and Helariutta, Y. (2005) Phloem and xylem specification: pieces of the puzzle emerge. Curr. Opin. Plant Biol. 8, 512–517. [DOI] [PubMed] [Google Scholar]
- Carraro, N. , Peaucelle, A. , Laufs, P. and Traas, J. (2006) Cell differentiation and organ initiation at the shoot apical Meristem. Plant Mol. Biol. 60, 811–826. [DOI] [PubMed] [Google Scholar]
- Chaffey, N. (1999) Wood formation in forest trees: from Arabidopsis to Zinnia . Trends Plant Sci. 4, 203–204. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Chen, B. and Zhang, D. (2015) Transcript profiling of Populus tomentosa genes in normal, tension, and opposite wood by RNA‐seq. BMC Genom. 16, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clamp, M. , Fry, B. , Kamal, M. , Xie, X. , Cuff, J. , Lin, M.F. , Kellis, M. et al. (2007) Distinguishing protein‐coding and noncoding genes in the human genome. Proc. Natl Acad. Sci. 104, 19428–19433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, S.E. , Jacobsen, S.E. , Levin, J.Z. and Meyerowitz, E.M. (1996) The CLAVATA and SHOOT MERISTEMLESS loci competitively regulate meristem activity in Arabidopsis . Development, 122, 1567–1575. [DOI] [PubMed] [Google Scholar]
- Cosgrove, D.J. (2015) Plant expansins: diversity and interactions with plant cell walls. Curr. Opin. Plant Biol. 25, 162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desprez, T. , Juraniec, M. , Crowell, E.F. , Jouy, H. , Pochylova, Z. , Parcy, F. , Höfte, H. et al. (2007) Organization of cellulose synthase complexes involved in primary cell wall synthesis in Arabidopsis thaliana . Proc. Natl Acad. Sci. 104, 15572–15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmawardhana, P. , Brunner, A.M. and Strauss, S.H. (2010) Genome‐wide transcriptome analysis of the transition from primary to secondary stem development in Populus trichocarpa . BMC Genom. 11, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner, T.J. , Sherr, I. and Scarpella, E. (2009) Regulation of preprocambial cell state acquisition by auxin signaling in Arabidopsis leaves. Development, 136, 3235–3246. [DOI] [PubMed] [Google Scholar]
- Du, J. and Groover, A. (2010) Transcriptional regulation of secondary growth and wood formation. J. Integr. Plant Biol. 52, 17–27. [DOI] [PubMed] [Google Scholar]
- Du, J. , Mansfield, S.D. and Groover, A.T. (2009) The Populus homeobox gene ARBORKNOX2 regulates cell differentiation during secondary growth. Plant J. 60, 1000–1014. [DOI] [PubMed] [Google Scholar]
- Duan, C.G. , Zhang, H. , Tang, K. , Zhu, X. , Qian, W. , Hou, Y.J. , Wang, B. et al. (2015) Specific but interdependent functions for Arabidopsis AGO4 and AGO6 in RNA‐directed DNA methylation. EMBO J., 34, 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchells, J.P. and Turner, S.R. (2010) The PXY‐CLE41 receptor ligand pair defines a multifunctional pathway that controls the rate and orientation of vascular cell division. Development, 137, 767–774. [DOI] [PubMed] [Google Scholar]
- Etchells, J.P. , Mishra, L.S. , Kumar, M. , Campbell, L. and Turner, S.R. (2015) Wood formation in trees is increased by manipulating PXY‐regulated cell division. Curr. Biol. 25, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, S. , Jacobsen, S.E. and Reik, W. (2010) Epigenetic reprogramming in plant and animal development. Science, 330, 622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R.D. , Clements, J. and Eddy, S.R. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, K. and Turner, S. (2007) PXY, a Receptor‐like kinase essential for maintaining polarity during plant vascular‐tissue development. Curr. Biol. 17, 1061–1066. [DOI] [PubMed] [Google Scholar]
- Flynn, R.A. and Chang, H.Y. (2014) Long noncoding RNAs in cell‐fate programming and reprogramming. Cell Stem Cell, 14, 752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foissac, S. and Sammeth, M. (2007) ASTALAVISTA: dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 35, W297–W299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendrel, A.‐V. , Lippman, Z. , Yordan, C. , Colot, V. and Martienssen, R.A. (2002) Dependence of heterochromatic histone H3 methylation patterns on the Arabidopsis gene DDM1 . Science, 297, 1871–1873. [DOI] [PubMed] [Google Scholar]
- Gordon, S.P. , Tseng, E. , Salamov, A. , Zhang, J. , Meng, X. , Zhao, Z. , Kang, D. et al. (2015) Widespread polycistronic transcripts in fungi revealed by single‐molecule mRNA sequencing. PLoS ONE, 10, e0132628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groover, A.T. , Mansfield, S.D. , DiFazio, S.P. , Dupper, G. , Fontana, J.R. , Millar, R. and Wang, Y. (2006) The Populus homeobox gene ARBORKNOX1 reveals overlapping mechanisms regulating the shoot apical meristem and the vascular cambium. Plant Mol. Biol. 61, 917–932. [DOI] [PubMed] [Google Scholar]
- Guo, Y. , Qin, G. , Gu, H. and Qu, L.‐J. (2009) Dof5.6/HCA2, a Dof transcription factor gene, regulates interfascicular cambium formation and vascular tissue development in Arabidopsis . Plant Cell, 21, 3518–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habu, Y. , Ando, T. , Ito, S. , Nagaki, K. , Kishimoto, N. , Taguchi‐Shiobara, F. , Numa, H. et al. (2015) Epigenomic modification in rice controls meiotic recombination and segregation distortion. Mol. Breed. 35, 103. [Google Scholar]
- Hamada, T. (2014) Microtubule organization and microtubule‐associated proteins in plant cells. Int. Rev. Cell Mol. Biol. 312, 1–52. [DOI] [PubMed] [Google Scholar]
- Havecker, E.R. , Wallbridge, L.M. , Hardcastle, T.J. , Bush, M.S. , Kelly, K.A. , Dunn, R.M. , Schwach, F. et al. (2010) The Arabidopsis RNA‐directed DNA methylation argonautes functionally diverge based on their expression and interaction with target loci. Plant Cell, 22, 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzberg, M. , Aspeborg, H. , Schrader, J. , Andersson, A. , Erlandsson, R. , Blomqvist, K. , Bhalerao, R. et al. (2001) A transcriptional roadmap to wood formation. Proc. Natl Acad. Sci. 98, 14732–14737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inzé, D. and Veylder, L.D. (2006) Cell cycle regulation in plant development. Annu. Rev. Genet. 40, 77–105. [DOI] [PubMed] [Google Scholar]
- Jackson, J.P. , Lindroth, A.M. , Cao, X. and Jacobsen, S.E. (2002) Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature, 416, 556. [DOI] [PubMed] [Google Scholar]
- Jacob, Y. , Feng, S. , LeBlanc, C.A. , Bernatavichute, Y.V. , Stroud, H. , Cokus, S. , Johnson, L.M. et al. (2009) ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat. Struct. Mol. Biol. 16, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson, S. and Douglas, C.J. (2007) Populus: a model system for plant biology. Annu. Rev. Plant Biol. 58, 435–458. [DOI] [PubMed] [Google Scholar]
- Jeon, J. , Cho, C. , Lee, M.R. , Van Binh, N. and Kim, J. (2016) CYTOKININ RESPONSE FACTOR2 (CRF2) and CRF3 regulate lateral root development in response to cold stress in Arabidopsis . Plant Cell, 28, 1828–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, J. , Zhang, H. , Kong, L. , Gao, G. and Luo, J. (2014) PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannangara, R. , Branigan, C. , Liu, Y. , Penfield, T. , Rao, V. , Mouille, G. , Höfte, H. et al. (2007) The transcription factor WIN1/SHN1 regulates cutin biosynthesis in Arabidopsis thaliana . Plant Cell, 19, 1278–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M. , Canio, W. , Kessler, S. and Sinha, N. (2001) Developmental changes due to long‐distance movement of a Homeobox fusion transcript in Tomato. Science, 293, 287–289. [DOI] [PubMed] [Google Scholar]
- Ko, J.H. , Kim, H.T. , Hwang, I. and Han, K.H. (2012a) Tissue‐type‐specific transcriptome analysis identifies developing xylem‐specific promoters in poplar. Plant Biotechnol. J. 10, 587–596. [DOI] [PubMed] [Google Scholar]
- Ko, J.H. , Kim, W.C. , Kim, J.Y. , Ahn, S.J. and Han, K.H. (2012b) MYB46‐mediated transcriptional regulation of secondary wall biosynthesis. Mol. Plant, 5, 961–963. [DOI] [PubMed] [Google Scholar]
- Kubo, M. , Udagawa, M. , Nishikubo, N. , Horiguchi, G. , Yamaguchi, M. , Ito, J. , Mimura, T. et al. (2005) Transcription switches for protoxylem and metaxylem vessel formation. Genes Dev. 19, 1855–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder, P. and Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, P.R. and Isebrands, J.G. (1971) The Plastochron Index as applied to developmental studies of cottonwood. Can. J. For. Res. 1, 1–11. [Google Scholar]
- Liu, L. , Shang‐Guan, K. , Zhang, B. , Liu, X. , Yan, M. , Zhang, L. , Shi, Y. et al. (2013) Brittle Culm1, a COBRA‐like protein, functions in cellulose assembly through binding cellulose microfibrils. PLoS Genet. 9, e1003704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Missirian, V. , Zinkgraf, M. , Groover, A. and Filkov, V. (2014) Evaluation of experimental design and computational parameter choices affecting analyses of ChIP‐seq and RNA‐seq data in undomesticated poplar trees. BMC Genom. 15(Suppl. 5), S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Zinkgraf, M. , Petzold, H.E. , Beers, E.P. , Filkov, V. and Groover, A. (2015) The Populus ARBORKNOX1 homeodomain transcription factor regulates woody growth through binding to evolutionarily conserved target genes of diverse function. New Phytol. 205, 682–694. [DOI] [PubMed] [Google Scholar]
- Long, J.A. and Barton, M.K. (1998) The development of apical embryonic pattern in Arabidopsis . Development, 125, 3027–3035. [DOI] [PubMed] [Google Scholar]
- Lynn, K. , Fernandez, A. , Aida, M. , Sedbrook, J. , Tasaka, M. , Masson, P. and Barton, M.K. (1999) The PINHEAD/ZWILLE gene acts pleiotropically in Arabidopsis development and has overlapping functions with the ARGONAUTE1 gene. Development, 126, 469–481. [DOI] [PubMed] [Google Scholar]
- Meyerowitz, E.M. (1997) Genetic control of cell division patterns in developing plants. Cell, 88, 299–308. [DOI] [PubMed] [Google Scholar]
- Miyashima, S. , Sebastian, J. , Lee, J.Y. and Helariutta, Y. (2013) Stem cell function during plant vascular development. EMBO J. 32, 178–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu, R.L. , Cao, Y.R. , Liu, Y.F. , Lei, G. , Zou, H.F. , Liao, Y. , Wang, H.W. et al. (2009) An R2R3‐type transcription factor gene AtMYB59 regulates root growth and cell cycle progression in Arabidopsis . Cell Res. 19, 1291–1304. [DOI] [PubMed] [Google Scholar]
- Numa, H. , Yamaguchi, K. , Shigenobu, S. and Habu, Y. (2015) Gene body CG and CHG methylation and suppression of centromeric CHH methylation are mediated by DECREASE IN DNA METHYLATION1 in rice. Mol. Plant 8, 1560–1562. [DOI] [PubMed] [Google Scholar]
- Ohashi‐Ito, K. and Fukuda, H. (2010) Transcriptional regulation of vascular cell fates. Curr. Opin. Plant Biol. 13, 670–676. [DOI] [PubMed] [Google Scholar]
- Otsuga, D. , DeGuzman, B. , Prigge, M.J. , Drews, G.N. and Clark, S.E. (2001) REVOLUTA regulates meristem initiation at lateral positions. Plant J. 25, 223–236. [DOI] [PubMed] [Google Scholar]
- Peng, L. , Skylar, A. , Chang, P.L. , Bisova, K. and Wu, X. (2014) CYCP2;1 integrates genetic and nutritional information to promote meristem cell division in Arabidopsis . Dev. Biol. 393, 160–170. [DOI] [PubMed] [Google Scholar]
- Peng, R. , Liu, Y. , Cai, Z. , Shen, F. , Chen, J. , Hou, R. and Zou, F. (2018) Characterization and analysis of whole transcriptome of giant panda spleens: implying critical roles of long non‐coding RNAs in immunity. Cell. Physiol. Biochem. 46, 1065–1077. [DOI] [PubMed] [Google Scholar]
- Pesquet, E. , Korolev, A.V. , Calder, G. and Lloyd, C.W. (2010) The microtubule‐associated protein AtMAP70‐5 regulates secondary wall patterning in Arabidopsis wood cells. Curr. Biol. 20, 744–749. [DOI] [PubMed] [Google Scholar]
- Pinosio, S. , Giacomello, S. , Faivre‐Rampant, P. , Taylor, G. , Jorge, V. , Le Paslier, M.C. , Zaina, G. et al. (2016) Characterization of the poplar pan‐genome by genome‐wide identification of structural variation. Mol. Biol. Evol. 33, 2706–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prassinos, C. , Ko, J.‐H. , Yang, J. and Han, K.‐H. (2005) Transcriptome profiling of vertical stem segments provides insights into the genetic regulation of secondary growth in hybrid Aspen Trees. Plant Cell Physiol. 46, 1213–1225. [DOI] [PubMed] [Google Scholar]
- Raines, T. , Shanks, C. , Cheng, C.‐Y. , McPherson, D. , Argueso, C.T. , Kim, H.J. , Franco‐Zorrilla, J.M. et al. (2016) The cytokinin response factors modulate root and shoot growth and promote leaf senescence in Arabidopsis . Plant J. 85, 134–147. [DOI] [PubMed] [Google Scholar]
- Rast, M.I. and Simon, R. (2008) The meristem‐to‐organ boundary: more than an extremity of anything. Curr. Opin. Genet. Dev. 18, 287–294. [DOI] [PubMed] [Google Scholar]
- Ronemus, M.J. , Galbiati, M. , Ticknor, C. , Chen, J. and Dellaporta, S.L. (1996) Demethylation‐induced developmental pleiotropy in Arabidopsis . Science, 273, 654–656. [DOI] [PubMed] [Google Scholar]
- Sabatini, S. , Heidstra, R. , Wildwater, M. and Scheres, B. (2003) SCARECROW is involved in positioning the stem cell niche in the Arabidopsis root meristem. Genes Dev. 17, 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampedro, J. , Carey, R.E. and Cosgrove, D.J. (2006) Genome histories clarify evolution of the expansin superfamily: new insights from the poplar genome and pine ESTs. J. Plant. Res. 119, 11–21. [DOI] [PubMed] [Google Scholar]
- Schoof, H. , Lenhard, M. , Haecker, A. , Mayer, K.F. , Jürgens, G. and Laux, T. (2000) The stem cell population of Arabidopsis shoot meristems is maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell, 100, 635–644. [DOI] [PubMed] [Google Scholar]
- Schrader, J. , Nilsson, J. , Mellerowicz, E. , Berglund, A. , Nilsson, P. , Hertzberg, M. and Sandberg, G. (2004) A high‐resolution transcript profile across the wood‐forming meristem of poplar identifies potential regulators of cambial stem cell identity. Plant Cell, 16, 2278–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafiq, S. , Berr, A. and Shen, W.‐H. (2014) Combinatorial functions of diverse histone methylations in Arabidopsis thaliana flowering time regulation. New Phytol. 201, 312–322. [DOI] [PubMed] [Google Scholar]
- Song, D. , Xi, W. , Shen, J. , Bi, T. and Li, L. (2011) Characterization of the plasma membrane proteins and receptor‐like kinases associated with secondary vascular differentiation in poplar. Plant Mol. Biol. 76, 97–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y. , Zhou, Q. , Zhang, W. , Fu, Y. and Huang, H. (2002) ASYMMETRIC LEAVES1, an Arabidopsis gene that is involved in the control of cell differentiation in leaves. Planta, 214, 694–702. [DOI] [PubMed] [Google Scholar]
- Sundell, D. , Street, N.R. , Kumar, M. , Mellerowicz, E.J. , Kucukoglu, M. , Johnsson, C. , Kumar, V. et al. (2017) AspWood: high‐spatial‐resolution transcriptome profiles reveal uncharacterized modularity of wood formation in Populus tremula. Plant Cell, 29, 1585–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, N.G. , Howells, R.M. , Huttly, A.K. , Vickers, K. and Turner, S.R. (2003) Interactions among three distinct CesA proteins essential for cellulose synthesis. Proc. Natl Acad. Sci. 100, 1450–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan, G.A. , DiFazio, S. , Jansson, S. , Bohlmann, J. , Grigoriev, I. , Hellsten, U. , Putnam, N. et al. (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science, 313, 1596–1604. [DOI] [PubMed] [Google Scholar]
- Waites, R. , Selvadurai, H.R.N. , Oliver, I.R. and Hudson, A. (1998) The PHANTASTICA gene encodes a MYB transcription factor involved in growth and dorsoventrality of lateral organs in Antirrhinum . Cell, 93, 779–789. [DOI] [PubMed] [Google Scholar]
- Walvoort, D.J.J. , Brus, D.J. and de Gruijter, J.J. (2010) An R package for spatial coverage sampling and random sampling from compact geographical strata by k‐means. Comput. Geosci. 36, 1261–1267. [Google Scholar]
- Wang, H.‐Z. and Dixon, R.A. (2012) On–off switches for secondary cell wall biosynthesis. Mol. Plant, 5, 297–303. [DOI] [PubMed] [Google Scholar]
- Wang, B. , Tseng, E. , Regulski, M. , Clark, T.A. , Hon, T. , Jiao, Y. , Lu, Z. et al. (2016) Unveiling the complexity of the maize transcriptome by single‐molecule long‐read sequencing. Nat. Commun. 7, 11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T. , Wang, H. , Cai, D. , Gao, Y. , Zhang, H. , Wang, Y. , Lin, C. et al. (2017) Comprehensive profiling of rhizome‐associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J. 91, 684–699. [DOI] [PubMed] [Google Scholar]
- Wang, M. , Wang, P. , Liang, F. , Ye, Z. , Li, J. , Shen, C. , Pei, L. et al. (2018) A global survey of alternative splicing in allopolyploid cotton: landscape, complexity and regulation. New Phytol. 217, 163–178. [DOI] [PubMed] [Google Scholar]
- Weigel, D. and Jurgens, G. (2002) Stem cells that make stems. Nature, 415, 751–754. [DOI] [PubMed] [Google Scholar]
- Wood, C.C. , Robertson, M. , Tanner, G. , Peacock, W.J. , Dennis, E.S. and Helliwell, C.A. (2006) The Arabidopsis thaliana vernalization response requires a polycomb‐like protein complex that also includes VERNALIZATION INSENSITIVE 3. Proc. Natl Acad. Sci. 103, 14631–14636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka‐Diller, J.W. , Helariutta, Y. , Fukaki, H. , Malamy, J.E. and Benfey, P.N. (2000) Molecular analysis of SCARECROW function reveals a radial patterning mechanism common to root and shoot. Development, 127, 595–603. [DOI] [PubMed] [Google Scholar]
- Xue, L.‐J. , Guo, W. , Yuan, Y. , Anino, E.O. , Nyamdari, B. , Wilson, M.C. , Frost, C.J. et al. (2013) Constitutively elevated salicylic acid levels alter photosynthesis and oxidative state but not growth in Transgenic Populus . Plant Cell, 25, 2714–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W. , Schuster, C. , Beahan, Cherie.T. , Charoensawan, V. , Peaucelle, A. , Bacic, A. , Doblin, Monika.S. et al. (2016) Regulation of meristem morphogenesis by cell wall synthases in Arabidopsis . Curr. Biol. 26, 1404–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L. , Lyu, L. , Wu, W. , Lei, D. , Tu, Y. , Xu, D. , Feng, J. et al. (2017) Genome‐wide identification of long non‐coding RNA and mRNA profiling using RNA sequencing in subjects with sensitive skin. Oncotarget, 8, 114894–114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yordanov, Y.S. , Regan, S. and Busov, V. (2010) Members of the LATERAL ORGAN BOUNDARIES DOMAIN transcription factor family are involved in the regulation of secondary growth in Populus . Plant Cell, 22, 3662–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Q. and Dixon, R.A. (2011) Transcriptional networks for lignin biosynthesis: more complex than we thought? Trends Plant Sci. 16, 227–233. [DOI] [PubMed] [Google Scholar]
- Zhao, Y. , Song, D. , Sun, J. and Li, L. (2013) Populus endo‐beta‐mannanase PtrMAN6 plays a role in coordinating cell wall remodeling with suppression of secondary wall thickening through generation of oligosaccharide signals. Plant J. 74, 473–485. [DOI] [PubMed] [Google Scholar]
- Zhong, R. , Demura, T. and Ye, Z.‐H. (2006) SND1, a NAC domain transcription factor, is a key regulator of secondary wall synthesis in fibers of Arabidopsis. Plant Cell, 18, 3158–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, R. , Richardson, E.A. and Ye, Z.‐H. (2007) The MYB46 transcription factor is a direct target of SND1 and regulates secondary wall biosynthesis in Arabidopsis . Plant Cell, 19, 2776–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, R. , Lee, C. and Ye, Z.‐H. (2010) Evolutionary conservation of the transcriptional network regulating secondary cell wall biosynthesis. Trends Plant Sci. 15, 625–632. [DOI] [PubMed] [Google Scholar]
- Zhu, Y. , Song, D. , Sun, J. , Wang, X. and Li, L. (2013) PtrHB7, a class III HD‐Zip gene, plays a critical role in regulation of vascular cambium differentiation in Populus . Mol. Plant, 6, 1331–1343. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Schematic of the computational and bioinformatics pipeline for joint analysis of PacBio Iso‐Seq and RNA‐seq reads.
Figure S2 RT‐PCR verification of alternative splicing events for three genes.
Figure S3 Analysis of expression correlation between samples.
Figure S4 Heatmaps of different transcription factor WGCNA modules.
Figure S5 Expression verification of isoforms in different sections of the Populus stem.
Table S1 Sequences of primers used for alternative splicing and gene expression verification
Data S1 PacBio Iso‐Seq output analysis
Data S2 Transcripts from new genes
Data S3 Alternative splicing events
Data S4 Long non‐coding RNAs
Data S5 Fusion transcripts
Data S6 Mapping ratios of RNA‐seq sequences
Data S7 Differently expressed transcripts
Data S8 K‐means clusters
Data S9 GO enrichment analysis
Data S10 Cell wall‐associated transcripts
Data S11 Transcription factors